

Abstract
TGF-β regulates pleiotropic cellular responses including cell growth, differentiation, migration, apoptosis, extracellular matrix production, and many other biological processes. Although non-Smad signaling pathways are being increasingly reported to play many roles in TGF-β-mediated biological processes, Smads, especially receptor-regulated Smads (R-Smads), still play a central mediatory role in TGF-β signaling for epithelial-mesenchymal transition. Thus, the biological activities of R-Smads are tightly regulated at multiple points. Inhibitory Smad (I-Smad also called Smad7) acts as a critical endogenous negative feedback regulator of Smad-signaling pathways by inhibiting R-Smad phosphorylation and by inducing activated type I TGF-β receptor degradation. Roles played by Smad7 in health and disease are being increasingly reported, but the molecular mechanisms that regulate Smad7 are not well understood. In this study, we show that E3 ubiquitin ligase Itch acts as a positive regulator of TGF-β signaling and of subsequent EMT-related gene expression. Interestingly, the Itch-mediated positive regulation of TGF-β signaling was found to be dependent on Smad7 ubiquitination and its subsequent degradation. Further study revealed Itch acts as an E3 ubiquitin ligase for Smad7 polyubiquitination, and thus, that Itch is an important regulator of Smad7 activity and a positive regulator of TGF-β signaling and of TGF-β-mediated biological processes. Accordingly, the study uncovers a novel regulatory mechanism whereby Smad7 is controlled by Itch.
Keywords: EMT, Itch, Smad7, TGF-β, ubiquitination
INTRODUCTION
Transforming growth factor-β (TGF-β) is a pleiotropic cytokine that regulates cellular responses, such as, proliferation, growth, and tumorigenesis (Derynck and Akhurst, 2007). TGF-β activates canonical Smad signaling via serine/threonine kinase receptors called TGF-β type I receptor (TβRI) and TGF-β type II receptor (TβRII) (Shi and Massague, 2003). Activated TβRI phosphorylates receptor-regulated Smads (R-Smads) such as Smad2 and Smad3, thereby activates and promotes nuclear translocations of Smads. Furthermore, the TGF-β-induced Smad signaling pathway is tightly regulated by inhibitory Smads (I-Smads) such as Smad6 and Smad7. In particular, Smad7 forms a stable complex with TβRI, which leads to the inhibition of the phosphorylations of R-Smads (Smad2 and Smad3) and of heteromeric complex formation between R-Smads and co-Smad (Smad4) (Kavsak et al., 2000). In addition to these protein interactions, Smad7 also directly binds to the Smad-binding element (SBE) of PAI-1 using its MH2 domain to inhibit TGF-β signaling (Zhang et al., 2007). Furthermore, TGF-β signaling can be regulated by the ubiquitination of signaling components. E3 ubiquitin ligases including Smad ubiquitination regulatory factor1 (Smurf1) and Smurf2 terminate TGF-β signaling by inducing the ubiquitination and degradation of TβRI and RSmads (Kwon et al., 2013; Shi and Massague, 2003)
The E3 ubiquitin ligases can be classified into three subfamilies, homologous to E6-AP carboxyl terminus (HECT) domain containing ligase, really interesting new gene (RING) domain containing ligase, and U-box domain containing ligase. The E3 ubiquitin ligase Itch [also known as AIP4 (atrophin-1 interacting protein 4)] belongs to the HECT type E3 family. Itch contains an N-terminal Ca2+ dependent phospholipid-binding C2 domain, a protein-protein interaction WW domain, and a C-terminal HECT domain (Schwarz et al., 1998). Itch was originally identified in non-agouti-lethal 18H or Itchy mice, which exhibit defective immune and inflammatory responses (Perry et al., 1998). Itch has been implicated in tumorigenesis and chemosensitivity (Wei et al., 2012), and its substrates include c-Jun and Jun B, which are important regulators of immune responses (Fang et al., 2002). It has also been established that Itch plays an important role in differentiation of regulatory T cells via the regulation of FoxP3 (Venuprasad et al., 2008), which is a transcription factor and master regulator of regulatory T cell differentiation and TGF-β-induced regulatory T cell development (Su and Liu, 2010). However, the molecular mechanism by which Itch regulates T cell development and TGF-β signaling has not been determined. Recent studies indicate that Itch positively regulates TGF-β signaling by modulating Smad2 phosphorylation in mouse embryonic fibroblasts (Bai et al., 2004). In contrast, Lallemand and colleagues reported that Itch negatively regulates TGF-β signaling despite mediating Smad7 ubiquitination (Lallemand et al., 2005). Therefore the physiological role of Itch in TGF-β signaling remains to be determined.
