

Abstract
Background and Purpose
Both cannabinoid CB1 and adenosine A2A receptors (CB1 receptors and A2A receptors) control synaptic transmission at corticostriatal synapses, with great therapeutic importance for neurological and psychiatric disorders. A postsynaptic CB1−A2A receptor interaction has already been elucidated, but the presynaptic A2A receptor-mediated control of presynaptic neuromodulation by CB1 receptors remains to be defined. Because the corticostriatal terminals provide the major input to the basal ganglia, understanding the interactive nature of converging neuromodulation on them will provide us with novel powerful tools to understand the physiology of corticostriatal synaptic transmission and interpret changes associated with pathological conditions.
Experimental Approach
Pharmacological manipulation of CB1 and A2A receptors was carried out in brain nerve terminals isolated from rats and mice, using flow synaptometry, immunoprecipitation, radioligand binding, ATP and glutamate release measurement. Whole-cell patch-clamp recordings were made in horizontal corticostriatal slices.
Key Results
Flow synaptometry showed that A2A receptors were extensively co-localized with CB1 receptor-immunopositive corticostriatal terminals and A2A receptors co-immunoprecipitated CB1 receptors in these purified terminals. A2A receptor activation decreased CB1 receptor radioligand binding and decreased the CB1 receptor-mediated inhibition of high-K+-evoked glutamate release in corticostriatal terminals. Accordingly, A2A receptor activation prevented CB1 receptor-mediated paired-pulse facilitation and attenuated the CB1 receptor-mediated inhibition of synaptic transmission in glutamatergic synapses of corticostriatal slices.
Conclusions and Implications
Activation of presynaptic A2A receptors dampened CB1 receptor-mediated inhibition of corticostriatal terminals. This constitutes a thus far unrecognized mechanism to modulate the potent CB1 receptor-mediated presynaptic inhibition, allowing frequency-dependent enhancement of synaptic efficacy at corticostriatal synapses.
Tables of Links
| TARGETS | |
|---|---|
| GPCRsa | |
| Adenosine A1 receptors | |
| Adenosine A2A receptors | |
| Cannabinoid CB1 receptors | |
| Dopamine D2 receptors | |
| Enzymesb | |
| ADA, adenosine deaminase | |
| Diacylglycerol lipase | |
| Ecto 5′-nucleotidase, NT5E | |
| Glutamate decarboxylase | |
| Transporterc | |
| vGluT1, vesicular glutamate transporter 1, SLC17A7 |
| LIGANDS | |
|---|---|
| [3H]SR141716A | |
| Adenosine | |
| AM251 | |
| Aminooxyacetic acid | |
| ATP | |
| Brain-derived neurotrophic factor | |
| CGS21680 | |
| Fibroblast growth factor | |
| Glial cell-derived neurotrophic factor | |
| Glutamate | |
| WIN55212-2 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a,b,c).
Introduction
The corticostriatal pathway is a massive projection linking virtually the entire neocortex with the striatum – the latter being considered as the major input site of the basal ganglia (Goldman-Rakic and Selemon, 1986; Bolam et al., 2000). The principal neurons of the striatum – medium spiny neurons (MSNs) – integrate synaptic information from functionally diverse cortical regions to process signals controlling goal-directed behaviours and habits (Graybiel, 1995; Yin and Knowlton, 2006). As a gateway to trigger the recruitment of striatal circuits, alterations in the strength of the synaptic connections between the cortex and striatum play a critical role in these adaptive behavioural changes (Di Filippo et al., 2009).
GPCRs, such as the cannabinoid CB1 receptor, are key determinants of synaptic efficacy changes in corticostriatal synapses (Lovinger, 2010). Accordingly, the manipulation of the endocannabinoid system has a profound impact on striatal-dependent behavioural responses (El Manira and Kyriakatos, 2010; Katona and Freund, 2012). Another major controller of striatal function is the adenosine A2A receptor (Schiffmann et al., 2007). These A2A receptors are abundantly located in the dendritic spines of MSNs (Svenningsson et al., 1999) and are also present presynaptically, controlling glutamate release (Ciruela et al., 2006; Quiroz et al., 2009) and corticostriatal plasticity (D'Alcantara et al., 2001; Flajolet et al., 2008). Interestingly, A2A receptors seem to mainly act as a fine-tuning system, adapting the efficiency of different other modulator systems (Sebastião and Ribeiro, 2000; Ferré et al., 2011). The activation of striatal A2A receptors results in Gs-mediated accumulation of cAMP (Golf in the MSNs), in contrast to the stimulation of the other abundant striatal adenosine receptor, the inhibitory Gi/o-coupled adenosine A1 receptors (Dunwiddie and Masino, 2001). In physiological conditions, low-frequency (0.1 < Hz) neuronal activity is accompanied with a modest generation of adenosine, most likely from the metabolism of ATP of astrocytic origin, which exerts tonic inhibition of neighbouring excitatory synapses via A1 receptors (Cunha, 2008). This dominant form of paracrine adenosinergic neuromodulation probably serves to decrease the noise of the system at resting state (Cunha, 2008). In contrast, under high-frequency discharge of the nerve terminals, the ecto-5′-nucleotidase-mediated degradation of ATP, co-released from synaptic vesicles (Sperlágh and Vizi, 1996), will build up synaptic adenosine levels that are sufficient for autocrine A2A receptor activation (Cunha, 2008; Augusto et al., 2013). Pathological conditions such as ischaemia can also increase extracellular adenosine levels via outward transport, which are enough to stimulate both A1 and A2A receptors (Gomes et al., 2011).
A2A receptors have been reported to tightly regulate the endocannabinoid neuromodulation system in the striatum, as shown by the A2A–CB1 receptor interactions in the control of motor dysfunction (Ferré et al., 2010; Lerner et al., 2010; Tozzi et al., 2012) and addiction (Soria et al., 2004; Yao et al., 2006; Rossi et al., 2010; Justinová et al., 2011). This is reinforced by the reported heteromerization of A2A receptors with CB1 receptors that was demonstrated in heterologous expression systems and in the striatum (Carriba et al., 2007). However, this A2A–CB1 receptor interaction is mostly interpreted as resulting from a postsynaptic interaction (Yao et al., 2006; Rossi et al., 2010; Cerri et al., 2014; Pinna et al., 2014), whereas the predominant localization of CB1 receptors is presynaptic in the striatum (Köfalvi et al., 2005; Uchigashima et al., 2007). A possible presynaptic interaction between A2A and CB1 receptors has also been proposed to control the motor-depressant and addictive effects of cannabinoids (Ferré et al., 2010; Martíre et al., 2011; Justinová et al., 2014), but detailed experimental evidence is lacking. Here, we set out to further expand our previous observations (Martíre et al., 2011) now using selective presynaptic techniques and combining refined immunological, radioligand binding and functional assays to directly investigate A2A–CB1 receptor interactions in glutamatergic nerve terminals of corticostriatal synapses.
Methods
Animals
All animal care and experimental procedures were in accordance with the principles and procedures outlined as replacement, refinement and reduction of animals in research ‘3Rs’ in the guidelines of the European Union (86/609/EEC), Federation for Laboratory Animal Science Associations and the National Centre for the 3Rs (Kilkenny et al, 2010) and were approved by the Animal Care Committee of the Center for Neuroscience and Cell Biology of Coimbra and by the Centre for Interdisciplinary Research in Biology in College de France. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al, 2010; McGrath et al., 2010). A total of 70 animals were used in the experiments described here.
Animals were housed under a 12 h light on/off cycle with ad libitum access to food and water. Forty-nine male Wistar rats (180–240 g, 8–10-week-old) were purchased from Charles River (Barcelona, Spain) and six Oncins France Strain A (OFA) rats (16–22 postnatal days) from Charles River (L'Arbresle, France). Five pairs of A2A receptor and CB1 receptor null-mutant (knockout, KO) male mice on CD-1 background (Ledent et al., 1997; 1999,) and their wild-type (WT) littermates (35–45 g, 8–12-week-old) were also used and were genotyped by tail snips.
Synaptosomal preparations
Experimental procedures were carried out as previously described by Ferreira et al., (2009). Briefly, the animals were decapitated under halothane anaesthesia, and their brains were quickly removed into ice-cold 0.32 M sucrose solution containing 10 mM HEPES, 1 mM EDTA and 1/500 v/v protease inhibitor cocktail (Sigma-Aldrich, St Louis, MO, USA), pH 7.4. The pair of striata was rapidly dissected and homogenized instantly with a glass potter and a teflon homogenizer, and centrifuged at 3000× g for 5 min. The supernatant was collected and centrifuged at 13 000× g for 10 min to obtain the P2 crude synaptosomal fraction. For immunolabelling and flow cytometry analysis, the P2 fraction was further purified on a discontinuous Percoll gradient (3, 10 and 23%), as described in Köfalvi et al. (2005). The purified synaptosomes were kept at −80°C until use.
