

Abstract
Osteoclasts are unique cells responsible for the resorption of bone matrix. MicroRNAs (miRNAs) are involved in the regulation of a wide range of physiological processes. Here, we examined the role of miR-26a in RANKL-induced osteoclastogenesis. The expression of miR-26a was up-regulated by RANKL at the late stage of osteoclastogenesis. Ectopic expression of an miR-26a mimic in osteoclast precursor cells attenuated osteoclast formation, actin-ring formation, and bone resorption by suppressing the expression of connective tissue growth factor/CCN family 2 (CTGF/CCN2), which can promote osteoclast formation via up-regulation of dendritic cell-specific transmembrane protein (DC-STAMP). On the other hand, overexpression of miR-26a inhibitor enhanced RANKL-induced osteoclast formation and function as well as CTGF expression. In addition, the inhibitory effect of miR-26a on osteoclast formation and function was prevented by treatment with recombinant CTGF. Collectively, our results suggest that miR-26a modulates osteoclast formation and function through the regulation of CTGF.
Keywords: connective tissue growth factor, osteoclast differentiation, microRNA, RANKL
INTRODUCTION
Bone remodeling is a lifelong process where old bone is resorbed by osteoclasts and new bone is formed by osteoblasts. Although these two processes are tightly regulated, an imbalance in bone metabolism can cause various bone diseases such as osteoporosis (Boyle et al., 2003).
Giant multinucleated osteoclasts are derived from hematopoietic precursor cells. Macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor κB ligand (RANKL) are essential cytokines involved in osteoclast differentiation (Walsh et al., 2006). RANKL binds its receptor, receptor activator of NF-κB (RANK), and promotes osteoclast formation via induction and activation of various transcription factors such as PU.1, NF-κB, c-Fos, and nuclear factor of activated T cells c1 (NFATc1) (Danks and Takayanagi, 2013). In addition, RANKL induces cell-cell fusion of tartrate-resistant acid phosphatase (TRAP)-positive mononuclear pre-osteoclasts (pre-OCs) to become multinuclear mature osteoclasts at a late stage of osteoclastogenesis. Various molecules have been reported to be involved in the fusion process during osteoclast differentiation (Miyamoto, 2011). Cell-cell fusion of osteoclasts was completely abrogated in mice lacking the dendritic cell-specific transmembrane protein (DC-STAMP) and the d2 isoform of the v-ATPase V0 domain (ATP6v0d2), suggesting that DC-STAMP and ATP6v0d2 are essential for cell-cell fusion of osteoclasts (Lee et al., 2006; Yagi et al., 2005). The absence of cell-cell fusion in osteoclasts resulted in the severe reduction of bone-resorbing activity, which in turn increased bone mass in DC-STAMP-deficient and ATP6v0d2-deficient mice. Thus, osteoclast fusion might be an important process in bone metabolism.
Connective tissue growth factor (CTGF), also known as CCN family member 2 (CCN2), is a member of the CCN protein family. CTGF promotes endochondral ossification by enhancing the proliferation and maturation of chondrocytes and osteoblasts and the survival of endothelial cells (Takigawa, 2013). CTGF was recently found to promote osteoclastogenesis via induction of and interaction with DC-STAMP (Nishida et al., 2011). Aberrant CTGF production induced by TNF-α might play a role in abnormal osteoclastic activation in rheumatoid arthritis patients (Nozawa et al., 2009).
MicroRNAs (miRNAs) are small, noncoding endogenous and conserved single stranded RNA molecules of 18 to 22 nucleotides that regulate gene expression at the post-transcriptional level by base-pairing with complementary sequences, primarily in the 3′-untranslated regions (3′-UTRs) of protein coding transcripts. Binding of an miRNA to its target sites results in the selective suppression of protein synthesis (Ambros, 2004; Bartel, 2004). Each miRNA may regulate hundreds of genes to control the response of the cell to developmental and other environmental cues. Recent studies have suggested that osteoclast differentiation could be regulated by several miRNAs, including miR-29b, miR-124, 148a, and miR155 (Cheng et al., 2013; Lee et al., 2013; Rossi et al., 2013; Zhang et al., 2012). Although several miRNAs play important roles in osteoclast differentiation, the role of miRNAs in osteoclast formation and function is poorly understood.
In this study, we investigated miRNA expression profiles during osteoclastogenesis and identified miR-26a as a negative regulator of osteoclast formation and function by inhibiting CTGF at the post-transcriptional level. Our results indicate that miR-26a is a novel regulator in osteoclastogenesis and a new therapeutic target for bone metabolic disorders with excessive osteoclast activity.