Here, we demonstrate that Itch regulates TGF-β-induced Smad7 ubiquitination and epithelial-mesenchymal transition (EMT). Knockdown of endogenous Itch by RNA interference significantly increased TGF-β-induced Smad7 expression. Furthermore, Itch regulated TGF-β-induced EMT gene expression. Thus, our results suggest that Itch is a positive regulator of the TGF-β-mediated Smad signaling pathway via Smad7 ubiquitination and protein degradation.
MATERIALS AND METHODS
Reagents and antibodies
Human recombinant TGF-β1 (Transforming growth factor) was purchased from R&D Systems (Germany). MG132 was purchased from Sigma (USA). Control siRNA was purchased from Bioneer (Korea), and siRNA against Itch was from Santa Cruz (USA). Mouse anti-HA, mouse anti-c-Myc, goat anti-Smad6/7, mouse anti-Ubiquitin, and rabbit anti-occludin were purchased from Santa Cruz. Rabbit anti-N-cadherin was from Cell Signaling Technology (USA). Goat anti-Snail was from Abcam, mouse anti-Tubulin from Sigma, and mouse anti-Itchα from BD Science.
Cell culture
Human lung epithelial A549 and Cos7 cells were obtained from the American Type Culture Collection (ATCC, USA). A549 cells were cultured in F12K medium (Invitrogen, USA) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 g/ml streptomycin. Cos7 cells were cultured in DMEM (Hyclone, USA) supplemented with 10 % FBS and antibiotics. Cells were maintained at 37°C in a humidified 5% CO2 in air atmosphere.
Plasmid constructs and transfection
pSBE-luc, pBIND-Smad3 (Gal4-fused Smad3), PAI-1 (type 1 plasminogen activator inhibitor) promoter p800-luc, pRL-tk (Renila luciferase), and pG5-luc reporter plasmid used have been previously described (Woo et al., 2008). pHA-Smad7, pGFP-Ubiquitin, pMyc-Itch, and pMyc-Itch-Mut were obtained from the Addgene plasmid repository (Addgene plasmid 11733; Hayashi H, Hospital for Sick Children, Canada; Dantuma NP, The Medical Nobel Institute, Sweden, 11427 and 11428; Magnifico, Center for Cancer Research, USA). pcDNA3.1/His C vector was from Invitrogen and used as a control. Cells were transfected with plasmids as indicated in figures using Lipofectamine (Invitrogen, USA).
Small interfering RNA (siRNA)
A549 cells were transiently transfected with 20 pM of control or Itch siRNA using Lipofectamine 2000 reagent (Invitrogen, USA), according to the manufacturer’s instruction. Cells transfected with Control siRNA or Itch siRNA were exposed to TGF-β1 (2 ng/ml) for 48 h after siRNA transfection, and expression levels of Itch protein were measured by immunoblotting.
Ubiquitination assay
Cells transfected with HA-tagged Smad7, GFP-tagged Ubiquitin and Myc-tagged Itch, or Myc-tagged Itch-mutant were treated with MG132 (10 μM) or DMSO as a control. Cells were collected in 1 ml RIPA buffer (50 mM Tris-HCl [pH7.4], 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 0.1% SDS (sodium dodecyl sulfate), 1 mM dithiothreitol containing 5 mM NEM (N-ethylmaleimide), 0.01 mM protease inhibitor cocktail (PIC), and 1 mM phenylmethanesulfonyl fluoride (PMSF), and centrifuged at 13,000 g for 10 min. Cell lysates were incubated with protein A/G agarose beads and anti-HA antibody at 4°C for 24 h. Beads were then collected, washed three times with RIPA buffer, and immunoprecipitates were separated by SDS-PAGE. Levels of ubiquitinated forms of Smad7 were determined by immunoblotting with anti-Ubiquitin antibody.
