

Abstract
Background and Purpose
In a recently conducted phase III clinical trial, RELAX-AHF, serelaxin infusion over 48 h improved short- and long-term clinical outcomes in patients with acute heart failure. In this study we used human primary cells from the umbilical vasculature to better understand the signalling mechanisms activated by serelaxin.
Experimental Approach
We examined the acute effects of serelaxin on signal transduction mechanisms in primary human umbilical vascular cells and its chronic actions on markers of cardiovascular function and disease.
Key Results
The RXFP1 receptor, the cognate serelaxin receptor, was expressed at the cell surface in HUVECs and human umbilical vein smooth muscle cells (HUVSMCs), human umbilical artery smooth muscle cells (HUASMCs) and human cardiac fibroblasts (HCFs), but not human umbilical artery endothelial cells. In HUVECs and HUVSMCs, serelaxin increased cAMP, cGMP accumulation and pERK1/2, and the concentration–response curves (CRCs) were bell-shaped. Similar bell-shaped CRCs for cGMP and pERK1/2 were observed in HCFs, whereas in HUASMCs, serelaxin increased cAMP, cGMP and pERK1/2 with sigmoidal CRCs. Gαi/o and lipid raft disruption, but not Gαs inhibition, altered the serelaxin CRC for cAMP and cGMP accumulation in HUVSMC but not HUASMC. Longer term serelaxin exposure increased the expression of neuronal NOS, VEGF, ETβ receptors and MMPs (gelatinases) in RXFP1 receptor-expressing cells.
Conclusions and Implications
Serelaxin caused acute and chronic changes in human umbilical vascular cells that were cell background dependent. Bell-shaped CRCs that were observed only in venous cells and fibroblasts involved Gαi/o located within membrane lipid rafts.
Tables of Links
| TARGETS | |
|---|---|
| GPCRsa | Enzymesc |
| β2-adrenoceptor | eNOS |
| ETB receptor | ERK1/2 |
| RXFP1 receptor | Guanylyl cyclase (GC) |
| RXFP2 receptor | iNOS |
| Ligand gated ion channelsb | MMP2 |
| P2X1 receptor | MMP9 |
| nNOS | |
| PI3K |
| LIGANDS | |
|---|---|
| cAMP | Isoprenaline |
| cGMP | NF023 |
| EGF | NF449 |
| FGF-2 | Nitric oxide (NO) |
| Forskolin | Relaxin |
| Hydrocortisone | Suramin |
| IGF-1 | VEGFA |
| Insulin | Wortmannin |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2009) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a,b,c).
Introduction
Acute heart failure (AHF) is a major global health challenge with high morbidity and mortality that represents a great burden on health care (Mosterd and Hoes, 2007). Along with predictions of increasing prevalence, treatment options for AHF have changed little over the last two decades and consequently patients continue to experience high morbidity and mortality. However, in the recent phase III clinical trial (RELAX-AHF), serelaxin (the recombinant form of human relaxin-2) produced a moderate improvement in one of the primary end points, dyspnoea, but also significantly reduced patient mortality at day 180 without any notable side effects (Teerlink et al., 2013). Further analysis of the RELAX-AHF findings showed fewer signs of cardiac, renal and liver damage with early administration of serelaxin, which may contribute to the long-term survival benefit (Metra et al., 2013).
Relaxin is a hormone that mediates cardiovascular adaptations observed during pregnancy and in particular systemic and renal vasodilatation (Conrad, 2010), although these effects are also observed in non-pregnant animals, ex vivo studies and in animal models of cardiovascular disease (Masini et al., 1997; 2006,; Bani et al., 1998b). There are two distinct actions of serelaxin that have been described in in vitro and in vivo studies. Rapid serelaxin-mediated responses, observed after stimulation of serelaxin for minutes to hours (<1 h), occur via a Gαi/PI3K/cAMP/Akt/eNOS-dependent mechanism in human subcutaneous and rodent renal and mesenteric arteries and also in human coronary artery and aortic endothelial cells (McGuane et al., 2011b). Sustained serelaxin responses, observed after stimulation of serelaxin for days (24–48 h), are seen in rodent small renal and human subcutaneous arteries involving MMPs (gelatinases), endothelin receptor B (ETB receptor), VEGF and NOS (Jeyabalan et al., 2003; McGuane et al., 2011a).
Although in vitro and in vivo studies support the potential benefits of serelaxin in humans in cardiovascular disease, there are knowledge gaps in our understanding of the mechanism of action. There is little information on the cells targeted by serelaxin and on signal transduction mechanisms in tissues relevant to the human cardiovascular system that endogenously express the RXFP1 receptor, the cognate serelaxin receptor. However, it is clear that serelaxin affects the tone of blood vessels. In rats, it was recently reported that the RXFP1 receptor is localized to endothelial and smooth muscle cells, although there are marked regional variations in distribution (Jelinic et al., 2014). However, very little is known on the expression of the RXFP1 receptor and the signalling pathways it activates in human arteries and veins. In rats, the effects of serelaxin on arteries have been described in detail (Jeyabalan et al., 2003; Conrad et al., 2004; Conrad and Shroff, 2011) but much less is known of effects in veins. In addition, rat mesenteric arteries and veins both express RXFP1 receptors, yet only the arteries show serelaxin-mediated vascular remodelling (Jelinic et al., 2014).
The effects of serelaxin on arteries and veins could be critical for the understanding of the clinical actions of serelaxin because nitrates, a classical therapy for heart failure, reduce congestion by causing venodilatation. Serelaxin, a known arteriodilator, also reduced congestion without causing hypotension in RELAX-AHF (Teerlink et al., 2013), suggesting that serelaxin could potentially have additional venodilator properties. Therefore, we examined whether serelaxin targets cells in both the arterial and venous vasculature to activate vasodilator signal transduction mechanisms.
In order to address these knowledge gaps, we examined signal transduction mechanisms in primary cells from the human arterial and venous umbilical vasculature and heart, including endothelial cells, smooth muscle cells and cardiac fibroblasts. These were examined for RXFP1 receptor expression and markers of cardiovascular function and disease: short-term effects of serelaxin (<1 h) on cAMP, cGMP and pERK1/2; and the longer term effects of serelaxin (1–48 h) on the expression of VEGF, ETB receptors, NOS isoforms and MMP activity.
Methods
Cell culture
Primary cultures of human umbilical artery endothelial cells (HUAECs), HUVECs, human umbilical artery smooth muscle cells (HUASMCs), human umbilical vein smooth muscle cells (HUVSMCs) and fetal human cardiac fibroblasts (HCFs: pooled from fetal atria and ventricles) were obtained from ScienCell Research Laboratories (San Diego, CA, USA). Endothelial cells were characterized by their expression of von Willebrand factor (Factor VIII), CD31 and by uptake of acetylated low-density lipoprotein (DiI-Ac-LDL). Smooth muscle cells were characterized by the presence of α-smooth muscle actin and desmin and fetal HCF were characterized by the presence of fibronectin (ScienCell). Cells were maintained in Medium 199 containing 5% FBS, penicillin (100 u·mL−1), streptomycin (100 μg·mL−1) and the relevant growth supplements for optimal growth of each cell type (ScienCell,). Endothelial cells were grown in endothelial cell growth supplement [BSA 10 μg·mL−1, apo-transferrin 10 μg·mL−1, insulin 5 μg·mL−1, EGF 10 ng·mL−1, fibroblast growth factor-2 (FGF-2) 2 ng·mL−1, VEGF 2 ng·mL−1, insulin-like growth factor-1 (IGF-1) 2 ng·mL−1, hydrocortisone 1 μg·mL−1, retinoic acid 10−7 M], smooth muscle cells in smooth muscle cell growth supplement (BSA 10 μg·mL−1, apo-transferrin 10 μg·mL−1, insulin 5 μg·mL−1, FGF-2 2 ng·mL−1, IGF-1 2 ng·mL−1, hydrocortisone 1 μg·mL−1) and fibroblasts in fibroblast cell growth supplement-2 (BSA 10 μg·mL−1, apo-transferrin 10 μg·mL−1, insulin 7.5 μg·mL−1, EGF 2 ng·mL−1, FGF-2 2 ng·mL−1, hydrocortisone 1 μg·mL−1) (information provided by ScienCell). Early passage cultures (2–5) were used for all experiments.
Reverse-transcription real-time PCR (RT-qPCR)
RNA was extracted using the TRIzol® reagent (Invitrogen, Mulgrave, Victoria, Australia) and cDNA synthesized using the iScript cDNA synthesis kit (BioRad, Gladesville, NSW, Australia) according to the manufacturer's instructions. RT-PCR was performed using the Taqman Assay for RXFP1 receptors (Invitrogen) according to the manufacturer's instructions.
Radioligand binding
Serelaxin was iodinated with Na125I (Perkin-Elmer, Glen Waverley, Victoria, Australia) using the chloramine-T method as described previously (van der Westhuizen et al., 2010). Whole-cell competition-binding assays were performed as described previously (Halls et al., 2005). Briefly, cells (0.8–1 × 106 cells) with [125I]-serelaxin (200 pM) were allowed to compete with unlabelled serelaxin (10 pM–0.1 μM) for 90 min at room temperature. Total binding was determined by radioligand alone whereas non-specific binding was determined by 0.1 μM unlabelled serelaxin.
cAMP and cGMP accumulation assays
cAMP accumulation was determined as previously described (Halls et al., 2006). Briefly, cells were plated into 24-well plates (1 × 105 cells per well) and grown overnight to achieve a confluent monolayer. Prior to stimulation, cells were serum starved in M199 medium for 4 h. Where appropriate, the cells were pre-incubated with the PI3K inhibitor wortmannin (100 nM, 30 min), the Gαi/o inhibitor PTX (50 ng·mL−1, 18 h), the Gαs inhibitor NF449 (10 μM, 30 min), the Gαi/o inhibitor NF023 (10 μM, 30 min), suramin (10 μM, 30 min) or filipin III (1 μg·mL−1, 1 h). Levels of cAMP and cGMP were detected according to the manufacturer's instructions (Perkin-Elmer).
pERK1/2 assay
pERK1/2 was measured using the Surefire ERK kit (TGR BioSciences, Hindmarsh, South Australia, Australia) as described previously (van der Westhuizen et al., 2007). Briefly, cells were plated into 24-well plates (1 × 105 cells per well) and grown overnight to achieve a confluent monolayer. Prior to stimulation, cells were serum starved in M199 medium for 4 h. Where appropriate, the cells were pre-incubated with wortmannin (100 nM, 30 min) or PTX (50 ng·mL−1, 18 h). Levels of pERK1/2 were detected according to the manufacturer's instructions (Perkin-Elmer).
