

Abstract
This study aimed to investigate the role of RIP1 and RIP3 in the pathogenesis of aplastic anemia (AA) induced by cyclophosphamide and busulphan in mice. Animals were randomly divided into three groups: the control group, the AA group, and the Nec-1 group. Mouse AA model was established by intraperitoneal injection of cyclophosphamide (40 mg/kg/d) and busulfan (20 mg/kg/d) for 12 days. The Nec-1 group mice received intraperitoneal injection of Nec-1 (2 mg/kg/d) for 12 days prior to intraperitoneal injection of cyclophosphamide (40 mg/kg/d) and busulfan (20 mg/kg/d) for 12 days. The control mice received intraperitoneal injection of equal volume of saline. At 12 h after the last intraperitoneal injection, blood and bone marrow tissues were collected from mice. Peripheral blood cells were analyzed using hematology analyzer and the histological changes of bone marrow tissues were examined using scanning electron microscopy (SEM). The levels of RIP3 and RIP3 in bone marrow were measured using Western blot analysis and the interaction of RIP1 and RIP3 proteins was investigated on the basis of immunoprecipitation analysis. ELISA was used to measure the levels of IL-6, TNF-α, and FLT-3L in bone marrow tissue supernatant. Apoptosis and necrosis of bone marrow cells were analyzed using flow cytometry. Western blot showed that the expression of RIP1 and RIP3 was significantly increases in AA mice compared to the normal controls. Immunoprecipitation detected the pro-necrotic RIP1-RIP3 complex, suggesting that RIP1 and RIP3 mediated necroptosis may involved in the damage of bone marrow cells. Compared to the AA mice, Nec-1 group mice exhibited significantly increase of peripheral blood cells and mononuclear cells in bone marrow tissues and decrease of the apoptosis/necrosis of bone marrow cells. In addition, we observed significant decrease of IL-6, TNF-α, and FLT-3L in bone marrow tissue supernatant in the Nec-1 group mice compared to AA mice. Our results suggest that Nec-1 can prevent the development of AA by inhibiting bone marrow cells necrosis and the production of inflammatory mediators. RIP1 and RIP3-mediated necroptosis may involve in the pathogenesis of AA induced by cyclophosphamide and busulfan in mice.
Keywords: RIP1, RIP3, necroptosis, bone marrow cells, aplastic anemia, mouse
Introduction
Aplastic anemia (AA) is a blood disorder characterized by hematopoietic stem/progenitor cell exhaustion, hematopoietic microenvironment damage, and peripheral blood pancytopenia. It has been reported that AA is caused by chemicals, biological factors, radiation, or some unknown factors, but the underlying mechanism is largely unknown [1]. Currently, experimental evidence supports that AA is an autoimmune diseases in which abnormally activated T cells target and damage bone marrow cells. Previous studies have reported that a number of negative regulators of bone marrow such as TNF-α, IFN-γ, and FasL were highly expression in the bone marrow tissue of AA patients, which activated the apoptosis of hematopoietic stem/progenitor cells and caused AA [2,3].
Apoptosis is an important mechanism to maintain physiological homeostasis by eliminating damaged or transformed cells without causing local inflammation. It has been reported that abnormal activation of apoptosis is involved in numerous human diseases [4]. A large number of proteins and molecules are involved in the apoptosis pathway. Among them, the cysteinyl aspartate specific proteinase (caspase) family plays a critical role in the initiation of apoptosis, signal transduction, and apoptosis effects [5]. The recent study has reported that some types of cells switched from caspase dependent apoptosis to caspase-independent cell death-necroptosis when caspase was inhibited by caspase specific inhibitor zVAD-fmk [6]. Necroptosis is a highly regulated process of cell death that is initiated by the death ligands including TNF-α and FasL and mediated by receptor interacting protein (RIP) such as RIP1 and RIP3. It has been reported that necroptosis contributed to the pathogenesis of a wide range of human diseases such as infectious diseases, neurodegenerative diseases, and autoimmune diseases [7-10]. The RIP family members RIP1 and RIP3 phosphorylate each other and the RIP1-RIP3 complex induces the formation of necrotic complex, which is the key step of necroptosis [11-13].
Whether RIP1 and RIP3-mediated necroptosis is involved in the pathogenesis of AA is still not clear. In this study, we investigated the role of RIP1 and RIP3 in the development of AA based on a mouse AA model induced by cyclophosphamide and busulphan.
