

Abstract
Aim: AQP4 in the brain is involved in the occurrence and development of a variety of encephalopathy. AQPs family changes in kidney were accompanied by altered UTs family. The aim of this study was to observe AQP4 and UT-A3 expression in CNS and to explore their role in the pathogenesis of endotoxemia encephalopathy following peripheral LPS injection in mice. Methods: Endotoxemia was induced in C57Bl/6 mice by intraperitoneal injection of LPS. The expression of UT-A3 and AQP4 in brain were detected by Western blot and immunohistochemistry, the level of cytokines were detected by ELISA, and the content of LDH, AST/ALT, BUN and CREA were detected by colorimetric method. Results: As compared with the control group, in model group, the brain weight/ body weight ratio increased by 13%. Meanwhile, a 2.5 fold increase in LDH and a 1.2 fold increase in AST/ALT were found in peripheral serum (P < 0.05), and also, BUN and CREA increased 2.5 fold (P < 0.01). In addition to severe CNS injury in response to lipopolysaccharide, the contents of cytokines and the expression of AQP4 protein in hippocampal is increased (P < 0.05), while the expression of UT-A3 protein in the hippocampus and cortical astrocytes decreased (P < 0.05). And, in part, Dexa pretreatment attenuated those effects. Conclusions: In endotoxemia encephalopathy, AQPs and UTs which regulate the functions of cell membrane are both altered. We suggested that the molecular mechanisms of regulation in endotoxemia may provide a new strategy for clinical treatment of the disease and drug binding sites.
Keywords: Lipopolysaccharide (LPS), urea transporters (UTs), aquaporins (AQPs), dexamethasone (Dexa), brain
Introduction
Endotoxins on the outer membrane of gram-negative bacteria are macromolecular proteins composed primarily of lipopolysaccharide (LPS), which activate effector cells to release a number of inflammatory mediators. Clinically, bacterial endotoxemia is a complicated syndrome, mediated by the opening and closing of channels [1,2]. Endotoxemia refers to gram-negative bacteria invading the blood, in which LPS degradation leads to a pathological process, and is usually caused by infections, burns, scalding, etc [3]. It is characterized by systemic inflammatory response syndrome (SIRS), septic shock, ischemia-reperfusion injury, diffuse intravascular coagulation (DIC), multiple organ dysfunction syndrome (MODS), and multiple organ failure syndrome (MOFS) [4]. These processes have a high mortality rate, with no effective treatment. Endotoxemia, also called septicemia (sepsis) is characterized by an increase in cytokine levels leading to endotoxic shock, of which IL-6, IL-1, and TNF-α levels increase the most. Mouse model studies of LPS disease pathology have shown that endotoxin acts directly on myocardial extracellular components to activate cardiovascular endothelial cells and release a variety of cytokines [3]. The local myocardial circulation produces proinflammatory cytokines, such as TNF-α and IL-1-β. Toxins play an important role in myocardial cardiac cells increase formation of oxygen free radicals and lipid peroxidation, changing cardiac structure and function of vascular endothelial cells, resulting in myocardial cell injury [4]. Dexamethasone (Dexa) is a synthetic glucocorticoid, which helps alleviate inflammation, allergic symptoms, and shock resistance, and is widely used in the clinic, which has been widely used in treatment of sepsis in last few decades. It also inhibits LPS-induced TNF-α by reducing TNF receptor affinity, and inhibits TNF-α mutant (TNF-m), endocytosis, degradation, and promotes its dissociation. Thus, Dexa counteracts the cytotoxic effect of TNF-α. Because TNF-α is an important inflammatory mediator, it may be clinically useful, providing a mechanism by which glucocorticoid therapy may alleviate endotoxin shock. The world mediates could be changed by attenuates, because it is well known that high-dose glucocorticoids had been used for approximately 3 decades to reduce inflammation in patients with various infectious diseases [5]. To better understand novel mechanisms by which glucocorticoid therapy mediates endotoxin shock, we explored the impact of Dexa on urea transporter (UT) proteins.
Aquaporins (AQPs) are integral membrane pore proteins. Preston et al. first identified them in erythrocyte membranes and renal tubular cells [6]. AQPs form channels to regulate cellular water content. Water transport through aquaporins is osmotically driven [7], and nearly 12 different types have been described in mammals [8]. Three of them, AQP1, AQP4, and AQP9 are expressed in the brain [9]. AQP1 is expressed in the epithelial cells of the choroid plexus [10]. The expression of AQPs adapts to exterior stimuli and the severity of the alteration [11]. AQP4 mRNA is upregulated in rats with focal cerebral ischemia, suggesting a role for AQP4 in brain edema [12]. In addition, mice lacking AQP4 are partially protected from brain edema in water intoxication and ischemic models of brain injury [13]. AQP4 was also recently found to play an important role in central nervous system (CNS) edema [14].