Luciferase assay
Cells were co-transfected with DNA plasmids including pRL-tk, pG5-luc, pBIND-Smad3, SBE-luc or p800-luc, Itch, Itch-Mut, or HA-Smad7 for 24 h as indicated in figures. Transfected cells were then treated with TGF-β1 (2 ng/ml) for 8 h and lysed in 5× passive lysis buffer. Luciferase activity was determined using a dual luciferase reporter assay system (Promega, USA) and a luminometer. All experiments were performed in triplicate and repeated at least three times.
Western blot analysis
Cells were lysed in lysis buffer containing 0.01mM PIC and 1 mM PMSF, incubated on ice for 15 min, and centrifuged (13,000× g, 4°C). Proteins were separated by SDS-PAGE, transferred to polyvinylidene difloride (PVDF) membranes and visualized by ECL (GE Health). Protein images were captured using a LAS 3000 image analyzer (Fujifilm).
RNA isolation and quantitative RT-PCR
Total RNAs were extracted using Trizol reagent (Invitrogen, USA), according to the manufacturer’s instructions. cDNA synthesis was performed using TaqMan reverse transcription reagent (Applied Biosystems, USA). Quantitative RT-PCR analyses were performed using the ABI PRISM 7500 system (Applied Biosystems, USA) and Power SYBR Green (Applied Biosystems). Quantification was performed using the efficiency-corrected ΔΔCq method. The primer sequences used were as follows: human GAPDH – forward 5′-GGAGCCAAAAGGGTCATCAT-3′ and reverse 5′-GTGATGGCATGGACTGTGGT-3′; human N-cadherin – forward 5′-TCAGTGGCGGAGATCCTACT-3′ and reverse 5′-TGCTTTGACCACGGTGACTA-3′; human Slug – forward 5′-GAGCATTTGCAGACAGGTCA-3′ and reverse 5′-TTGGAGCAGTTTTTGCACTG-3′; and human Snail – forward 5′-TGTCTGCGTGGGTTTTTGTA-3′ and reverse 5′-GGAGCTTCCCAGTGACTCTG-3′.
Statistical analysis
Results in bar graphs are presented as means ± SDs and are representative of three independent experiments. Statistical analysis was performed using the Student’s t-test, and p values of less than 0.05 were considered statistically significant.
RESULTS AND DISCUSSION
Regulation of TGF-β-induced Smad3 transcriptional activity by Itch
To determine the role of Itch in TGF-β singling, wild-type Itch (Itch-WT) or ubiquitin ligase activity-deficient mutant Itch (Itch-Mut) was overexpressed, and the effects of Itch-WT and Itch-Mut on TGF-β-induced Smad binding element (SBE) promoter activity were investigated. As shown in Fig. 1A, Itch-WT over-expression significantly enhanced TGF-β-induced SBE promoter activity, whereas Itch-Mut overexpression inhibited this activity. Additionally, TGF-β-induced Smad3 transcriptional activity, which was assessed using a GAL4-fused Smad3 construct was significantly enhanced by co-transfection with wild-type Itch, but not by co-transfection with mutant Itch, suggesting that Itch has a positive effect on the transcriptional activity of Smad3 (Fig. 1B). In-line with these results, knockdown of Itch with specific siRNA against Itch greatly inhibited the TGF-β-induced promoter activity of type 1 plasminogen activator inhibitor (PAI-1), one of the most well-known TGF-β- and Smad response genes, and SBE promoter activity (Figs. 1C and 1D). To address the involvement of serum containing factors in effect of Itch on Smad3 transcriptional activity, we performed the same experiments with SBE-luc and p800-luc which encompasses −800 to +71 bp of the human PAI-1 promoter under serum free conditions, and the same tendency was observed under these conditions (data not shown). These observations indicate that Itch positively regulates TGF-β-mediated Smad3 transcriptional activity in a serum-independent manner.
Fig. 1.