Materials and methods
Reagents
Busulphan and cyclophosphamide were purchased from GlaxoSmithKline and Jiangsu Hengrui Medicine Co. (Jiangsu, China). Nec-1 and Rabbit Anti-RIPK3 antibody were from Santa Cruz Biotechnology Inc (California, USA) and Bioworld Technology, Inc (Minnesota, USA). Rabbit anti-GAPDH and Goat Anti-Rabbit IgG were purchased form Hangzhou Goodhere Biotechnology, Co. (Wuhan, China) and Biosharp (China). Fluor 488 annexin V and PI kit were from Invitrogen (USA). Immunoprecipitation SiO2-MagBeads Kit was purchased from BioCanal (China). Rabbit Anti-RIPK1, IL-6, TNF-α, and FLT-3L ELISA kit and protein extraction kit were purchased from the Boster Company (Wuhan, China). BCA Protein Assay Kit, SDS-PAGE Gel preparation kit, and BeyoECL Plus were purchased from the BiosharpBeyotime Institute of Biotechnology, China.
Animals and grouping
Healthy male ICR mice (18-22 g, 6-8 weeks) were provided by the Experimental Animal Center of ZheJiang Province, China (Certification No. CSDZG-7). A total of 150 mice were randomly divided into three groups (n=50 for each group): the normal control, the AA model, and Nec-1 groups.
Establish of aplastic anemia model
The AA group mice received intraperitoneal injection of cyclophosphamide (40 mg/kg) and busulfan (20 mg/kg) for 12 days (one time per day). The control mice received intraperitoneal injection of saline of same volume.
Blood test and histopathological examination of bone marrow
Twenty-four hours after the last intraperitoneal injection, tail vein blood was collected from eight mice randomly selected from each group for blood test. Subsequently, the mice were sacrificed by cervical dislocation and one femur was surgically dissected. After removing epiphysis from the femur, bone marrow cells were washed off using 1 ml PBS to prepare bone marrow cell suspension. Nucleated cells in 20 μl bone marrow cell suspension were counted using the blood cell analyzer. The remaining bone marrow cell suspension was stored for follow-up experiments.
Another femur were dissected and fixed in 10% formalin solution for histopathological examination. The femur was further decalcified in 5% nitric acid solution for 7~12 h. Then, paraffin-embedded sections were prepared routinely for hematoxylin and eosin (HE) staining and histopathological examination.
Nec-1 administration
The Nec-1 group mice first received intraperitoneal injection of Nec-1 (2 mg/kg) and then received intraperitoneal injection of cyclophosphamide (40 mg/kg) and busulfan (20 mg/kg) for 12 days (one time per day). The AA group mice first received intraperitoneal injection of saline of same volume as Nec-1 and then received intraperitoneal injection of Nec-1 (2 mg/kg) and then received intraperitoneal injection of cyclophosphamide (40 mg/kg) and busulfan (20 mg/kg) for 12 days (one time per day).
Hoechst and propidium iodide (PI) staining
After washed with PBS, 5 μl hoechst were added into bone marrow cells (1 × 106~1 × 107) and incubated at room temperature for 10-15 mins. After washed with PBS, 5 μl PI were added and incubated at room temperature in dark room for 10-15 mins. Then, the cells were washed with PBS and examined under fluorescence microscope.
Examination of bone marrow cells using scanning electron microscopy (SEM)
After washed with PBS, bone marrow cells were centrifugated for 5 mins at 600-800 rpm to precipitate bone marrow cells. The cell pallet was resuspended and fixed in 2.5% glutaraldehyde for SEM examination.
Western blotting to measure the expression of RIP1 and RIP3
Eight mice were randomly selected on the 3, 6, 9, and 12 days after the intraperitoneal injection of cyclophosphamide and busulfan to examine the expression of RIP1 and RIP3. Bone marrow cells were collected from one femur as mentioned above and washed for three times with PBS. The protein concentration was determined using the Bradford method. Protein isolation solution was added to bone marrow cells for total protein extraction. The protein samples were boiled in water bath for 3 min and 50 μg total proteins were separated in 10% SDS-PAGE and transferred to PVDF membrane. After blocked with 5% skim milk for 1 hr, first antibodies were added and incubated overnight at 4°C. The PVDF membrane was then rinsed with TBST for three times and incubated with relative secondary antibodies (anti-RIP1 and anti-RIP3, respectively) for 1 hr at room temperature. The PVDF membrane was washed with TBST for three times and stained with ECL chemiluminescence. The expression of RIP1 and RIP3 was analyzed with the ImageQuant LAS 4000.