Immunolabelling and flow cytometric analysis of purified nerve terminals
Immunochemical labelling was performed according to a method for staining of intracellular antigens (Schmid et al., 1991; Gylys et al., 2000), with little modification. Briefly, purified nerve terminals were fixed in 1 mL of 0.25% paraformaldehyde in PBS (135 mM NaCl, 1.3 mM KCl, 3.2 mM NaH2PO4 and 0.5 mM KH2PO4) for 1 h at 4°C and then centrifuged at 3000× g for 3 min at 4°C. For permeabilization, the pellets were incubated in PBS with 0.2% Tween-20 for 15 min at 37°C and then centrifuged at 3000× g for 3 min. The pellets were then resuspended in PBS for immunolabelling. Primary and secondary antibodies (Supporting Information Table S1) were diluted in PBS containing 2% normal goat serum (Vector Laboratories, Burlingame, CA, USA). For validation/titration of the primary antibodies, see Supporting Information Fig. S1. Incubation volume was 100 μL and incubation time was 30 min at 4°C for both the primary and the secondary antibodies. Each incubation was followed by three washes in PBS with 0.2% Tween-20 and centrifugation at 3000× g for 3 min. The samples were resuspended in filtered PBS for flow-synaptometric analysis.
Analysis was performed using a FACSCalibur flow cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ, USA – equipped with a 488 nm argon-ion laser). Sample flow was set at 350 events per second; 50 000 ungated events were collected for analysis. A threshold was set on forward light scatter to exclude debris. To correct for spectral overlap during multicolour flow cytometry experiments, colour compensation was performed. Offline data analysis was performed using BD Cell Quest Pro software (Becton, Dickinson and Company). For detailed description see Supporting Information Fig. S1.
Receptor binding
Synaptosomal membranes were prepared as previously described (Rebola et al., 2005) upon resuspensions of P2 synaptosomes in 2 mL of ice-cold assay solution [50 mM Tris HCl, 3 mM MgCl2, 1 μM CaCl2, 2 mM EDTA and protease inhibitor cocktail (Sigma-Aldrich), pH 7.4]. Single point CB1 receptor binding experiment with 3.82 ± 0.29 nM (n = 7) of the CB1 receptor antagonist/inverse agonist, [3H]SR141716A, was carried out as before (Ferreira et al., 2012), with 30 min pre-incubation in the presence of adenosine deaminase (ADA, 2 U·mL−1) and of the diacylglycerol lipase inhibitor, OMDM188 (300 nM, a kind gift of Dr Vincenzo Di Marzo). Non-specific binding was determined using the CB1 receptor antagonist/inverse agonist AM251 (1 μM). Each of the seven independent assays was carried out on synaptic membranes derived from two rats, altogether 14 rats, and assayed in quadruplicate (28 filters per condition). The tritium content of each sample was counted using a Tricarb 2900TR β-counter (Perkin Elmer, Lisbon, Portugal). The specific binding was expressed as amount of ligand specifically bound per milligram of protein.
Immunoprecipitation
Immunoprecipitation assays were carried out in both crude and Percoll-purified rat striatal synaptosomal fractions (pooled from three rats to obtain enough material), as previously described (Marques et al., 2013). Briefly, protein extracts were incubated with 50% protein G-Sepharose bead slurry (GE Healthcare, Little Chalfont, UK) for 3 h at 4°C to eliminate non-specific binding. After incubation, the pre-cleared supernatants containing 1 mg protein were incubated overnight with rotation at 4°C with a mouse anti-A2A receptor antiserum (Merck-Millipore, Darmstadt, Germany) pre-coupled covalently to protein G-sepharose (GE Healthcare), in the presence of 1% BSA (Sigma-Aldrich) and protease inhibitors (Roche Diagnostics, Amadora, Portugal). The beads were washed three times with isolation buffer containing 150 mM KCl, 20 mM MOPS and 1% Triton X-100 (pH 7.4) and resuspended in six-times diluted SDS-PAGE sample buffer [0.35 M Tris, 30% glycerol, 10% SDS, 0.6 M dithiothreitol, 0.012% bromophenol blue (pH 6.8)]. Bound proteins eluted from the immune complexes were denatured by heating to 95°C for 5 min and then separated by electrophoresis on SDS-PAGE gels. Proteins were then electrotransferred onto nitrocellulose membranes (Amersham, Little Chalfont, UK) and probed with rabbit anti-CB1 receptor (Supporting Information Table S1) and mouse anti-A2A receptor (Supporting Information Table S1) antibodies, diluted in Tris-buffered saline supplemented with Tween-20 (0.1% v/v) and BSA (5% m·v−1). Immunoreactivity was visualized using HRP-conjugated goat anti-rabbit or anti-mouse secondary antibodies (Thermo Scientific Pierce, Rockford, IL, USA) with a subsequent incubation with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific Pierce) and the images were acquired using Versadoc3000 apparatus and analysed with ImageLab software (Bio-Rad, Amadora, Portugal). A negative control containing the same amount of mouse IgG2A instead of the mouse anti-A2A receptor antibody was run in parallel for each experiment.
[14C]glutamate release
Experiments were carried out with slight modifications to previous publications (Köfalvi et al., 2005), which are: the synaptosomes were loaded with [14C]-U-glutamate (20 μM) for 10 min and the superfused synaptosomes, trapped in the 16 microvolume chamber release system, were stimulated with 30 mM KCl twice for 1 min (S1, S2), with a 10 min interval. All Krebs-HEPES solutions used for this assay contained the glutamate decarboxylase inhibitor aminooxyacetic acid (100 μM) to prevent [14C]glutamate degradation. For detailed description, see Köfalvi et al. (2005) and Ferreira et al. (2009). The validation of the A1 receptor, A2A receptor as well as CB1 receptor -mediated neuromodulation in Wistar rat and CD-1 mouse striatal synaptosomes is summarized in Supporting Information Table S2.
ATP release assay from striatal synaptosomes
ATP quantification was carried out in 96-well plates, using a Perkin Elmer Victor3 multi-label plate reader in luminometer mode. The ATP assay mix (Sigma-Aldrich) used by us allows quantitative bioluminescent determination of very low ATP levels ranging from 2 × 10−12 to 8 × 10−5 M, according to Navizet et al. (2011). Solutions used were (i) basal saline medium (in mM): 115 NaCl, 3 KCl, 1.2 KH2PO4, 25 HEPES, 10 glucose, 1.2 MgSO4, 1 CaCl2, pH = 7.4; and (ii) potassium saline medium (in mM): 118 KCl, 1.2 KH2PO4, 25 HEPES, 10 glucose, 1.2 MgSO4, 1 CaCl2, pH = 7.4. A 150 μL aliquot of basal saline medium with compounds to be tested or their vehicle, DMSO (0.1% v/v), 15 μL of ATP assay mix and 35 μL synaptosomal suspension (∼1 mg·mL−1) provided the 200 μL final reaction volume. This mixture was incubated at 25°C for 3 min in an Eppendorf tube to ensure functional recovery of the sample and then it was transferred into a well of the plate at 25°C, inside the reader. Afterwards, a kinetic protocol was initiated with the duration of 140 s. During the first 60 s, a stable baseline was recorded corresponding to the basal extrasynaptic ATP level. Subsequently, synaptosomes were stimulated with KCl (30 mM) or were challenged only with the same amount of NaCl serving as osmotic control. Average readings in the presence of high NaCl were subtracted from KCl-stimulated average readings.