MATERIALS AND METHODS
Reagents
All cell culture media and supplements were obtained from HyClone Laboratories (USA). Soluble recombinant mouse RANKL was purified from insect cells and human M-CSF was a gift from D. Fremont (Washington University, USA). The synthetic mmu-miR-26a mimic, mmu-miR-26a inhibitor, and negative control were purchased from Bioneer Corporation (Korea). miRNeasy Mini Kit, miScript Reverse Transcription Kit, miScript SYBR Green PCR Kit, and miScript Primer Assay Kit were purchased from Qiagen (QIAGEN GmbH, Germany). Recombinant human CTGF was purchased from PeproTech (USA). Primary antibodies included CTGF (Santa Cruz Biotechnology, USA) and actin (Sigma-Aldrich, USA).
Osteoclast formation and TRAP staining
Murine osteoclasts were prepared from bone marrow cells as previously described (Youn et al., 2013). Murine bone marrow cells were cultured in α-MEM (HyClone Laboratories) containing 10% FBS (HyClone Laboratories) with M-CSF (20 ng/ml) for 3 days. Floating cells were removed and adherent cells were used as osteoclast precursors. To generate osteoclasts, bone marrow-derived macrophage-like cells (BMMs) were cultured with M-CSF (30 ng/ml) and RANKL (100 ng/ml) for 4 days. Cultured cells were fixed and stained for TRAP. TRAP-positive multinuclear cells (MNCs) containing more than three nuclei were counted as osteoclasts. Cells were observed using the Leica DMIRB microscope equipped with an N plan 10 × 0.25 numerical aperture objective lens (Leica Microsystems, Germany). Images were obtained with a ProgRes CFscan (Jenoptik, Germany) camera using ProgRes Capture Pro (Jenoptik).
Actin ring staining
For the generation of mononuclear pre-osteoclasts (pre-OCs), BMMs were cultured for 2 days in the presence of M-CSF (30 ng/ml) and RANKL (100 ng/ml). Pre-OCs were cultured for an additional 24 h with M-CSF (30 ng/ml) and RANKL (50 ng/ml). After fixation, cells were incubated in 0.1% Triton X-100 in PBS for 5 min and stained with Texas Red®-X Phalloidin (Molecular probes, USA). Images were obtained with a ProgRes CFscan (Jenoptik) camera and ProgRes Capture Pro (Jenoptik).
Pit formation assay
BMMs were cultured on Osteo Assay plates (Corning, USA) in the presence of M-CSF (30 ng/ml) and RANKL (100 ng/ml) for 2 days. Pre-OCs were cultured for an additional 36 h with MCSF (30 ng/ml) and RANKL (50 ng/ml). Resorption was quantified based on the number of pits. Resorption lacunae were visualized by bright-field light using the Leica DMIRB microscope with a ProgRes CFscan (Jenoptik) camera and ProgRes Capture Pro (Jenoptik).
MiRNA microarray analysis
BMMs were cultured with M-CSF (30 ng/ml) and RANKL (100 ng/ml) for the indicated times and cultured cells were lysed to isolate total RNA. The expression levels in each sample were measured using the mouse miRNA Expression Profiling array V.2 (Illumina Inc., USA). The assay panel contains 656 assays for mouse miRNAs described in the miRBase database v9.1. As indicated in the manufacturer’s instructions, miRNAs were polyadenylated and converted to complementary DNA after hybridization with miRNA-specific oligos and PCR-amplified. BeadStudio v3.1.3 (Illumina) was used to extract the array data. The signal value was log transformed and normalized by the quantile method. Hierarchical clustering was used to determine similarity using complete linkage and euclidean distance. MiRNAs with at least a twofold difference in expression level in RANKL-stimulated BMMs for the indicated times compared to control BMMs were selected to determine the expression profile.
Transfection
The synthetic miR-26a mimics, miR-26a inhibitors, or negative control were transfected into pre-OCs using the Lipofectamine™ RNAiMAX (Life Technologies, USA) according to the manufacturer’s instructions. In brief, 50 nM miRNA mimic, miRNA inhibitor, or negative control was transfected into 1.2 × 104 cells/well in 96-well plates or 3.2 × 105 cells/well in 6-well plates. Cell culture media was changed after 4 h and transfected cells were cultured for an additional 24–36 h with M-CSF (30 ng/ml) and RANKL (50 ng/ml). Cells were used for osteoclastogenesis and other analyses as indicated.
RNA isolation and quantitative reverse transcription PCR
To detect mRNA and miRNA during osteoclastogenesis, total RNA was isolated using the miRNeasy Kit (Qiagen) following the manufacturer’s protocol. Purified RNA was reverse transcribed into cDNA using the miScript Reverse Transcription Kit (Qiagen). Quantitative reverse transcription PCR (qRT-PCR) analysis was performed in triplicate with a Rotor-Gene Q instrument (Qiagen). For the quantification of mature miRNA, amplification was performed using the miScript Primer Assay and the miScript Universal Primer. For mRNA quantification, expression levels of specific genes were measured in triplicate using gene-specific primers with the QuantiTect SYBR Green PCR Kit (Qiagen). The thermal cycling conditions were as follows: 15 min at 95°C, followed by 40 cycles of 94°C for 15 s, 55°C for 30 s, and 72°C for 30 s. Amounts of mRNAs were normalized to an endogenous housekeeping gene, Gapdh. The relative quantitation value for each target gene compared with the calibrator for that target was expressed as 2−(Ct−Cc) (where Ct and Cc are the mean threshold cycle differences after normalizing to Gapdh). The relative expression levels of samples were presented by semi-log plot.
