

Abstract
Background and Purpose
Neuroinflammation through microglial activation is involved in the pathogenesis of neurodegenerative diseases including Parkinson's disease (PD), a major neurodegenerative disorder characterized by dopaminergic neuronal death in the substantia nigra. We examined our novel synthetic compound VSC2 for its anti-inflammatory properties towards development of a PD therapy.
Experimental Approach
We tested the effects of VSC2 on production of various NF-κB-dependent proinflammatory molecules and Nrf2-dependent antioxidant enzymes in BV-2 microglia and in vivo.
Key Results
The vinyl sulfone compound, VSC2, most effectively suppressed the production of NO in LPS-activated microglia. It also down-regulated expression of inducible NOS (iNOS), COX-2, IL-1β and TNF-α and inhibited nuclear translocalization and transcriptional activity of NF-κB. VSC2 increased total and nuclear Nrf2 levels, induced Nrf2 transcriptional activity and was bound to Keap1 with high affinity. Expression of the Nrf2-regulated antioxidant enzyme genes NAD(P)H quinone oxidoreducase-1 (NQO-1), haem oxygenase-1 (HO-1) and glutamylcysteine ligase (GCL) were up-regulated by VSC2. In the MPTP mouse model of PD, oral administration of VSC2 decreased the number of activated microglia in the substantia nigra, lowered the levels of iNOS, COX-2 and IL-1β, and protected the dopaminergic neurons. VSC2 also elevated the levels of NQO1, HO-1, GCL and Nrf2 in the nigrostriatal area.
Conclusions and Implications
VSC2 has both anti-inflammatory and antioxidant properties and prevented neuroinflammation in microglia and in an animal model of PD. This suggests VSC2 as a potential candidate for PD therapy.
Tables of Links
| TARGETS |
|---|
| Enzymes |
| COX-2 |
| HO-1, haem oxygenase 1 |
| iNOS, inducible NOS |
| LIGANDS |
|---|
| Curcumin |
| Dopamine |
| IL-1β |
| NO |
| TNF-α |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Neuroinflammation is commonly observed in several neurodegenerative disorders including Parkinson's disease (PD) and Alzheimer's disease and is mainly caused by microglial activation. Although neuroinflammation is a mechanism that protects the organism from infection and injury, if not turned off in a timely manner, it can become chronic and contribute to neurodegeneration (see Tansey and Goldberg, 2010; Blandini, 2013).
The primary neuropathological feature of PD is the extensive loss of dopaminergic neurons in the substantia nigra (SN). The currently available treatments for PD only relieve the symptoms without delaying the degeneration itself, and drugs that can modify the disease progression are being actively sought. While the reasons for the selective loss of the nigral dopaminergic neurons still remain to be clarified, oxidative stress and neuroinflammation are believed to be major contributors. Increased reactive oxygen species (ROS) production within the microglia by enzymes such as NADPH oxidase, as well as ROS produced by nearby cells, are known to elicit activation of the MAPK pathways and IκB kinase and nuclear translocalization of the transcription factor NF-κB (Gloire et al., 2006). This in turn leads to increased production of proinflammatory enzymes such as inducible NOS (iNOS), COX-2, the cytokines TNF-α and IL-1β and NO, all of which can contribute to neurodegeneration. Indeed, we and others have previously reported that ways to suppress production of these proinflammatory molecules can protect the nigral dopaminergic neurons in animal models of PD (Teismann and Ferger, 2001; Zhou et al., 2005; Cho et al., 2009; Son et al., 2007; Stuckenholz et al., 2013).
The transcription factor Nrf2 has been shown to down-regulate inflammatory responses. For example, TNF-α-induced lung inflammation and LPS-induced NF-κB activation were greatly increased in Nrf2 knockout mice (Thimmulappa et al., 2006). Inflammatory mediators such as TNF-α, IL-1β, COX-2 and iNOS were overexpressed in the Nrf2 knockout mice and these mice were hypersensitive to neuroinflammation induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP; Rojo et al., 2010) and LPS (Innamorato et al., 2008). NF-κB activation (Jeong et al., 2004) and microglial activation (Innamorato et al., 2008) induced by LPS could be attenuated by diverse Nrf2 activators, such as phenethyl isothiocyanate, sulforaphane and curcumin. It has also been reported that Nrf2 activation is closely related with down-regulation of the NF-κB-associated inflammatory activity in macrophages and microglia (Lin et al., 2008; Koh et al., 2011; Lee et al., 2011; Kang et al., 2013). Although the mechanism by which Nrf2 down-regulates neuroinflammation awaits elucidation, it is generally believed that the mechanism involves the Nrf2-regulated antioxidant enzyme activities that eliminate intracellular ROS within microglia. These enzymes include NAD(P)H: quinone oxidoreductase 1 (NQO1), haem oxygenase-1 (HO-1) and glutamate-cysteine ligase (GCL).
Chalcone has a unique α,β-unsaturated ketone, a structure which is thought to elicit anti-inflammatory properties, among others (see Bukhari et al., 2013). We have recently discovered that introduction of a vinyl sulfone to this α,β-unsaturated carbonyl moiety leads to a significant increase in the ability to activate the Nrf2 signalling pathway. We have subsequently generated 56 different vinyl sulfone derivatives (Woo et al., 2014), some of which are shown in Table 1. In the present study, we demonstrate that one of these compounds, a vinyl sulfone with a methoxy group at the 2′ position (VSC2; (E)-1-(2-((2-methoxyphenyl)sulfonyl)vinyl)-2-chlorobenzene) has an excellent anti-inflammatory and antioxidant profile in microglia both in vitro and in vivo.
Table 1.