UTs are expressed in the kidney, and the hippocampus, olfactory bulb, and caudate nucleus of the brain [15], heart [16,17], testicular tissue [18], and on red blood cell membranes [19]. The urea fast channel protein mediates urea transport across membrane channels. Urea channel proteins have been cloned, including UT-A and B, two families encoded by the slc14a1 and 2 genes, located on chromosome 18 q12-21 [16]. The UT-A family is composed of five subtypes, expressed mainly in the kidney. The UT-B family is widely distributed and plays an important role in urine concentration in the kidney [20]. UT expression is controlled by vasopressin, plasma osmotic pressure, and adjustment of dietary protein content. Long-term changes in the internal cellular environment influences UT mRNA and protein expression. Limiting water for 1-3 days adjusts animal UT mRNA and protein, as the inner and outer concentrations of UT-A2 mRNA and protein increase. Diuresis also affects urea permeability and UT-A mRNA and protein expression and such change selectively impacts nephron segments [21]. Lithium therapy administered to psychiatric patients results in concentrated urine. Standard breeding of animal models indicated that after 25 days, and adding lithium to treat intramedullary collecting duct (IMCD) suspension, found marrow UT-A1 fell by 50%, base fell by 25%, and the UT-B pulp base by 40% [21]. Glucocorticoids increase segmental urea excretion, and adrenal resection reduces urea excretion and increases urea permeability. Following IMCD in these resections, UT-A1 protein levels change [22]. Rat experiments have shown that long-term application of cyclosporine A causes kidney UT-A2, UT-A3, and UT-B reductions [23]. During osmotic diuresis, urea raises the expression of UTs [24]. Normal rat heart contains a variety of UT-A proteins, including one at 39, 51, and 56 kd. In uremia and hypertension, expression of the 56 kd protein increases, and both the 51 and 56 kd proteins are increased during heart failure. These proteins are also involved in the regulation of heart function [24]. Cloning of the two closely related mammalian urea transporters UT-A and UT-B by Serena et al. helped characterize the molecular mechanisms regulating expression of UTs in the kidney, including those modulating transcriptional control of UT-A abundance [19]. Fenton et al. used immunoblotting to show that kidney and brain expressed the highest levels of UT-A protein [25]. Schmidt et al. demonstrated that renal UTs are downregulated by severe inflammation, which likely accounts for tubular dysfunction, suggesting that downregulation of renal UTs during LPS-induced acute renal failure (ARF) is mediated by proinflammatory cytokines and is independent of sepsis-induced hypotension [26]. In this study, we report an endotoxemia model created by intraperitoneal injection of endotoxin that stimulates UT protein expression in the brain. In addition, we administered Dexa to test whether brain inflammation is caused by UT regulated expression of endotoxin in the blood. This may provide a new possibility to research and treatment endotoxin blood disease of drug binding sites [27].
Materials and methods
Materials
Male C57BL/6 mice (12 week-old) were purchased from the experimental animal center of Jilin University. Immunostaining used the Vectastain Elite ABC avidin/biotin staining kit (Vector Laboratories Inc., Burlingame, CA, USA). Antibodies to UT-A3, and AQP4 were kindly provided by Yang Baoxue (University of California, Los Angeles, USA). The anti-beta-actin antibodies were obtained from Sanjian Inc. (Tianjin, China). The enhanced chemiluminescence (ECL) western blot kit was purchased from Amersham (Buckinghamshire, UK). The study was approved by the Institutional Ethics Board of School of Medicine, Jilin University.
Animal models
24 male C57BL/6 mice were randomly divided into four groups (n = 8 per group) as follows: control group, LPS group, and Dexamethasone plus LPS group. LPS was phenol extracted from Escherichia coli serotype O111:B4 (Sigma, Poole, UK). The control group was injected with 0.5 ml 0.85% Nacl intraperitoneally. LPS group, and Dexamethasone plus LPS groups mice were injected intraperitoneally with LPS (10 mg/kg). In Dexamethasone plus LPS groups, mice were injected intraperitoneally with Dexamethasone (2.5 mg) at 1 h before LPS injection.
Determination of blood biochemistry parameters
Mice were sacrificed under deep anesthesia with pentobarbital 12 h after LPS injection for blood collection (centrifugation at 3000 g, 10 min), and the following blood biochemistry parameters measured for all groups: lactate dehydrogenase (also called lactic acid dehydrogenase, or LDH) is an enzyme found in almost all body tissues. It plays an important role in cellular respiration, the process by which glucose (sugar) from food is converted into usable energy for our cells. Although LDH is abundant in tissue cells, blood levels of the enzyme are normally low. However, when tissues are damaged by injury or disease, they release more LDH into the bloodstream. Conditions that can cause increased LDH in the blood include liver disease, heart attack, anemia, muscle trauma, bone fractures, cancers, and infections [40]. The ratio of AST to ALT sometimes can help determine whether the liver or another organ has been damaged. The aspartate aminotransferase/alanine aminotransferase (AST/ALT) ratio is sometimes useful in differentiating between causes of liver damage [41]. Blood urea nitrogen (BUN) is an indication of renal health. If Glomerular Filtration Rate (GFR) and blood volume decrease (hypovolemia) then BUN will increase. Other factors responsible for its increment are fever, increased catabolism, high protein diet and gastrointestinal bleeding. Serum creatinine (a blood measurement) is an important indicator of renal health because it is an easily-measured by-product of muscle metabolism that is excreted unchanged by the kidneys. Creatinine itself is produced via a biological system involving creatine, phosphocreatine (also known as creatine phosphate), and adenosine triphosphate (ATP, the body’s immediate energy supply). Measuring serum creatinine is a simple test, and it is the most commonly used indicator of renal function [42].
Determination of TNFα, IL-1β, IL-6, and INF-γ protein by ELISA
Four major proinflammatory cytokines, TNFα, IL-1β, IL-6, and INF-γ, were determined by ELISA as described previously [43]. Briefly, brain tissues from each pup were collected 12 h after LPS injection, when inflammatory cytokines in the brain peak [44]. Brains were homogenized by sonication in 1.5 ml ice-cold serum-free DMEM medium and centrifuged at 10,000 × g for 15 min at 4°C. Supernatants were collected and protein concentration determined by the Bradford method. ELISA was performed following the manufacturer’s instructions and data were acquired using a 96-well plate reader (Bio-Rad Laboratories, Inc. Hercules, CA, USA). The cytokine levels are expressed as pg cytokine/mg protein.