Western blot analysis
Cultured cells were washed with ice-cold PBS and lysed in extraction buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40, and protease inhibitors). Cell lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blot analysis. Signals were detected and analyzed with a LAS3000 Luminescent Image Analyzer (GE Healthcare Japan, Japan).
RESULTS
miR-26a is up-regulated in RANKL-induced osteoclast differentiation
To find the novel modulators that can affect osteoclastogenesis, we investigated miRNA expression profiles during osteoclast differentiation by miRNA microarray analysis. BMMs were cultured with M-CSF and RANKL for 3 days prior to analysis. We found that the expression of 80 miRNAs was elevated and 59 miRNAs was down-regulated during RANKL-induced osteoclastogenesis (Fig. 1A). Among them, we chose to study miR-26a and examined its expression pattern during osteoclast differentiation by qRT-PCR. Consistent with miRNA microarray results, expression of miR-26a was gradually increased during RANKL-induced osteoclastogenesis (Fig. 1B).
Fig. 1.
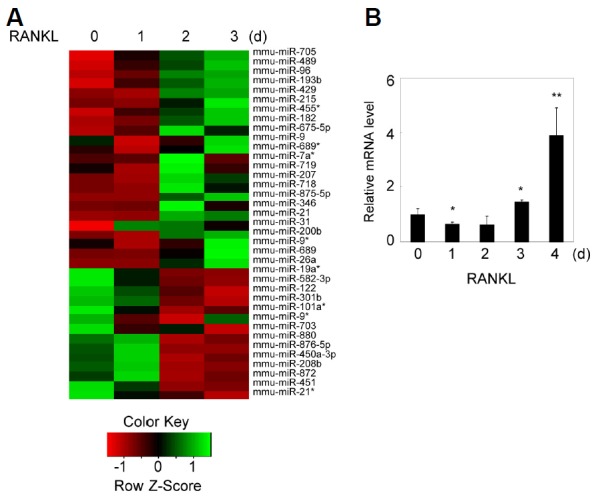
miR-26a is up-regulated during RANKL-induced osteoclastogenesis. (A) Microarray analysis was performed using RNAs isolated during RANKL-induced osteoclast differentiation. High expression levels of miRNAs are indicated in green, whereas low expression levels of miRNAs are depicted in red. (B) Validation of microarray data by qRT-PCR analysis of miR-26a expression. Data represent means ± SD of triplicate samples. Results are representative of at least three independent sets of similar experiments. *p < 0.05, **p < 0.01 vs. control BMMs.
miR-26a negatively regulates RANKL-mediated osteoclastogenesis
Next, we investigated the role of miR-26a in osteoclastogenesis. Since miR-26a was strongly expressed at the late stage of osteoclastogenesis, we examined the effect of miR-26a in osteoclasts using preosteoclasts (pre-OCs) instead of BMMs. BMMs were cultured for 2 days with M-CSF and RANKL to generate pre-OCs. Pre-OCs were transfected with an miR-26a mimic, which acts like endogenous mature miR-26a, resulting in a significant increase in mature miR-26a when measured by qRT-PCR analysis (Fig. 2A). The introduction of the miR-26a mimic in pre-OCs strongly inhibited RANKL-induced osteoclast formation (Figs. 2B and 2C). Next, we examined the effect of miR-26a on osteoclastogenesis using an miR-26a inhibitor. We confirmed the down-regulation of mature miR-26a by transfection of the miR-26a inhibitor into pre-OCs (Fig. 2D). Conversely, the miR-26a inhibitor significantly enhanced osteoclast formation (Figs. 2E and 2F). These results suggest that miR-26a negatively regulates RANKL-induced osteoclast formation.
Fig. 2.