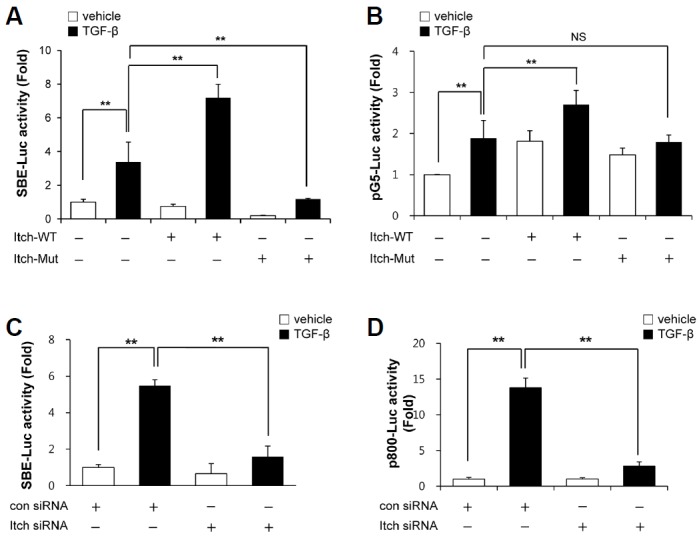
Regulation of TGF-β1-induced Smad3 transcriptional activity by Itch. (A) A549 cells were co-transfected with pSBE-luc, pRL-tk, plus pMyc-Itch or pMyc-Itch-mut. Transfected cells were then treated with TGF-β1 (2 ng/ml) for 9 h, and relative luciferase activities were determined using a dual luciferase reporter assay system and a luminometer. (B) A549 cells were co-transfected with pG5-luc, pBIND-Smad3 plus pRL-tk with or without Itch, and then were stimulated with TGF-β1 (2 ng/ml) for 9 h. Relative luciferase activities were determined using a dual luciferase reporter assay system. (C) A549 cells were cotransfected with pSBE-luc, pRL-tk, plus scrambled RNA or siRNA against Itch (20 pM). Control scrambled or Itch siRNA transfected cells were then treated with TGF-β1 (2 ng/ml) for 9 h and lysed to determine luciferase activity. (D) A549 cells were co-transfected with a luciferase construct containing PAI-1 promoter region (p800-luc), pRL-tk, plus scrambled RNA or siRNA against Itch (20 pM). Control scrambled or Itch siRNA transfected cells were then treated with TGF-β1 (2 ng/ml) for 9 h and lysed to measure luciferase activity. Results are expressed as means ± SDs and are representative of three independent experiments. *p < 0.05 (n = 3). NS indicates non-significant.
Regulation of Smad7 ubiquitination by Itch
Because Itch was found to act as a positive regulator of the TGF-β-induced Smad signaling pathway and the E3 ubiquitin ligase activity of Itch was required for this positive regulatory function, we hypothesized that Itch may target endogenous negative regulators in the TGF-β signaling pathway. It has been previously reported that Smad7 (an important endogenous negative regulator of the TGF-β signaling pathway) is polyubiquitinated and its ubiquitination-dependent degradation is an important regulator of Smad7 activity. Thus, we first investigated whether Itch targets Smad7 to positively regulate TGF-β signaling. As shown in Fig. 2A, TGF-β-induced SBE promoter activity was significantly inhibited by overexpressing Smad7, and the overexpression of wild-type Itch ameliorated the inhibitory effect of Smad7, whereas the overexpression of E3 ligase activity-deficient mutant Itch (Itch-mut) did not. Consistent with these results, TGF-β-induced PAI-1 promoter activity was also significantly inhibited by overexpressing Smad7, and this inhibitory effect of Smad7 was ameliorated by wild-type Itch but not by Itch-mut (Fig. 2B). Taken together, these results suggest Itch targets the negative regulator Smad7 to positively regulate TGF-β signaling.
Fig. 2.