Immunoblotting analysis
Protein expression of UT-A3 in C57 mouse brain was determined by western blot analysis as described previously [45], with modifications. Briefly, 12 hours after LPS injection, brains were quickly removed and flash frozen in liquid nitrogen and stored at -80°C. Tissues were homogenized in extraction buffer (Biosource, Camarillo, CA, USA) containing a mixture of protease inhibitors (Merck Millipore, Billerica, MA, USA), and 1 mM PMSF applied with a Sonic Dismembrator (Fisher Scientific, Suwanee, GA, USA), 3 times for 10 s each. Homogenate protein levels were determined by the Bradford method. The homogenates were diluted with Laemmli sample buffer (1:2, v/v) and boiled for 5 min. Equal quantities of protein (10 g/10 L) were loaded into each well of a 12% SDS-polyacrylamide gradient gel (MINI-PROTEAN TGX, 4-20%, Bio-Rad Laboratories). The separated proteins were transferred electrophoretically to PVDF membranes (Merck Millipore) at 100 V for 1 h. The blots were then incubated with blocking solution containing 5% non-fat milk and 0.1% Tween-20 in Tris-buffered saline (TBS) for 1 h, before incubating with primary antibody (1:1000) in blocking solution overnight at 4°C. Blots were then incubated with peroxidase-conjugated antibodies in blocking solution (1:2000) for 1 h at room temperature. Immunoreactivity was detected using the enhanced chemiluminescence plus (ECL) system (GE Healthcare, Piscataway, NJ, USA) and visualized with Chemidoc MP Imaging System, and quantified with Image Lab software (both from Bio-Rad Laboratories). To ensure equal amounts of protein were applied to the immunoblot, membranes were treated with a stripping buffer (Thermo Fisher Scientific, Rockford, IL, USA) and reprobed for β-actin (1:1000, TianJing SanJian) to normalize results.
Immunohistochemistry
Immunohistochemistry was performed as described previously [46]. Brain sections were immunostained to visualize UT-A3 and AQP4. Diaminobenzidine (DAB) was used for color development. The microvessel density (MVD) count method refers to the Weidner correction method [47]. Positive reactions were determined in five random fields of each sample with image processing software Image-Pro Plus 6.0 (Media Cybernetics, Rockville, MD, USA).
Statistics analysis
Data were analyzed with SPSS 17.0 software. Unless noted otherwise, data are given as the mean ± SEM. Groups were compared by one-way analysis of variance (ANOVA) followed by Bonferroni multiple comparisons test and Student’s t-test. A value of P < 0.05 was considered significant.
Results
Model validation using LPS and combination of LPS + dexamethasone
Schmidt et al. determined that LPS (10 mg/kg) caused significant decrease in the expression of UTs and aggravated renal function. Therefore, we performed experiments with 10 mg/kg LPS to induce inflammation. We found that the brain weight/body weight ratio increased by 13% in the LPS group (Figure 1), indicating that LPS injection causes edema [28]. Interestingly, Blood biochemical parameters were tested 12 hours following LPS administration and 1 hr following Dexa treatment, since LPS increases cellular lactate dehydrogenase (LDH) leakage. Serum LDH concentration increased 2.5 fold in the LPS group relative to the control group (P < 0.05) and was near normal levels after Dexa intervention (P < 0.05) (Figure 2A). Serum aspartate aminotransferase/alanine aminotransferase (AST/ALT) increased 1.2 fold relative to LPS treatment and was near normal levels after Dexa intervention (P < 0.05) (Figure 2B).
Figure 1.
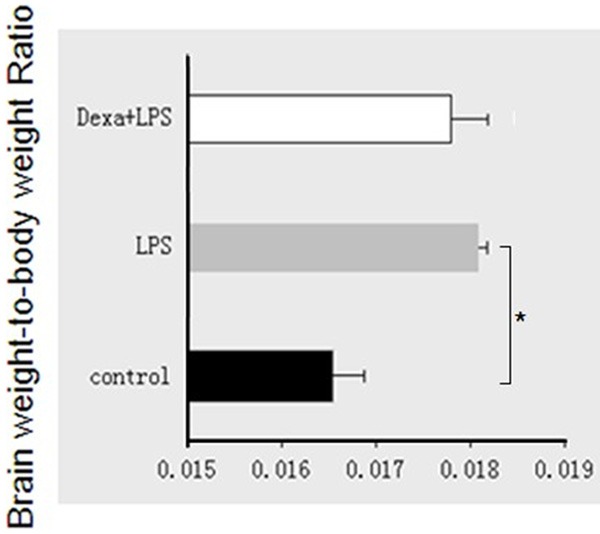
Brain weight/body weight ratio following LPS treatment. The combination of LPS + dexamethasone was examined in endotoxemia after 12 h of treatment. The results are expressed as the mean ± SE of six animals in each group, *P < 0.05 represents a significant difference.
Figure 2.
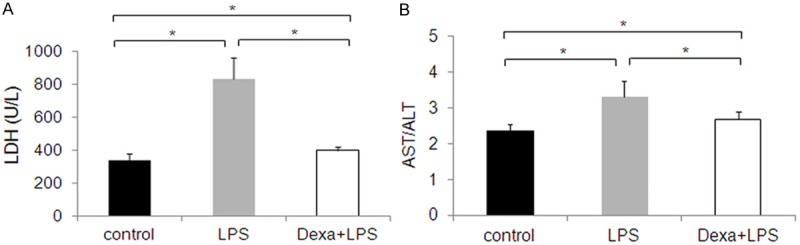
Mice treated with LPS for 12 h exhibited a 2.5-fold increase in LDH levels (A), and a 1.2-fold increase in AST/ALT levels (B), compared to controls. The results are expressed as the mean ± SE of six animals in each group, *P < 0.05 represents a significant difference.