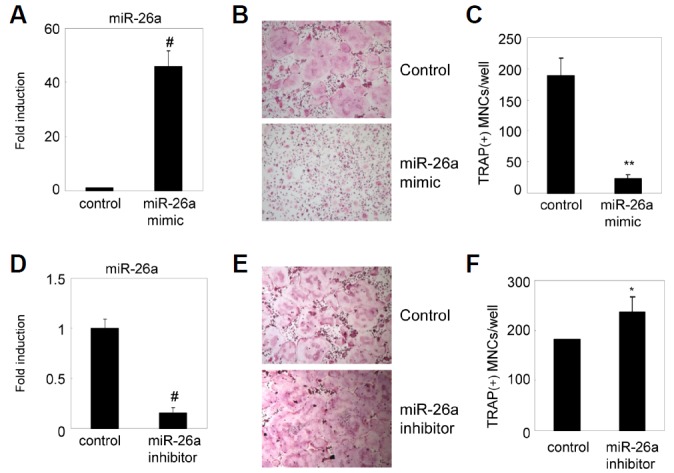
miR-26a inhibits RANKL-induced osteoclast formation. BMMs were cultured for 2 days in the presence of MCSF and RANKL. (A–C) Two-day-old pre-OCs were transfected with the miR-26a mimic or a negative control. (A) Expression level of miR-26a was detected by qRT-PCR. Data represent means ± SD of triplicate samples. #p < 0.0005 vs. control. (B, C) Transfected pre-OCs were cultured for an additional 24 h with M-CSF and RANKL. (B) Cultured cells were fixed and stained for TRAP. (C) Numbers of TRAP + MNCs were counted. Data represent means ± SD of triplicate samples. **p < 0.001 vs. control. Results are representative of at least three independent sets of similar experiments. (D–E) Pre-OCs were transfected with miR-26a inhibitor or negative control. (D) Expression level of miR-26a was detected by qRT-PCR. Data represent means ± SD of triplicate samples. #p < 0.0005 vs. control. (E, F) Transfected pre-OCs were cultured for an additional 24 h with M-CSF and RANKL. (E) Cultured cells were fixed and stained for TRAP. (F) Numbers of TRAP+MNCs were counted. Data represent means ± SD of triplicate samples. **p < 0.05 vs. control. Results are representative of at least three independent sets of similar experiments.
miR-26a regulates osteoclast function
Because miR-26a attenuated osteoclast formation, we investigated the role of miR-26a in osteoclast function. To examine the effect of miR-26a on actin ring formation, pre-OCs transfected with miR-26a mimic or miR-26a inhibitor were further cultured with M-CSF and RANKL and stained with phalloidin. Compared with the control, the miR-26a mimic significantly inhibited peripheral actin ring formation in osteoclasts, whereas the miR-26a inhibitor significantly increased actin ring formation (Figs. 3A and 3B). Next, we investigated the effect of miR-26a on bone resorption activity using a synthetic calcium phosphate matrix. Compared to the control, the miR-26a mimic markedly blocked resorption pit formation, whereas the miR-26a inhibitor significantly increased pit formation (Figs. 3C and 3D). Taken together, these results suggest that miR-26a could regulate osteoclast formation and function during RANKL-induced osteoclastogenesis.
Fig. 3.
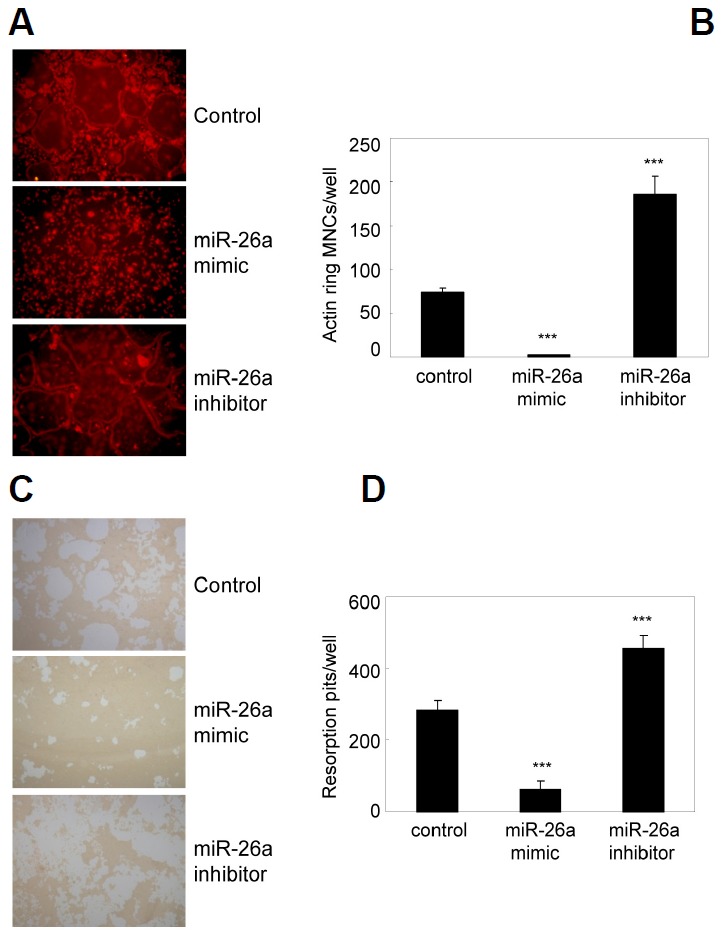
miR-26a attenuates actin ring formation and bone resorption. (A, B) BMMs were cultured for 2 days in the presence of M-CSF and RANKL. Two-day-old pre-OCs were transfected with a miR-26a mimic, miR-26a inhibitor, or negative control as indicated. Transfected cells were further cultured in the presence of M-CSF and RANKL. (A) Cells were fixed and stained with phalloidin. (B) Numbers of MNCs containing an actin ring were counted. Data represent means ± SD of triplicate samples. ***p < 0.001 vs. control. Results are representative of at least three independent sets of similar experiments. (C–D) BMMs were cultured on Osteo Assay plates for 2 days in the presence of M-CSF and RANKL. Two-dayold pre-OCs were transfected with a miR-26a mimic, miR-26a inhibitor, or negative control as indicated. Transfected cells were further cultured for 36 h in presence with M-CSF and RANKL. (C) Resorption lacunae were visualized by bright-field microscopy. (D) Numbers of resorption pits were counted. Data represent means ± SD of triplicate samples. ***p < 0.001 vs. control. Results are representative of at least three independent sets of similar experiments.