Gelatin zymography
Gelatin zymography was performed as described previously to determine changes in levels of MMP2 (gelatinase A) and MMP9 (gelatinase B) in cultured medium samples (Chow et al., 2012).
Western blotting
Western blotting was performed as described previously (Chow et al., 2012). Briefly, 20–30 μg of protein was separated on a polyacrylamide gel, transferred to a PVDF membrane and probed with the following primary antibodies: anti-eNOS antibody (R&D Systems; 1:1000, overnight at 4°C), anti-nNOS antibody (R&D Systems; 1:500, overnight at 4°C), anti-iNOS antibody (Cell Signalling, Beverley, MA, USA; 1:1000, overnight at 4°C), anti-VEGF antibody (Abcam, Cambridge, MA, USA; 1:1000, 2 h at room temperature), anti-ETB receptor antibody (Pierce, Rockford, IL, USA; 1:1000, overnight at 4°C), anti-β-actin antibody (Cell Signalling; 1:1000, 2 h at room temperature). Membranes were then probed with secondary antibodies (Alexa Fluor 647 anti-rabbit IgG antibody, Alexa Fluor 647 anti-mouse IgG antibody; 2 μg·mL−1; Invitrogen) and scanned using the Typhoon Trio (GE Healthcare, Melbourne, Victoria, Australia), and densitometry was conducted on each band using the ImageJ software (NIH, Bethesda, MD, USA).
Statistical analysis
Data were analysed using GraphPad Prism v6.0 (La Jolla, CA, USA). All data represent the mean ± SEM of ‘n’ independent experiments. Signalling assays were conducted in duplicate with each point being taken as the mean of the replicates. Concentration–response curves (CRCs) were fitted using either a sigmoidal or Gaussian model. Binding data were fitted using a one-site competition-binding model to obtain pIC50 values that were used to calculate pKi. Densitometry on bands from gel zymography and Western blotting was conducted using the ImageJ software. Statistical differences among treatment groups were determined by a one-way anova with a Dunnett's post hoc test for each cell type studied and statistical significance accepted at P < 0.05.
Materials
Serelaxin was kindly provided by Dr D.R. Stewart (Novartis, Basel, Switzerland). Pertussis toxin (PTX), wortmannin, filipin III and suramin were purchased from Sigma (Castle Hill, NSW, Australia). NF023 and NF449 were purchased from Calbiochem (Alexandria, NSW, Australia). TGF-β1 was purchased from R&D Systems (Gymea, NSW, Australia).
Results
Cell surface RXFP1 receptors expression occurs in HUVECs, HUVSMCs, HUASMCs and HCFs, but not HUAECs
RXFP1 receptor mRNA, measured by qPCR, was present in HUAECs, HUVECs, HUVSMCs, HUASMCs, HCFs and human testis (positive control; Figure 1A). Cell surface RXFP1 receptor expression, measured by competition binding of [125I]-serelaxin with unlabelled serelaxin, was detected in HUASMCs, HUVECs, HUVSMCs and HCFs, but not in HUAECs (Figure 1B). Binding affinity correlated well with that observed in HEK cells recombinantly expressing RXFP1 receptors (Supporting Information Table S1). The lack of cell surface expression of RXFP1 receptors in HUAECs was supported by the failure of serelaxin to cause cAMP accumulation (Supporting Information Fig. S1a), cGMP accumulation (Supporting Information Fig. S1b) or pERK1/2 (Supporting Information Fig. S1c) in these cells.
Figure 1.
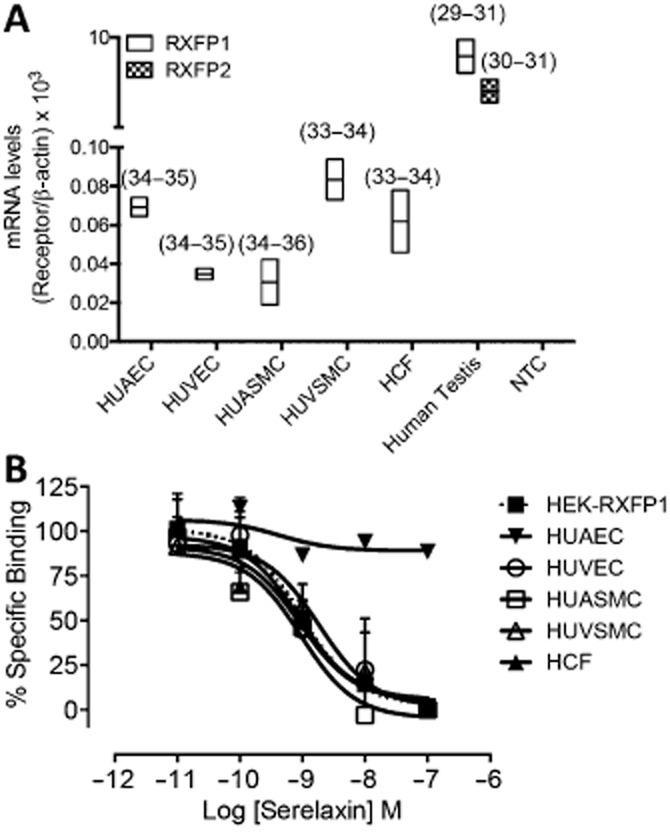
The expression of RXFP1 and RXFP2 receptor mRNA and RXFP1 receptor protein in human primary umbilical vascular cells and human primary cardiac fibroblasts. qPCR (A) was utilized to show expression levels of RXFP1 and RXFP2 receptor mRNA in HUAECs, HUVECs, HUASMCs, HUVSMCs and HCFs relative to β-actin (n = 2). RXFP2 receptor mRNA was only measureable in the positive control. Cell surface RXFP1 receptor protein expression was determined by radioligand binding (B) utilizing [125I]-serelaxin and showed specific serelaxin binding in HEK-RXFP1 cells (n = 6), HUASMCs (n = 4), HUVECs (n = 4), HUVSMCs (n = 3) and HCFs (n = 3), but not in HUAECs (n = 2).
cAMP accumulation in response to acute serelaxin administration
Serelaxin increases cAMP in vitro (Halls et al., 2009b) and stimulates inotropy in human atrial myocardium (Dschietzig et al., 2011). In this study, serelaxin (30 nM) increased cAMP accumulation in HUVECs, HUVSMCs and HUASMCs with the response peaking by 30 min (Supporting Information Fig. S2a). In HUVECs (Figure 2A) and HUVSMCs (Figure 2B), serelaxin (30 min) concentration- dependently increased cAMP accumulation to 5% of the forskolin response respectively. CRCs for cAMP accumulation in both cell types were bell-shaped with pEC50s of 9.1 ± 0.4 and 9.6 ± 0.4, respectively, for the initial part of the curve. As previous studies have shown that serelaxin-mediated cAMP accumulation in HEK-RXFP1 receptor cells involves Gαi proteins and PI3K (Halls et al., 2009b), we investigated the effect of PTX (Gαi/o inhibitor) and wortmannin (PI3K inhibitor) on the cAMP response. Both PTX (50 ng·mL−1, 18 h) and wortmannin (100 nM, 30 min) significantly inhibited serelaxin-mediated (30 nM) cAMP accumulation in HUVECs (Figure 2A) and HUVSMCs (Figure 2B). In HUASMCs, however, serelaxin (30 min) increased cAMP accumulation but the response was unaffected by PTX (50 ng·mL−1, 18 h) or wortmannin (100 nM, 30 min) pretreatment, suggesting that the response predominantly involved Gαs (Figure 2C). Most interestingly, and in contrast to HUVECs and HUVSMCs, the CRC to serelaxin in HUASMCs was sigmoidal with a pEC50 of 9.0 ± 0.3. In HCFs, serelaxin failed to cause cAMP accumulation (Figure 2D), reflecting previous findings in rat atrial and ventricular fibroblasts (Samuel et al., 2004; Mosterd and Hoes, 2007).
Figure 2.