Effect of LPS alone, and LPS + dexamethasone on Serum BUN and CREA levels
To study the effects of LPS infection on BUN and CREA serum samples were collected. BUN and CREA was assayed using spectrophotometer. Serum BUN and CREA levels were increased 2.5 fold (Figure 3A, 3B) after LPS injection, and that Dexa treatment modulates these changes. The result showed significant difference in serum levels of BUN and CREA in LPS infected mice. At the same time, urea channel proteins are expressed in kidney and tissues such as the brain, heart, and testicles, where urea levels are adjusted to maintain normal cell function. Importantly, substantial changes to serum BUN and CREA concentrations regulate changes in gene and protein levels [29].
Figure 3.
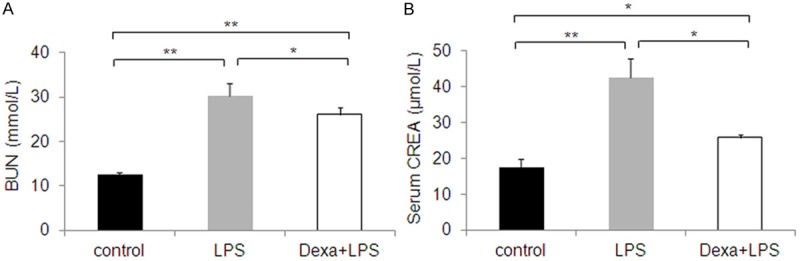
Effect of dexamethasone supplementation on serum (A) BUN and (B) CREA response to endotoxin. Serum BUN and CREA concentrations increase following LPS treatment, but decrease following LPS + dexamethasone treatment, compared with LPS alone. The results are expressed as the mean ± SE of six animals in each group, *P < 0.05, **P < 0.01 represents a significant difference.
Detection of brain cytokines following LPS alone, or LPS + dexamethasone
As shown in Figure 4, TNF-α, IL-1β, IFN-γ, and IL-6 concentrations in the brain were determined in mice injected with LPS, and concomitant treatment with dexamethasone markedly attenuated brain tissue cytokine concentration after LPS injection. At baseline, circulating levels of TNF-α were undetectable by ELISA. However, there was a 1.5 fold increase in TNF-α, IL-1β, IL-6, and INF-γ 2 hr following LPS injection. We used this model to study the increase in cytokines associated with a systemic inflammatory response mediated by UTs and how this correlated with AQP protein expression.
Figure 4.
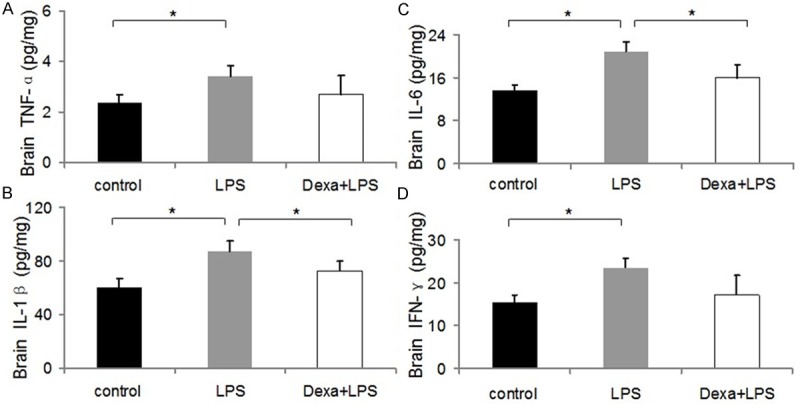
Dexamethasone-attenuated systemic LPS-stimulated increases in inflammatory cytokines (A, TNF-α, B, IL-1β, C, IL-6, and D, INF-γ) in brain 12 h after injection. Cytokine levels were elevated compared with control, but were attenuated by dexamethasone. The results are expressed as the mean ± SE of six animals in each group, *P < 0.05, **P < 0.01 represents a significant difference for the LPS or LPS + Dexamethasone groups compared with control.
Effect of LPS alone, and LPS + dexamethasone on AQP4 and UT-A3 expression
We sought to further characterize the mechanism by which brain cytokine concentrations are upregulated during endotoxemia. Blood endotoxin causes cerebral edema owing to a number of factors, including the increased expression of TNF-α, IL-1β, IL-6, and INF-γ [30]. To determine whether edema occurred in brains of mice administered LPS, we measured the brain weight/body weight ratio of control and LPS-treated mice. Given that AQP4 is the primary water channel protein in brain, we studied its expression in endotoxemic mice. AQP4 expression increased by 42% at the protein level in LPS-treated mice compared with controls, as determined by western blot analysis (Figure 5A, 5B). Similarly, UT-A3 protein expression was also examined by Western blot analysis. UT-A expression decreased following LPS treatment. UT-A3 protein fell by 38% (Figure 5A, 5B). Mice were additionally treated with Dexa (3 mg/kg ip). Mice treated with LPS + Dexa exhibited significant downregulation of AQP4 and upregulation of UT-A3 protein expression compared with LPS only treatment (Figure 5B). These data indicate that glucocorticoid treatment attenuates the LPS effect on UT-A3 membrane proteins, and protects cell membrane stability.
Figure 5.