miR-26a directly targets CTGF in osteoclasts
Using bioinformatics tools for miRNA target prediction (Target Scan, PicTar, and miRanda), we identified a putative miR-26a binding site in the 3′-UTR of CTGF. The miR-26a binding site in CTGF 3′-UTR was highly conserved in mammals (Fig. 4A). To examine whether miR-26a directly targets CTGF and regulates translational repression, CTGF protein levels were examined by western blot analysis. Introduction of miR-26a into pre-OCs down-regulated CTGF expression, whereas miR-26a inhibitor strongly induced the expression of CTGF in osteoclasts (Fig. 4B). Next, we assessed the levels of fusion molecules such as DC-STAMP and ATP6v0d2. We found that the expression of DC-STAMP and ATP6v0d2 was significantly downregulated by miR-26a, whereas the miR-26a inhibitor strongly increased the levels of both fusion molecules (Fig. 4C). These results indicate that miR-26a can directly induce CTGF translational repression, hereby regulating the expression of fusion molecules.
Fig. 4.
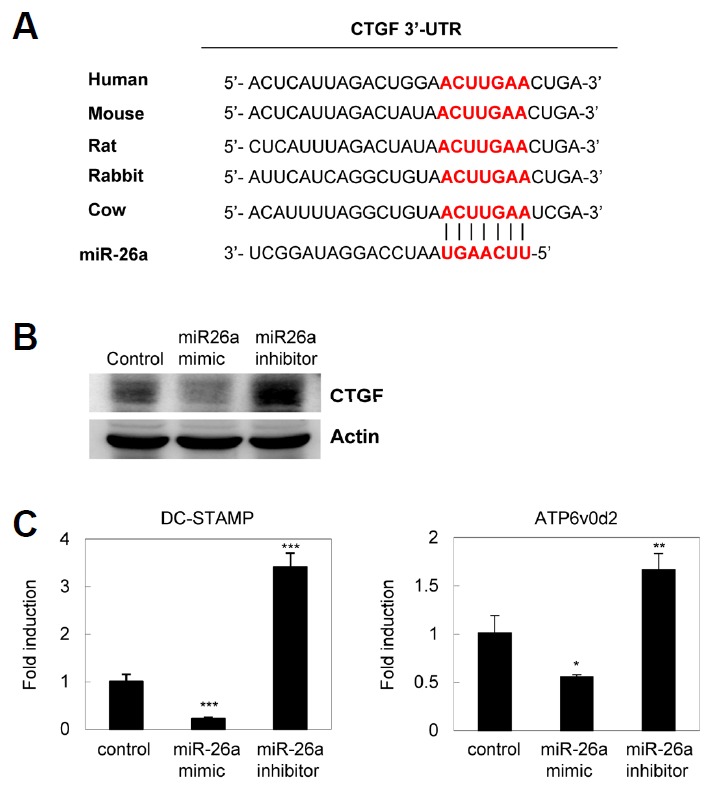
CTGF is a target of miR-26a. (A) The alignment between CTGF and miR-26a, as predicted by bioinformatics analysis of sequences from various animals, is indicated by vertical bars. The sequence alignment of putative miR-26a and its targeting site on the 3′-UTR of CTGF reveals a high level of complementarily and sequence conservation in vertebrates. (B, C) Pre-OCs were transfected with the miR-26a mimic, inhibitor, or negative control as indicated. Transfected pre-OCs were cultured for an additional 24 h in presence with M-CSF and RANKL. (B) Whole cell lysates were harvested from cultured cells and were immunoblotted with anti-CTGF and anti-actin Abs. (C) mRNA levels of DC-STAMP and ATP6v0d2 were assessed by quantitative real-time PCR. Data represent means ± SD of triplicate samples. Results are representative of at least three independent sets of similar experiments. *p < 0.05, **p < 0.01, ***p < 0.001 vs. control.
Recombinant CTGF averts the inhibitory effect of miR-26a on osteoclast formation and function
To investigate whether the inhibitory effect of miR-26a on osteoclast formation could be rescued by the addition of recombinant CTGF, pre-OCs transfected with the miR-26a mimic or miR-26a inhibitor were cultured with M-CSF and RANKL in the absence or presence of recombinant CTGF. Addition of recombinant CTGF strongly blocked miR-26a-mediated down-regulation of osteoclast formation (Figs. 5A and 5B), actin ring formation (Figs. 5C and 5D), and bone resorption (Figs. 5E and 5F). These results indicate miR-26a regulates osteoclast formation and function through CTGF.