Electrophysiology
Whole-cell patch-clamp recordings from MSNs were made in horizontal brain slices from OFA rats (Figure 5A) (postnatal days 15–22), as previously described (Fino et al., 2005). Briefly, the artificial CSF contained (in mM): 125 NaCl, 2.5 KCl, 25 glucose, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2 and 10 μM pyruvic acid bubbled with 95% O2 and 5% CO2; the borosilicate glass pipettes (6–8 MΩ) contained (in mM): 105 potassium gluconate, 30 KCl, 10 HEPES, 10 phosphocreatine, 4 ATP-Mg, 0.3 GTP-Na, 0.3 EGTA (adjusted to pH 7.35 with KOH). All the experiments were carried in the presence of 50 μM picrotoxin (Sigma-Aldrich). Electrical stimulation was performed with a bipolar electrode (Phymep, Paris, France), placed at the layer 5 of the somatosensory cerebral cortex, by applying a monophasic and constant current (duration: 100–150 μs) (ISO-Flex stimulator controlled by a Master-8, A.M.P.I., Jerusalem, Israel). All recordings were performed at 32°C using a temperature control system (Bath-controller V, Luigs & Neumann, Ratingen, Germany). Individual neurons were identified using infrared differential interference contrast microscopy with CCD camera (Hamamatsu C2400-07; Hamamatsu, Japan). Signals were amplified using an EPC9-2 amplifier (HEKA Elektronik, Lambrecht, Germany). The range of access resistance was 80–200 MΩ. The liquid junction potential was calculated and corrected. Voltage-clamp recordings were filtered at 5 kHz and sampled at 10 kHz using the program Pulse-8.53 (HEKA Elektronik). The series resistance was compensated at 75–80% and variation of series resistance above 20% led to the rejection of the experiment. Offline analysis was performed using Igor-Pro 6.0.3 (Wavemetrics, Lake Oswego, OR, USA).
Figure 5.
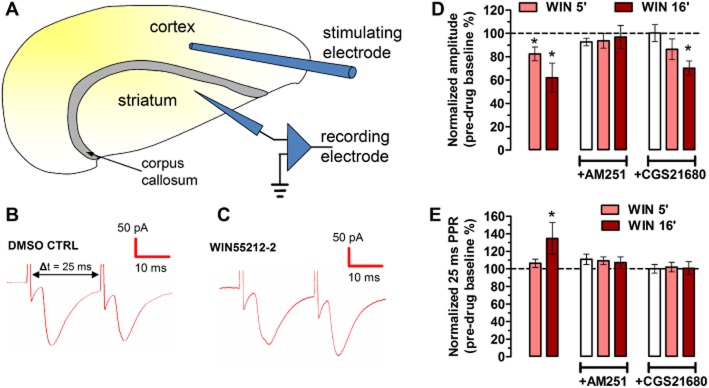
CB1 receptor activation decreases basal synaptic transmission and increases PPR in rat corticostriatal afferents, which is attenuated or prevented by the A2A receptor agonist CGS21680 (30 nM). (A) The whole-cell patch-clamp configuration in horizontal corticostriatal slices from 15–22-day-old rats, with stimulation in the layer V of the adjacent neocortex and recording in the dorsolateral striatum. (B, C) Representative paired-pulse traces (25 ms interpulse interval) in the presence of WIN55212-2 (500 nM) and its vehicle, DMSO. (D) Bar graphs representing the EPSC amplitude values normalized to the pretreatment period after 5 and 16 min of WIN55212-2 perfusion. WIN55212-2 decreased EPSC amplitude at both time points, which was sensitive to AM251, which per se had no effect. Ten minutes of pretreatment with CGS21680 attenuated the CB1 receptor-mediated inhibition of EPSC amplitudes. CGS21680 did not produce effects on its own. All bars are mean ± SEM derived from n ≥ 6 animals. *P < 0.05 versus 100% (pretreatment CTRL). (E) Bar graphs representing the normalized PPR (the second response in relation to the first response) at 5 and 16 min after the beginning of WIN55212-2 perfusion. The WIN55212-2-induced increase (*P < 0.05) in the PPR was prevented both by AM251 and CGS21680, which had no effects per se.
Data analysis
Raw effect data from [14C]glutamate and ATP release assays and from electrophysiology were normalized to the appropriate control of the same experiment. These normalized data were tested for normality by the Kolmogorov–Smirnov normality test. Statistical significance was calculated by one-sample t-test against the hypothetical value of 100 (as 100%, i.e. vehicle control). Pairs of treatment or condition groups were compared with Student's paired t-test, while the antibody titration curves were compared with two-way anova between the WT and the KO mice. P < 0.05 was accepted as significant difference.
Materials
1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-(1-piperidyl)pyrazole-3-carboxamide (AM251), (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-napthalenylmethanone (WIN55212-2) and (6aR,10aR)-6a,7,10,10a-tetrahydro-3-[5-(1H-imidazol-1-yl)-1,1-dimethylpentyl]-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-1-ol (O-2545) were purchased from Abcam Biochemicals, Cambridge, UK; 3-[4-[2-[[6-amino-9-[(2R,3R,4S,5S)-5-(ethylcarbamoyl)-3,4-dihydroxy-oxolan-2-yl]purin-2-yl]amino]ethyl]phenyl]-propanoic acid (CGS21680) was purchased from Tocris Bioscience, Bristol, UK; (DMSO, MOPS, aminooxyacetic acid, halothane, HEPES, Percoll, ADA, BSA and sucrose were purchased from Sigma-Aldrich. [3H]SR141716A (specific activity: 60 Ci·mmol−1; 1 mCi·mL−1) and [14C]-U-glutamate (specific activity: 200 mCi·mmol−1; 0.1 mCi·mL−1) were purchased from American Radiolabeled Chemicals (St Louis, MO, USA). All other reagents were purchased from Merck Biosciences (Darmstadt, Germany).
Results
A2A and CB1 receptors co-localize in corticostriatal glutamatergic nerve terminals
A basic piece of evidence in a study of the functional cross-talk between CB1 and A2A receptors in corticostriatal glutamatergic terminals is to demonstrate their simultaneous co-localization at these sites. Thus, we analysed striatal synaptosomes via flow synaptometry (Figure 1A): we double-labelled them for synaptophysin and the vesicular glutamate transporter vGluT1 (Figure 1B) or triple-labelled for vGluT1, CB1 and A2A receptors (Figure 1C–E). Concerning the double labelling, 85.2 ± 2.5% (n = 3) of the analysed particles were positive for synaptophysin. Of these particles, 58.5 ± 3.5% were positive for vGluT1 (Figure 1B). Triple labelling allowed to precisely quantify that 49.4 ± 3.3% of the vGluT1-positive terminals also stained for CB1 receptors (n = 3) (Figure 1C) and 30.9 ± 1.7% bore A2A receptors (n = 3) (Figure 1D). Additionally, 47.1 ± 2.7% of the CB1 receptor-positive terminals were also positive for A2A receptors (n = 3) (Figure 1E). In other words, as supported by Figure 1E, 75.1 ± 3.6% (n = 3) of the A2A receptor-positive terminals were also positive for CB1 receptors in vGlut1-positive nerve terminals.
Figure 1.

Flow-synaptometric analysis of immunolabelled Percoll-gradient purified striatal synaptosomes. (A) Representative flow synaptometry plot of striatal synaptosomes for size (forward scatter is proportional to the particle diameter) and for complexity/granularity (side scatter). (B) Representative plot and statistics of synaptosomes double-labelled for synaptophysin (a marker of synaptosomes) and vGluT1 (a marker of corticostriatal terminals). (C and D) Representative plots and statistics of synaptosomes labelled for vGluT1/CB1 receptor (C) and vGluT1/A2A receptor (D) respectively (the two graphs are derived from the same triple-labelled sample). (E) Representative plot and statistics of vGluT1-positive synaptosomes expressing CB1 and A2A receptors. Note that most of the nerve terminals staining positive for A2A receptors (A2AR) were also positive for CB1 receptors (CB1R).
Figure 2.
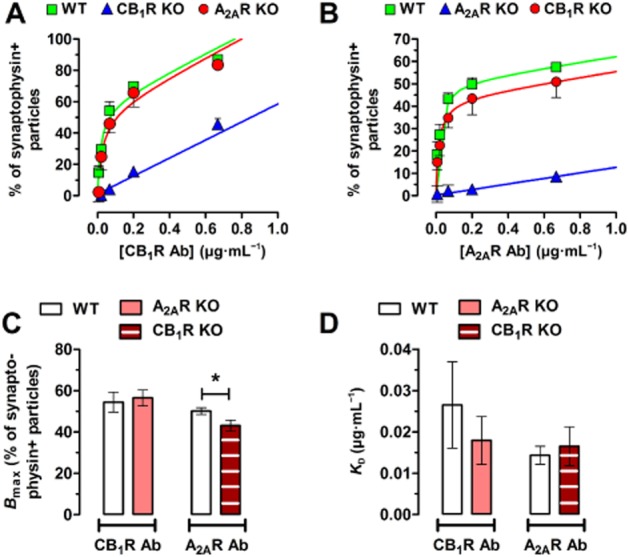
(A) Total binding isotherms of the anti-CB1 receptor antibody in the corticostriatal terminals of the WT versus the A2A receptor KO mice (A2AR KO), while the CB1 receptor KO mice (CB1 KO) display antibody binding of non-specific nature only. (B) Total binding isotherms of the anti-A2A receptor antibody in the corticostriatal terminals of the WT versus the CB1 receptor KO mice, while the A2A receptor KO mice display antibody binding of non-specific nature only. (C) Bar graphs representing the mean values of the maximum binding sites (Bmax) of the anti-CB1 and anti-A2A receptor antibodies. *P < 0.05. (D) Bar graphs representing the mean changes of the KD of the anti-CB1 and anti-A2A receptor antibodies. Bars represent the mean ± SEM of five individual experiments.