Vinyl sulfone compounds with their respective chemical names
| Compounda | Chemical names |
|---|---|
| VSC2 | (E)-1-(2-((2-Methoxyphenyl)sulfonyl)vinyl)-2-chlorobenzene |
| VSC3 | (E)-1-(2-((3-Methoxyphenyl)sulfonyl)vinyl)-2-chlorobenzene |
| VSC4 | (E)-1-(2-((4-Methoxyphenyl)sulfonyl)vinyl)-2-chlorobenzene |
| VSC2a | (E)-1-(2-((2-Methoxyphenyl)sulfonyl)vinyl)-2-trifluoromethylbenzene |
| VSC2b | (E)-1-(2-((2-Methoxyphenyl)sulfonyl)vinyl)-3-trifluoromethylbenzene |
| VSC2c | (E)-1-(2-((2-Methoxyphenyl)sulfonyl)vinyl)-4-trifluoromethylbenzene |
| VSC2d | (E)-1-(2-((2-Methoxyphenyl)sulfonyl)vinyl)-2-fluorobenzene |
| VSC2e | (E)-1-(2-((2-Methoxyphenyl)sulfonyl)vinyl)-3-fluorobenzene |
| VSC2f | (E)-1-(2-((2-Methoxyphenyl)sulfonyl)vinyl)-4-fluorobenzene |
| VSC2g | (E)-1-(2-((2-Methoxyphenyl)sulfonyl)vinyl)-3-chlorobenzene |
| VSC2h | (E)-1-(2-((2-Methoxyphenyl)sulfonyl)vinyl)-4-chlorobenzene |
Syntheses of these compounds have previously been reported by us (Woo et al., 2014).
Methods
Syntheses of vinyl sulfone compounds
The vinyl sulfone compounds were prepared by the methods previously reported by us (Woo et al., 2014) and their structures were confirmed by NMR. The full chemical names are listed in Table 1.
Cell culture
BV-2 mouse microglial cells were cultured in DMEM containing 10% FBS, 100 IU·L−1 penicillin and 10 μg·mL−1 streptomycin at 37°C, 95% air and 5% CO2 in humidified atmosphere.
Preparation of proteins for Western blot
Nuclear fractions and cell lysates were obtained using the protocol described previously (Woo et al., 2014). To obtain in vivo samples, the striatal tissue was dissected out on ice and immediately homogenized in fourfold (w/v) 10 mM phosphate buffer (pH 7.0) containing 0.1% NP-40. After centrifugation at 14 000× g for 15 min, the supernatant was obtained.
Western blot analyses
As previously described (Son et al., 2007; Woo et al., 2014), equal amounts (30 μg) of protein were separated, transferred onto a membrane and blocked. The membranes were incubated overnight with primary antibody against NF-κB (1:1000), iNOS (1:1000), COX-2 (1:200), Nrf2 (1:2000), HO-1 (1:1000), GCLC (1:3000), GCLM (1:200), NQO1 (1:1000), lamin B (1:200), hsp90 (1:1000) or β-actin (1:20000) at 4°C and then with HRP-conjugated secondary antibodies for 1 h at room temperature. Protein bands were visualized using chemiluminescence substrate and quantitatively analysed by densitometry.
RT-PCR
Total RNA was isolated from cells using TRIzol reagent. Reverse transcription was carried out using M-MuLV reverse transcriptase and random primer at 25°C for 10 min, 37°C for 60 min and 70°C for 10 min. RT-PCR was performed on the cDNA obtained from 4 μg of total RNA as described previously (Woo et al., 2014). Real-time PCR was performed using TOPreal qPCR 2 × PreMix with SYBR green (Enzynomics, Daejeon, Korea) and a CFX real-time PCR detection system (Bio-Rad) according to the manufacturer's instructions. The sequences of primers used were as follows: NQO1 (forward, 5′-GCGAGAAGAGCCCTGATTGTACTG-3′; reverse, 5′-TCTCAAACCAGCCTTTCAGAATGG-3′), HO-1 (forward, 5′-CAAGCCGAGAATGCTGAGTTCATG-3′; reverse, 5′-GCAAGGGATGATTTCCTGCCAG-3′), GCLC (forward, 5′-ACATCTACCACGCAGTCAAGGACC-3′; reverse, 5′-CTCAAGAACATCGCCTCCATTCAG-3′), GCLM (forward, 5′-GCCACCAGATTTGACTGCCTTTG-3′); reverse, 5′-TGCTCTTCACGATGACCGAGTACC-3′), IL-1β (forward, 5′-TGTAATGAAAGACGGCACACC-3′; reverse, 5′-TCTTCTTTGGGTATTGCTTGG-3′), TNF-α (forward, 5′-CTGTAGCCCACGTCGTAGC-3′; reverse, 5′-TTGAGATCCATGCCGTTG-3′), iNOS (forward, 5′-GGCAGCCTGTGAGACCTTTG-3′; reverse, 5′-GCATTGGAAGTGAAGCGTTTC-3′), COX-2 (forward, 5′-TGAGTACCGCAAACGCTTCTC-3′; reverse, 5′-TGGACGAGGTTTTTCCACCAG-3) and GADPH (forward, 5′-CGACTTCAACAGCAACTCCCACTCTTCC-3′; reverse, 5′-TGGGTGGTCCAGGGTTTCTTACTCCTT-3′).
Antioxidant response element (ARE)-luciferase activity assay
BV-2 cells were seeded on 12-well plates and transiently transfected with 1 μg of ARE-pGL4 plasmid (Hara et al., 2006) and 0.2 μg pRL-TK plasmid using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) for 48 h. Cells were treated with various concentrations of VSC2 for 6 h. Luciferase activities in the cell lysates were measured using dual-luciferase reporter assay system (Promega, Madison, WI, USA). The luciferase activity was then normalized against the control pRL-TK.
NF-κB binding element-luciferase assay
BV-2 cells were seeded on 12-well plates and transiently transfected with 1 μg of NF-κB-luciferase plasmid and 0.2 μg pRL-TK plasmid using Lipofectamine 2000 reagent for 48 h. Cells were treated with various concentrations of VSC2 with 0.2 μg·mL−1 LPS for 6 h. Luciferase activities in the cell lysates were measured using dual-luciferase reporter assay system (Promega). The luciferase activity was then normalized against the control pRL-TK.
Cytotoxicity assay
BV-2 cells were seeded on 96-well plates and treated with various concentrations of VSC2 for 24h. The intracellular ATP levels were measured using the CellTiter-Glo luminescent cell viability assay kit (Promega).
Griess assay
BV-2 cells were seeded on 96-well plates and treated with various concentrations of VSC2 with 0.2 μg·mL−1 LPS. After 24 h, the NO2 level in the medium was measured as described previously (Cho et al., 2009). The concentration of compounds causing 50% inhibition (IC50) of LPS-induced NO induction was calculated by sigmoidal non-linear regression analysis (GraphPad Prism 4.0, La Jolla, CA, USA).