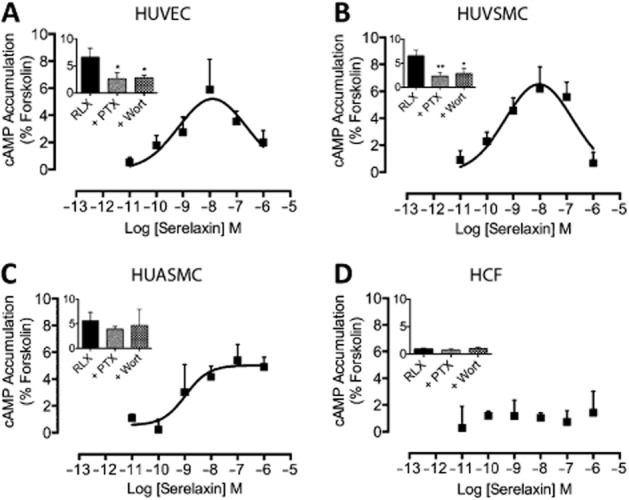
The effect of serelaxin on cAMP accumulation in human primary umbilical vascular cells and cardiac fibroblasts. Serelaxin treatment (30 min) increased cAMP accumulation in (A) HUVECs (n = 6), (B) HUVSMCs (n = 6) and (C) HUASMCs (n = 4), but not in (D) HCFs (n = 3). The serelaxin CRC was bell-shaped for HUVECs and HUVSMCs but sigmoidal for HUASMCs. For each cell type, the effect of PTX (50 ng·mL−1, 18 h) and wortmannin (100 nM, 30 min) pretreatment was determined after exposure to serelaxin (30 nM) for 30 min to determine the role of Gαi and PI3K. Statistical significance was assessed using a one-way anova with a Dunnett's post hoc test compared with serelaxin alone: *P < 0.05 and **P < 0.01.
cGMP accumulation in response to acute serelaxin stimulation
Serelaxin-mediated vasodilatation, in vitro (Bani et al., 1998a) and ex vivo (Jeyabalan et al., 2003; McGuane et al., 2011a,b,), occurs via a NOS/NO/cGMP-dependent mechanism. In human vascular cells, serelaxin (30 nM) time dependently increased cGMP accumulation in HUVECs, HUVSMCs, HUASMCs and HCFs with the response peaking by 30 min (Supporting Information Fig. S2b). Serelaxin increased cGMP accumulation concentration-dependently in HUVECs (Figure 3A: pEC50 = 8.9 ± 0.4), HUVSMCs (Figure 3B: pEC50 = 9.2 ± 0.4), HUASMCs (Figure 3C: pEC50 = 9.1 ± 0.3) and HCFs (Figure 3D: pEC50 = 9.1 ± 0.3). The maximum serelaxin response was higher in HUVECs and HCFs (75 and 60% of DEA) but lower in HUASMCs (40% of DEA) and HUVSMCs (25% of DEA). PTX (50 ng·mL−1, 18 h) and wortmannin (100 nM, 30 min) pretreatment significantly inhibited serelaxin-mediated (30 nM) cGMP accumulation, confirming the involvement of Gαi and PI3K, but these responses were qualitatively different in different cell types. In HUVECs, HUVSMCs and HCFs, serelaxin produced bell-shaped CRCs whereas in HUASMCs, as observed for cAMP, it was sigmoidal.
Figure 3.
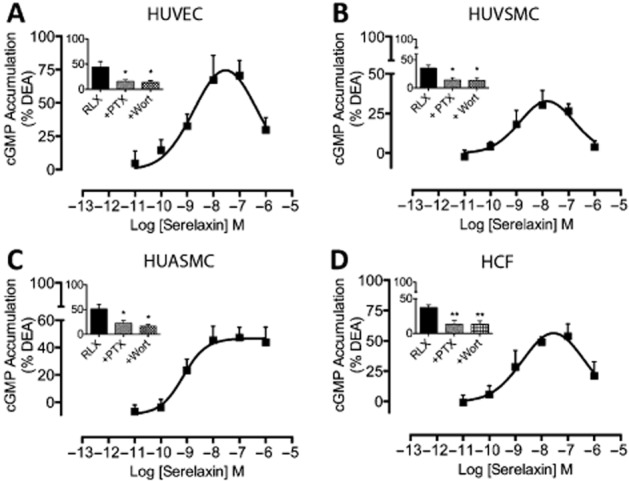
The effect of serelaxin on cGMP accumulation in human primary umbilical vascular cells and cardiac fibroblasts. Serelaxin treatment (30 min) increased cGMP accumulation in (A) HUVECs (n = 7), (B) HUVSMCs (n = 5), (C) HUASMCs (n = 6) and (D) HCFs (n = 5). The serelaxin CRC was bell-shaped for HUVECs, HUVSMCs and HCFs but sigmoidal for HUASMCs. PTX (50 ng·mL−1, 18 h) and wortmannin (100 nM, 30 min) pretreatment significantly inhibited serelaxin (30 nM)-mediated cGMP accumulation in each cell type. Statistical significance was assessed using a one-way anova with a Dunnett's post hoc test compared with serelaxin alone: *P < 0.05 and **P < 0.01.
ERK1/2 phosphorylation in response to acute serelaxin stimulation
ERK1/2, a kinase that promotes cell survival and inhibits apoptosis, is phosphorylated following treatment with serelaxin in HUVECs and vascular smooth muscle cells (Bani et al., 1998b; Zhang et al., 2002; Dschietzig et al., 2003; Masini et al., 2006), but the mechanism involved is not known. Our study is the first to show that serelaxin causes an increase in pERK1/2 in human vascular cells and cardiac fibroblasts in a Gαi- and PI3K-dependent manner. Serelaxin (30 nM) treatment rapidly increased pERK1/2 in HUVECs (15% of FBS), HUVSMCs (15% of FBS) and HCFs (50% of FBS) peaking at 10 min, and in HUASMCs (25% of FBS) at 5 min with responses that declined to basal after 15 min, except in HUVECs where pERK1/2 levels plateaued after 10 min (Supporting Information Fig. S2c). Responses in all cell types were inhibited by PTX and wortmannin pretreatment suggesting the involvement of Gαi and PI3K in pERK1/2 responses (Supporting Information Fig. S3a–d). Serelaxin concentration dependently increased pERK1/2 in HUVECs (Supporting Information Fig. S3a, pEC50: 9.2 ± 0.3), HUVSMCs (Supporting Information Fig. S3b, pEC50: 9.2 ± 0.4), HUASMCs (Supporting Information Fig. S3c, pEC50: 9.1 ± 0.4) and HCFs (Supporting Information Fig. S3d, pEC50: 9.1 ± 0.3). As in the case of cAMP and cGMP accumulation, CRCs were bell-shaped in HUVECs, HUVSMCs and HCFs, and sigmoidal in HUASMCs.
Role of G proteins in the regulation of serelaxin concentration–response relationships
Previous studies have suggested that differential coupling to G proteins can explain biphasic or bell-shaped concentration–response relationships (Baker and Hill, 2006). As the RXFP1 receptor is known to couple to Gαs, GαO/B and Gαi/o (Halls et al., 2006), we used pharmacological inhibitors to selectively disrupt coupling of Gαs, GαO/B and Gαi/o to RXFP1 receptors. In both HUASMC (Figure 4A) and HUVSMC (Figure 4B), pretreatment with the Gαs inhibitor NF449 (10 μM, 30 min) reduced the E-max and right shifted the serelaxin CRC for cAMP accumulation. Similar effects were observed on the concentration–response relationships for cGMP accumulation (Figure 5A, B). In HUASMC, pretreatment with the Gαi/o inhibitor NF023 (10 μM, 30 min) reduced the E-max of cAMP (Figure 4C) and cGMP (Figure 4C) accumulation without affecting serelaxin potency. In HUVSMC, the Gαi/o inhibitor NF023 (10 μM, 30 min) reduced the E-max for cAMP (Figure 4D) and cGMP (Figure 5D) accumulation, but also altered the shape of the CRC from bell-shaped to sigmoidal. These data suggest that Gαs and Gαi/o regulate distinct phases of the serelaxin CRC for cAMP and cGMP accumulation in primary human vascular cells and that the G-protein coupling is influenced by the cellular background. This was further supported by co-incubation with NF449 and NF023 that totally abolished serelaxin-mediated cAMP (Figure 4E, F) and cGMP (Figure 5E, F) responses in both HUASMC and HUVSMC. As NF449 and NF023 are analogues of suramin, a generic purine receptor antagonist, and as such these peptides are selective antagonists for P2X1 receptors (Soto et al., 1999; Rettinger et al., 2005), we conducted control experiments that showed that pretreatment with suramin (10 μM) had no effect on serelaxin-mediated cAMP (Supporting Information Fig. S4a, b) and cGMP (Supporting Information Fig. S3c, d) accumulation in HUASMC and HUVSMC. Previous studies showed that Gαi can be localized to specific regions of the plasma membrane and RXFP1 receptor coupling to Gαi3, but not Gαs or GαO/B, is dependent on membrane lipid rafts in HEK-RXFP1 receptor cells (Halls et al., 2009a). Therefore, we disrupted lipid rafts in HUASMC by filipin III pretreatment (1 μg·mL−1, 1 h) that caused a drop in E-max for cAMP (Figure 4G) and cGMP (Figure 5G) accumulation without any change in serelaxin potency. Pretreatment of HUVSMC with filipin III (1 μg·mL−1, 1 h) reduced E-max, and converted the bell-shaped CRCs for cAMP (Figure 4H) and cGMP (Figure 5H) accumulation to sigmoidal CRCs. Thus, the effects of filipin III mimicked the effects of the Gαi/o inhibitor (NF023), suggesting that both the presence and the location of Gαi/o critically shape serelaxin concentration–response relationships.
Figure 4.