Interestingly, the selectivity analysis of the CB1 receptor (Figure 2A) and the A2A receptor (Figure 2B) antibodies in the CB1 receptor and A2A receptor KO mice and their WT littermates (Supporting Information Figure S1) suggested that CB1 receptors help the localization of A2A receptors to the presynapse. We found that the titration (saturation binding) curve of the anti-A2A receptor antibody showed a decrease of the number of binding sites in vGluT1-positive terminals of CB1 receptor KO mice, compared with WT mice (n = 5; P < 0.05; Figure 2C), which was not accompanied by any change in antibody affinities to its respective receptor (Figure 2D).
A2A receptors co-immunoprecipitate with CB1 receptors in striatal nerve terminals
The extensive co-localization of CB1 and A2A receptors in nerve terminals hints at a possible physical interaction of A2A and CB1 receptors, as reported in cultured cells and in striatal homogenates (Carriba et al., 2007). To test this hypothesis, we performed immunoprecipitation with an anti-A2A receptor antibody in both crude (the so-called P2 fraction) and in Percoll-purified striatal nerve terminals. Subsequent immunoblot analyses of the composition of the immunoprecipitates demonstrated CB1 receptors at the expected ∼51 kDa MW, with particular enrichment in the A2A receptor-immunoprecipitated fractions, compared with the initial (non-immunoprecipitated) homogenate (Figure 3).
Figure 3.
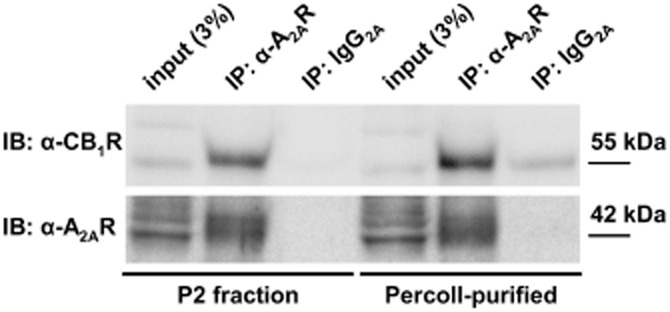
Co-immunoprecipitation (IP) of A2A and CB1 receptors in crude (P2 fraction) and Percoll-purified striatal synaptosomes. CB1 receptors were readily detected and enriched in complexes immunoprecipitated with the anti-A2A receptor antibody, but not with mouse IgG2A, either in P2 or Percoll-purified fractions of rat striatal synaptosomes. The label ‘input (3%)’ refers to the protein load corresponding to 3% of the protein quantity used for the IP.
The strong co-localization and co-immunoprecipitation data prompted us to test if A2A receptor activation affected CB1 receptor binding. Experiments were carried out under conditions of minimal levels of endogenous adenosine and 2-arachidonoyl-glycerol levels, after treatment with ADA and OMDM188 (see Methods section). A single-point receptor binding assay showed that CGS21680 (30 nM) decreased the binding of the radiolabelled CB1 receptor-selective ligand [3H]SR141716A to synaptosomal membranes from 3.23 ± 0.17 to 2.68 ± 0.18 pmol·mg−1 of protein (n = 7 in quadruplicates, P < 0.05) (figure not shown).
A2A receptor activation decreases the potency of CB1 receptor agonists in striatal glutamatergic terminals
Repetitive (S1 and S2) stimulation with high K+ (30 mM for 1 min) triggered the release of similar amounts of [14C]glutamate (Figure 4A) in a Ca2+-dependent manner: the first stimulation-evoked release (S1) was 54.1 ± 5.8% smaller in Ca2+-free condition (10 mM MgCl2 combined with 100 nM CaCl2; n = 6, P < 0.001) when compared with that under normal condition. The CB1 receptor agonists, WIN55212-2 (0.1–3 μM; n ≥ 6) or O-2545 (300 nM; n = 5), added 4 min before the second stimulus (S2), decreased the S2/S1 ratio (i.e. the second stimulus-evoked release) between 10.8 ± 3.8 to 45.4 ± 4.5%, depending on the concentration used (P < 0.05) (Figure 4A and B). The effect of WIN55212-2 was prevented by the selective CB1 receptor antagonist/inverse agonist AM251 (1 μM; n = 6) present during the pre-perfusion period (Figure 4B). In contrast, neither the A2A receptor agonist, CGS21680 (30 nM) (Figure 4B), nor two A2A receptor antagonists had significant effect on the release of [14C]glutamate evoked by either 15 or 30 mM K+ either in the rat or in the CD-1 mouse striatal synaptosomes (see Supporting Information Table S2). This indicates that a possible occlusion of the effects of CGS21680, by either an excessive K+ stimulus or by endogenous activation of A2A receptors did not occur, in these conditions.
Figure 4.
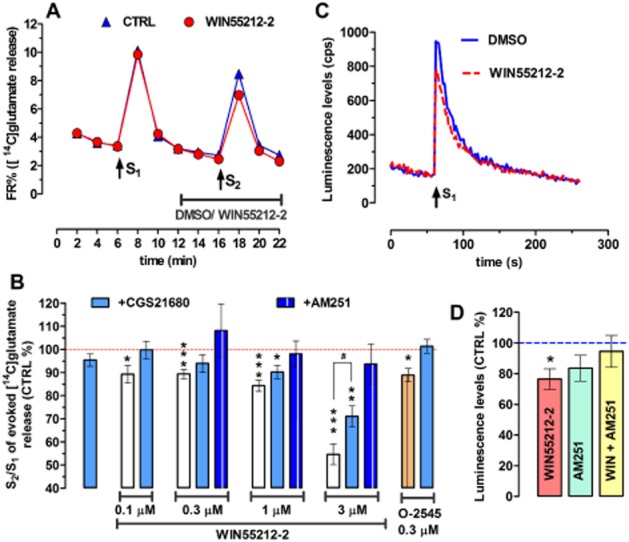
(A) Time course of [14C]glutamate release (as fractional release; FR%) from rat striatal synaptosomes following treatment with WIN55212-2 (1 μM) and the respective control (CTRL). Release was stimulated by high-K+ (30 mM; 2 × 1 min), marked as S1 and S2. WIN55212-2 was added as indicated by the horizontal line. Data are mean ± SEM of n = 21 independent observations in duplicate. Missing error bars are inside the symbols. (B) Bar graph representing the effect of A2A receptor activation on the CB1 receptor-induced inhibition of high-K+-evoked release of [14C]glutamate. The Y axis represents the effect of the treatment on the S2/S1 ratio, normalized to the vehicle control. WIN55212-2 per se significantly inhibited the release of [14C]glutamate in all concentrations (0.1, 0.3, 1 and 3 μM). The inhibitory effect of WIN55212-2 on the S2/S1 ratio was prevented by the CB1 receptor antagonist AM251 (1 μM, applied during the preperfusion period, i.e. before S1). The selective A2A receptor agonist CGS216880 (30 nM, co-applied with WIN55212-2), which per se had no effect on the high-K+-evoked release of [14C]glutamate, prevented the action of WIN55212-2 at 0.1 and 0.3 μM and significantly attenuated the action of WIN55212-2 at 3 μM. Similarly, O-2545 (0.3 μM), another CB1 receptor agonist, inhibited the release of [14C]glutamate and CGS21680 (30 nM) prevented that action. All bars are mean ± SEM derived from n ≥ 6 animals. *P < 0.05; **P < 0.01; ***P < 0.001 versus DMSO control (red dashed line) and #P < 0.05 versus without CGS21680. (C) Representative time course of high-K+-evoked (32 mM) release of ATP from striatal nerve terminals. Stimulus with KCl is marked as S1. Consistent with ATP being co-released with glutamate, activation of the CB1 receptors by WIN55212-2 (1 μM) also inhibits the KCl-evoked release of ATP. (D) As represented in the bar graphs, the CB1 receptor-mediated inhibition of ATP release (n = 6; *P < 0.05) was prevented by the CB1 receptor antagonist, AM251 (1 μM), which per se had no effect.