Measurement of TNF-α
BV-2 cells were seeded on 6-well plates and treated with various concentrations of VSC2 with 0.2 μg·mL−1 LPS. After 24 h, TNF-α in cell culture medium was measured by elisa using TNF-α elisa kit.
Measurement of IL-1β
BV-2 cells were seeded on 6-well plates and treated with various concentrations of VSC2 with 0.2 μg·mL−1 LPS for 24 h, and the cell lysate was obtained. The nigral tissue was dissected out on ice and immediately homogenized in fourfold (w/v) 10 mM phosphate buffer (pH 7.0) containing 0.1% NP-40. After centrifugation at 14 000× g for 15 min, the supernatant was obtained. IL-1β was measured by elisa using IL-1β elisa kit.
Production of an animal model of PD and its treatment
The procedures for the model of PD have been described earlier (Woo et al., 2014). Male C57Bl/6 mice (n = 13 per group) were given VSC2 by oral gavage, suspended in N-methyl-2-pyrrolidone and 20% Tween 80 in saline, at 10 mg·kg−1 per day, every day for 3 consecutive days. To make the PD model, MPTP (20 mg·kg−1, dissolved in saline) was injected intraperitoneally four times, 2 h apart, in a single day. The first MPTP injection was made 24 h after the first VSC2 administration.
Immunohistochemistry
Seven days after the first MPTP injection, the animals were anaesthetized with 30 mg·kg−1 zoletil and 1 mg·kg−1 xylazine and transcardially fixed in 4% paraformaldehyde. Brains were taken out, postfixed in 4% paraformaldehyde, cryoprotected and then cut into 20 μm sections. The substantia nigra (SN) pars compacta area was delineated according to the mouse brain atlas (Franklin and Paxinos, 1997). A total of five sections (every fourth section, 80 μm apart) from each animal was taken and we made sure that the nigral sections from the different treatment groups represented similar anatomical regions by carefully checking with the mouse brain atlas. They were subjected to tyrosine hydroxylase (TH) immunostaining. Additional five sections, each posteriorly adjacent to those immunostained against TH, were subjected to Iba-1 immunostaining. After incubation with either anti-TH (1:2000) or anti-Iba-1 (1:300) antibodies, the sections were exposed to biotinylated secondary antibody followed by avidin/biotinylated HRP using the Vectastain ABC kit. The immunoreactivities were visualized by incubation in 0.05% 3,3′-diaminobenzidine and 0.003% H2O2. Iba-1 immunopositive cells were quantitated by densitometry. Images of SN region from each section were captured using a Zeiss Observer Z1 Microscope operated by the ZEN software (Carl Zeiss, Göttingen, Germany). The immunodensity value of each region was measured after converting the image to grey scale using Image Guage 4.0 (Fuji Photo Film Co., Tokyo, Japan). The final immunodensity values were calculated by subtracting background density, obtained from regions lacking immunoreactions. All values for each group were averaged and represented % of vehicle-treated group ± SEM.
TH-Iba-1 double immunofluorescence staining
For double immunofluorescence staining against TH and Iba-1, five nigral sections (every fourth section, 80 μm apart) that were anteriorly adjacent to those immunostained against TH (explained earlier) were taken from all animals. They were blocked with PBS containing 1% BSA, 0.2% Triton X-100 and 0.05% sodium azide and then incubated overnight with anti-TH (1:2000) and anti-Iba-1 (1:300) antibodies, followed by incubation for 1 h with Fluor Alexa 546-labelled anti-mouse IgG (1:200) and Fluor Alexa 488-labelled anti-rabbit IgG (1:200). They were viewed under a confocal microscope (LSM 710; Carl Zeiss Microscopy GmbH, Jena, Germany).
Surface plasmon resonance
Surface plasmon resonance experiments were performed on a Biacore T100 instrument (GE Healthcare, Uppsala, Sweden) at 25°C to test if VSC2 binds to Keap1 protein. Anti-His antibody was first immobilized on CM5 chips using the amine coupling kit and His capture kit (GE Healthcare). At a flow rate of 10 μL·min−1, each flow cell was activated for 7 min with an equal mixture of 0.4 M 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride and 0.1 M N-hydroxysuccinimide. The anti-His antibody (50 μg·mL−1) in 10 mM sodium acetate (pH 4.5) was injected for 7 min, resulting in immobilization level of about 4000 resonance units. The surface on the CM5 chip was then deactivated for 7 min with 1 M ethanolamine (pH 8.5). Purified His-tagged Keap1 was then captured by injecting the protein (100 μg·mL−1) at a flow rate of 10 μL·min−1 for 150 s over the immobilized anti-His antibody. VSC2, prepared as 0.5–10 mM stock solutions in 10% DMSO, was diluted in HBS-EP+ (GE Healthcare) to make the final DMSO concentration of 1%. Binding experiments were performed by passing various concentrations of VSC2 in running buffer (1% DMSO in HBS-EP+) at a flow rate of 30 μL·min−1 for 180 s, followed by dissociation for 300 s. At each cycle, the sensor chip was regenerated by passing 10 mM glycine-Cl (pH 1.5) at a flow rate of 30 μL·min−1 for 60 s. The recorded sensograms were then analysed (BIAevaluation software, GE Healthcare). To correct for non-specific interactions, the reference surface data (running buffer only) was subtracted from the reaction surface data. The KD was calculated after globally fitting the data using a 1:1 binding model.
Data analyses
Data are expressed as mean ± SEM. Data were analysed using the two-tailed Student's t-test or one-or two-way anova, followed by post Dunnett's multiple comparison test and Tukey's multiple comparison test. Statistical tests were carried out using Prism. A value of P < 0.05 was considered statistically significant for all analyses.