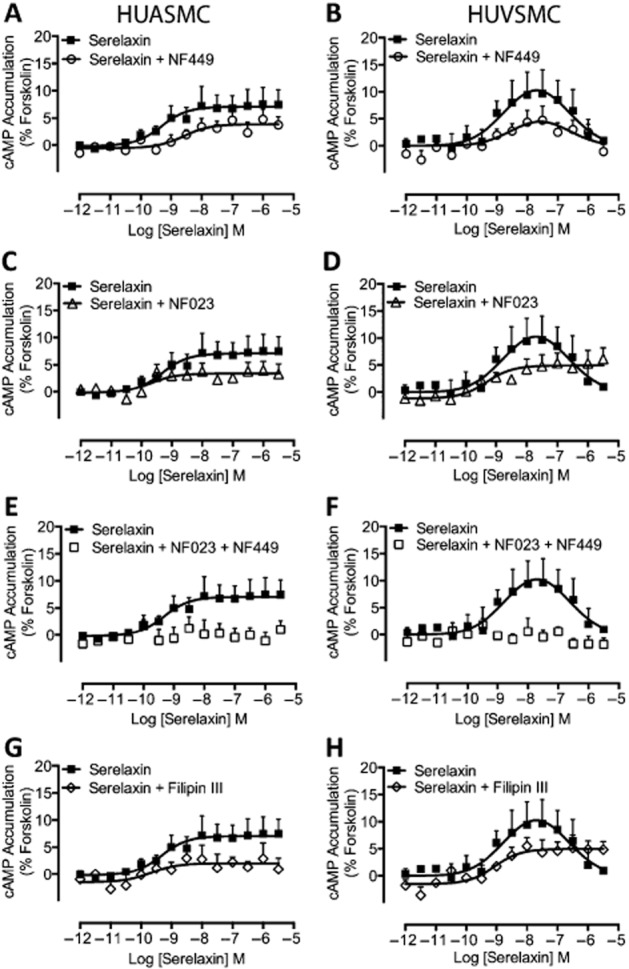
The role of G-protein coupling in serelaxin-mediated cAMP accumulation in human primary umbilical vascular cells. Pretreatment of HUASMC (n = 6) in (A) and of HUVSMC (n = 6) in (B) with the selective Gαs inhibitor NF449 (10 μM, 30 min) caused a rightward shift and reduced the E-max of the cAMP CRC to serelaxin without modifying the shape of the curve. Pretreatment of HUASMC (n = 7) in (C) and of HUVSMC (n = 6) in (D) with the selective Gαi/o inhibitor NF023 (10 μM, 30 min) reduced the E-max of the cAMP CRC to serelaxin and changed the shape of the curve observed with HUVSMC (D) from bell-shaped to sigmoidal. In both (E) HUASMC (n = 6) and (F) HUVSMC (n = 6), pretreatment with both NF449 and NF023 completely abolished serelaxin-mediated cAMP responses, showing that the responses result entirely from RXFP1 receptors interaction with G proteins. In (G) HUASMC (n = 6) and in (H) HUVSMC (n = 6), pretreatment with filipin III (1 μg·mL−1, 1 h), which disrupts lipid rafts, mimicked the effect of the Gαi/o inhibitor NF023 – reducing E-max and converting CRCs from bell-shaped to sigmoidal in HUVSMC.
Figure 5.
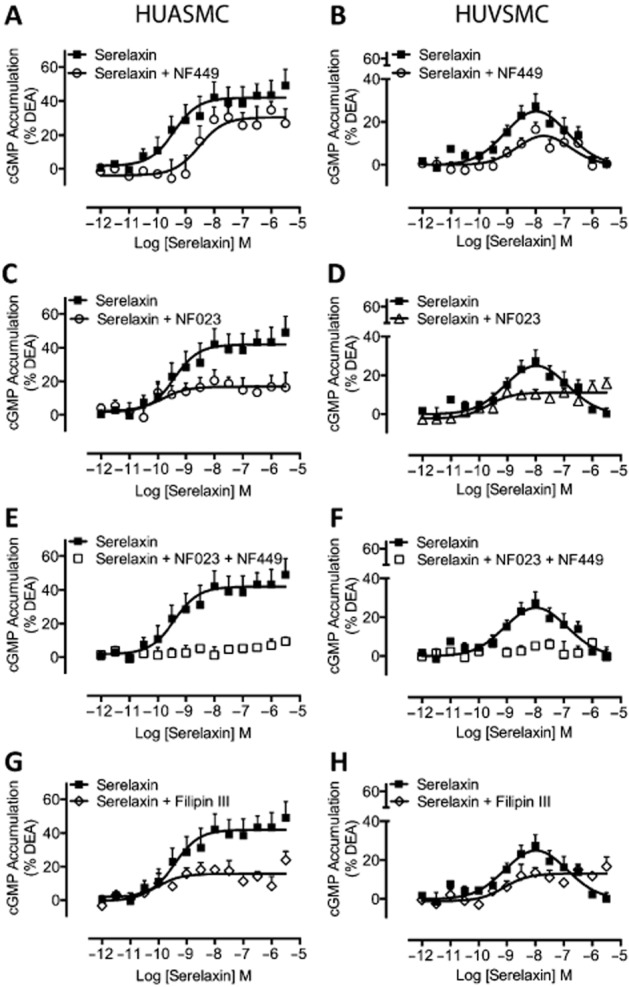
The role of G-protein coupling in serelaxin-mediated cGMP accumulation in human primary umbilical vascular cells. Pretreatment of HUASMC (n = 6) in (A) and of HUVSMC (n = 6) in (B) with the selective Gαs inhibitor NF449 (10 μM, 30 min) caused a rightward shift and reduced the E-max of the cGMP CRC to serelaxin without modifying the shape of the curve. Pretreatment of HUASMC (n = 7) in (C) and of HUVSMC (n = 6) in (D) with the selective Gαi/o inhibitor NF023 (10 μM, 30 min) reduced the E-max of the cGMP CRC to serelaxin and changed the shape of the curve observed with HUVSMC (D) from bell-shaped to sigmoidal. In both (E) HUASMC (n = 6) and (F) HUVSMC (n = 6), pretreatment with both NF449 and NF023 completely abolished serelaxin-mediated cGMP responses, showing that the responses result entirely from RXFP1 receptors interaction with G proteins. In (G) HUASMC (n = 6) and in (H) HUVSMC (n = 6), pretreatment with filipin III (1 μg·mL−1, 1 h), which disrupts lipid rafts, mimicked the effect of the Gαi/o inhibitor NF023 – reducing E-max and converting CRCs from bell-shaped to sigmoidal in HUVSMC.
Changes in expression of VEGF, ETB and nNOS in human vascular cells following 48 h serelaxin administration
The effects of 48 h serelaxin treatment on protein expression of VEGF, ETB and NOS are implicated in its vasodilator effects (Dschietzig et al., 2003; Conrad, 2010; McGuane et al., 2011b). Serelaxin (10 ng·mL–1; 1.68 nM) caused a twofold increase in nNOS expression in HUVECs, HUVSMCs and HCFs, and a threefold increase in HUASMCs (Figure 6A–D) that parallels our findings previously observed in rat renal myofibroblasts (Mookerjee et al., 2009). Similarly, and also consistent with previous findings (Alexiou et al., 2013), serelaxin increased the expression of iNOS in HUVECs (∼2-fold induction) and had no effect on the expression of eNOS (data not shown). We were unable to detect and measure iNOS and eNOS in HUASMCs, HUVSMCs and HCFs (data not shown). Similar to previous studies in human endometrial cells (Unemori et al., 2000), serelaxin also elevated VEGF165 (referred to as VEGF-A) expression by 1.5-fold in HUVECs, HUASMCs and HCFs (Figure 6A, C, D). VEGF-A is the most predominant isoform expressed in the vasculature and it has a crucial role in angiogenesis. There are four different isoforms of VEGF-A (VEGF121, VEGF165, VEGF189, VEGF206) of which VEGF165 is the most predominant (Ferrara, 2004). Although VEGF expression has been reported to be associated with cAMP accumulation (Unemori et al., 2000), it was unaltered in HUVSMCs (Figure 6B) despite increased cAMP accumulation in these cells (Figure 2B). Furthermore, in HCFs, serelaxin increased VEGF expression but had no effect on cAMP accumulation (Figure 2D). The changes may involve the ERK1/2 (Milanini et al., 1998; van der Westhuizen et al., 2007) or NO (Dulak et al., 2000) pathways that are activated in all cell types. Furthermore, and consistent with previous studies (Dschietzig et al., 2003), serelaxin increased ETB receptor expression in HUVECs and HUVSMCs by two- to threefold, and in HUASMCs and HCFs by 1.5-fold (Figure 6A–D). ETB receptor expression has been reported to be downstream of MMP and ERK1/2 signalling (Dschietzig et al., 2003; Jeyabalan et al., 2003; Chow et al., 2012), and these pathways were activated in all cell types where ETB receptor expression was increased (see Figure 7 and Supporting Information Fig. S3).
Figure 6.
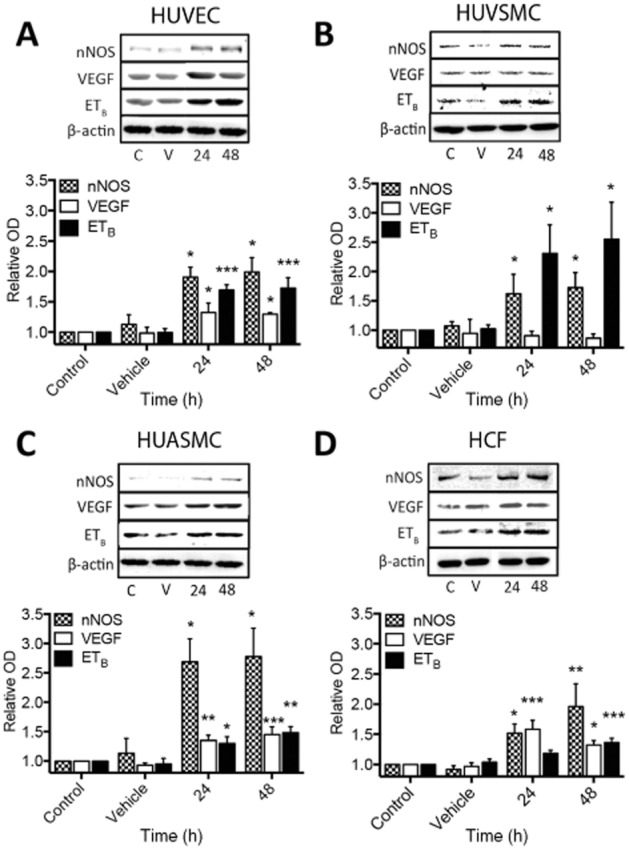
Changes in the expression of nNOS, VEGF and ETB receptors in human primary umbilical vascular cells and cardiac fibroblasts after serelaxin (1.68 nM) exposure for 24 and 48 h. In HUVECs (A), serelaxin treatment increased the expression of nNOS (n = 5), ETB receptors (n = 6) and VEGF (n = 7). In HUVSMCs (B), serelaxin treatment increased the expression of nNOS (n = 5) and ETB (n = 5) but not VEGF (n = 4); however, in HUASMC (C), serelaxin treatment increased the expression of nNOS (n = 6), ETB receptors (n = 7) and VEGF (n = 5). In HCFs (D), similar to HUVECs and HUASMCs, serelaxin treatment increased the expression of nNOS (n = 5), ETB receptors (n = 5) and VEGF (n = 5). A representative blot of each protein and β-actin, a loading control, is shown with the densitometry in each figure. Statistical significance was assessed using a one-way anova with a Dunnett's post hoc test compared with vehicle alone: *P < 0.05 and **P < 0.01.