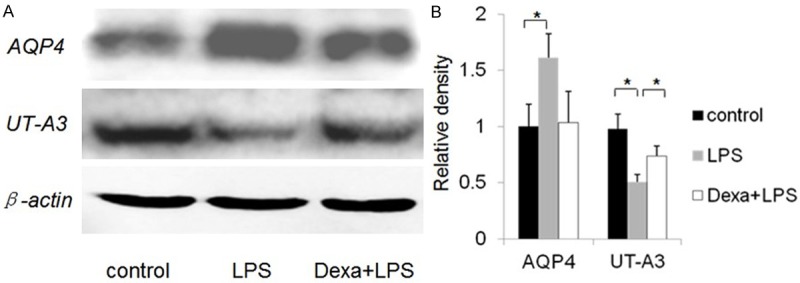
Western blot analysis of AQP4, and UT-A3 in brain. A: Each lane, corresponding to a single mouse, was loaded with 30 μg of protein. B: Quantification of band densities (mean ± SE of three mice). *P < 0.05 **P < 0.01 vs. the control group. The expression of AQP4 was significantly higher in mice with experimental endotoxemia than in control mice. Expression of UT-A3 was lower in mice with experimental endotoxemia than in control mice. AQP4 expression was downregulated in mice with experimental endotoxemia relative to mice treated with LPS. In contrast, UT-A3 expression was upregulated.
The expression of UT-A3, and AQP4 in hippocampus and cortex was further analyzed by IHC staining. As shown in Figure 6, AQP4 expression in brain was up-regulated in the LPS only group compared to control. In contrast, UT-A3 expression was down-regulated. These changes were partly improved by Dexa treatment, as observed for UT-A3 and AQP4 (Figure 6).
Figure 6.
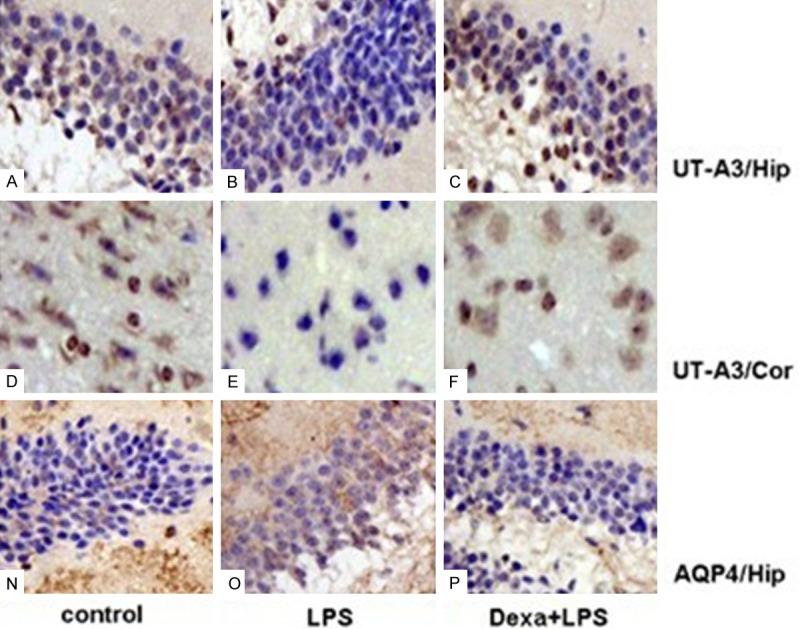
Immunohistochemical staining of AQP4 and UT-A3 in brain tissues. Tissue sections were stained using polyclonal antibody for AQP4 and UT-A3, Antibody dilution = 1:1,000. A-F: immunostaining using polyclonal antibody to UT-A3. Shown are hippocampal (Hip) cells (A-C); cortical cells (D-F). N-P: immunostaining using polyclonal antibody to AQP4. Shown are hippocampal (Hip) cells (N-P), magnification, 400 ×.
Discussion
The present study clearly demonstrates that LPS treatment significantly increases the expression of AQPs and decreases the expression of UT-As in different regions of the brain. These phenotypic changes were associated with an increase in BUN and CREA. Our observations suggest that the increase in the expression of AQP4 and decreases the expression of UT-A3 in the brain endotoxemia may be one of the mechanisms for the impairment of endotoxin blood disease model. To evaluate the degree of damage in LPS-induced endotoxemia, we measured serum LDH and AST/ALT concentrations and observed the effect of Dexa treatment. We speculated that Dexa maybe have a stabilizing effect on the cell membrane, maintaining cell membrane integrity, and to a certain extent reducing the release of intracellular enzymes. Brain damage may be caused by endotoxemia, but Dexa also has a protective effect, limiting brain endotoxemia.