Materials
FBS, horse serum, RPMI 1640, DMEM, penicillin-streptomycin, trypsin/EDTA and TRIzol reagent were from Life Technologies (Seoul, Korea). LPS, PMSF and MPTP were obtained from Sigma-Aldrich (St. Louis, MO, USA). Primary antibodies used were: rabbit polyclonal anti-mouse Nrf2, GCL catalytic subunit (GCLC) (Novus Biologicals, Littleton, CO, USA), GCL modulatory subunit (GCLM), NF-κB and Iba-1 (Wako Chemicals, Osaka, Japan) antibodies; goat polyclonal anti-mouse COX-2, lamin B (Santa Cruz Biotechnology, Santa Cruz, CA, USA), HO-1 (Enzo Life Sciences, Farmingdale, NY, USA), Hsp90 (Cell Signaling, Danvers, MA, USA) and NQO1 (Ab Frontier, Seoul, South Korea) antibodies; and mouse monoclonal anti-mouse β-actin and tyrosine hydroxylase (TH) antibodies (Sigma-Aldrich). Anti-rabbit IgG, anti-goat IgG and anti-mouse IgG were from Sigma-Aldrich. Alexa Fluor 546 goat anti-mouse IgG and Alexa Fluor 488 goat anti-rabbit IgG were purchased from Molecular Probe (Eugene, OR, USA). Recombinant His-tagged Keap1 protein was provided by Bioprogen (Daejeon, Korea). First strand cDNA synthesis kit for reverse transcription (RT)-PCR was purchased from MBI Fermentas (Ontario, Canada). elisa kits for IL-1β and TNF-α were from R&D Systems (Minneapolis, MN, USA) and eBioscience (San Diego, CA, USA) respectively. Bradford protein assay kit was from Bio-Rad (Hempstead, UK). Vectastain ABC kit and biotinylated secondary antibodies were purchased from Vector Laboratories (Burlingame, CA, USA). Enhanced luminal-based chemiluminescence Western blotting detection system was obtained from Pierce Chemical (Rockford, IL, USA).
Results
VSC2 down-regulates NO production in activated microglia that correlates with Nrf2 activity
To determine whether vinyl sulfone derivatives might exhibit anti-inflammatory properties, we selected three methoxy vinyl sulfone compounds having a chloro group at 2′ position. VSC2, VSC3 and VSC4. These compounds differed in that a methoxy group was substituted at 2′, 3′ and 4′ positions of the phenyl group respectively (Figure 1A). BV-2 microglia cells were activated by LPS and the amount of NO produced in the presence of each compound was assessed. The data showed that all three compounds suppressed NO production in a dose-dependent manner, with VSC2 demonstrating the highest activity (Figure 1B). Significant inhibition was achieved at 1 μM VSC2 and almost complete inhibition at 5 μM (Figure 1B), with an IC50 of 1.52 μM. VSC3 and VSC4 were relatively less active (IC50 = 2.96 and 16.22 μM respectively). Curcumin, used as a positive control compound, had an IC50 of 13.24 μM. Interestingly, we had previously noted that VSC2 had the highest potency in inducing Nrf2-dependent gene expression (3.8-fold compared with vehicle-treated control for HO-1 at 20 μM), followed by VSC3 (2.8-fold) and VSC4 (1.6-fold) (Woo et al., 2014). Thus, it was possible that a correlation existed between the NO-suppressing and Nrf2-activating activities of these compounds. For confirmation, we investigated the effects of nine different VSC2 derivatives on NO production and compared them with their Nrf2-activating activities (Table 2). The results revealed a fairly good linearity (Figure 1C). The data taken together suggested that the NO-suppressing activity of the vinyl sulfone compounds was likely to be associated with Nrf2 signalling. The subsequent experiments were performed on VSC2, the compound with the highest potency.
Figure 1.
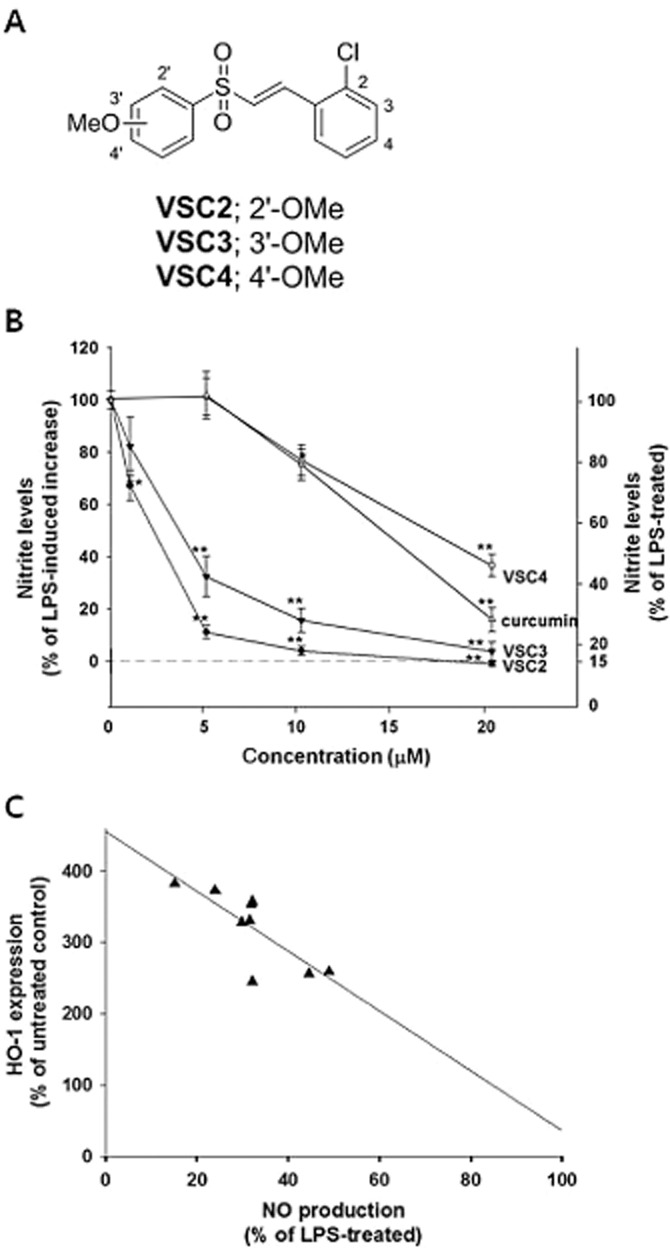
VSC2 down-regulates NO production in activated microglia that correlates with Nrf2 activity. (A) Chemical structures of VSC2, VSC3 and VSC4; (B) BV-2 cells were exposed to various concentrations of vinyl sulfones and 0.2 μg mL−1 LPS. After 24 h, the NO in the cell culture medium was measured. Curcumin was used as a positive control compound. The data are expressed as % of LPS-induced increase ± SEM and % of LPS-treated ± SEM; *P < 0.05, **P < 0.01 versus LPS-treated. (C) BV-2 cells were exposed to 20 μM of various vinyl sulfones (listed in Table 1) and 0.2 μg·mL−1 LPS. After 24 h, NO in the cell culture medium was measured. % of LPS-treated was plotted against degrees of HO-1 induction, and the best-fit line was generated by Sigma Plot software.