Figure 7.
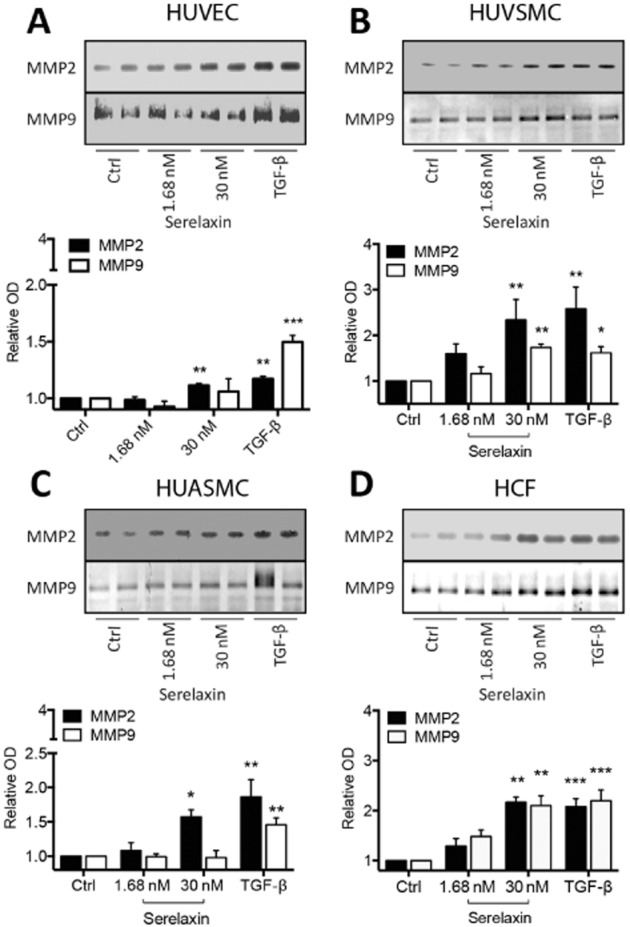
Changes in the activity of MMPs in human primary umbilical vascular cells and cardiac fibroblasts using zymography to assess changes in activity of MMP2 and MMP9 after long-term serelaxin exposure (1.68 and 30 nM) for 48 h. Serelaxin treatment increased the activity of MMP2 in (A) HUVECs (n = 5), (B) HUVSMCs (n = 6), (C) HUASMCs (n = 7) and (D) HCFs (n = 5). Serelaxin also significantly increased the activity of MMP9 but only in (B) HUVSMCs (n = 5) and (D) HCFs (n = 5) and not in (A) HUVECs (n = 5) and (C) HUASMCs (n = 5). Furthermore, and consistent with the lack of RXFP1 receptor expression (Figure 1), serelaxin had no effect on MMP activity in HUAECs (n = 2). A representative scan of each zymography is shown along with the densitometry in each figure. Statistical significance was assessed using a one-way anova with a Dunnett's post hoc test compared with control alone: *P < 0.05 and **P < 0.01.
Changes in MMP activity in human vascular cells following 48 h serelaxin administration
The matrix/collagen-degrading enzymes MMP2 and MMP9 have roles in the remodelling of organs and blood vessels with clear influences on organ function and blood haemodynamics (Jeyabalan et al., 2003). In addition, they convert big ET to ET1-32 that activates ETB receptors to enhance vasodilatation (Fernandez-Patron et al., 1999). In vivo, serelaxin increases vascular MMP activity to inhibit myogenic reactivity and promote vasodilatation (Jeyabalan et al., 2006a,b,). Our study supports these findings as serelaxin (1.68 and 30 nM for 48 h) increased MMP2 activity in HUVECs, HUVSMCs, HUASMCs and HCFs (Figure 7A–D). Serelaxin (30 nM) caused a three- and twofold increase in MMP2 activity in HUVSMCs and HCFs, but only a 1.2- and 1.5-fold increase in activity in HUVECs and HUASMCs. Serelaxin (30 nM) doubled MMP9 activity in HUVSMCs and HCFs (Figure 7B, D) but had no effect in HUVECs and HUASMCs (Figure 7A, C). In HUAECs, serelaxin had no effect on MMP2 and MMP9 activity (data not shown), consistent with the lack of cell surface RXFP1 receptors expression in HUAECs. Gelatinase activity and/or expression is controlled through a pERK/nNOS/NO/cGMP-dependent pathway (Chow et al., 2012), consistent with our findings as these signalling pathways were activated in all cell types expressing RXFP1 receptors (Figure 3 and Supporting Information Fig. S3).
Discussion and conclusions
In the RELAX-AHF clinical trial, serelaxin infusion over 48 h improved short- and long-term outcomes in patients with acute cardiac failure. Here we have examined the acute (<1 h) and chronic (24–48 h) effects of serelaxin on signalling in human vascular cells in order to better understand the mechanism of serelaxin action in humans experimentally and clinically. This study demonstrates that serelaxin had different effects on human vascular cells dependent on the cellular background. We showed that the bell-shaped CRCs that are a hallmark of serelaxin activity experimentally (Unemori et al., 1996; Danielson and Conrad, 2003; Halls et al., 2006) and clinically (Teerlink et al., 2009) can be demonstrated in human venous cells and cardiac fibroblasts and that they are dependent on the type and location of G proteins present.
Acute serelaxin administration causes rapid dilation of human subcutaneous arteries and rodent renal and mesenteric arteries in a Gαi/PI3K/Akt/eNOS-dependent manner (McGuane et al., 2011b). The present study demonstrates in human vascular cells that serelaxin increases cAMP, cGMP and pERK1/2 in a Gαi- and PI3K-dependent manner (Figure 8). Longer term serelaxin effects (up to 48 h) reported include changes in expression of MMPs, ETB receptors, VEGF and NOS (McGuane et al., 2011a) that were similar to our findings in human vascular cells showing that 48 h serelaxin exposure increased the expression of ETB receptors, VEGF and nNOS and increased the activity of MMP2, albeit in a cell-dependent manner (Figure 8). Although it is difficult to extrapolate these findings to cells of the systemic vasculature, these changes in human umbilical cells may reflect the haemodynamic changes observed during pregnancy and in vivo, particularly the increase in global arterial compliance and decrease in vascular resistance (thereby improving cardiac output) observed in pregnant (Debrah et al., 2006) and non-pregnant animals (Conrad et al., 2004; Xu et al., 2010).
Figure 8.

Signal transduction mechanisms employed by serelaxin in human umbilical vascular cells and HCFs and their potential physiological effects. Short-term (<1 h) serelaxin stimulation produces cAMP/cGMP accumulation in vascular cells and pERK1/2 in all cells that is differentially regulated by Gαs, Gαi, GαOB and PI3K, and these pathways are likely to be involved in the vasodilator and anti-apoptotic effects of serelaxin respectively. In HUASMCs, the serelaxin-mediated cAMP or VEGF response did not involve GαOB and PI3K, whereas in HCFs, the RXFP1 receptor was not coupled to cAMP production. Longer term (24–48 h) serelaxin treatment increased VEGF expression involving both cAMP-dependent and cAMP-independent mechanisms, and these pathways are likely to be involved in angiogenesis. In addition, serelaxin treatment increased the activity of MMP2 and MMP9 to mediate its remodelling actions, but these enzymes are also secreted to convert big ET to ET1-32 that activates ETB receptors to further enhance vasodilatation (Conrad, 2010). Solid lines indicate mechanisms identified in the current study whereas dashed lines indicate previously established mechanisms.
Serelaxin had different effects on human primary cells isolated from the arterial and venous circulations which have implications for our understanding of its clinical actions. Although serelaxin is an arteriodilator (Jeyabalan et al., 2003; Conrad and Shroff, 2011; McGuane et al., 2011b), our study suggests that serelaxin may also have potent venodilating properties in humans. Studies on the rat vasculature suggest that serelaxin has predominantly arteriodilating effects. Rat mesenteric and femoral arteries and veins expressed RXFP1 receptors in endothelial and smooth muscle cells, but serelaxin-mediated remodelling was only observed in the mesenteric arteries (Jelinic et al., 2014) while serelaxin-mediated dilation in rat mesenteric veins has also been observed (Li et al., 2005).
Interestingly, the effects of serelaxin on the expression of NOS isoforms were consistent across the different vascular cells but varied in magnitude. Increased nNOS expression in HUASM and HUVSM cells paralleled previous findings in rat vascular and microvascular smooth muscle cells (Boulanger et al., 1998; Kavdia, 2004), rat renal microvasculature (Ichihara et al., 1998), rabbit cardiomyocytes (Xu et al., 1999) and porcine carotid endothelial cells (Buchwalow et al., 2002). However, serelaxin also increased nNOS expression in umbilical endothelial and smooth muscle cells and cardiac fibroblasts. iNOS was not detected in HUASM and HUVSM cells and was not induced by serelaxin treatment, but in accord with previous findings (Quattrone et al., 2004) serelaxin treatment increased expression of iNOS in HUVECs (data not shown). The findings from HUASM and HUVSM cells contrast with previous studies in vascular smooth muscle cells from rat carotid artery and rat renal cells (Mohaupt et al., 1994; Boulanger et al., 1998).