Fig. 5.
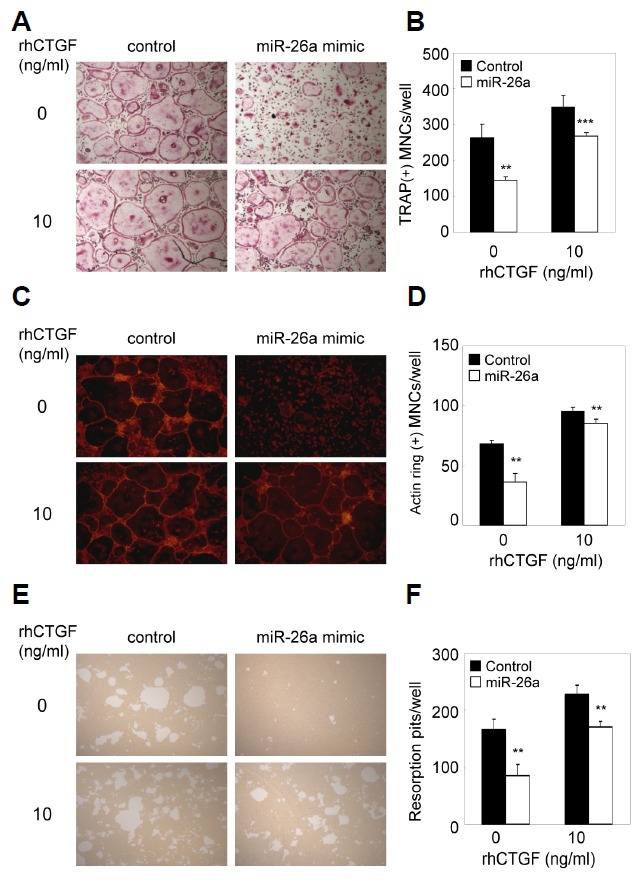
Inhibitory effect of miR-26a is rescued by CTGF. (A–D) BMMs were cultured for 2 days in the presence of M-CSF and RANKL. Two day-old pre-OCs were transfected with miR-26a mimic, miR-26a inhibitor, or negative control as indicated. Transfected cells were further cultured with M-CSF and RANKL in the presence or absence of CTGF (10 ng/ml). (A) Cultured cells were fixed and stained for TRAP. (B) Numbers of TRAP + MNCs were counted. (C) Cultured cells were fixed and stained with phalloidin. (D) Numbers of MNCs containing an actin ring were counted. (E, F) BMMs were cultured on Osteo Assay plates for 2 days in the presence of M-CSF and RANKL. Two-day-old pre-OCs were transfected with the miR-26a mimic, miR-26a inhibitor, or negative control as indicated. Transfected cells were cultured for 36 h with M-CSF and RANKL in the presence or absence of CTGF (10 ng/ml). (E) Resorption lacunae were visualized by bright-field microscopy. (F) Numbers of resorption pits were counted. Data represent means ± SD of triplicate samples. **p < 0.05, ***p < 0.001 vs. control. Results are representative of at least three independent sets of similar experiments
DISCUSSION
RANKL induces osteoclast formation through the regulation of various genes such as NF-κB, c-Fos, and NFATc1 (Asagiri and Takayanagi, 2007). RANKL-mediated osteoclast formation requires co-stimulatory signaling through FcRγ and DAP12 (Walsh et al., 2006). Besides, RANKL potentiates osteoclastogenesis via down-regulation of negative modulators including MafB, IRF-8, and Bcl6 (Kim et al., 2007; Miyauchi et al., 2010; Zhao et al., 2009). In addition to transcriptional regulation, accumulated evidence has shown that post-transcriptional and post-translational regulations might be important for RANKL-mediated osteoclastogenesis (Kim et al., 2011; Lee et al., 2013). Since miRNAs can regulate differentiation of various cells, we speculated that miRNAs might regulate genes involved in osteoclast differentiation. In an effort to find a novel regulatory mechanism in osteoclast differentiation, we investigated the expression profiles of miRNA during osteoclastogenesis by miRNA microarray analysis and the role of miR-26a in osteoclast differentiation. This study provides the first demonstration that miR-26a might fine-tune osteoclast formation and function at the post-transcriptional level.
Previous studies have shown that miR-26a acts as an important regulator in cell proliferation and differentiation. miR-26a plays a crucial role in the regulation of mouse hepatocyte proliferation during liver regeneration (Zhou et al., 2012), and it could modulate osteogenic differentiation of human adipose tissue-derived stem cells by SMAD1 transcription factor (Luzi et al., 2008). In addition, miR-26a promotes myogenesis by post-transcriptional repression of Ezh2, a known suppressor of skeletal muscle cell differentiation (Wong and Tellam, 2008). Our findings demonstrate that miR-26a acts as a negative regulator in osteoclast formation via suppression of CTGF.