Su et al. [31] have shown that LPS induced during acute lung injury (ALI) reduces AQP1 expression in pulmonary capillary endothelial cells. However, AQP1 loss does not change the pulmonary inflammatory reaction induced by LPS and pulmonary edema, and Dexa stabilizes AQP1 expression. AQP1 is expressed mainly in the liver sinus and blood vessel endothelial cell membranes [32]. LPS inhibits AQP1 mRNA expression in the liver, and is inhibited to various degrees by Dexa. We believe the results of AQP4 expression on the cell membrane helps explain how Dexa stabilizes LPS induced brain damage, and expression of aquaporins on the cell membrane reduces damage, and inflammation. TNF-α expression and the drop in IL-1β is mediated by Dexa regulating aquaporin expression on the cell membrane, and to a certain extent, maintaining the stability of the cell membrane [33]. We also found that UT-A3 protein expression and not just AQPs play a role in LPS inflammation. Dexa treatment increases UT-A3 protein expression, which maintains cell membrane stability. LPS treatment also increases BUN and CREA, which is important for renal function [34]. In addition, liver is the largest organ for urea synthesis, and organs such as brain and heart transport urea through membrane channel proteins in the kidney into the blood. Increased BUN increases cellular protein synthesis [35], increases UTs, which maintain stable guanylic acid in the cell cycle, maintaining normal cellular metabolism. Interestingly, Li et al. [36] observed that BUN and CREA levels of both UT-A or UT-B null mice increase. We infer from this that endotoxemia caused by metabolic disorders may adjust expression of UT proteins on the cell membrane. Metabolic disorders are not directly related to endotoxin, but may be regulated through metabolic hormones. Therefore, we sought to determine if UT might impact endotoxemia in the blood. In LPS-mediated sepsis, brain AQP4 expression decreased following Dexa treatment at 3 mg/ml [28], whereas UT-A3 expression increased. The mechanism by which this occurs may be related to inhibiting release of TNF-α, and IL-1β. However, Dexa did not exhibit any effect on TNF and IFN; We speculate that this model might help us understand the complex pathogenesis of brain endotoxemia. IHC analyses suggest that brain UT-A3 play specific roles in hippocampal and cortical cells [37]. However, until we have a better understanding of how the brain metabolizes urea, the role of UT-A3 in hippocampal and cortical cells remains speculative. UT-A expression in the brain of endotoxemic mice suggests that UT-A plays a key role in regulating LPS-induced endotoxemia. Northern blot analysis has shown that UT-B mRNA is expressed in neurons and various other cell types throughout the brain [37]. Our western blot and IHC analysis revealed that neuronal endotoxin concentrations in mice correlates with UT-A3 expression. Dexa injections increased neuronal UT-A3 expression and decreased AQP4 expression, understanding the molecular mechanisms regulating this process may bring new insights to endotoxemia. Our findings suggest that Dexa inhibits water reabsorption by downregulating AQPs and increases delivery of concentrated urea to the IM interstitium and its recycling by promoting UT expression. These molecular changes account, at least in part, for the endotoxin-induced brain injury. It is the other way to describe their role in the pathogenesis of endotoxin-induced brain edema and injury; the inflammatory cytokines alter the expression of membrane transporters, such as UTs or AQPs. Dexa treatment can prevent endotoxemia caused by brain injury, and may increase protein channel expression, and subsequently inhibit the synthesis of inflammatory mediators. This regulates the body’s defense, and may improve its ability to repair tissue damage and play a protective role. Glucocorticoids have protective effects on the cell membrane, as they improve resistance to poison, stabilize lysosomal membranes in macrophages, reduce lysosomal enzyme release, reduce structural damage,increase the density of vascular endothelium, prevent water leakage, and reduce tissue edema. There is a clear role for endotoxin [38] and membrane AQP expression in different tissues during endotoxemia [39]. Collectively, these observations suggest that LPS induces endotoxemia through a series of changes. These include changes in serum biochemical indicators and tissue factors, and a change in urea that impacts cell membrane stability and expression of cell membrane channel proteins. These results indicate that UTs are involved in immune activation, but this topic will need further study to fully elucidate their roles.
Acknowledgements
This work was funded by the National Natural Science Foundation of China (No. 30900518, No. 81000271, No. 81170304, No. 81370240). Project 2014030 Supported by Graduate Innovation Fund of Jilin University.
Disclosure of conflict of interest
None.
References
- 1.Buckley JF, Singer M, Clapp LH. Role of KATP channels in sepsis. Cardiovasc Res. 2006;72:220–230. doi: 10.1016/j.cardiores.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 2.Japiassu AM, Salluh JI, Bozza PT, Bozza FA, Castro-Faria-Neto HC. Revisiting steroid treatment for septic shock: molecular actions and clinical effects--a review. Mem Inst Oswaldo Cruz. 2009;104:531–548. doi: 10.1590/s0074-02762009000400001. [DOI] [PubMed] [Google Scholar]