Interestingly, we demonstrated that serelaxin produced bell-shaped CRCs in HUVECs, HUVSMCs and HCFs, but not in HUASMCs. Our findings suggest that Gαi/o associated with lipid rafts has an important role to play in determining the shape of the concentration–response relationship. The pattern of G proteins involved in responses to serelaxin differs with cell type. In HEK cells recombinantly expressing RXFP1 receptors and in human THP-1 cells, serelaxin activation of RXFP1 receptors results in coupling to Gαs, GαOB and Gαi3, whereas in rat cardiac fibroblasts there is coupling to Gαs and Gαi3, in colo16 cells and rat renal fibroblasts to Gαs and GαOB, and in T47D cells solely to Gαs (Halls et al., 2009b; Mookerjee et al., 2009). We have also shown that disruption of membrane rafts in HEK-RXFP1 receptor cells eliminates serelaxin signalling via the Gαi3 pathway (Halls et al., 2009a). In HUASMC, cAMP and cGMP concentration–response relationships were sigmoidal, compatible with responses involving Gαs and Gαi3 as inhibition of Gαs with NF449 or Gαi with NF023 desensitized the response but did not change the shape of the curves (Figures 4 and 5A, B). Blockade of both Gαs and Gαi completely abrogated cAMP and cGMP responses (Figures 4 and 5C). Disruption of lipid rafts with filipin III had a similar effect to Gαi inhibition (Figures 4 and 5D). In HUVSMC, cAMP and cGMP CRCs were bell-shaped, suggesting that the responses involved not only Gαs and Gαi3, but GαOB, and that the latter coupled less efficiently to RXFP1 receptors. Inhibition of Gαs with NF449 desensitized the responses but did not change the shape of the curves (Figures 4 and 5E). In contrast, inhibition of Gαi with NF023 both desensitized (blockade of Gαi3) and changed the shape of the curve to sigmoid (Figures 4 and 5F). Again, blockade of Gαs, Gαi3 and GαOB completely abrogated responses (Figures 4 and 5G). Disruption of lipid rafts with filipin III had the same effect as inhibition of inhibitory G proteins (Figures 4 and 5H). The results suggest that both Gαs and Gαi3 contribute to the increases in cAMP (Halls et al., 2006; 2009a) and cGMP observed in both HUASMC and HUVSMC and that GαOB inhibits these responses but only operates in HUVSMC and interacts with RXFP1 receptors at lower affinity than the other G proteins. Association with lipid rafts seems to be obligatory for the operation of Gi/o proteins as disruption with filipin prevented their action. Differential G-protein coupling to RXFP1 receptors has physiological and pathological implications as Gαi expression is increased, whereas Gαs expression is unaffected during heart failure. Additionally, the positive inotropic effects of serelaxin are partially inhibited by Gαi/o and PI3K inhibitors in the non-failing human atrial myocardium, whereas in the failing human atrial myocardium, Gαi/o and PI3K inhibition completely abolished the inotropic effects (Dschietzig et al., 2011).
Another important observation in our study was that different G proteins mediated distinct phases of the serelaxin CRC with Gαs regulating the initial phase, whereas Gαi/o (and possibly Gαo/B) mediating the latter phase. This bears resemblance to the activation of other family A GPCRs. There is a concentration-dependent switch from G-protein-dependent to G-protein-independent signalling by the β2-adrenoceptor because low agonist concentrations activated ERK1/2 via Gαs, whereas higher agonist concentrations activated ERK1/2 via Src tyrosine kinase (Sun et al., 2007). Similarly, in a human breast cell line MCF-10A, low concentrations of isoprenaline target a β2-adrenoceptor population localized in membrane rafts and stimulate Gαs to enhance cell adhesion, while higher concentrations activate a β2-adrenoceptor population outside membrane rafts to inhibit cell proliferation by a Gαs and PKA-dependent signalling pathway (Bruzzone et al., 2014). Likewise, the adenosine A1 receptor also exists in multiple states that couple to different G proteins. Adenosine A1 receptor-stimulated cAMP response element-gene transcription produced a biphasic CRC. The inhibitory component was inhibited by PTX whereas the stimulatory component was enhanced, suggesting Gαi-coupled inhibition followed by Gαs-coupled stimulation (Baker and Hill, 2006).
Despite the exciting findings obtained, there were some limitations to this study. The cells were obtained from human umbilical cords. The umbilical vessels have unique functions in that the umbilical vein transports oxygenated blood to the fetus whereas the artery returns deoxygenated blood to the maternal circulation. The umbilical artery lacks the elastic lamina and adventitia found in other arteries, these being replaced by connective tissue, whereas the umbilical vein contains an unusually thick muscle layer (Spurway et al., 2012), and therefore acts much like an artery. Lack of RXFP1 receptor expression in the umbilical artery endothelial cells appears to be a regional effect as serelaxin causes signal transduction in endothelial cells from human coronary arteries (McGuane et al., 2011b). However, cells from the umbilical cord do provide model systems that allow the study of serelaxin responses in human cells and can provide novel insights into the signal transduction mechanisms activated by serelaxin in the vasculature. Although it is likely that cells from the systemic and renal circulation play a more important role in AHF, the relative lack of availability of such human cells poses a problem. However, we used primary human cells that endogenously express RXFP1 receptors, that are readily available, are relevant to the cardiovascular system and are likely to represent a good model to study physiologically relevant responses. Furthermore, HCFs used in the current study were obtained from human hearts and these cells did not show responses that were markedly different from cells from the umbilical cord. Nonetheless, future studies will extend the number and types of human cardiovascular cells studied. Furthermore, in physiology, cellular crosstalk is likely to impact intra- and extracellular serelaxin signalling. Therefore, future studies investigating serelaxin responses in co-culture systems may establish better models that reflect more closely the pharmacodynamic effects of serelaxin in the treatment of AHF.
Acknowledgments
We thank Corthera, Inc. (San Mateo, CA) for the supply of serelaxin. This study was supported by the Australian Research Council Linkage Grant (LP110100288 to R. J. S., C. S. S., R. A. B. and Industry Partner Corthera Inc., a Novartis Company) and the National Health & Medical Research Council (NHMRC) of Australia Senior Research Fellowships to C. S. S. (APP1041766) and R. A. D. B. (APP1042650).
Glossary
- AHF
acute heart failure
- CRC
concentration–response curve
- DEA
diethylamine NONOate
- eNOS
endothelial NOS
- ETB receptor
endothelin type B receptor
- HCF
human cardiac fibroblast
- HUAEC
human umbilical artery endothelial cell
- HUASMC
human umbilical artery smooth muscle cell
- HUVSMC
human umbilical vein smooth muscle cell
- iNOS
inducible NOS
- nNOS
neuronal NOS
- pERK1/2
phosphorylated ERK 1 and 2
- PI3K
phosphoinositide 3-kinase
- PTX
pertussis toxin
- RXFP1 receptor
relaxin family peptide receptor 1
Author contributions
M. S., C. S. S., R. A. B., D. R. S. and R. J. S. participated in research design. M. S. and C. S. S. conducted the experiments. R. A. B. and D. R. S. contributed reagents or tools. M. S. and R. J. S. performed data analysis. M. S., C. S. S., R. A. B., D. R. S. and R. J. S. wrote or contributed to writing of manuscript.
Conflict of interest
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Serelaxin stimulation did not cause a functional response in HUAECs. HUAECs do not express RXFP1 receptors at the cell surface (Figure 1), and in accord with this finding, addition of serelaxin (30 nM) did not affect (a) cAMP accumulation (n = 4), (b) cGMP accumulation (n = 4) or (c) pERK1/2 (n = 3). Each point represents the mean ± SEM of four to six independent experiments performed in duplicate.
Figure S2 Time course studies of serelaxin in human primary vascular and cardiac cells. Serelaxin (30 nM) treatment increased (a) cAMP accumulation in HUVEC (n = 7), HUVSMC (n = 3) and HUASMC (n = 4), but not in HCF (n = 3). Serelaxin (30 nM) treatment also increased cGMP accumulation but in all cells including HUVEC (n = 6), HUVSMC (n = 5), HUASMC (n = 3) and HCF (n = 3). CRCs were conducted at 30 min for each cell type. Similarly, serelaxin (30 nM) increased pERK1/2 in HUVEC (n = 9), HUVSMC (n = 4), HUASMC (n = 4) and HCF (n = 3) that peaked between 5 and 10 min after which it returned to baseline. However, in HUVEC, pERK1/2 plateaued after 10 min and remained at that level for up to 60 min. CRCs for pERK1/2 were conducted at 10 min for HUVEC, HUVSMC and HCF but at 5 min for HUASMC. Each point represents the mean ± SEM performed in duplicate.
Figure S3 The effects of serelaxin stimulation on pERK1/2 in human primary vascular cells. Serelaxin stimulation (10 min for HUVECs, HUVSMCs, HCFs and 5 min for HUASMCs) increased pERK1/2 in (a) HUVECs (n = 5), (b) HUVSMCs (n = 6), (c) HUASMCs (n = 6) and (d) HCFs (n = 5). The serelaxin CRCs were bell-shaped for HUVECs, HUVSMCs and HCFs, but sigmoidal for HUASMCs. PTX (50 ng·mL−1, 18 h) and wortmannin (100 nM, 30 min) pretreatment significantly inhibited serelaxin (30 nM)-mediated pERK1/2 in each cell type. Statistical significance was assessed using a one-way anova with a Dunnett's post hoc test compared with serelaxin alone: *P < 0.05 and **P < 0.01.
Figure S4 The effect of suramin on the dose–response relationship of serelaxin in primary human smooth vascular cells. Pretreatment of suramin (10 μM, 30 min) to HUASMC had no effect on (a) cAMP (n = 3) or (c) cGMP accumulation (n = 3). Similarly, suramin (10 μM, 30 min) pretreatment to HUVSMC had no effect on the dose–response relationship of serelaxin-mediated (b) cAMP (n = 3) or (d) cGMP accumulation (n = 3). Similarly, pretreatment of HUASMC and HUVSMC with 100 μM suramin (30 min) reduced the E-max of (a, b) cAMP (n = 3) and (c, d) cGMP accumulation (n = 3).