To find possible target genes for miR-26a in osteoclastogenesis, we used a bioinformatics approach including Pictar, Targetscan and miRanda. Since our data showed that miR-26a regulates osteoclast maturation such as cell-cell fusion at the late stage of osteoclastogenesis, we speculated that the target gene of miR-26a might regulate cell-cell fusion of osteoclasts. It has been reported that CTGF potentiates RANKL-induced osteoclastogenesis in the late stage of osteoclastogenesis (Nishida et al., 2011). In addition, RANKL-induced osteoclastogenesis is impaired in fetal liver cells of CTGF-/- mice, and this phenotype can be rescued by the addition of exogenous recombinant CTGF or forced expression of DC-STAMP. Furthermore, CTGF promotes osteoclast formation via induction of and interaction with DC-STAMP (Nishida et al., 2011), which is an essential molecule for osteoclast fusion (Yagi et al., 2005). These results strongly suggest that CTGF might regulate osteoclast fusion by modulating DC-STAMP in the late stage of osteoclastogenesis. Forced expression of miR-26a in pre-OCs resulted in down-regulation of CTGF expression, while inhibition of miR-26a significantly augmented CTGF expression (Fig. 4). In addition, recombinant CTGF averted the inhibitory effect of miR-26a on osteoclast formation and bone resorption (Fig. 5), suggesting that miR-26a regulates osteoclast formation and function through the modulation of CTGF expression. Consistent with our data, Wei et al. (2013) reported that CTGF and collagen I are targets of miR-26a in cardiac fibroblasts. Here, we suggest that miR-26a down-regulates osteoclast formation via suppression of the CTGF/DC-STAMP axis at the late stage of osteoclastogenesis.
miR-26a was strongly expressed in the late stage of osteoclastogenesis. The related isoform, miR-26b, which differs from the miR-26a mature sequence by three nucleotides, was not differentially expressed (data not shown). Although the regulatory mechanism of miR-26a gene expression in osteoclasts has not been revealed, it appears that miR-26a provides negative feedback regulation in osteoclast formation. Further study of the detailed regulatory mechanism of miR-26a gene expression will provide a better understanding on the role of miR-26a in bone homeostasis.
In summary, the present study proposes a novel role of miR-26a in osteoclast formation and function by regulating CTGF expression. Our finding provides new insights into bone metabolism and the possibility for the development of a new therapeutic target to control various bone diseases.
Acknowledgments
This work was supported by a Grant (2014R1A2A2A01003966) from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning, a Grant (CRI13905-23) Chonnam National University Hospital Biomedical Research Institute and a Grant HI11C0674 of the Korean Health Technology R&D Project, Ministry of Health & Welfare (to K.K.).
REFERENCES
- Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- Asagiri M., Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40:251–264. doi: 10.1016/j.bone.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Boyle W.J., Simonet W.S., Lacey D.L. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- Cheng P., Chen C., He H.B., Hu R., Zhou H.D., Xie H., Zhu W., Dai R.C., Wu X.P., Liao E.Y., et al. miR-148a regulates osteoclastogenesis by targeting V-maf musculoaponeurotic fibrosarcoma oncogene homolog B. J. Bone Miner. Res. 2013;28:1180–1190. doi: 10.1002/jbmr.1845. [DOI] [PubMed] [Google Scholar]
- Danks L., Takayanagi H. Immunology and bone. J. Biochem. 2013;154:29–39. doi: 10.1093/jb/mvt049. [DOI] [PubMed] [Google Scholar]
- Kim K., Kim J.H., Lee J., Jin H.M., Kook H., Kim K.K., Lee S.Y., Kim N. MafB negatively regulates RANKL-mediated osteoclast differentiation. Blood. 2007;109:3253–3259. doi: 10.1182/blood-2006-09-048249. [DOI] [PubMed] [Google Scholar]
- Kim J.H., Kim K., Youn B.U., Jin H.M., Kim J.Y., Moon J.B., Ko A., Seo S.B., Lee K.Y., Kim N. RANKL induces NFATc1 acetylation and stability via histone acetyltransferases during osteoclast differentiation. Biochem. J. 2011;436:253–262. doi: 10.1042/BJ20110062. [DOI] [PubMed] [Google Scholar]
- Lee S.H., Rho J., Jeong D., Sul J.Y., Kim T., Kim N., Kang J.S., Miyamoto T., Suda T., Lee S.K., et al. v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat. Med. 2006;12:1403–1409. doi: 10.1038/nm1514. [DOI] [PubMed] [Google Scholar]