- 3.Comstock KL, Krown KA, Page MT, Martin D, Ho P, Pedraza M, Castro EN, Nakajima N, Glembotski CC, Quintana PJ, Sabbadini RA. LPS-induced TNF-alpha release from and apoptosis in rat cardiomyocytes: obligatory role for CD14 in mediating the LPS response. J Mol Cell Cardiol. 1998;30:2761–2775. doi: 10.1006/jmcc.1998.0851. [DOI] [PubMed] [Google Scholar]
- 4.Wang X, Evans RD. Effect of endotoxin and platelet-activating factor on lipid oxidation in the rat heart. J Mol Cell Cardiol. 1997;29:1915–1926. doi: 10.1006/jmcc.1997.0430. [DOI] [PubMed] [Google Scholar]
- 5.Suffredini AF, Munford RS. Novel therapies for septic shock over the past 4 decades. JAMA. 2011;306:194–199. doi: 10.1001/jama.2011.909. [DOI] [PubMed] [Google Scholar]
- 6.Preston GM, Carroll TP, Guggino WB, Agre P. Appearance of water channels in Xenopus oocytes expressing red cell CHIP28 protein. Science. 1992;256:385–387. doi: 10.1126/science.256.5055.385. [DOI] [PubMed] [Google Scholar]
- 7.Zeuthen T. Water-transporting proteins. J Membr Biol. 2010;234:57–73. doi: 10.1007/s00232-009-9216-y. [DOI] [PubMed] [Google Scholar]
- 8.Yool AJ. Aquaporins: multiple roles in the central nervous system. Neuroscientist. 2007;13:470–485. doi: 10.1177/1073858407303081. [DOI] [PubMed] [Google Scholar]
- 9.Arcienega II, Brunet JF, Bloch J, Badaut J. Cell locations for AQP1, AQP4 and 9 in the non-human primate brain. Neuroscience. 2010;167:1103–1114. doi: 10.1016/j.neuroscience.2010.02.059. [DOI] [PubMed] [Google Scholar]
- 10.Speake T, Freeman LJ, Brown PD. Expression of aquaporin 1 and aquaporin 4 water channels in rat choroid plexus. Biochim Biophys Acta. 2003;1609:80–86. doi: 10.1016/s0005-2736(02)00658-2. [DOI] [PubMed] [Google Scholar]
- 11.Masseguin C, Corcoran M, Carcenac C, Daunton NG, Güell A, Verkman AS, Gabrion J. Altered gravity downregulates aquaporin-1 protein expression in choroid plexus. J Appl Physiol (1985) 2000;88:843–850. doi: 10.1152/jappl.2000.88.3.843. [DOI] [PubMed] [Google Scholar]
- 12.Taniguchi M, Yamashita T, Kumura E, Tamatani M, Kobayashi A, Yokawa T, Maruno M, Kato A, Ohnishi T, Kohmura E, Tohyama M, Yoshimine T. Induction of aquaporin-4 water channel mRNA after focal cerebral ischemia in rat. Brain Res Mol Brain Res. 2000;78:131–137. doi: 10.1016/s0169-328x(00)00084-x. [DOI] [PubMed] [Google Scholar]
- 13.Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- 14.Saadoun S, Papadopoulos MC. Aquaporin-4 in brain and spinal cord oedema. Neuroscience. 2010;168:1036–1046. doi: 10.1016/j.neuroscience.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Chen G, Yang B. Urea transporter physiology studied in knockout mice. Front Physiol. 2012;3:217. doi: 10.3389/fphys.2012.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu H, Meng Y, Zhu J, Kang L, Jia X, Guo L, Zhang L, Ye M, Hu L, Zhao X, Gu J, Yang B, Zou H. Differential protein expression in heart in UT-B null mice with cardiac conduction defects. Proteomics. 2009;9:504–511. doi: 10.1002/pmic.200701079. [DOI] [PubMed] [Google Scholar]
- 17.Du Y, Meng Y, Zhu J, Kang L, Jia X, Guo L, Zhang L, Ye M, Hu L, Zhao X, Gu J, Yang B, Zou H. Quantitative proteomic study of myocardial mitochondria in urea transporter B knockout mice. Proteomics. 2014;14:2072–83. doi: 10.1002/pmic.201400123. [DOI] [PubMed] [Google Scholar]
- 18.Guo L, Zhao D, Song Y, Zhao H, Zhao X, Yang B. Reduced urea flux across the blood-testis barrier and early maturation in the male reproductive system in UT-B-null mice. Am J Physiol Cell Physiol. 2007;293:C305–C312. doi: 10.1152/ajpcell.00608.2006. [DOI] [PubMed] [Google Scholar]
- 19.Bagnasco SM. Gene structure of urea transporters. Am J Physiol Renal Physiol. 2003;284:F3–F10. doi: 10.1152/ajprenal.00260.2002. [DOI] [PubMed] [Google Scholar]
- 20.Smith CP, Rousselet G. Facilitative urea transporters. J Membr Biol. 2001;183:1–14. doi: 10.1007/s00232-001-0048-7. [DOI] [PubMed] [Google Scholar]
- 21.Hu MC, Bankir L, Trinh-Trang-Tan MM. mRNA expression of renal urea transporters in normal and Brattleboro rats: effect of dietary protein intake. Exp Nephrol. 1999;7:44–51. doi: 10.1159/000020583. [DOI] [PubMed] [Google Scholar]
- 22.Meng Y, Zhao C, Zhang X, Zhao H, Guo L, Lü B, Zhao X, Yang B. Surface electrocardiogram and action potential in mice lacking urea transporter UT-B. Sci China C Life Sci. 2009;52:474–478. doi: 10.1007/s11427-009-0047-y. [DOI] [PubMed] [Google Scholar]
- 23.Lim SW, Li C, Sun BK, Han KH, Kim WY, Oh YW, Lee JU, Kador PF, Knepper MA, Sands JM, Kim J, Yang CW. Long-term treatment with cyclosporine decreases aquaporins and urea transporters in the rat kidney. Am J Physiol Renal Physiol. 2004;287:F139–F151. doi: 10.1152/ajprenal.00240.2003. [DOI] [PubMed] [Google Scholar]
- 24.Kim D, Klein JD, Racine S, Murrell BP, Sands JM. Urea may regulate urea transporter protein abundance during osmotic diuresis. Am J Physiol Renal Physiol. 2005;288:F188–F197. doi: 10.1152/ajprenal.00200.2004. [DOI] [PubMed] [Google Scholar]
- 25.Fenton RA, Stewart GS, Carpenter B, Howorth A, Potter EA, Cooper GJ, Smith CP. Characterization of mouse urea transporters UT-A1 and UT-A2. Am J Physiol Renal Physiol. 2002;283:F817–F825. doi: 10.1152/ajprenal.00263.2001. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt C, Hocherl K, Bucher M. Cytokine-mediated regulation of urea transporters during experimental endotoxemia. Am J Physiol Renal Physiol. 2007;292:F1479–F1489. doi: 10.1152/ajprenal.00460.2006. [DOI] [PubMed] [Google Scholar]