Immunoprecipitation to investigate the interaction between RIP1 and RIP3
After removing supernatant from the bone marrow cell suspension by centrifugation, cell lysis buffer including protease inhibitor was added to cell pellet and incubated on ice for 30 minutes. The cell lysate was centrifugated at 4°C for 30 min at the maximum speed. A small amount lysates supernant was stored for Western blotting assay. Corresponding antibody (1 μg) was added to lysates supernant and incubated at room temperature overnight. G Sepharose beads (10 μl) were rinsed with cell lysis buffer and centrifugated at 3000 rpm for 3 mins for three times. Then, the 10 μ G Sepharose beads mixed with the antibody-incubated lysates supernant and incubated at 4°C for 2-4 hrs with gentle shake to allow G Sepharose beads to be well coupled with antibody. The mixture was then centrifuged at 4°C and 3000 rpm for 3 minutes to collect beads on the bottom. After rinsed with 1 ml cell lysis buffer for 3 to 4 times, 15 μl 2 × SDS loading buffer was added to the beads and boiled in a water bath for 5 minutes, and centrifugated to collect the supernatant for Immunoprecipitation.
Flow cytometry for evaluation of cell death and apoptosis
100 μl bone marrow cells (1 × 106/ml) were washed with PBS and centrifugated to remove supernatant. The cell pellet was resuspended in cell staining buffer and with the addition of 5 μl FITC-Annexin V and 1 μl PI. After incubation at room temperature for 10-15 mins, the bone marrow cells were analyzed with flow cytometry.
Enzyme-linked immunosorbent assay (ELISA) for the levels of IL-6, TNF-α, FLT-3L in bone marrow cells
The stored bone marrow cell suspension was used in ELISA for the measurement of levels of IL-6, TNF-α, and FLT-3L. Briefly, after the ELISA plate wells were coated with antibodies against IL-6, TNF-α, and FLT-3L, respectively, bone marrow cell suspension samples were added to allow the binding between cytokines with antibodies. Then, the unbound cytokines were washed away and secondary antibody was added for incubation. After washing unbound antibodies and cytokines, the chromogenic substrate was added and the OD value was determined using the microplate reader. Standard curve of each cytokine was determined for the evaluation of the levels of IL-6, TNF-α, and FLT-3L in bone marrow suspension.
Statistical analysis
All data were expressed as mean ± standard deviation (SD) and the t-test was used to evaluate the difference between groups. A P value less than 0.05 considered to be statistically significant.
Results
Changes of the peripheral blood cells and bone marrow nucleated cells
Compared with the control group, the white blood cells (WBC), red blood cell (RBC), hemoglobin (Hb), platelet (PLT), and bone marrow nucleated cell count (BMNC) in the AA group mice significantly decreased by 64.32%, 42.33%, 20.04%, 61.53%, and 68.25%, respectively (Figure 1). Compared with the AA group, WBC, RBC, PLT, and BMNC in the Nec-1 group mice significantly increased.
Figure 1.
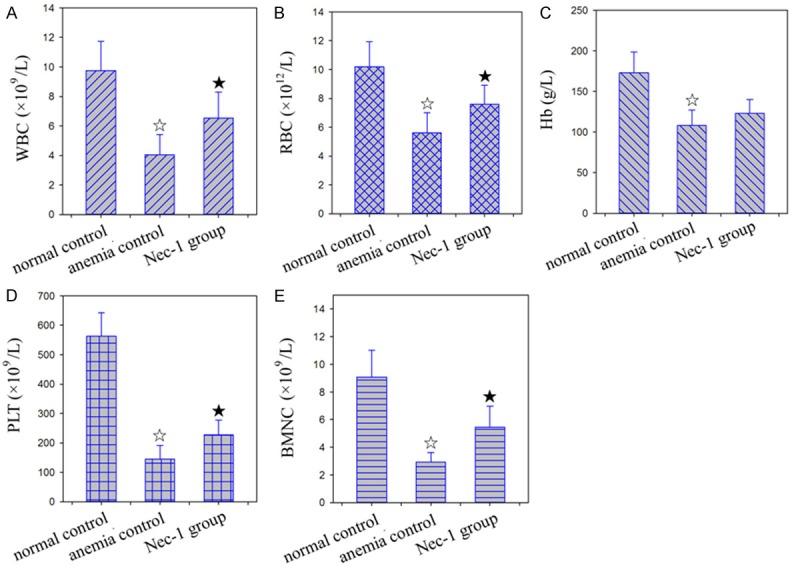
The results of peripheral blood examination. A: WBC; B: RBC; C: Hb; D: PLT; E: BMNC. ☆: P < 0.05, compared with the normal control. ★: P < 0.05, compared with the anemia control.
Histopathological changes of bone marrow
Compared to the control group, the bone marrow tissues of AA group mice exhibited inhibition of proliferation of bone marrow tissues, replacement of hematopoietic tissues with fat tissues, significant decreases of hematopoietic area, megakaryocytes and hematopoietic cells, and significant increases of endothelial cells, fat cells and other non-hematopoietic cells. In addition, the bone marrow of AA group mice had significantly increased cell degeneration, focal necrosis, congestion of stromal sinus, hemorrhage, and edema (Figure 2).