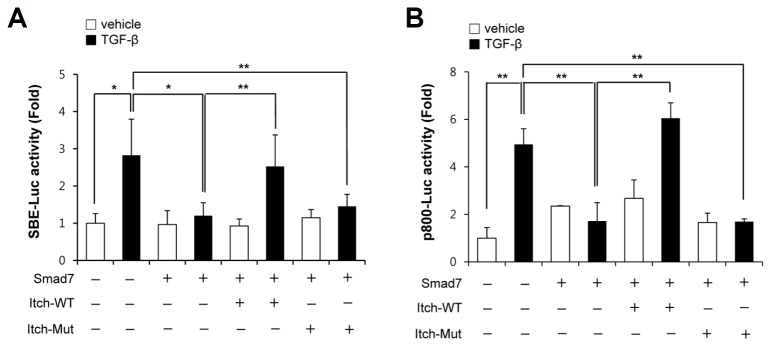
Itch regulated TGF-β1-induced Smad3 transcriptional activity by inhibiting Smad7. (A) A549 cells cotransfected with pSBE-luc, pRL-tk, pHA-Smad7 plus pMyc-Itch or pMyc-Itch-mut were not treated (□) or treated (▪) with TGF-β1 (2 ng/ml) for 9 h. Relative luciferase activities were determined by dual luciferase reporter assay using a luminometer. (B) A549 cells were co-transfected with p800-luc, pRL-tk, pMyc-Itch, pMyc-Itch-mut, plus pHA-Smad7 as indicated in the figure, and then treated TGF-β1 (2 ng/ml) for 9 h and lysed to determine luciferase activity. Results are expressed as means ± SDs and are representative of three independent experiments. *p < 0.05, **p < 0.01.
Itch induced the ubiquitination and subsequent degradation of Smad7
Since Smad7 was found to be a regulatory target of Itch, we investigated the mechanism involved. Because our results suggested that the Itch-mediated regulations of TGF-β signaling and Smad7 activity were dependent on the E3 ubiquitin ligase activity of Itch (Figs. 1 and 2), we tested whether Itch acts as an ubiquitin E3 ligase for Smad7 polyubiquitination. As shown in Fig. 3A, Itch overexpression enhanced Smad7 polyubiquitination, whereas the overexpression of E3 ubiquitin ligase activity-deficient mutant Itch (Itch-mut) inhibited Smad7 polyubiquitination. The observation that Itch overexpression did not affect the amount of poly-ubiquitination induced by proteasome inhibition with MG132 in whole cell lysates, suggested that Itch specifically regulated the ubiquitination-dependent protein degradation of Smad7 but not total amount of polyubiquitination.
Fig. 3.
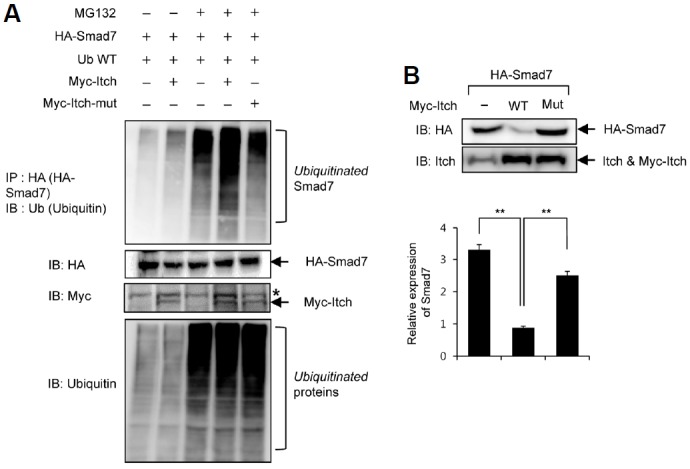
Itch induced the ubiquitination and subsequent degradation of Smad7. (A) Cos7 cells were co-transfected with pHA-Smad7, pUbiquitin, pMyc-Itch or pMyc-Itch-mut as indicated in the figure and then treated with 10 μM MG132 for 8 h. Protein lysates were subjected to immunoprecipitation with anti-HA antibody and then immunoblotted with anti-ubiquitin, anti-HA, and anti-Myc antibodies, respectively. Amounts of polyubiquitinated protein in cell lysates were determined by immunoblotting with anti-ubiquitin antibody. *indicates a non-specific band. (B) A549 cells were transfected with pHA-Smad7 plus pMyc-Itch or pMyc-Itch-mut as indicated in the figure. Expression levels of Smad and Itch were determined by Western blotting with specific antibodies as indicated.
In addition to the ubiquitination of Smad7, the overexpression of wild-type Itch inhibited the protein expression of Smad7, whereas the overexpression of Itch-mut did not (Fig. 3B). Notably, the inhibitory effect of Itch on Smad7 was specific for an inhibitory Smad7, because the protein stability of Smad2 (a receptor Smad) was not affected by Itch or Itch-mut (data not shown). Taken together, these findings suggest that Itch induces the polyubiquitination of Smad7 and its subsequent degradation, and thereby acts as a positive regulator of TGF-β signaling.