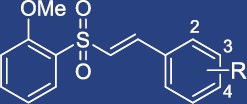
Table 2.
Effects of VSC2-derivatives on HO-1 and NO production
| |||
|---|---|---|---|
| Compound | R | HO-1 (%)a | NO (%)b |
| VSC2a | 2-CF3 | 328 ± 31.8 | 29.9 ± 3.1 |
| VSC2b | 3-CF3 | 256 ± 39.8 | 44.6 ± 1.7 |
| VSC2c | 4-CF3 | 357 ± 11.7 | 32.1 ± 2.9 |
| VSC2d | 2-F | 373 ± 11.1 | 24.0 ± 0.7 |
| VSC2e | 3-F | 353 ± 23.4 | 31.9 ± 0.5 |
| VSC2f | 4-F | 259 ± 19.6 | 48.9 ± 1.5 |
| VSC2 | 2-Cl | 382 ± 12.6 | 15.2 ± 0.5 |
| VSC2g | 3-Cl | 245 ± 17.6 | 32.1 ± 1.7 |
| VSC2h | 4-Cl | 330 ± 19.8 | 31.5 ± 2.4 |
| Vehiclec | 100 | 16.2 ± 0.6 | |
| LPS-treated | 100.0 | ||
HO-1-inducing activity at 20 μM in BV-2 cells, determined previously by Woo et al., (2014), expressed as % of untreated control ± SEM.
NO production at 20 μM in LPS-treated BV-2 cells, expressed as % of LPS-treated control ± SEM.
Vehicle: 0.04% DMSO.
VSC2 suppresses expression of proinflammatory enzymes in activated microglia
We tested whether VSC2 might alter the gene expression of iNOS, the NO-synthesizing enzyme in microglia. As shown in Figure 2A and 2B, iNOS gene expression, dramatically enhanced by LPS, was dose-dependently suppressed by VSC2 at both mRNA and protein levels. Production of the proinflammatory enzyme COX-2 was also suppressed in a similar pattern (Figure 2C and 2D). VSC2 had no cytotoxicity by itself in the same concentration range (Figure 2E), demonstrating that the reductions were not due to cell death. The data from densitometric analyses of RT-PCR (gel photos on top, Figure 2A and 2C) and real-time RT-PCR (histograms on bottom) corresponded with each other very well, indicating that the two methods could be utilized interchangeably at least under our experimental conditions. Such findings were consistently observed in all of the subsequent experiments that measured mRNA levels (Figures 3A, 3B and 6A–6D).
Figure 2.
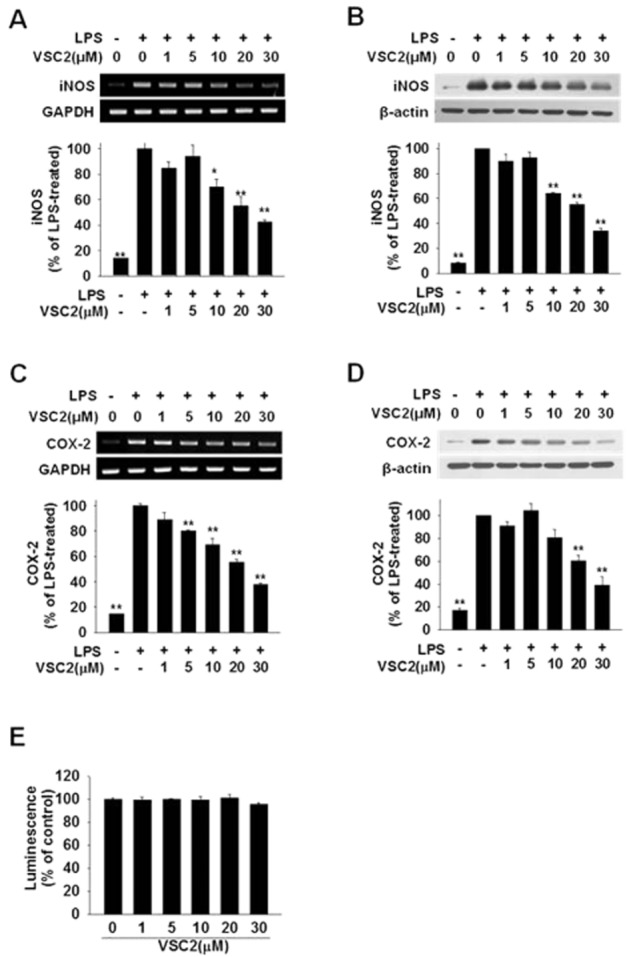
VSC2 suppresses expression of proinflammatory enzymes in activated microglia. BV-2 cells were exposed to various concentrations of VSC2 and 0.2 μg·mL−1 LPS. (A, C) The cells were harvested after 6 h and RT-PCR (gel photos above) and real-time RT-PCR (histograms below) were performed against iNOS and COX-2, using GAPDH as an internal control. (B,D) The cells were harvested after 24 h and Western blot analyses were performed against iNOS and COX-2, using β-actin as an internal control. The data are expressed as % of LPS-treated ± SEM; *P < 0.05, **P < 0.01 versus LPS-treated. (E) Viability of BV-2 cells that had been exposed to various concentrations of VSC2 for 24 h was assessed by intracellular ATP assay. No changes in viability were observed.
Figure 3.