Table S1 The binding affinity of serelaxin (pKi) and the potency (pEC50) of serelaxin for cAMP accumulation, cGMP accumulation and pERK1/2 in human primary vascular cells. Values in parentheses refer to n experiments.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ligand-gated ion channels. Br J Pharmacol. 2013b;170:1582–1606. doi: 10.1111/bph.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013c;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexiou K, Wilbring M, Matschke K, Dschietzig T. Relaxin protects rat lungs from ischemia-reperfusion injury via inducible NO synthase: role of ERK-1/2, PI3K, and forkhead transcription factor FKHRL1. PLoS ONE. 2013;8:e75592. doi: 10.1371/journal.pone.0075592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG, Hill SJ. A comparison of the antagonist affinities for the Gi- and Gs-coupled states of the human adenosine A1-receptor. J Pharmacol Exp Ther. 2006;320:218–228. doi: 10.1124/jpet.106.113589. [DOI] [PubMed] [Google Scholar]
- Bani D, Failli P, Bello MG, Thiemermann C, Bani Sacchi T, Bigazzi M, et al. Relaxin activates the L-arginine-nitric oxide pathway in vascular smooth muscle cells in culture. Hypertension. 1998a;31:1240–1247. doi: 10.1161/01.hyp.31.6.1240. [DOI] [PubMed] [Google Scholar]
- Bani D, Masini E, Bello MG, Bigazzi M, Sacchi TB. Relaxin protects against myocardial injury caused by ischemia and reperfusion in rat heart. Am J Pathol. 1998b;152:1367–1376. [PMC free article] [PubMed] [Google Scholar]
- Boulanger CM, Heymes C, Benessiano J, Geske RS, Lévy BI, Vanhoutte PM. Neuronal nitric oxide synthase is expressed in rat vascular smooth muscle cells: activation by angiotensin II in hypertension. Circ Res. 1998;83:1271–1278. doi: 10.1161/01.res.83.12.1271. [DOI] [PubMed] [Google Scholar]
- Bruzzone A, Saulière A, Finana F, Sénard J-M, Lüthy I, Galés C. Dosage-dependent regulation of cell proliferation and adhesion through dual β2-adrenergic receptor/cAMP signals. FASEB J. 2014;28:1342–1354. doi: 10.1096/fj.13-239285. [DOI] [PubMed] [Google Scholar]
- Buchwalow IB, Podzuweit T, Böcker W, Samoilova VE, Thomas S, Wellner M, et al. Vascular smooth muscle and nitric oxide synthase. FASEB J. 2002;16:500–508. doi: 10.1096/fj.01-0842com. [DOI] [PubMed] [Google Scholar]
- Chow BSM, Chew EGY, Zhao C, Bathgate RAD, Hewitson TD, Samuel CS. Relaxin signals through a RXFP1-pERK-nNOS-NO-cGMP-dependent pathway to up-regulate matrix metalloproteinases: the additional involvement of iNOS. PLoS ONE. 2012;7:e42714. doi: 10.1371/journal.pone.0042714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KP. Unveiling the vasodilatory actions and mechanisms of relaxin. Hypertension. 2010;56:2–9. doi: 10.1161/HYPERTENSIONAHA.109.133926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KP, Shroff SG. Effects of relaxin on arterial dilation, remodeling, and mechanical properties. Curr Hypertens Rep. 2011;13:409–420. doi: 10.1007/s11906-011-0231-x. [DOI] [PubMed] [Google Scholar]
- Conrad KP, Debrah DO, Novak J, Danielson LA, Shroff SG. Relaxin modifies systemic arterial resistance and compliance in conscious, nonpregnant rats. Endocrinology. 2004;145:3289–3296. doi: 10.1210/en.2003-1612. [DOI] [PubMed] [Google Scholar]
- Danielson L, Conrad KP. Time course and dose response of relaxin-mediated renal vasodilation, hyperfiltration, and changes in plasma osmolality in conscious rats. J Appl Physiol. 2003;95:1509–1514. doi: 10.1152/japplphysiol.00545.2003. [DOI] [PubMed] [Google Scholar]
- Debrah DO, Novak J, Matthews JE, Ramirez RJ, Shroff SG, Conrad KP. Relaxin is essential for systemic vasodilation and increased global arterial compliance during early pregnancy in conscious rats. Endocrinology. 2006;147:5126–5131. doi: 10.1210/en.2006-0567. [DOI] [PubMed] [Google Scholar]
- Dschietzig T, Bartsch C, Richter C, Laule M, Baumann G, Stangl K. Relaxin, a pregnancy hormone, is a functional endothelin-1 antagonist: attenuation of endothelin-1-mediated vasoconstriction by stimulation of endothelin type-B receptor expression via ERK-1/2 and nuclear factor-kappaB. Circ Res. 2003;92:32–40. doi: 10.1161/01.res.0000051884.27117.7e. [DOI] [PubMed] [Google Scholar]
- Dschietzig T, Alexiou K, Kinkel H-T, Baumann G, Matschke K, Stangl K. The positive inotropic effect of relaxin-2 in human atrial myocardium is preserved in end-stage heart failure: role of G(i)-phosphoinositide-3 kinase signaling. J Card Fail. 2011;17:158–166. doi: 10.1016/j.cardfail.2010.08.011. [DOI] [PubMed] [Google Scholar]
- Dulak J, Józkowicz A, Dembinska-Kiec A, Guevara I, Zdzienicka A, Zmudzinska-Grochot D, et al. Nitric oxide induces the synthesis of vascular endothelial growth factor by rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2000;20:659–666. doi: 10.1161/01.atv.20.3.659. [DOI] [PubMed] [Google Scholar]
- Fernandez-Patron C, Radomski MW, Davidge ST. Vascular matrix metalloproteinase-2 cleaves big endothelin-1 yielding a novel vasoconstrictor. Circ Res. 1999;85:906–911. doi: 10.1161/01.res.85.10.906. [DOI] [PubMed] [Google Scholar]
- Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- Halls ML, Bond CP, Sudo S, Kumagai J, Ferraro T, Layfield S, et al. Multiple binding sites revealed by interaction of relaxin family peptides with native and chimeric relaxin family peptide receptors 1 and 2 (LGR7 and LGR8) J Pharmacol Exp Ther. 2005;313:677–687. doi: 10.1124/jpet.104.080655. [DOI] [PubMed] [Google Scholar]
- Halls ML, Bathgate RAD, Summers RJ. Relaxin family peptide receptors RXFP1 and RXFP2 modulate cAMP signaling by distinct mechanisms. Mol Pharmacol. 2006;70:214–226. doi: 10.1124/mol.105.021691. [DOI] [PubMed] [Google Scholar]
- Halls ML, van der Westhuizen ET, Wade JD, Evans BA, Bathgate RAD, Summers RJ. Relaxin Family Peptide Receptor (RXFP1) Coupling to Gαi3 Involves the C-Terminal Arg752 and localization within membrane raft microdomains. Mol Pharmacol. 2009a;75:415–428. doi: 10.1124/mol.108.051227. [DOI] [PubMed] [Google Scholar]
- Halls ML, Hewitson TD, Moore X-L, Du X-J, Bathgate RAD, Summers RJ. Relaxin activates multiple cAMP signaling pathway profiles in different target cells. Ann N Y Acad Sci. 2009b;1160:108–111. doi: 10.1111/j.1749-6632.2008.03814.x. [DOI] [PubMed] [Google Scholar]
- Ichihara A, Inscho EW, Imig JD, Navar LG. Neuronal nitric oxide synthase modulates rat renal microvascular function. Am J Physiol. 1998;274:F516–F524. doi: 10.1152/ajprenal.1998.274.3.F516. [DOI] [PubMed] [Google Scholar]
- Jelinic M, Leo C-H, Post Uiterweer ED, Sandow SL, Gooi JH, Wlodek ME, et al. Localization of relaxin receptors in arteries and veins, and region-specific increases in compliance and bradykinin-mediated relaxation after in vivo serelaxin treatment. FASEB J. 2014;28:275–287. doi: 10.1096/fj.13-233429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyabalan A, Novak JJ, Danielson LA, Kerchner LJ, Opett SL, Conrad KP. Essential role for vascular gelatinase activity in relaxin-induced renal vasodilation, hyperfiltration, and reduced myogenic reactivity of small arteries. Circ Res. 2003;93:1249–1257. doi: 10.1161/01.RES.0000104086.43830.6C. [DOI] [PubMed] [Google Scholar]
- Jeyabalan A, Kerchner LJ, Fisher MC, McGuane JT, Doty KD, Conrad KP. Matrix metalloproteinase-2 activity, protein, mRNA, and tissue inhibitors in small arteries from pregnant and relaxin-treated nonpregnant rats. J Appl Physiol. 2006a;100:1955–1963. doi: 10.1152/japplphysiol.01330.2005. [DOI] [PubMed] [Google Scholar]
- Jeyabalan A, Novak J, Doty KD, Matthews J, Fisher MC, Kerchner LJ, et al. Vascular matrix metalloproteinase-9 mediates the inhibition of myogenic reactivity in small arteries isolated from rats after short-term administration of relaxin. Endocrinology. 2006b;148:189–197. doi: 10.1210/en.2006-0989. [DOI] [PubMed] [Google Scholar]
- Kavdia M. Contribution of nNOS- and eNOS-derived NO to microvascular smooth muscle NO exposure. J Appl Physiol. 2004;97:293–301. doi: 10.1152/japplphysiol.00049.2004. [DOI] [PubMed] [Google Scholar]
- Li Y, Brookes ZL, Kaufman S. Acute and chronic effects of relaxin on vasoactivity, myogenic reactivity and compliance of the rat mesenteric arterial and venous vasculature. Regul Pept. 2005;132:41–46. doi: 10.1016/j.regpep.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Masini E, Bani D, Bello MG, Bigazzi M, Mannaioni PF, Sacchi TB. Relaxin counteracts myocardial damage induced by ischemia-reperfusion in isolated guinea pig hearts: evidence for an involvement of nitric oxide. Endocrinology. 1997;138:4713–4720. doi: 10.1210/endo.138.11.5520. [DOI] [PubMed] [Google Scholar]