However, CGS21680, when co-applied with WIN55212-2 (n ≥ 6) or O-2545 (n = 5), prevented the inhibition of the 30 mM K+-evoked release of [14C]glutamate by nanomolar concentrations of these CB1 receptor agonists (Figure 4B) [P > 0.05 for CGS21680+WIN55212-2 (0.1 and 0.3 μM) vs. DMSO control]. CGS21680 also significantly attenuated the inhibition by WIN55212-2 at 3 μM (n = 6) (Figure 4B) [P < 0.05 for CGS21680+WIN55212-2 (3 μM) vs. WIN55212-2 (3 μM) alone].
CB1 receptor activation attenuates ATP release in nerve terminals of the rat striatum
Besides being a source of phasic adenosine levels which activate the A2A receptors (Augusto et al. 2013), ATP is also a co-transmitter to glutamate (Burnstock, 2013). Based on the earlier data on [14C]glutamate release, we expected CB1 receptor activation to also suppress the release of ATP from striatal nerve terminals. Indeed, we observed that WIN55212-2 (1 μM) inhibited the high-K+-evoked release of ATP by 24.4 ± 6.7% (n = 6, P < 0.05) (Figure 4C and D), which was again prevented by the CB1 receptor antagonist, AM251 (1 μM), which per se did not alter the evoked release of ATP (P > 0.05) (Figure 4D).
A2A receptor activation inhibits CB1 receptor-mediated depression of glutamatergic transmission in the dorsolateral striatum
It is well known that CB1 receptors depress corticostriatal glutamatergic transmission (Gerdeman and Lovinger, 2001). Accordingly, the CB1 receptor agonist WIN55212-2 (500 nM) reduced the amplitude of evoked monosynaptic EPSCs by 17.5 ± 5.0% after 5 min and by 37.9 ± 12.5% after 16 min (n = 8, P < 0.05) (Figure 5B-D). The CB1 receptor antagonist AM251 (500 nM) prevented this WIN55212-2-induced depression of synaptic transmission while having no effect alone (n = 5) (Figure 5D).
A2A receptor activation by CGS21680 (30 nM) also prevented the WIN55212-2-induced synaptic depression in the first 5 min (n = 6, P > 0.05) but not at the later time point, although the CB1 receptor-mediated inhibition was smaller in the absence (37.9 ± 12.5%) than in the presence of CGS21680 (29.7 ± 6.1%, P < 0.05) (Figure 5D). Of note, CGS21680 per se did not alter basal synaptic transmission (P > 0.05) (Figure 5D).
A2A receptor activation inhibits CB1 receptor-mediated increase in paired-pulse ratio (PPR)
While changes in synaptic transmission can be a result of both presynaptic and postsynaptic events, an increase in the PPR reflects presynaptic mechanisms (Schulz et al., 1994; Gerdeman and Lovinger, 2001). Thus, we analysed the monosynaptic EPSC ratios of paired stimuli, delivered with a 25 ms interval (Figure 5A-C,E). The drug-naïve PPR mean value was 0.76 ± 0.03 (n = 22 cells from six rats) (Figure 5B and E). As shown in Figure 5C and E, application of WIN55212-2 (500 nM) for 16 min significantly increased the PPR (by 34.6 ± 17.9%, n = 9, P < 0.05), while a 5 min superfusion period with WIN55212-2 was not enough to cause a significant PPR increase (by 6.4 ± 4.7%, n = 9, P > 0.05). The CB1 receptor antagonist AM251 (500 nM) prevented the WIN55212-2-induced increase in PPR while having no effect alone (n = 5) (Figure 5E).
In accordance with the previous findings, CGS21680 (30 nM, added 10 min before WIN55212-2) prevented WIN55212-2 from increasing the PPR (n ≥ 6), while per se did not alter basal synaptic transmission (P > 0.05) (Figure 5E).
Discussion
The present study provides direct evidence for the physical and functional interaction of A2A and CB1 receptors in corticostriatal terminals. Indeed, we now provide for the first time direct evidence for the co-localization of both A2A and CB1 receptors in the same, individually identified corticostriatal glutamatergic nerve terminal. It is important to note that the selectivity of antibodies raises increasing concern in the scientific community (see Grimsey et al., 2008). Here, we carefully titrated our primary antibodies and validated them in the KO mice and their WT littermates whenever possible. This allowed us to avoid common mistakes, such as false co-localizations or mis-estimation of the frequency of labelling.
Furthermore, we showed that these A2A and CB1 receptors form presynaptic heteromers in purified striatal nerve terminals, which is novel information, as this heterodimer was first identified in heterologous expression systems and was reported to be also present in total striatal extracts (Carriba et al., 2007). Heterodimers can interact either at the level of intracellular signalling, or by modulating G protein availability, or simply by physically altering the conformation of the partner receptor (Franco et al., 2006; 2008,). Our findings do not directly indicate the molecular nature of interaction but, from the binding data and the fact that the two receptors on their own utilize different pools of G proteins, it is possible that the interaction involves physical modulation of conformation, rather that occurring at the level of G proteins.
This physical association of A2A and CB1 receptors suggests a tight functional interplay in the control of glutamatergic nerve terminals in the striatum. The functional consequences of this finding were demonstrated here with a combination of direct presynaptic tools of increasing complexity (radioligand binding in nerve terminal membranes, glutamate release assay in acutely isolated nerve terminals and PPR measurements in isolated monosynaptic contacts in corticostriatal slices), all of which showed that A2A receptor activation significantly attenuated CB1 receptor function. In particular, we showed that activation of A2A receptors decreased the robust presynaptic CB1 receptor-mediated inhibition of corticostriatal glutamate release (Gerdeman and Lovinger, 2001; Köfalvi et al., 2005). This observation per se does not directly argue for a presynaptic location of the A2A receptors involved. However, as the CB1 receptors mediating the increase in the PPR are presynaptic, we can indirectly infer that those A2A receptors co-localize presynaptically with these CB1 receptors, as also strongly suggested by the neurochemical data.
The physiological role of this presynaptic A2A–CB1 receptor complex is likely to be associated with the well-known high-pass filter phenomenon for corticostriatal activity (Bamford et al., 2004). In fact, it is well established that increased synaptic activity is directly coupled to an increased release of two of the most potent substances acting as presynaptic inhibitory feedback signals, the release of adenosine acting through inhibitory A1 receptors (Fredholm et al., 2005) and endocannabinoids acting through presynaptic CB1 receptors (Lovinger, 2010). The efficiency of these two presynaptic inhibitory systems is best illustrated by the observations that A1 and CB1 receptors are highly abundant GPCRs in the brain. During high-frequency discharge, it is necessary to overcome these efficient presynaptic inhibitory systems to allow the passage of salient information. Therefore, high-pass filters become essential to implement long-term increases of corticostriatal activity with relevant stimuli. The present results add a critical piece of evidence to suggest that A2A receptors participate in this high-pass filtering in response to phasic changes in synaptic adenosine levels (Cunha, 2008). Indeed, it has been previously shown that ATP is co-released with glutamate (Pankratov et al., 2006) in a frequency-dependent manner (Wieraszko et al., 1989; Cunha et al., 1996). Furthermore, Augusto et al. (2013) have reported that adenosine, generated from ATP by ecto-5′-nucleotidase, constitutes the particular source that activates striatal A2A receptor. In the case of a low-frequency discharge, the corticostriatal terminals will not produce enough ATP-derived adenosine to activate presynaptic A2A receptor and the extracellular adenosine levels will be enough only to activate the inhibitory A1 receptor, as previously shown (Ciruela et al., 2006). Moreover, the glutamatergic activity will also produce retrograde inhibitory endocannabinoid signalling (Castillo et al., 2012; Katona and Freund, 2012). By contrast, salient and relevant information that should be encoded as increases of synaptic plasticity are associated with a higher frequency of discharge of corticostriatal afferents. Under such conditions, ATP-derived adenosine would now be enough to activate A2A receptors, which will play a double role of attenuating both presynaptic CB1 receptor inhibition, as shown here, as well as presynaptic A1 receptor inhibition (Ciruela et al., 2006). The engagement of A2A receptors has the additional potential of bolstering the function of different neurotrophins, such as brain-derived neurotrophic factor (Sebastião and Ribeiro, 2009), glial cell-derived neurotrophic factor (Gomes et al., 2009) as well as fibroblast growth factor (Flajolet et al., 2008), which further assist the implementation of long-term plastic changes in corticostriatal synapses. Notably, A2A receptors are selectively engaged to control synaptic plasticity rather than basal synaptic transmission in different synapses (Rebola et al., 2008; Costenla et al., 2011), particularly in corticostriatal synapses (D'Alcantara et al., 2001; Flajolet et al., 2008). This is in agreement with the observed failure of CGS21680 to alter basal synaptic transmission under whole-cell patch-clamp configuration in the dorsolateral (somatosensory) striatum (present study) as well as in extracellular recording in the dorsomedial (associative) striatum (Martíre et al., 2011). Notably, Ciruela et al. (2006) reported a significant increase following CGS21680 administration in the high-K+-induced release of glutamate in striatal synaptosomes, which was not observed in the present study, in agreement with a previous study where 4-aminopyridine stimulation was used to provoke [14C]glutamate release in striatal synaptosomes (Martíre et al., 2011) and with another report showing the lack of CGS21680 modulation of the high-K+-induced release of [3H]glutamate in hippocampal synaptosomes (Lopes et al., 2002). There are differences in the composition of the assay medium and in the execution of the experiments that may account for the differences in the effects of CGS21680 in these assays.