Previous studies have indicated a deficiency of Itch in mouse embryonic fibroblasts reduced sensitivity to TGF-β-induced cell growth arrest and decreased Smad2 phosphorylation. The authors suggested that Itch mediated TGF-β-induced Smad2 ubiquitination and enhanced the interaction between Smad2 and TβRI in a Smad7-independent manner (Bai et al., 2004). However, under our experimental conditions using A549 cells, no significant difference in TGF-β-induced Smad2 phosphorylation levels was observed in the presence or absence of Itch siRNA. Furthermore, Lallemand et al. (2005). also reported that Itch did not affect the Smad2 phosphorylation induced by TGF-β in 293 cells, and found that depletion of Itch with Itch siRNA slightly increased TGF-β-induced Smad2 phosphorylation without affecting total Smad2 protein expression. In our view, this disparity was caused by the use of different cell types and the knockdown techniques used. Nonetheless, the present study provides evidence that Itch acts as a negative regulator of Smad7 by inducing its ubiquitination and protein degradation.
Itch regulated TGF-β-induced EMT via Smad7 inhibition
Since we found that the overexpression of Itch induced the ubiquitination and degradation of Smad7, we sought to confirm the endogenous role of Itch in TGF-β-induced Smad7 ubiquitination using siRNA Itch. An immunoprecipitation assay showed TGF-β stimulated Smad7 ubiquitination and that this induction was markedly reduced by Itch depletion (Fig. 4A), thus suggesting that Itch plays a critical role in Smad7 ubiquitination under TGF-β stimulation. Next, to address the physiological role of the Itch-mediated inhibition of Smad7 in TGF-β signaling, we examined the effect of Itch depletion on TGF-β-induced EMT-related gene expression. A549 cells transfected with control RNA or Itch siRNA were exposed to TGF-β for 48 h. As shown in Fig. 4B, we achieved ∼80% knockdown of Itch protein expression using Itch siRNA. Furthermore, TGF-β-induced Smad7 protein expression was significantly enhanced by Itch knockdown (Fig. 4B). In addition, the role of Itch on TGF-β-induced EMT was determined by examining the effects of Itch knockdown on the mRNA and protein expressions of EMT-related genes. As shown in Fig. 4C, Itch knockdown significantly inhibited the inductions of the protein expressions of the mesenchymal markers N-cadherin and Snail by TGF-β. On the other hand, the expression of the epithelial marker occludin was markedly reduced by TGF-β and this downregulation was impaired by Itch depletion. Consistent with these results, the TGF-β-induced mRNA expressions of EMT-regulated genes were significantly inhibited by Itch knockdown (Figs. 4D–4F). A similar effect on EMT-regulated genes after Itch knockdown was observed in Panc1 cells (a human pancreatic carcinoma cell line; data not shown). Taken together, these findings indicate that Itch is an E3 ubiquitin ligase of Smad7, and acts as an important positive regulator of TGF-β-induced EMT.
Fig. 4.