Figure 2.
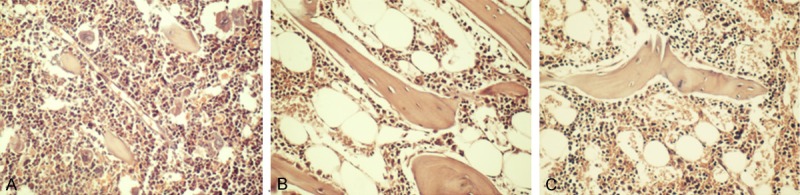
Effects of Nec-1 on bone marrow of mice with AA (200×). A: The normal control; B: The anemia control; C: The Nec-1 group.
Compared with the AA group, hematopoietic cells and megakaryocytes in the bone marrow tissues were significantly increased in Nec-1 group mice. Endothelial cells in the bone marrow tissues significantly reduced in Nec-1 group mice compared to the AA group mice. In addition, hematopoietic area increased and edema was relieved in Nec-1 group mice compared with the AA group mice (Figure 2).
Morphological changes of bone marrow cells
Both PI and hoechst can bind with nucleus DNA (or RNA). PI cannot penetrate the normal cell membrane whereas hoechst is a fluorescent dye with cell membrane permeability. The cell membrane of normal cells and early apoptotic cells are intact, and can be stained blue by hoechst. When the cells are at the middle/late stage of apoptosis or necrosis, the cell membrane is damaged, and then the nuclei were stained with red PI. As shown in Figure 3A, the red PI staining suggests that middle/late stage apoptosis or necrosis of bone marrow cells in the AA group mice. SEM further demonstrated the presence of both apoptotic and necrotic cells in the bone marrow of AA mice. Under SEM, apoptotic cells exhibited shrinkage and retraction of cell membrane, but necrotic cells swelled with the formation of blisters (Figure 3C and 3D).
Figure 3.
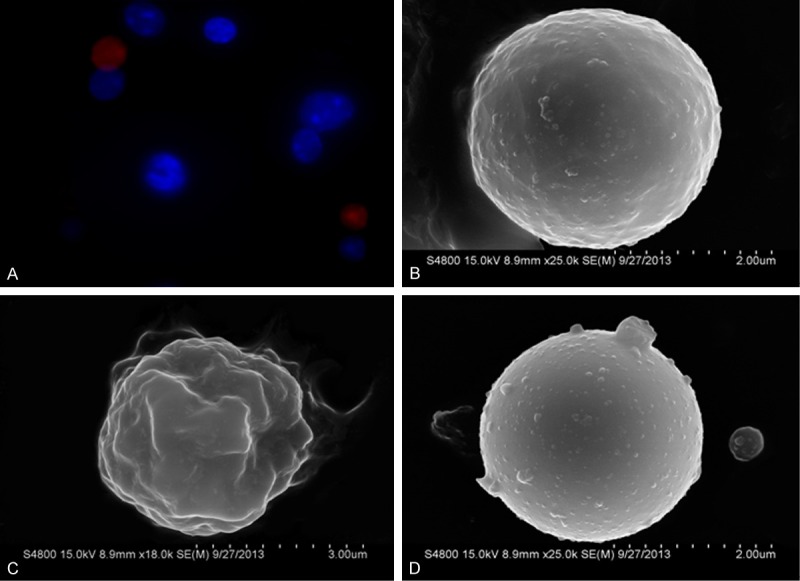
Morphological change of bone marrow cells in mice with AA. A: PI dyeing positive bone marrow cells (red stained cells), indicated the middle and late stage apoptotic and necrotic cells (400 ×, Fluorescence microscope); B: Normal bone marrow cell (SEM); C: Apoptotic bone marrow cell, cell membrane showed considerable invaginations (SEM); D: Necrotic bone marrow cell, cell swelling accompanied with cell membrane blebbing (SEM).
In the early stage of apoptosis, posphatidylserine (PS) shifts from the inner side to the outside of cell membrane. Annexin V is a Ca2+-dependent phospholipid-binding protein that specifically binds with PS high affinity. Using fluorescein-labeled Annexin V as a probe, flow cytometry can distinguish cells at early stage of apoptosis from dead cells (include middle/late stage of apoptosis cells and necrosis cells) when combined with PI staining. Flow cytometry results demonstrated that, the ratio of cells at early stage of apoptosis and dead cells was significantly higher in the AA and Nec-1 group mice compared to the control mice. However, cells at early stage of apoptosis and dead cells significantly reduced to 29.47% and 35.76% in the bone marrow tissues of Nec-1 group mice compared to the AA group mice (Figure 4).