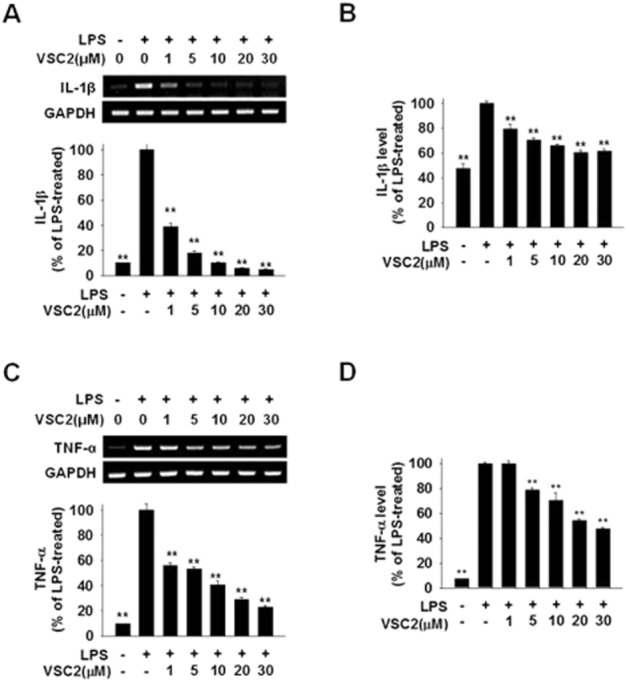
VSC2 suppresses production of proinflammatory cytokines in activated microglia. BV-2 cells were exposed to various concentrations of VSC2 and 0.2 μg·mL−1 LPS. (A, C) The cells were harvested after 6 h and RT-PCR (gel photos above) and real-time RT-PCR (histograms below) were performed against IL-1β and TNF-α; (B, D) elisa was performed against IL-1β and TNF-α after 24 h. The data are expressed as % of LPS-treated ± SEM; **P < 0.01 versus LPS-treated.
Figure 6.

VSC2 induces gene expression of antioxidant enzymes in microglial cells. BV-2 cells were exposed to various concentrations of VSC2. (A–D) The cells were harvested after 6 h for RT-PCR (gel photos above) and real-time RT-PCR (histograms below) against the antioxidant enzymes. (E–H) The cells were harvested after 24 h, the cell lysates were subjected to Western blot, densitometric analyses were performed and the data were normalized against the internal control β-actin. The data are expressed as fold induction of untreated control ± SEM; *P < 0.05, **P < 0.01 versus untreated control.
VSC2 suppresses production of proinflammatory cytokines in activated microglia
VSC2 was examined for its effects on production of the proinflammatory cytokines. LPS-induced elevation of IL-1β was suppressed by VSC2 (Figure 3A and 3B), with 1 μM already dramatically lowering both mRNA and protein levels. TNF-α was also down-regulated in a dose-dependent manner, with significant down-regulation observed at 1–5 μM VSC2 (Figure 3C and D).
VSC2 suppresses NF-κB signalling in activated microglia
Effects of VSC2 on NF-κB, the transcription factor responsible for expression of proinflammatory genes, were tested. As shown in Figure 4A, the NF-κB p65 subunit, which was dramatically increased in the nuclear fraction upon LPS treatment, was lowered in a VSC2-dose-dependent manner. The nuclear fraction was not contaminated by cytosolic proteins as confirmed by the absence of immunoreactivity to the cytosolic marker hsp90. The transcriptional activity of nuclear NF-κB was also tested after transfecting the cells with a plasmid containing the NF-κB binding element and luciferase gene. Determination of the luciferase activity revealed that the VSC2 treatment efficiently lowered the NF-κB-dependent gene transcription (Figure 4B).
Figure 4.
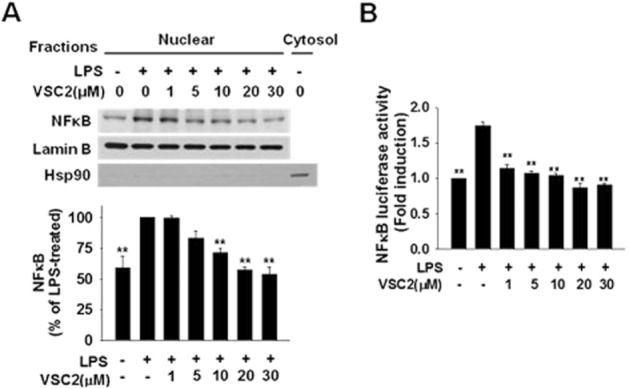
VSC2 suppresses NF-κB signalling in activated microglia. (A) BV-2 cells were treated with various concentrations of VSC2 for 1 h and co-treated with 0.2 μg·mL−1 LPS for an additional hour. NF-κB in the nuclear fraction was estimated by Western blot analysis. The same blot was subjected to Western blot against lamin B as an internal control and the cytosolic marker protein hsp90 as a negative control. After densitometry, the data were normalized against lamin B. Data are expressed as % of LPS-treated control ± SEM. (B) BV-2 cells were transfected with a plasmid containing NF-kB-luciferase reporter construct. After 48 h, the cells were treated with various concentrations of VSC2 and 0.2 μg·mL−1 LPS for 6 h. Luciferase activity in the cell lysate was measured. Data are expressed as induction fold of untreated control ± SEM; **P < 0.01 versus LPS-treated.
VSC2 binds Keap1 and activates Nrf-2 signalling in microglia
Whether VSC2 might activate the microglial Nrf2 signalling was determined. As shown in Figure 5A, nuclear Nrf2 was dose-dependently increased by VSC2, with a significant increase at 1 μM. Because the activated cytosolic Nrf2 is not subjected to degradation by the ubiquitin-proteasome system (Fourquet et al., 2010), the total level would be expected to increase. Indeed, VSC2 caused elevation of total cellular Nrf2 protein (Figure 5B). We also tested whether VSC2 might lead to elevation of the Nrf2 transcriptional activity by transfecting the cells with a plasmid containing ARE, the Nrf2-binding element, and determining the luciferase reporter activity. The results showed that VSC2 was indeed able to increase the ARE-dependent luciferase activity in a dose-dependent manner (Figure 5C).
Figure 5.