This central role of A2A receptors as a high-pass filter is likely to be further assisted by postsynaptic A2A receptors, which inhibit dopamine D2 receptor-mediated endocannabinoid production in the MSN dendrites (Lerner et al., 2010; Tozzi et al., 2012). Hence, adenosine will exert a double inhibition of endocannabinoid signalling to ensure the rescue of salient corticostriatal activity. Indeed, Bamford et al. (2004) noted that postsynaptic D2 receptor-mediated presynaptic inhibition of glutamate release is frequency-dependent, sparing only the most active corticostriatal terminals. This strongly corroborates our hypothesis.
As the principal input, the ‘driver’, to the whole basal ganglia is the corticostriatal pathway, the modulation of these afferents by presynaptic receptors will have profound effect on all basal ganglia-related lower- and higher-order brain functions including motor coordination, psychomotor drive, emotions, memory or decision making (Nakano et al., 2000). For instance, it is believed that the lack of D2 receptor-stimulated endocannabinoid synthesis in Parkinson's disease hampers presynaptic CB1 receptor-mediated control of corticostriatal afferents, leading to dyskinesias (Brotchie, 2003; Kreitzer and Malenka, 2007; Shen et al., 2008a). Additionally, we envisage an increased synaptic adenosine production and a further impairment of presynaptic CB1 receptor activity, even after L-DOPA administration. This could be one reason why A2A receptor-blocking strategies are interesting as a palliative strategy in addition to L-DOPA (Gomes et al., 2011). Another involvement of this newly described presynaptic receptor heterodimer may be in drug addiction. It was recently found by some of these authors that the presynaptic A2A receptors facilitate the cocaine-induced psychomotor drive in corticostriatal terminals (Shen et al., 2008b; 2013,), which once again can be explained by the negative control by A2A receptors on the CB1 receptor-mediated inhibition of these afferents.
Another example of the importance of this interaction derives from the finding that CB1 receptors exert neuroprotection both in vitro and in vivo in focal and global ischaemia models (Nagayama et al., 1999; Melis et al., 2006). Hence, one can further speculate that ischaemic activation of A2A receptors would hamper CB1 receptor-mediated neuroprotection. Indeed, A2A receptor blockade has been shown to be neuroprotective in several models (Gomes et al., 2011).
Altogether, the presynaptic A2A–CB1 receptor complex in corticostriatal terminals emerges as a novel mode of optimizing corticostriatal information processing. Additionally, the identification of this functional heteromer presynaptically, in the corticostriatal terminals, further strengthens the rationale of simultaneously targeting these two receptors rather than each individually, to achieve more efficient palliative therapies to alleviate striatal pathophysiology in motor and addictive diseases.
Acknowledgments
This work was supported by FEDER/COMPETE (PEst-C/SAU/LA0001/2014), QREN (09-68-ESR-FP-010 and W911NF-10-1-0059), FCT (PTDC/SAU-NSC/122254/2010 to R. A. C., PTDC/SAU-NEU/100729/2008 to A. K. and SFRH/BPD/85738/2012 to S. G. F.), DARPA (09-68-ESR-FP-010) and INSERM and Collège de France (to L. V.). We are grateful to K.H. Gylys (UCLA School of Medicine) for the help with flow synaptometry protocol.
Glossary
- 3Rs
replacement, refinement and reduction of animals in research
- ADA
adenosine deaminase
- MSN(s)
medium spiny neuron(s)
- OFA
Oncins France Strain A
- PPR
paired-pulse ratio
- vGlut1
vesicular glutamate transporter 1
Author contributions
A. K., S. G. F. and R. A. C. were responsible for the study design. S. G. F., I. N-C. and T. H. performed the flow synaptometry experiments. S. G. F., J. M. M. and R. J. R. were responsible for co-immunoprecipitation. S. G. F. and A. K. were responsible for the CB1 receptor binding assay. S. G. F. and A. K. performed the glutamate release experiments. F. Q. G. and Â. R. T. were responsible for ATP measurement. S. G. F. and L. V. were responsible for electrophysiology. C. L. was responsible for sourcing and genotyping of A2A and CB1 receptor knockout mice and their wild-type littermates. A. K., R. A. C., L. V. and T. H. obtained financial support. S. G. F. was responsible for the first draft.
Conflicts of interest
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Data analysis and validation of the A2A and CB1 receptor antibodies/ titrations. (A and B) Examples of fluorescence histograms (for anti-synaptophysin and anti-CB1 receptor antibodies respectively. Specific signal for single-labelled synaptosomes was calculated by subtracting the percentage of labelling (M2 region) by the secondary antibodies alone (histogram filled with violet colour) from the percentage of labelling with the antibody of interest (histogram in red colour). M1 region represents the unlabelled synaptosomes. (C) Dot plot showing double-labelled synaptosomes for synaptophysin and vGlut1. Double-labelled particles appear in the upper right quadrant. Titration curve of the A2A receptor antibody (D) and CB1 receptor antibody (E) in the wild-type (blue curve with filled circles) and knockout mice (red curve with filled squares). Green boxes indicate the working concentration/dilution of each antibody used in the rat synaptosomes. (F) Bar graph exhibiting the virtually identical labelling by the two different synaptophysin antibodies (n = 3).
Table S1 Antibodies used (abbreviations: FS, flow synaptometry; IP, immunoprecipitation).