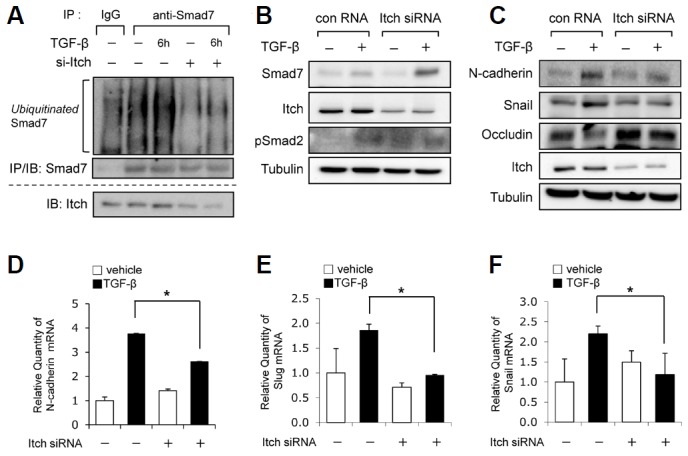
Itch regulated TGF-β signaling for EMT by inhibiting Smad7. (A) A549 cells were transfected with control siRNA or Itch siRNA, as indicated in the figure, and then pretreated with 10 μM MG132 for 1 h. Transfected cells were exposed to TGF-β1 (2 ng/ml) for 6 h and cell lysates were subjected to an immunoprecipitation assay using anti-Smad7 antibodies and anti-IgG, respectively. Ubiquitinated Smad7 levels were determined by immunoblotting with anti-Ubiquitin antibody. (B) A549 cells were transfected with scrambled RNA or Itch siRNA and then treated with TGF-β1 (2 ng/ml) for 48 h. The expression levels of Smad7 and Itch were determined by Western blotting with anti-Smad7 and anti-Itch antibodies, respectively. (C) A549 cells were transfected with scrambled RNA or siRNA against Itch, and then treated with TGF-β1 (2 ng/ml) for 48 h. The expression levels of EMT-related genes, that is, N-cadherin, Snail, and Occludin were determined using specific antibodies as indicated. The protein expressions of Itch and tubulin were determined by immunoblotting with specific antibodies. The transcriptional levels of N-cadherin (D), Slug (E), and Snail (F) were determined by quantitative RT-PCR. A549 cells were transfected with scrambled RNA or Itch siRNA, and then treated with (▪) or without (□) TGF-β1 (2 ng/ml) for 48 h. The relative levels of N-cadherin, Slug, and Snail mRNAs in TGF-β1-treated cells versus those in non-treated cells are shown. Values were normalized versus GAPDH mRNA. Results are representative of three independent experiments and are expressed as means ± SDs. *p < 0.05 (n = 3).
Itch, also called Atrophin-1 interacting protein 4 (AIP4), was originally identified as a protein that interacted with atrophin-1, the protein implicated in denatrorural pallidoluysian atrophy, a neurogenic disease (Wood et al., 1998). Itch plays a critical role in modulating immune response and tumorigenesis (Lohr et al., 2010), and reportedly, negatively regulates Wnt signaling and the hippo pathway (Salah et al., 2011; Wei et al., 2012), and positively regulates the stability of several substrates, including p73 and p63 (Ho et al., 2011; Rossi et al., 2006). The present study shows that Smad7 ubiquitination by Itch regulates TGF-β-mediated EMT, which suggests Itch positively regulates the TGF-β-mediated Smad signaling pathway and EMT.
Acknowledgments
This study was supported by a grant from the Korean Health Technology R&D Project, Korean Ministry of Health & Welfare (#A111520) and the Korean National Research Foundation (#NRF-2013R1A1A1011264).
REFERENCES
- Bai Y., Yang C., Hu K., Elly C., Liu Y.C. Itch E3 ligase-mediated regulation of TGF-beta signaling by modulating smad2 phosphorylation. Mol. Cell. 2004;15:825–831. doi: 10.1016/j.molcel.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Derynck R., Akhurst R.J. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nat. Cell Biol. 2007;9:1000–1004. doi: 10.1038/ncb434. [DOI] [PubMed] [Google Scholar]
- Fang D., Elly C., Gao B., Fang N., Altman Y., Joazeiro C., Hunter T., Copeland N., Jenkins N., Liu Y.C. Dysregulation of T lymphocyte function in itchy mice: a role for Itch in TH2 differentiation. Nat. Immunol. 2002;3:281–287. doi: 10.1038/ni763. [DOI] [PubMed] [Google Scholar]
- Ho K.C., Zhou Z., She Y.M., Chun A., Cyr T.D., Yang X. Itch E3 ubiquitin ligase regulates large tumor suppressor 1 stability [corrected] Proc. Natl. Acad. Sci. USA. 2011;108:4870–4875. doi: 10.1073/pnas.1101273108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavsak P., Rasmussen R.K., Causing C.G., Bonni S., Zhu H., Thomsen G.H., Wrana J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol. Cell. 2000;6:1365–1375. doi: 10.1016/s1097-2765(00)00134-9. [DOI] [PubMed] [Google Scholar]