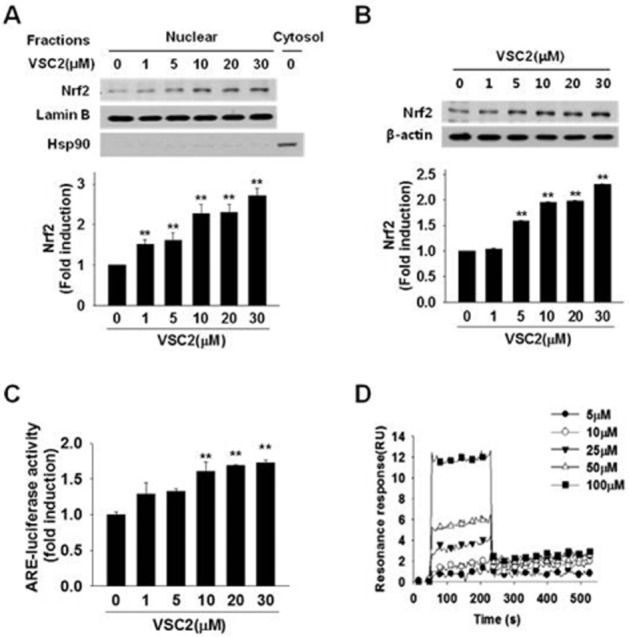
VSC2 binds Keap1 and activates Nrf2 signalling in microglia. BV-2 cells were exposed to various concentrations of VSC2. (A) The cells were harvested after 3 h and Nrf2 Western blot analysis was performed on nuclear fraction. The same blot was subjected to Western blot against lamin B as an internal control and the cytosolic marker protein hsp90 as a negative control. (B) The cells were harvested after 24 h and Nrf2 Western blot analysis was performed on cell lysate, using β-actin as an internal control. (C) BV-2 cells were transfected with a plasmid containing an ARE-luciferase reporter construct. After 48 h, the cells were treated with various concentrations of VSC2 for 6 h and luciferase activity in the cell lysate was measured. The data are expressed as fold induction of untreated control ± SEM; **P < 0.01 versus untreated control. (D) Surface plasmon resonance analysis was performed to assess binding kinetics between VSC2 and Keap1. The data are expressed as resonance units (RU) as a function of time.
Because Nrf2 activation is known to require release from its binding protein Keap1, we tested whether VSC2 might directly act on Keap1. This was done by detecting changes in surface plasmon resonance of Keap1 in the presence of VSC2. For this, Keap1 was immobilized on a sensor chip and VSC2 in free solution was allowed to interact with Keap1. Analysis of the sensograms indicating real-time interaction showed a dose-dependent increase (Figure 5D) and the KD was determined to be 5.05 × 10−11. The data together indicated the presence of a direct binding between VSC2 and Keap1.
VSC2 induces gene expression of antioxidant enzymes in microglial cells
Because the Nrf2-dependent antioxidant enzymes have been associated with anti-inflammatory properties (Rushworth et al., 2008; Paine et al., 2010), we asked whether these enzyme genes might be induced in microglial cells by VCS2 in the same concentration range. The NQO1 mRNA level appeared to be dramatically and dose-dependently elevated by VSC2, as its level in untreated BV-2 cells was very low (Figure 6A). Western blot analysis also revealed the elevation of NQO1 protein that was already evident at 1 μM (Figure 6E). HO-1, the enzyme that converts haem to biliverdin and carbon monoxide, was also increased dose-dependently at both mRNA and protein levels by VSC2, with significant increases observed at 5 μM (Figure 6B and 6F). GCL, an enzyme in the biosynthetic pathway for the major cellular antioxidant glutathione, consists of two subunits, the modulatory (GCLM) and catalytic (GCLC) subunits (Botta et al., 2008; Lu, 2009). RT-PCR against GCLM and GCLC showed that both subunits were effectively induced by VSC2 (Figure 6C and 6D). Dose-dependent increases were also observed at the protein level (Figure 6G and 6H).
VSC2 inhibits microglial activation in an animal model of PD
We determined whether VSC2 could suppress neuroinflammation in vivo by utilizing the MPTP-elicited model of PD in mice. Iba-1, a microglial marker (Sierra et al., 2007), was used to visualize microglial cells in the SN, the region of the dopaminergic neurons that undergo degeneration in PD. As seen in Figure 7A, an increase in the Iba-1-immunoreactive cells was evident in the SN region in the MPTP-treated animals, but this was not observed in the VSC2-co-treated animals. Higher magnification revealed that the MPTP treatment caused the microglia cells to assume amoeboid morphology typical of activated microglia (Loane and Byrnes, 2010). In comparison, the microglia in the VSC2-co-treated animals and vehicle-treated animals had a ramified morphology of resting microglia. Quantitative analysis showed that the Iba-1 immunodensity was doubled in the MPTP animals (196 ± 20% of vehicle-treated control), whereas the level remained low in the VSC2-co-treated (124 ± 21%). Double immunofluorescent staining for TH and Iba-1 (Figure 7B) also confirmed that the number of microglia is greatly reduced in the VSC2-co-treated group. Upon merging, the TH-positive dopaminergic neurons were detected only where microglia was absent, indirectly confirming the role of activated microglia in dopaminergic neurodegeneration. VSC2 alone had no apparent effect on Iba-1 or TH staining in the SN (data not shown).
Figure 7.