Table S2 Comparative pharmacological analysis of A1 and A2A adenosine and CB1 cannabinoid receptors on striatal synaptosomal glutamate release.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The concise guide to pharmacology 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol. 2013c;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augusto E, Matos M, Sévigny J, El-Tayeb A, Bynoe MS, Müller CE, et al. Ecto-5′-nucleotidase (CD73)-mediated formation of adenosine is critical for the striatal adenosine A2A receptor functions. J Neurosci. 2013;33:11390–11399. doi: 10.1523/JNEUROSCI.5817-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford NS, Shang H, Schmitz Y, Wu NP, Cepeda C, Levine MS, et al. Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron. 2004;42:653–663. doi: 10.1016/s0896-6273(04)00265-x. [DOI] [PubMed] [Google Scholar]
- Bolam JP, Hanley JJ, Booth PA, Bevan MD. Synaptic organization of the basal ganglia. J Anat. 2000;196:527–542. doi: 10.1046/j.1469-7580.2000.19640527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brotchie JM. CB1 cannabinoid receptor signalling in Parkinson's disease. Curr Opin Pharmacol. 2003;3:54–61. doi: 10.1016/s1471-4892(02)00011-5. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Introduction to purinergic signalling in the brain. Adv Exp Med Biol. 2013;986:1–12. doi: 10.1007/978-94-007-4719-7_1. [DOI] [PubMed] [Google Scholar]
- Carriba P, Ortiz O, Patkar K, Justinova Z, Stroik J, Themann A, et al. Striatal adenosine A2A and cannabinoid CB1 receptors form functional heteromeric complexes that mediate the motor effects of cannabinoids. Neuropsychopharmacology. 2007;32:2249–2259. doi: 10.1038/sj.npp.1301375. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76:70–81. doi: 10.1016/j.neuron.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerri S, Levandis G, Ambrosi G, Montepeloso E, Antoninetti GF, Franco R, et al. Neuroprotective potential of adenosine A2A and cannabinoid CB1 receptor antagonists in an animal model of Parkinson disease. J Neuropathol Exp Neurol. 2014;73:414–424. doi: 10.1097/NEN.0000000000000064. [DOI] [PubMed] [Google Scholar]
- Ciruela F, Casadó V, Rodrigues RJ, Luján R, Burgueño J, Canals M, et al. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J Neurosci. 2006;26:2080–2087. doi: 10.1523/JNEUROSCI.3574-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costenla AR, Diógenes MJ, Canas PM, Rodrigues RJ, Nogueira C, Maroco J, et al. Enhanced role of adenosine A2A receptors in the modulation of LTP in the rat hippocampus upon ageing. Eur J Neurosci. 2011;34:12–21. doi: 10.1111/j.1460-9568.2011.07719.x. [DOI] [PubMed] [Google Scholar]
- Cunha RA. Different cellular sources and different roles of adenosine: A1 receptor-mediated inhibition through astrocytic-driven volume transmission and synapse-restricted A2A receptor-mediated facilitation of plasticity. Neurochem Int. 2008;52:65–72. doi: 10.1016/j.neuint.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Vizi ES, Ribeiro JA, Sebastião AM. Preferential release of ATP and its extracellular catabolism as a source of adenosine upon high- but not low-frequency stimulation of rat hippocampal slices. J Neurochem. 1996;67:2180–2187. doi: 10.1046/j.1471-4159.1996.67052180.x. [DOI] [PubMed] [Google Scholar]
- D'Alcantara P, Ledent C, Swillens S, Schiffmann SN. Inactivation of adenosine A2A receptor impairs long term potentiation in the accumbens nucleus without altering basal synaptic transmission. Neuroscience. 2001;107:455–464. doi: 10.1016/s0306-4522(01)00372-4. [DOI] [PubMed] [Google Scholar]
- Di Filippo M, Picconi B, Tantucci M, Ghiglieri V, Bagetta V, Sgobio C, et al. Short-term and long-term plasticity at corticostriatal synapses: implications for learning and memory. Behav Brain Res. 2009;199:108–118. doi: 10.1016/j.bbr.2008.09.025. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- El Manira A, Kyriakatos A. The role of endocannabinoid signaling in motor control. Physiology (Bethesda) 2010;25:230–238. doi: 10.1152/physiol.00007.2010. [DOI] [PubMed] [Google Scholar]
- Ferré S, Lluís C, Justinová Z, Quiroz C, Orrú M, Navarro G, et al. Adenosine-cannabinoid receptor interactions. Implications for striatal function. Br J Pharmacol. 2010;160:443–453. doi: 10.1111/j.1476-5381.2010.00723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Quiroz C, Orru M, Guitart X, Navarro G, Cortès A, et al. Adenosine A2A receptors and A2A receptor heteromers as key players in striatal function. Front Neuroanat. 2011;5:1–8. doi: 10.3389/fnana.2011.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira SG, Lomaglio T, Avelino A, Oliveira CR, Cunha RA, Köfalvi A. N-arachidonoyldopamine induces glutamate and dopamine release via a novel Ca2+ channel. Neuropharmacology. 2009;56:676–683. doi: 10.1016/j.neuropharm.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Ferreira SG, Teixeira FM, Garção P, Agostinho P, Ledent C, Cortes L, et al. Presynaptic CB1 cannabinoid receptors control frontocortical serotonin and glutamate release – species differences. Neurochem Int. 2012;61:219–226. doi: 10.1016/j.neuint.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Glowinski J, Venance L. Bidirectional activity-dependent plasticity at corticostriatal synapses. J Neurosci. 2005;25:11279–11287. doi: 10.1523/JNEUROSCI.4476-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flajolet M, Wang Z, Futter M, Shen W, Nuangchamnong N, Bendor J, et al. FGF acts as a co-transmitter through adenosine A2A receptor to regulate synaptic plasticity. Nat Neurosci. 2008;11:1402–1409. doi: 10.1038/nn.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Casadó V, Mallol J, Ferrada C, Ferré S, Fuxe K, et al. The two-state dimer receptor model: a general model for receptor dimers. Mol Pharmacol. 2006;69:1905–1912. doi: 10.1124/mol.105.020685. [DOI] [PubMed] [Google Scholar]
- Franco R, Casadó V, Cortés A, Mallol J, Ciruela F, Ferré S, et al. G-protein-coupled receptor heteromers: function and ligand pharmacology. Br J Pharmacol. 2008;153:S90–S98. doi: 10.1038/sj.bjp.0707571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. Adenosine and brain function. Int Rev Neurobiol. 2005;63:191–270. doi: 10.1016/S0074-7742(05)63007-3. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Lovinger DM. Cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J Physiol. 2001;85:468–471. doi: 10.1152/jn.2001.85.1.468. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Selemon LD. Topography of corticostriatal projections in nonhuman primates and implications for functional parcellation of the neostriatum. In: Jones EG, Peters A, editors. Cerebral Cortex. New York: Plenum; 1986. pp. 447–466. . In: (eds). [Google Scholar]
- Gomes CA, Simões PF, Canas PM, Quiroz C, Sebastião AM, Ferré S, et al. GDNF control of the glutamatergic cortico-striatal pathway requires tonic activation of adenosine A receptors. J Neurochem. 2009;108:1208–1219. doi: 10.1111/j.1471-4159.2009.05876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes CV, Kaster MP, Tomé AR, Agostinho PM, Cunha RA. Adenosine receptors and brain diseases: neuroprotection and neurodegeneration. Biochim Biophys Acta. 2011;1808:1380–1399. doi: 10.1016/j.bbamem.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Graybiel AM. Building action repertoires: memory and learning functions of the basal ganglia. Curr Opin Neurobiol. 1995;5:733–741. doi: 10.1016/0959-4388(95)80100-6. [DOI] [PubMed] [Google Scholar]
- Grimsey NL, Goodfellow CE, Scotter EL, Dowie MJ, Glass M, Graham ES. Specific detection of CB1 receptors; cannabinoid CB1 receptor antibodies are not all created equal! J Neurosci Methods. 2008;171:78–86. doi: 10.1016/j.jneumeth.2008.02.014. [DOI] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Cole GM. Quantitative characterization of crude synaptosomal fraction (P-2) components by flow cytometry. J Neurosci Res. 2000;61:186–192. doi: 10.1002/1097-4547(20000715)61:2<186::AID-JNR9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Justinová Z, Ferré S, Redhi GH, Mascia P, Stroik J, Quarta D, et al. Reinforcing and neurochemical effects of cannabinoid CB1 receptor agonists, but not cocaine, are altered by an adenosine A2A receptor antagonist. Addict Biol. 2011;16:405–415. doi: 10.1111/j.1369-1600.2010.00258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justinová Z, Redhi GH, Goldberg SR, Ferré S. Differential effects of presynaptic versus postsynaptic adenosine A2A receptor blockade on Δ9-tetrahydrocannabinol (THC) self-administration in squirrel monkeys. J Neurosci. 2014;34:6480–6484. doi: 10.1523/JNEUROSCI.5073-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Freund TF. Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci. 2012;35:529–558. doi: 10.1146/annurev-neuro-062111-150420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG NC3Rs Reporting Guidelines Working Group. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köfalvi A, Rodrigues RJ, Ledent C, Mackie K, Vizi ES, Cunha RA, et al. Involvement of cannabinoid receptors in the regulation of neurotransmitter release in the rodent striatum: a combined immunochemical and pharmacological analysis. J Neurosci. 2005;25:2874–2884. doi: 10.1523/JNEUROSCI.4232-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Ledent C, Vaugeois JM, Schiffmann SN, Pedrazzini T, El Yacoubi M, Vanderhaeghen JJ, et al. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2A receptor. Nature. 1997;388:674–678. doi: 10.1038/41771. [DOI] [PubMed] [Google Scholar]