- Masini E, Cuzzocrea S, Mazzon E, Muià C, Vannacci A, Fabrizi F, et al. Protective effects of relaxin in ischemia/reperfusion-induced intestinal injury due to splanchnic artery occlusion. Br J Pharmacol. 2006;148:1124–1132. doi: 10.1038/sj.bjp.0706811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuane JT, Danielson LA, Debrah JE, Rubin JP, Novak J, Conrad KP. Angiogenic growth factors are new and essential players in the sustained relaxin vasodilatory pathway in rodents and humans. Hypertension. 2011a;57:1151–1160. doi: 10.1161/HYPERTENSIONAHA.110.165027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuane JT, Debrah JE, Sautina L, Jarajapu YPR, Novak J, Rubin JP, et al. Relaxin induces rapid dilation of rodent small renal and human subcutaneous arteries via PI3 kinase and nitric oxide. Endocrinology. 2011b;152:2786–2796. doi: 10.1210/en.2010-1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metra M, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, et al. Effect of serelaxin on cardiac, renal, and hepatic biomarkers in the Relaxin in Acute Heart Failure (RELAX-AHF) development program: correlation with outcomes. J Am Coll Cardiol. 2013;61:196–206. doi: 10.1016/j.jacc.2012.11.005. [DOI] [PubMed] [Google Scholar]
- Milanini J, Viñals F, Pouysségur J, Pagès G. p42/p44 MAP kinase module plays a key role in the transcriptional regulation of the vascular endothelial growth factor gene in fibroblasts. J Biol Chem. 1998;273:18165–18172. doi: 10.1074/jbc.273.29.18165. [DOI] [PubMed] [Google Scholar]
- Mohaupt MG, Elzie JL, Ahn KY, Clapp WL, Wilcox CS, Kone BC. Differential expression and induction of mRNAs encoding two inducible nitric oxide synthases in rat kidney. Kidney Int. 1994;46:653–665. doi: 10.1038/ki.1994.318. [DOI] [PubMed] [Google Scholar]
- Mookerjee I, Hewitson TD, Halls ML, Summers RJ, Mathai ML, Bathgate RAD, et al. Relaxin inhibits renal myofibroblast differentiation via RXFP1, the nitric oxide pathway, and Smad2. FASEB J. 2009;23:1219–1229. doi: 10.1096/fj.08-120857. [DOI] [PubMed] [Google Scholar]
- Mosterd A, Hoes AW. Clinical epidemiology of heart failure. Heart. 2007;93:1137–1146. doi: 10.1136/hrt.2003.025270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42:D1098–D1106. doi: 10.1093/nar/gkt1143. (Database Issue): [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quattrone S, Chiappini L, Scapagnini G, Bigazzi B, Bani D. Relaxin potentiates the expression of inducible nitric oxide synthase by endothelial cells from human umbilical vein in in vitro culture. Mol Hum Reprod. 2004;10:325–330. doi: 10.1093/molehr/gah040. [DOI] [PubMed] [Google Scholar]
- Rettinger J, Braun K, Hochmann H, Kassack MU, Ullmann H, Nickel P, et al. Profiling at recombinant homomeric and heteromeric rat P2X receptors identifies the suramin analogue NF449 as a highly potent P2X1 receptor antagonist. Neuropharmacology. 2005;48:461–468. doi: 10.1016/j.neuropharm.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Samuel CS, Unemori EN, Mookerjee I, Bathgate RAD, Layfield SL, Mak J, et al. Relaxin modulates cardiac fibroblast proliferation, differentiation, and collagen production and reverses cardiac fibrosis in vivo. Endocrinology. 2004;145:4125–4133. doi: 10.1210/en.2004-0209. [DOI] [PubMed] [Google Scholar]
- Soto F, Lambrecht G, Nickel P, Stühmer W, Busch AE. Antagonistic properties of the suramin analogue NF023 at heterologously expressed P2X receptors. Neuropharmacology. 1999;38:141–149. doi: 10.1016/s0028-3908(98)00158-0. [DOI] [PubMed] [Google Scholar]
- Spurway J, Logan P, Pak S. The development, structure and blood flow within the umbilical cord with particular reference to the venous system. Aust J Ultrasound Med. 2012;15:97–102. doi: 10.1002/j.2205-0140.2012.tb00013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Huang J, Xiang Y, Bastepe M, Jüppner H, Kobilka BK, et al. Dosage-dependent switch from G protein-coupled to G protein-independent signaling by a GPCR. EMBO J. 2007;26:53–64. doi: 10.1038/sj.emboj.7601502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teerlink JR, Metra M, Felker GM, Ponikowski P, Voors AA, Weatherley BD, et al. Relaxin for the treatment of patients with acute heart failure (Pre-RELAX-AHF): a multicentre, randomised, placebo-controlled, parallel-group, dose-finding phase IIb study. Lancet. 2009;373:1429–1439. doi: 10.1016/S0140-6736(09)60622-X. [DOI] [PubMed] [Google Scholar]
- Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, et al. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet. 2013;381:29–39. doi: 10.1016/S0140-6736(12)61855-8. [DOI] [PubMed] [Google Scholar]
- Unemori EN, Pickford LB, Salles AL, Piercy CE, Grove BH, Erikson ME, et al. Relaxin induces an extracellular matrix-degrading phenotype in human lung fibroblasts in vitro and inhibits lung fibrosis in a murine model in vivo. J Clin Invest. 1996;98:2739–2745. doi: 10.1172/JCI119099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unemori EN, Lewis M, Constant J, Arnold G, Grove BH, Normand J, et al. Relaxin induces vascular endothelial growth factor expression and angiogenesis selectively at wound sites. Wound Repair Regen. 2000;8:361–370. doi: 10.1111/j.1524-475x.2000.00361.x. [DOI] [PubMed] [Google Scholar]
- van der Westhuizen ET, Werry TD, Sexton PM, Summers RJ. The relaxin family peptide receptor 3 activates extracellular signal-regulated kinase 1/2 through a protein kinase C-dependent mechanism. Mol Pharmacol. 2007;71:1618–1629. doi: 10.1124/mol.106.032763. [DOI] [PubMed] [Google Scholar]
- van der Westhuizen ET, Christopoulos A, Sexton PM, Wade JD, Summers RJ. H2 relaxin is a biased ligand relative to H3 relaxin at the relaxin family peptide receptor 3 (RXFP3) Mol Pharmacol. 2010;77:759–772. doi: 10.1124/mol.109.061432. [DOI] [PubMed] [Google Scholar]
- Xu KY, Huso DL, Dawson TM, Bredt DS, Becker LC. Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proc Natl Acad Sci U S A. 1999;96:657–662. doi: 10.1073/pnas.96.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Chakravorty A, Bathgate RAD, Dart AM, Du X-J. Relaxin therapy reverses large artery remodeling and improves arterial compliance in senescent spontaneously hypertensive rats. Hypertension. 2010;55:1260–1266. doi: 10.1161/HYPERTENSIONAHA.109.149369. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Liu S-H, Erikson M, Lewis M, Unemori E. Relaxin activates the MAP kinase pathway in human endometrial stromal cells. J Cell Biochem. 2002;85:536–544. doi: 10.1002/jcb.10150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Serelaxin stimulation did not cause a functional response in HUAECs. HUAECs do not express RXFP1 receptors at the cell surface (Figure 1), and in accord with this finding, addition of serelaxin (30 nM) did not affect (a) cAMP accumulation (n = 4), (b) cGMP accumulation (n = 4) or (c) pERK1/2 (n = 3). Each point represents the mean ± SEM of four to six independent experiments performed in duplicate.
Figure S2 Time course studies of serelaxin in human primary vascular and cardiac cells. Serelaxin (30 nM) treatment increased (a) cAMP accumulation in HUVEC (n = 7), HUVSMC (n = 3) and HUASMC (n = 4), but not in HCF (n = 3). Serelaxin (30 nM) treatment also increased cGMP accumulation but in all cells including HUVEC (n = 6), HUVSMC (n = 5), HUASMC (n = 3) and HCF (n = 3). CRCs were conducted at 30 min for each cell type. Similarly, serelaxin (30 nM) increased pERK1/2 in HUVEC (n = 9), HUVSMC (n = 4), HUASMC (n = 4) and HCF (n = 3) that peaked between 5 and 10 min after which it returned to baseline. However, in HUVEC, pERK1/2 plateaued after 10 min and remained at that level for up to 60 min. CRCs for pERK1/2 were conducted at 10 min for HUVEC, HUVSMC and HCF but at 5 min for HUASMC. Each point represents the mean ± SEM performed in duplicate.
Figure S3 The effects of serelaxin stimulation on pERK1/2 in human primary vascular cells. Serelaxin stimulation (10 min for HUVECs, HUVSMCs, HCFs and 5 min for HUASMCs) increased pERK1/2 in (a) HUVECs (n = 5), (b) HUVSMCs (n = 6), (c) HUASMCs (n = 6) and (d) HCFs (n = 5). The serelaxin CRCs were bell-shaped for HUVECs, HUVSMCs and HCFs, but sigmoidal for HUASMCs. PTX (50 ng·mL−1, 18 h) and wortmannin (100 nM, 30 min) pretreatment significantly inhibited serelaxin (30 nM)-mediated pERK1/2 in each cell type. Statistical significance was assessed using a one-way anova with a Dunnett's post hoc test compared with serelaxin alone: *P < 0.05 and **P < 0.01.
Figure S4 The effect of suramin on the dose–response relationship of serelaxin in primary human smooth vascular cells. Pretreatment of suramin (10 μM, 30 min) to HUASMC had no effect on (a) cAMP (n = 3) or (c) cGMP accumulation (n = 3). Similarly, suramin (10 μM, 30 min) pretreatment to HUVSMC had no effect on the dose–response relationship of serelaxin-mediated (b) cAMP (n = 3) or (d) cGMP accumulation (n = 3). Similarly, pretreatment of HUASMC and HUVSMC with 100 μM suramin (30 min) reduced the E-max of (a, b) cAMP (n = 3) and (c, d) cGMP accumulation (n = 3).
Table S1 The binding affinity of serelaxin (pKi) and the potency (pEC50) of serelaxin for cAMP accumulation, cGMP accumulation and pERK1/2 in human primary vascular cells. Values in parentheses refer to n experiments.