- 27.Li F, Lei T, Zhu J, Wang W, Sun Y, Chen J, Dong Z, Zhou H, Yang B. A novel small-molecule thienoquinolin urea transporter inhibitor acts as a potential diuretic. Kidney Int. 2013;83:1076–1086. doi: 10.1038/ki.2013.62. [DOI] [PubMed] [Google Scholar]
- 28.Alexander JJ, Jacob A, Cunningham P, Hensley L, Quigg RJ. TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor-1. Neurochem Int. 2008;52:447–456. doi: 10.1016/j.neuint.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spector DA, Yang Q, Wade JB. High urea and creatinine concentrations and urea transporter B in mammalian urinary tract tissues. Am J Physiol Renal Physiol. 2007;292:F467–F474. doi: 10.1152/ajprenal.00181.2006. [DOI] [PubMed] [Google Scholar]
- 30.Xiong L, Yang L. Effects of alkaloid sinomenine on levels of IFN-gamma, IL-1beta, TNF-alpha and IL-6 in a rat renal allograft model. Immunotherapy. 2012;4:785–791. doi: 10.2217/imt.12.80. [DOI] [PubMed] [Google Scholar]
- 31.Su X, Song Y, Jiang J, Bai C. The role of aquaporin-1 (AQP1) expression in a murine model of lipopolysaccharide-induced acute lung injury. Respir Physiol Neurobiol. 2004;142:1–11. doi: 10.1016/j.resp.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 32.Jiao G, Li E, Yu R. Decreased expression of AQP1 and AQP5 in acute injured lungs in rats. Chin Med J (Engl) 2002;115:963–967. [PubMed] [Google Scholar]
- 33.Rutkovskiy A, Mariero L H, Nygard S, Stensløkken KO, Valen G, Vaage J. Transient hyperosmolality modulates expression of cardiac aquaporins. Biochem Biophys Res Commun. 2012;425:70–75. doi: 10.1016/j.bbrc.2012.07.052. [DOI] [PubMed] [Google Scholar]
- 34.Schrier RW. Blood urea nitrogen and serum creatinine: not married in heart failure. Circ Heart Fail. 2008;1:2–5. doi: 10.1161/CIRCHEARTFAILURE.108.770834. [DOI] [PubMed] [Google Scholar]
- 35.Kalhan SC. Protein metabolism in pregnancy. Am J Clin Nutr. 2000;71(Suppl 5):1249S–1255S. doi: 10.1093/ajcn/71.5.1249s. [DOI] [PubMed] [Google Scholar]
- 36.Li X, Chen G, Yang B. Urea transporter physiology studied in knockout mice. Front Physiol. 2012;3:217. doi: 10.3389/fphys.2012.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doran JJ, Klein JD, Kim YH, Smith TD, Kozlowski SD, Gunn RB, Sands JM. Tissue distribution of UT-A and UT-B mRNA and protein in rat. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1446–R1459. doi: 10.1152/ajpregu.00352.2004. [DOI] [PubMed] [Google Scholar]
- 38.Eefsen M, Jelnes P, Schmidt LE, Vainer B, Bisgaard HC, Larsen FS. Brain expression of the water channels aquaporin-1 and -4 in mice with acute liver injury, hyperammonemia and brain edema. Metab Brain Dis. 2010;25:315–323. doi: 10.1007/s11011-010-9213-y. [DOI] [PubMed] [Google Scholar]
- 39.Trinh-Trang-Tan MM, Cartron JP, Bankir L. Molecular basis for the dialysis disequilibrium syndrome: altered aquaporin and urea transporter expression in the brain. Nephrol Dial Transplant. 2005;20:1984–1988. doi: 10.1093/ndt/gfh877. [DOI] [PubMed] [Google Scholar]
- 40.Broz P, Monack DM. Measuring inflammasome activation in response to bacterial infection. Methods Mol Biol. 2013;1040:65–84. doi: 10.1007/978-1-62703-523-1_6. [DOI] [PubMed] [Google Scholar]
- 41.Nyblom H, Berggren U, Balldin J, Olsson R. High AST/ALT ratio may indicate advanced alcoholic liver disease rather than heavy drinking. Alcohol Alcohol. 2004;39:336–339. doi: 10.1093/alcalc/agh074. [DOI] [PubMed] [Google Scholar]
- 42.Allen PJ. Creatine metabolism and psychiatric disorders: Does creatine supplementation have therapeutic value? Neurosci Biobehav Rev. 2012;36:1442–1462. doi: 10.1016/j.neubiorev.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fan LW, Kaizaki A, Tien LT, Pang Y, Tanaka S, Numazawa S, Bhatt AJ, Cai Z. Celecoxib attenuates systemic lipopolysaccharide-induced brain inflammation and white matter injury in the neonatal rats. Neuroscience. 2013;240:27–38. doi: 10.1016/j.neuroscience.2013.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pang Y, Cai Z, Rhodes PG. Disturbance of oligodendrocyte development, hypomyelination and white matter injury in the neonatal rat brain after intracerebral injection of lipopolysaccharide. Brain Res Dev Brain Res. 2003;140:205–214. doi: 10.1016/s0165-3806(02)00606-5. [DOI] [PubMed] [Google Scholar]
- 45.Yin D, Li Y, Lin H, Guo B, Du Y, Li X, Jia H, Zhao X, Tang J, Zhang L. Functional graphene oxide as a plasmid-based Stat3 siRNA carrier inhibits mouse malignant melanoma growth in vivo. Nanotechnology. 2013;24:105102. doi: 10.1088/0957-4484/24/10/105102. [DOI] [PubMed] [Google Scholar]
- 46.Zhang L, Gao L, Zhao L, Guo B, Ji K, Tian Y, Wang J, Yu H, Hu J, Kalvakolanu DV, Kopecko DJ, Zhao X, Xu DQ. Intratumoral delivery and suppression of prostate tumor growth by attenuated Salmonella enterica serovar typhimurium carrying plasmid-based small interfering RNAs. Cancer Res. 2007;67:5859–5864. doi: 10.1158/0008-5472.CAN-07-0098. [DOI] [PubMed] [Google Scholar]
- 47.Weidner N, Folkman J, Pozza F, Bevilacqua P, Allred EN, Moore DH, Meli S, Gasparini G. Tumor angiogenesis: a new significant and independent prognostic indicator in early-stage breast carcinoma. J Natl Cancer Inst. 1992;84:1875–1887. doi: 10.1093/jnci/84.24.1875. [DOI] [PubMed] [Google Scholar]