- Ledent C, Valverde O, Cossu G, Petitet F, Aubert JF, Beslot F, et al. Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science. 1999;283:401–404. doi: 10.1126/science.283.5400.401. [DOI] [PubMed] [Google Scholar]
- Lerner TN, Horne EA, Stella N, Kreitzer AC. Endocannabinoid signalling mediates psychomotor activation by adenosine A2A antagonists. J Neurosci. 2010;30:2160–2164. doi: 10.1523/JNEUROSCI.5844-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes LV, Cunha RA, Kull B, Fredholm BB, Ribeiro JA. Adenosine A2A receptor facilitation of hippocampal synaptic transmission is dependent on tonic A1 receptor inhibition. Neuroscience. 2002;112:319–329. doi: 10.1016/s0306-4522(02)00080-5. [DOI] [PubMed] [Google Scholar]
- Lovinger DM. Neurotransmitter role in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology. 2010;58:951–961. doi: 10.1016/j.neuropharm.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques JM, Rodrigues RJ, Valbuena S, Rozas JL, Selak S, Marin P, et al. CRMP2 tethers kainate receptor activity to cytoskeleton dynamics during neuronal maturation. J Neurosci. 2013;33:18298–18310. doi: 10.1523/JNEUROSCI.3136-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martíre A, Tebano MT, Chiodi V, Ferreira SG, Cunha RA, Köfalvi A, et al. Pre-synaptic adenosine A2A receptors control cannabinoid CB1 receptor-mediated inhibition of striatal glutamatergic neurotransmission. J Neurochem. 2011;116:273–280. doi: 10.1111/j.1471-4159.2010.07101.x. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis M, Pillolla G, Bisogno T, Minassi A, Petrosino S, Perra S, et al. Protective activation of the endocannabinoid system during ischemia in dopamine neurons. Neurobiol Dis. 2006;24:15–27. doi: 10.1016/j.nbd.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin K, et al. Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J Neurosci. 1999;19:2987–2995. doi: 10.1523/JNEUROSCI.19-08-02987.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K, Kayahara T, Tsutsumi T, Ushiro H. Neural circuits and functional organization of the striatum. J Neurol. 2000;247:1–15. doi: 10.1007/pl00007778. [DOI] [PubMed] [Google Scholar]
- Navizet I, Liu YJ, Ferré N, Roca-Sanjuán D, Lindh R. The chemistry of bioluminescence: an analysis of chemical functionalities. Chemphyschem. 2011;12:3064–3076. doi: 10.1002/cphc.201100504. [DOI] [PubMed] [Google Scholar]
- Pankratov Y, Lalo U, Verkhratsky A, North RA. Vesicular release of ATP at central synapses. Pflugers Arch. 2006;452:589–597. doi: 10.1007/s00424-006-0061-x. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42:D1098–1106. doi: 10.1093/nar/gkt1143. (Database Issue): [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinna A, Bonaventura J, Farré D, Sánchez M, Simola N, Mallol J, et al. L-DOPA disrupts adenosine A2A-cannabinoid CB1-dopamine D2 receptor heteromer cross-talk in the striatum of hemiparkinsonian rats: biochemical and behavioral studies. Exp Neurol. 2014;253:180–191. doi: 10.1016/j.expneurol.2013.12.021. [DOI] [PubMed] [Google Scholar]
- Quiroz C, Luján R, Uchigashima M, Simoes AP, Lerner TN, Borycz J, et al. Key modulatory role of presynaptic adenosine A2A receptors in cortical neurotransmission to the striatal direct pathway. Scientificworldjournal. 2009;9:1321–1344. doi: 10.1100/tsw.2009.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebola N, Canas PM, Oliveira CR, Cunha RA. Different synaptic and subsynaptic localization of adenosine A2A receptors in the hippocampus and striatum of the rat. Neuroscience. 2005;132:893–903. doi: 10.1016/j.neuroscience.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Rebola N, Lujan R, Cunha RA, Mulle C. Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron. 2008;57:121–134. doi: 10.1016/j.neuron.2007.11.023. [DOI] [PubMed] [Google Scholar]
- Rossi S, De Chiara V, Musella A, Mataluni G, Sacchetti L, Siracusano A, et al. Effects of caffeine on striatal neurotransmission: focus on cannabinoid CB1 receptors. Mol Nutr Food Res. 2010;54:525–531. doi: 10.1002/mnfr.200900237. [DOI] [PubMed] [Google Scholar]
- Schiffmann SN, Fisone G, Moresco R, Cunha RA, Ferré S. Adenosine A2A receptors and basal ganglia physiology. Prog Neurobiol. 2007;83:277–292. doi: 10.1016/j.pneurobio.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid I, Uittenbogaart CH, Giorgi JV. A gentle fixation and permeabilization method for combined cell surface and intracellular staining with improved precision in DNA quantification. Cytometry. 1991;12:279–285. doi: 10.1002/cyto.990120312. [DOI] [PubMed] [Google Scholar]
- Schulz PE, Cook EP, Johnston D. Changes in paired-pulse facilitation suggest presynaptic involvement in long-term potentiation. J Neurosci. 1994;14:5325–5337. doi: 10.1523/JNEUROSCI.14-09-05325.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastião AM, Ribeiro JA. Fine-tuning neuromodulation by adenosine. Trends Pharmacol Sci. 2000;21:341–346. doi: 10.1016/s0165-6147(00)01517-0. [DOI] [PubMed] [Google Scholar]
- Sebastião AM, Ribeiro JA. Triggering neurotrophic factor actions through adenosine A2A receptor activation: implications for neuroprotection. Br J Pharmacol. 2009;158:15–22. doi: 10.1111/j.1476-5381.2009.00157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HY, Coelho JE, Ohtsuka N, Canas PM, Day YJ, Huang QY, et al. A critical role of the adenosine A2A receptor in extrastriatal neurons in modulating psychomotor activity as revealed by opposite phenotypes of striatum and forebrain A2A receptor knock-outs. J Neurosci. 2008b;28:2970–2975. doi: 10.1523/JNEUROSCI.5255-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HY, Canas PM, Garcia-Sanz P, Lan JQ, Boison D, Moratalla R, et al. Adenosine A2A receptors in striatal glutamatergic terminals and GABAergic neurons oppositely modulate psychostimulant action and DARPP-32 phosphorylation. PLoS ONE. 2013;8:e80902. doi: 10.1371/journal.pone.0080902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008a;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria G, Castañé A, Berrendero F, Ledent C, Parmentier M, Maldonado R, et al. Adenosine A2A receptors are involved in physical dependence and place conditioning induced by THC. Eur J Neurosci. 2004;20:2203–2213. doi: 10.1111/j.1460-9568.2004.03682.x. [DOI] [PubMed] [Google Scholar]
- Sperlágh B, Vizi ES. Neuronal synthesis, storage and release of ATP. Semin Neurosci. 1996;8:175–186. [Google Scholar]
- Svenningsson P, Le Moine C, Fisone G, Fredholm BB. Distribution, biochemistry and function of striatal adenosine A2A receptors. Prog Neurobiol. 1999;59:355–396. doi: 10.1016/s0301-0082(99)00011-8. [DOI] [PubMed] [Google Scholar]
- Tozzi A, de Iure A, Marsili V, Romano R, Tantucci M, Di Filippo M, et al. A2A adenosine receptor antagonism enhances synaptic and motor effects of cocaine via CB1 cannabinoid receptor activation. PLoS ONE. 2012;7:e38312. doi: 10.1371/journal.pone.0038312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchigashima M, Narushima M, Fukaya M, Katona I, Kano M, Watanabe M. Subcellular arrangement of molecules for 2-arachidonoyl-glycerol-mediated retrograde signaling and its physiological contribution to synaptic modulation in the striatum. J Neurosci. 2007;27:3663–3676. doi: 10.1523/JNEUROSCI.0448-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieraszko A, Goldsmith G, Seyfried TN. Stimulation-dependent release of adenosine triphosphate from hippocampal slices. Brain Res. 1989;485:244–250. doi: 10.1016/0006-8993(89)90567-2. [DOI] [PubMed] [Google Scholar]
- Yao L, McFarland K, Fan P, Jiang Z, Ueda T, Diamond I. Adenosine A2a blockade prevents synergy between mu-opiate and cannabinoid CB1 receptors and eliminates heroin-seeking behavior in addicted rats. Proc Natl Acad Sci U S A. 103:7877–7882. doi: 10.1073/pnas.0602661103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Knowlton BJ. The role of the basal ganglia in habit formation. Nature Rev Neurosci. 2006;7:464–476. doi: 10.1038/nrn1919. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Data analysis and validation of the A2A and CB1 receptor antibodies/ titrations. (A and B) Examples of fluorescence histograms (for anti-synaptophysin and anti-CB1 receptor antibodies respectively. Specific signal for single-labelled synaptosomes was calculated by subtracting the percentage of labelling (M2 region) by the secondary antibodies alone (histogram filled with violet colour) from the percentage of labelling with the antibody of interest (histogram in red colour). M1 region represents the unlabelled synaptosomes. (C) Dot plot showing double-labelled synaptosomes for synaptophysin and vGlut1. Double-labelled particles appear in the upper right quadrant. Titration curve of the A2A receptor antibody (D) and CB1 receptor antibody (E) in the wild-type (blue curve with filled circles) and knockout mice (red curve with filled squares). Green boxes indicate the working concentration/dilution of each antibody used in the rat synaptosomes. (F) Bar graph exhibiting the virtually identical labelling by the two different synaptophysin antibodies (n = 3).
Table S1 Antibodies used (abbreviations: FS, flow synaptometry; IP, immunoprecipitation).
Table S2 Comparative pharmacological analysis of A1 and A2A adenosine and CB1 cannabinoid receptors on striatal synaptosomal glutamate release.