Figure 4.
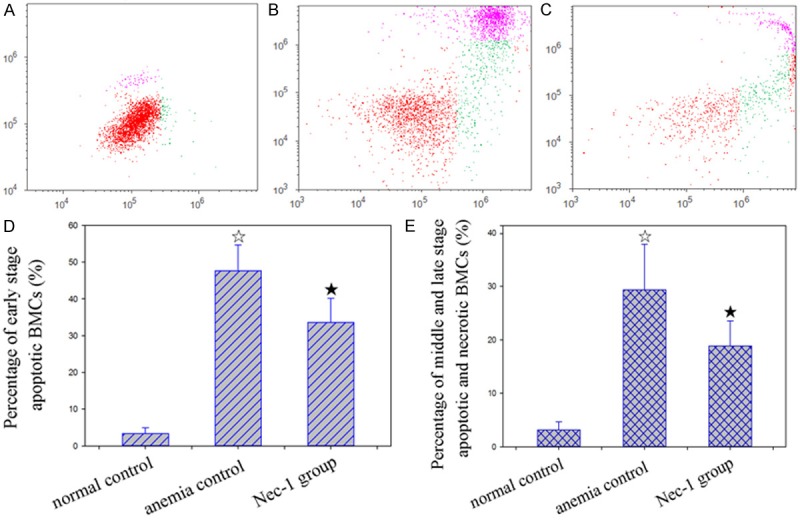
Effect of Nec-1 on death of bone marrow cells in mice with AA. A, B, C: Gated region of flow cytometric analysis. Green scatter dots indicates the early stage apoptotic population, pink scatter dots indicates the middle and late stage apoptotic and necrotic population. A: Normal control, B: Anemia control, C: Nec-1 group. D: Percentage of early stage apoptotic BMCs, E: Percentage of middle and late stage apoptotic and necrotic BMCs. ☆: P < 0.05, compared with normal control. ★: P < 0.05, compared with anemia control.
The expression of RIP1 and RIP3 in bone marrow cells of mice
Based on Western blot assay, the expression of RIP1 and RIPs started to increase on the 9th day and 3rd day post of intraperitoneal injection of cyclophosphamide and busulphan, and then the proteins of RIP1 and RIP3 in bone marrow were kept at a high level (Figure 5).
Figure 5.
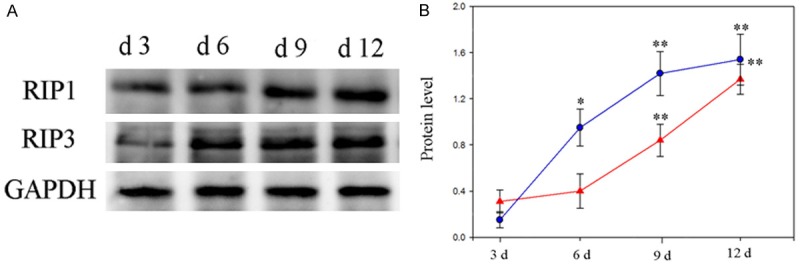
A: Western blot analysis of expression of RIP1, RIP3 proteins in bone marrow of mice with AA. B: The expression trends of RIP1(red line), RIP3(blue line). *: P < 0.05, **: P < 0.01, compared with 3 d group.
Immunoprecipitation of RIP1 and RIP3 proteins in bone marrow cells
After immunoprecipitation of RIP1, Western blotting results suggested that RIP3 was co-precipitated with RIP1 based on the observation of the pro-necrotic RIP1-RIP3 complex in bone marrow cells of AA mice induced by cyclophosphamide and busulphan. The pro-necrotic RIP1-RIP3 complex was not detected in bone marrow cells of control mice (Figure 6). In addition, the RIP1 and RIP3 proteins in bone marrow cells of AA mcie without immunoprecipitation were significantly higher than that in control mice, especially the level of RIP3 protein (Figure 6).
Figure 6.
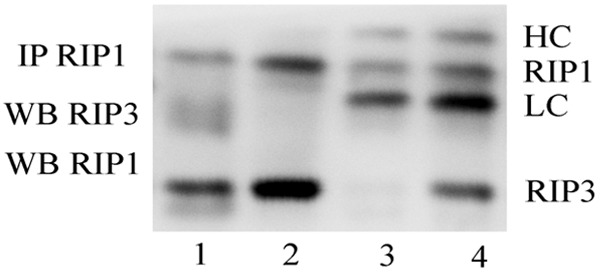
Formation of pro-necrotic RIP1-RIP3 complex in bone marrow cells of aplastic anemia mice induced by cyclophosphamide and busulphan. RIP1 was immunoprecipitated and the presence of RIP3 in the immunecomplex was determined by Western blot. lanes 1: Unimmunoprecipitated control (normal group); lanes 2: Unimmunoprecipitated control (anemia group); lanes 3: Normal control; lanes 4: Anemia. HC: Heavy chain of antigen; LC: Light chain of antigen.