- Kwon A., Lee H.L., Woo K.M., Ryoo H.M., Baek J.H. SMURF1 plays a role in EGF-induced breast cancer cell migration and invasion. Mol. Cells. 2013;36:548–555. doi: 10.1007/s10059-013-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemand F., Seo S.R., Ferrand N., Pessah M., L’Hoste S., Rawadi G., Roman-Roman S., Camonis J., Atfi A. AIP4 restricts transforming growth factor-beta signaling through a ubiquitination-independent mechanism. J. Biol. Chem. 2005;280:27645–27653. doi: 10.1074/jbc.M500188200. [DOI] [PubMed] [Google Scholar]
- Lohr N.J., Molleston J.P., Strauss K.A., Torres-Martinez W., Sherman E.A., Squires R.H., Rider N.L., Chikwava K.R., Cummings O.W., Morton D.H., et al. Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease. Am. J. Hum. Genet. 2010;86:447–453. doi: 10.1016/j.ajhg.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry W.L., Hustad C.M., Swing D.A., O’Sullivan T.N., Jenkins N.A., Copeland N.G. The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a18. H mice. Nat. Genet. 1998;18:143–146. doi: 10.1038/ng0298-143. [DOI] [PubMed] [Google Scholar]
- Rossi M., Aqeilan R.I., Neale M., Candi E., Salomoni P., Knight R.A., Croce C.M., Melino G. The E3 ubiquitin ligase Itch controls the protein stability of p63. Proc. Natl. Acad. Sci. USA. 2006;103:12753–12758. doi: 10.1073/pnas.0603449103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salah Z., Melino G., Aqeilan R.I. Negative regulation of the Hippo pathway by E3 ubiquitin ligase ITCH is sufficient to promote tumorigenicity. Cancer Res. 2011;71:2010–2020. doi: 10.1158/0008-5472.CAN-10-3516. [DOI] [PubMed] [Google Scholar]
- Schwarz S.E., Rosa J.L., Scheffner M. Characterization of human hect domain family members and their interaction with UbcH5 and UbcH7. J. Biol. Chem. 1998;273:12148–12154. doi: 10.1074/jbc.273.20.12148. [DOI] [PubMed] [Google Scholar]
- Shi Y., Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Su J., Liu Y.C. Foxp3 positive regulatory T cells: a functional regulation by the E3 ubiquitin ligase Itch. Semin. Immunopathol. 2010;32:149–156. doi: 10.1007/s00281-009-0192-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venuprasad K., Huang H., Harada Y., Elly C., Subramaniam M., Spelsberg T., Su J., Liu Y.C. The E3 ubiquitin ligase Itch regulates expression of transcription factor Foxp3 and airway inflammation by enhancing the function of transcription factor TIEG1. Nat. Immunol. 2008;9:245–253. doi: 10.1038/niXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W., Li M., Wang J., Nie F., Li L. The E3 ubiquitin ligase ITCH negatively regulates canonical Wnt signaling by targeting dishevelled protein. Mol. Cell. Biol. 2012;32:3903–3912. doi: 10.1128/MCB.00251-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo C.H., Shishido T., McClain C., Lim J.H., Li J.D., Yang J., Yan C., Abe J. Extracellular signal-regulated kinase 5 SUMOylation antagonizes shear stress-induced antiinflammatory response and endothelial nitric oxide synthase expression in endothelial cells. Circ. Res. 2008;102:538–545. doi: 10.1161/CIRCRESAHA.107.156877. [DOI] [PubMed] [Google Scholar]
- Wood J.D., Yuan J., Margolis R.L., Colomer V., Duan K., Kushi J., Kaminsky Z., Kleiderlein J.J., Sharp A.H., Ross C.A. Atrophin-1, the DRPLA gene product, interacts with two families of WW domain-containing proteins. Mol. Cell. Neurosci. 1998;11:149–160. doi: 10.1006/mcne.1998.0677. [DOI] [PubMed] [Google Scholar]
- Zhang S., Fei T., Zhang L., Zhang R., Chen F., Ning Y., Han Y., Feng X.H., Meng A., Chen Y.G. Smad7 antagonizes transforming growth factor beta signaling in the nucleus by interfering with functional Smad-DNA complex formation. Mol. Cell. Biol. 2007;27:4488–4499. doi: 10.1128/MCB.01636-06. [DOI] [PMC free article] [PubMed] [Google Scholar]