- Lee Y., Kim H.J., Park C.K., Kim Y.G., Lee H.J., Kim J.Y., Kim H.H. MicroRNA-124 regulates osteoclast differentiation. Bone. 2013;56:383–389. doi: 10.1016/j.bone.2013.07.007. [DOI] [PubMed] [Google Scholar]
- Luzi E., Marini F., Sala S.C., Tognarini I., Galli G., Brandi M.L. Osteogenic differentiation of human adipose tissue-derived stem cells is modulated by the miR-26a targeting of the SMAD1 transcription factor. J. Bone Miner. Res. 2008;23:287–295. doi: 10.1359/jbmr.071011. [DOI] [PubMed] [Google Scholar]
- Miyamoto T. Regulators of osteoclast differentiation and cell-cell fusion. Keio J. Med. 2011;60:101–105. doi: 10.2302/kjm.60.101. [DOI] [PubMed] [Google Scholar]
- Miyauchi Y., Ninomiya K., Miyamoto H., Sakamoto A., Iwasaki R., Hoshi H., Miyamoto K., Hao W., Yoshida S., Morioka H., et al. The Blimp1-Bcl6 axis is critical to regulate osteoclast differentiation and bone homeostasis. J. Exp. Med. 2010;207:751–762. doi: 10.1084/jem.20091957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida T., Emura K., Kubota S., Lyons K.M., Takigawa M. CCN family 2/connective tissue growth factor (CCN2/CTGF) promotes osteoclastogenesis via induction of and interaction with dendritic cell-specific transmembrane protein (DC-STAMP) J. Bone Miner. Res. 2011;26:351–363. doi: 10.1002/jbmr.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa K., Fujishiro M., Kawasaki M., Kaneko H., Iwabuchi K., Yanagida M., Suzuki F., Miyazawa K., Takasaki Y., Ogawa H., et al. Connective tissue growth factor promotes articular damage by increased osteoclastogenesis in patients with rheumatoid arthritis. Arthritis Res. Ther. 2009;11:R174. doi: 10.1186/ar2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M., Pitari M.R., Amodio N., Di Martino M.T., Conforti F., Leone E., Botta C., Paolino F.M., Del Giudice T., Iuliano E., et al. miR-29b negatively regulates human osteoclastic cell differentiation and function: implications for the treatment of multiple myeloma-related bone disease. J. Cell. Physiol. 2013;228:1506–1515. doi: 10.1002/jcp.24306. [DOI] [PubMed] [Google Scholar]
- Takigawa M. CCN2: a master regulator of the genesis of bone and cartilage. J. Cell Commun. Signal. 2013;7:191–201. doi: 10.1007/s12079-013-0204-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh M.C., Kim N., Kadono Y., Rho J., Lee S.Y., Lorenzo J., Choi Y. Osteoimmunology: interplay between the immune system and bone metabolism. Annu. Rev. Immunol. 2006;24:33–63. doi: 10.1146/annurev.immunol.24.021605.090646. [DOI] [PubMed] [Google Scholar]
- Wei C., Kim I.K., Kumar S., Jayasinghe S., Hong N., Castoldi G., Catalucci D., Jones W.K., Gupta S. NF-kappaB mediated miR-26a regulation in cardiac fibrosis. J. Cell. Physiol. 2013;228:1433–1442. doi: 10.1002/jcp.24296. [DOI] [PubMed] [Google Scholar]
- Wong C.F., Tellam R.L. MicroRNA-26a targets the histone methyltransferase enhancer of zeste homolog 2 during myogenesis. J. Biol. Chem. 2008;283:9836–9843. doi: 10.1074/jbc.M709614200. [DOI] [PubMed] [Google Scholar]
- Yagi M., Miyamoto T., Sawatani Y., Iwamoto K., Hosogane N., Fujita N., Morita K., Ninomiya K., Suzuki T., Miyamoto K., et al. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J. Exp. Med. 2005;202:345–351. doi: 10.1084/jem.20050645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn B.U., Kim K., Kim J.H., Lee J., Moon J.B., Kim I., Park Y.W., Kim N. SLAT negatively regulates RANKL-induced osteoclast differentiation. Mol. Cells. 2013;36:252–257. doi: 10.1007/s10059-013-0159-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Zhao H., Chen J., Xia B., Jin Y., Wei W., Shen J., Huang Y. Interferon-beta-induced miR-155 inhibits osteoclast differentiation by targeting SOCS1 and MITF. FEBS Lett. 2012;586:3255–3262. doi: 10.1016/j.febslet.2012.06.047. [DOI] [PubMed] [Google Scholar]
- Zhao B., Takami M., Yamada A., Wang X., Koga T., Hu X., Tamura T., Ozato K., Choi Y., Ivashkiv L.B., et al. Interferon regulatory factor-8 regulates bone metabolism by suppressing osteoclastogenesis. Nat. Med. 2009;15:1066–1071. doi: 10.1038/nm.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Ju W., Wang D., Wu L., Zhu X., Guo Z., He X. Down-regulation of microRNA-26a promotes mouse hepatocyte proliferation during liver regeneration. PLoS One. 2012;7:e33577. doi: 10.1371/journal.pone.0033577. [DOI] [PMC free article] [PubMed] [Google Scholar]