Compared to control mice, the protein levels of IL-6, TNF-α, and FLT-3L was significantly increased in the bone marrow tissuss of AA group mice. Among them, the increase of FLT-3L was the most significant by 4.46 times of increase. Compared to the AA group mice, the the protein levels of IL-6, TNF-α, and FLT-3L were significantly decreased by 7.91%, 13.73% and 20.36% in the bone marrow tissues of Nec-1 group mice (Figure 7).
Figure 7.
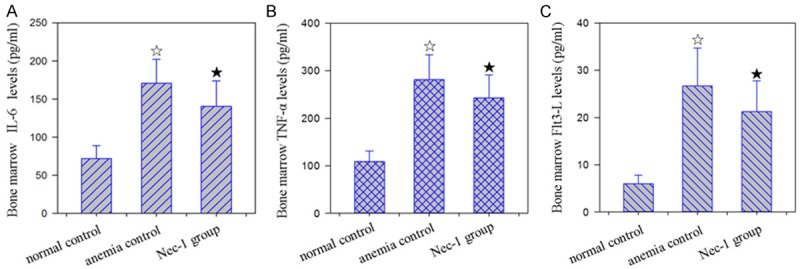
Effect of Nec-1 on bone marrow IL-6, TNF-α, FLT-3L levels in mice with AA. A: Normal group, B: anemia group, C: Nec-1 group. ☆: P < 0.05, compared with normal control. ★: P < 0.05, compared with anemia control. Effect of Nec-1 on bone marrow IL-6, TNF-α, FLT-3L levels in mice with AA.
Discussion
Apoptosis and necrosis, two distinct types of cell death, play a vital role in the development and tissue homeostasis. Compared to necrosis, apoptosis is an active and programmed cell death that is controlled by multiple gene products. Apoptosis is involved in numerous human diseases including AA. Scopes et al. have observed that hematopoietic stem/progenitor cells of CD34+ positive in the bone marrow of AA patients were significantly less than that in healthy individuals based on immunofluorescence using CD34 monoclonal antibody [14]. The results of Philpott and colleagues have suggested that the apoptosis of CD34+ hematopoietic stem/progenitor cells in the bone marrow of AA patients contributed to their reduction and the pathogenesis of AA [15]. Necrosis is caused by physical, chemical, or biological factors and morphologically is characterized by disruption of the cell membrane and a swelling of the cytoplasm and mitochondria. Recent study has found a novel type of necrosis namely necroptosis that is regulated by the immune system, similarly as apoptosis [16]. Both necroptosis and apoptosis are biological processes that need energy and synthesis of new proteins. In addition, necroptosis and apoptosis are active process of self-regulation, which are triggered by the binding and activation of receptors and ligands and the involvement of RIP1 and RIP3. However, unlike necrosis, necroptosis is followed by the lysis of its mitochondria, lysosomes and cell membrane, therefore, causing significant inflammation and infiltration of inflammatory cells. Interestingly, necroptosis is different from common necrosis because no changes of the nuclear chromatin were observed in necroptosis and necroptosis can be reversed by apoptosis inhibitors including Nec-1.
In the present study, based on the mouse AA model induced by cyclophosphamide and busulphan, we investigated the role of RIP1/RIP3-mediaed necroptosis in the pathogenesis of AA using the apoptosis-specific inhibitor Nec-1. After the administration of cyclophosphamide and busulphan, the peripheral blood cells, hemoglobin, and bone marrow nucleated cells decreased significantly. In addition, histopathological examination demonstrated that the proliferation of bone marrow hematopoietic tissues and cells was inhibited, and non-hematopoietic cells (fat cells) were significantly increased. These observations suggest the success of mouse model of AA. In addition, our PI and hoechst staining and SEM results detected both apoptosis and necrosis in the bone marrow tissues of AA mice.
In this study, we examined the expression of RIP1 and RIP3 in the mouse bone marrow cells using Western blotting. Our results showed that the expression of both RIP1 and RIP3, especially the expression of RIP3, were significantly increased in the bone marrow tissues of AA mice compared to the control animals. RIP1 and RIP3 are members of the receptor-interacting protein (RIP) kinase family. Both RIP1 and RIP3 have RIP homotypic interaction motif (RHIM) at the N- and C-terminals, which enable them to interact with each other [19]. It has been reported that under the stimulation of necroptosis signal, RIP1 binds with RIP3 via the RHIM, which leads to RIP1 phosphorylation. RIP3 can also present self-phosphorylation under the stimulation of apoptosis signal. Phosphorylated RIP1 and RIP3 form pro-necrotic RIP1-RIP3 complex that promotes the necroptosis of cells [20]. It has been confirmed that the RIP1-RIP3 complex is necessary and specific in the development of necroptosis and has not been observed in other cell death events other than necroptosis [21]. Therefore, identification of the RIP1-RIP3 complex can be used as a specific index of necroptosis. In the present study, we detected the formation of pro-necrotic RIP1-RIP3 complex in bone marrow cells of AA mice, which were not observed in the normal control animals, suggesting that RIP1 and RIP3 are involved in the development of AA induced by cyclophosphamide and busulphan by promoting the necroptosis of bone marrow cells.
Nec-1 specifically inhibits necroptosis by preventing RIP1 and RIP3 from phosphorylation [16,22]. Previous studies have shown that Nec-1 prevented hypoxic-ischemic/reperfusion injury [23], prevented retinal detachment resulted from retinal photoreceptor cells apoptosis and ischemia/reperfusion injury of retina [24,25], reduced acute myocardial ischemia-reperfusion-induced myocardial injury [26], reduced peroxide-induced cell death and myocardial infarction area [27]. In the present study, we investigated the role of Nec-1 in mouse AA induced by cyclophosphamide and busulphan. By intraperitoneal injection of Nec-1, we found that Nec-1 significantly increased peripheral blood cells and bone marrow nucleated cells by inhibit bone marrow cell necroptosis to reduce the severity of bone marrow tissue damage of AA mouse.
In addition, we also measured the levels of IL-6, TNF-α, and FLT-3L level in bone marrow tissue supernatant. TNF-α, one of the major inflammatory cytokines secreted primarily by T cell, can arrest cell cycle and promote apoptosis of bone marrow cells [28]. IL-6 is among a variety of cytokines secreted by bone marrow stromal cells, which interact with each other to activate immune effector cells to damage hematopoietic stem/progenitor cell [29]. FLT-3L is a hematopoietic growth factor that is involved in the early stage of hematopoietic regulation. Specific binding of FLT-3L with its receptor Flt3 activated intracellular signal transduction to regulate hematopoiesis. The previous study provided evidence that the FLT-3L level can be used as an indicator to evaluate the biological status of bone marrow tissue damage [30]. Our results demonstrated that the levels of IL-6, TNF-α, and FLT-3L, especially the level of FLT-3L in the supernatant of bone marrow of cyclophosphamide and busulphan induced AA mice was significantly increased compared to the control mice. However, the levels of IL-6, TNF-α, and FLT-3L were significantly reduced by the necroptosis inhibitor Nec-1. Necrosis is an important source of inflammation, and production and release of inflammatory cytokines can further promote necrosis. Therefore, we hypothesized that Nec-1 downregulated inflammation and necroptosis to protection of bone marrow tissues by inhibiting the production of inflammatory mediators.
In conclusion, our results demonstrated that RIP1 and RIP3 mediated necroptosis was involved in the development of AA in mice induced by busulphan and cyclophosphamide. Therefore, RIP1 and RIP3 may be novel targets for the treatment of AA. However, the detailed mechanism and other molecules involved need to be further investigated.
Acknowledgements
This work was supported by Science and technology program project of Taizhou under Grant No. 1301ky19, Zhejiang, China. National natural science foundation of China under Grant no. 81373139. Zhejiang Provincial Natural Science Foundation of China under Grant No. LY12C05002, Zhejiang, China.
Disclosure of conflict of interest
None.
References
- 1.Gewirtz AM, Hoffman R. Current considerations of the etiology of aplastic anemia. Crit Rev Oncol Hematol. 1985;4:1–30. doi: 10.1016/s1040-8428(85)80018-4. [DOI] [PubMed] [Google Scholar]
- 2.Li JP, Zheng CL, Han ZC. Abnormal immunity and stem/progenitor cells in acquired aplastic anemia. Crit Rev Oncol Hematol. 2010;75:79–93. doi: 10.1016/j.critrevonc.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Risitano AM, Maciejewski JP, Selleri C, Rotoli B. Function and malfunction of hematopoietic stem cells in primary bone marrow failure syndromes. Curr Stem Cell Res Ther. 2007;2:39–52. doi: 10.2174/157488807779316982. [DOI] [PubMed] [Google Scholar]
- 4.Caroppi P, Sinibaldi F, Fiorucci L, Santucci R. Apoptosis and human diseases: mitochondrion damage and lethal role of released cytochrome C as proapoptotic protein. Curr Med Chem. 2009;16:4058–4065. doi: 10.2174/092986709789378206. [DOI] [PubMed] [Google Scholar]
- 5.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 6.Pétrilli V, Herceg Z, Hassa PO, Patel NS, Di Paola R, Cortes U, Dugo L, Filipe HM, Thiemermann C, Hottiger MO, Cuzzocrea S, Wang ZQ. Noncleavable poly (ADP-ribose) polymerase-1 regulates the inflammation response in mice. J Clin Invest. 2004;114:1072–1081. doi: 10.1172/JCI21854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010;7:302–313. doi: 10.1016/j.chom.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Günther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF, Becker C. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–339. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 10.Bonnet MC, Preukschat D, Welz PS, van Loo G, Ermolaeva MA, Bloch W, Haase I, Pasparakis M. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity. 2011;35:572–582. doi: 10.1016/j.immuni.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 11.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 12.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, Declercq W, Libert C, Cauwels A, Vandenabeele P. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35:908–918. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 14.Scopes J, Bagnara M, Gordon-Smith EC, Ball SE, Gibson FM. Haemopoietic progenitor cells are reduced in aplastic anaemia. Br J Haematol. 1994;86:427–430. doi: 10.1111/j.1365-2141.1994.tb04761.x. [DOI] [PubMed] [Google Scholar]
- 15.Philpott NJ, Scopes J, Marsh JC, Gordon-Smith EC, Gibson FM. Increased apoptosis in aplastic anemia bone marrow progenitor cells: possible pathophysiologic significance. Exp Hematol. 1995;23:1642–1648. [PubMed] [Google Scholar]
- 16.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 17.Dunai Z, Bauer PI, Mihalik R. Necroptosis: biochemical, physiological and pathological aspects. Pathol Oncol Res. 2011;17:791–800. doi: 10.1007/s12253-011-9433-4. [DOI] [PubMed] [Google Scholar]
- 18.Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, Kroemer G. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14:1237–1243. doi: 10.1038/sj.cdd.4402148. [DOI] [PubMed] [Google Scholar]
- 19.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 20.Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3:re4. doi: 10.1126/scisignal.3115re4. [DOI] [PubMed] [Google Scholar]
- 21.Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, Chan FK, Wu H. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–350. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu X, Chua KW, Chua CC, Liu CF, Hamdy RC, Chua BH. Synergistic protective effects of humanin and necrostatin-1 on hypoxia and ischemia/reperfusion injury. Brain Res. 2010;1355:189–194. doi: 10.1016/j.brainres.2010.07.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trichonas G, Murakami Y, Thanos A, Morizane Y, Kayama M, Debouck CM, Hisatomi T, Miller JW, Vavvas DG. Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc Natl Acad Sci U S A. 2010;107:21695–21700. doi: 10.1073/pnas.1009179107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J, Savitz SI. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res. 2010;88:1569–1576. doi: 10.1002/jnr.22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim SY, Davidson SM, Mocanu MM, Yellon DM, Smith CC. The cardioprotective effect of necrostatin requires the cyclophilin-D component of the mitochondrial permeability transition pore. Cardiovasc Drugs Ther. 2007;21:467–469. doi: 10.1007/s10557-007-6067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith CC, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM. Necrostatin: a potentially novel cardioprotective agent? Cardiovasc Drugs Ther. 2007;21:227–233. doi: 10.1007/s10557-007-6035-1. [DOI] [PubMed] [Google Scholar]
- 28.Hara T, Ando K, Tsurumi H, Moriwaki H. Excessive production of tumor necrosis factor-alpha by bone marrow T lymphocytes is essential in causing bone marrow failure in patients with aplastic anemia. Eur J Haematol. 2004;73:10–16. doi: 10.1111/j.1600-0609.2004.00259.x. [DOI] [PubMed] [Google Scholar]
- 29.Chen YF, Wu ZM, Xie C, Bai S, Zhao LD. Expression level of IL-6 secreted by bone marrow stromal cells in mice with aplastic anemia. ISRN Hematol. 2013:986219. doi: 10.1155/2013/986219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bertho JM, Demarquay C, Frick J, Joubert C, Arenales S, Jacquet N, Sorokine-Durm I, Chau Q, Lopez M, Aigueperse J, Gorin NC, Gourmelon P. Level of Flt3-ligand in plasma: a possible new bio-indicator for radiation-induced aplasia. Int J Radiat Biol. 2001;77:703–712. doi: 10.1080/09553000110043711. [DOI] [PubMed] [Google Scholar]