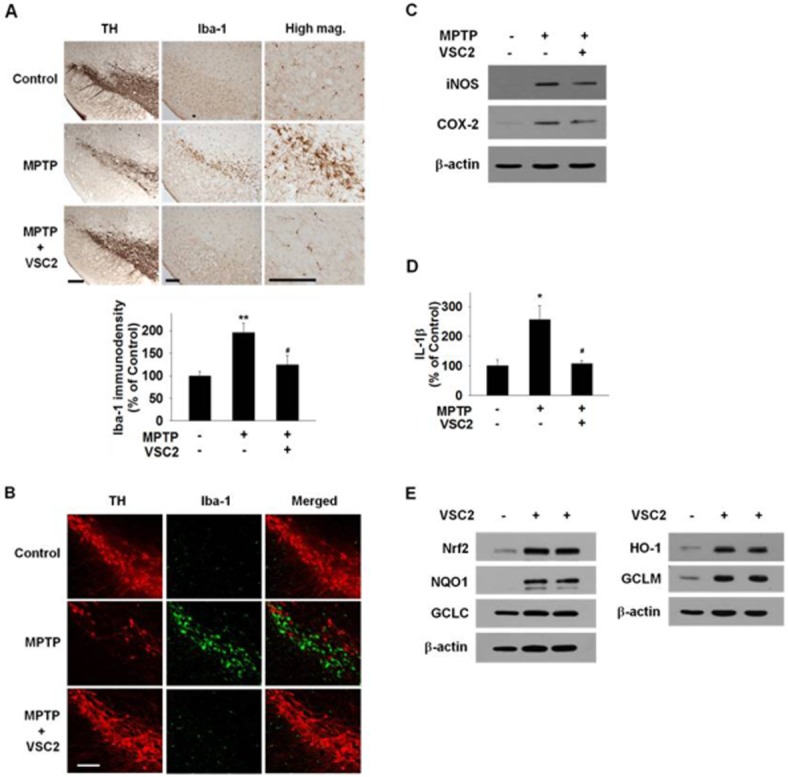
VSC2 has both anti-inflammatory and antioxidant properties in the brain nigrostriatal region. Mice were treated with MPTP only or co-treated with 10 mg·kg−1 VSC2. (A) Immunostaining was conducted against TH (left) and Iba-1 (middle and right) on adjacent nigral sections and they were developed in diaminobenzidine. The density of Iba-1-immunoreactivity was determined (below). (B) Double immunofluorostaining was conducted against TH (left) and Iba-1 (middle) on nigral sections and the two immunofluorographs were merged (right). Scale bars = 200 μm. (C) Striatal tissues were subjected to Western blot against iNOS and COX-2 using β-actin as an internal control. (D) Nigral tissues were subjected to elisa against IL-1β. (E) Striatal tissue samples of two different mice treated with 10 mg·kg−1 VSC2 were subjected to Western blot against Nrf2 and the antioxidant enzymes, using β-actin as an internal control. (A, D) Data are expressed as % of vehicle-treated ± SEM; *P < 0.05 and **P < 0.01 versus vehicle-treated; #P < 0.05 versus MPTP-treated.
We also determined whether the proinflammatory system might also be suppressed by VSC2 in the same in vivo system. Western blot analysis revealed that, as expected, iNOS and COX-2 were elevated in the MPTP-treated PD model compared with the vehicle-treated model. These increases were attenuated when the animals were co-treated with VSC2 (by 33% and 25% for iNOS and COX-2 respectively) (Figure 7C). The cytokine IL-1β, up-regulated in the MPTP animals, was also abolished in the VSC2-co-treated animals (Figure 7D). Whether the Nrf2-dependent system is induced in the same VSC2-treated animals was tested. As expected, Western blot analyses on the striatal tissues revealed dramatic elevations in the levels of Nrf2 (435% of untreated control) as well as the Nrf2-dependent antioxidant enzymes HO-1 (378%) and GCLM (378%) and NQO1 (Figure 7E). Among the antioxidant enzymes, NQO1 appeared to be most dramatically increased, but the degree of induction could not be accurately assessed due to the non-detectable level in the untreated control.
Discussion and conclusions
Currently, there is no therapy available that suppresses the progression of neurodegeneration in PD and neuroinflammation is believed to play a significant role in the pathogenesis of the disease (see Tansey and Goldberg, 2010; Blandini, 2013). Hence, anti-inflammatory agents that modify the disease progress would be highly beneficial. In the present study, we introduce a novel synthetic compound VSC2 that has a potent activity in down-regulating the production of proinflammatory mediators both in vitro and in vivo. VSC2 interacted with Keap1, caused Nrf2 activation and led to expression of the Nrf2-dependent antioxidant enzymes in microglia.
Compounds with anti-inflammatory properties have been shown to provide neuroprotection in animal models of PD. We have previously reported that doxycycline (Cho et al., 2009) and a novel synthetic compound 7-hydroxy-6-methoxy-2-propionyl-1,2,3,4-tetrahydro-isoquinoline (Son et al., 2007) suppressed the production of proinflammatory mediators and protected the nigral dopaminergic neurons in the mouse model of PD. Natural compounds such as licochalcone E (Kim et al., 2012) and sulforaphane (Jazwa et al., 2011; Morroni et al., 2013) and a new synthetic triterpenoid (Kaidery et al., 2013) have also been observed to possess similar properties. Herein, we introduce another compound that can effectively down-regulate the production of inflammatory molecules NO, iNOS, COX-2, TNF-α and IL-1β and the transcriptional activity of NF-κB in activated microglia, suppress microglial activation and protect the nigral neurons.
Evidence shows that Nrf2 is involved in the regulation of neuroinflammation in various PD models (Innamorato et al., 2008; 2010,; Rojo et al., 2010). In other systems, existence of a cross-talk between the Nrf2 system and NF-κB pathway has been reported (Wakabayashi et al., 2010) and NF-κB activation induced by LPS could be attenuated by natural Nrf2 activators (Jeong et al., 2004). We demonstrate in the present study that VSC2 induces Nrf2 activation by the following observations: (i) the compound bound to Keap1, the Nrf2-inhibiting protein whose modification releases and activates Nrf2 (Villeneuve et al., 2010); (ii) the compound increased Nrf2 in the nucleus, the site of action for the transcription factor; (iii) the compound led to the stabilization of Nrf2 (i.e. increased total Nrf2 level), indicative of liberation of Nrf2 from Keap1; (iv) the compound increased the transcriptional activity of Nrf2; and (v) the compound led to induction of expression of the Nrf2-regulated genes.
It is likely that the anti-inflammatory property of VSC2 is at least in part obtained via Nrf2 activation. The correlation between Nrf2-activating and anti-inflammatory activities among the various vinyl sulfones also supports this view. The vinyl sulfone compounds do not directly scavenge free radicals (data not shown), eliminating the possibility that VSC2 might directly remove ROS. Therefore, it is most likely that the Nrf2-regulated antioxidant enzymes within microglia, induced by VSC2, remove ROS and prevent the ROS-induced NF-κB activation and the subsequent production of proinflammatory molecules. Additionally, it is possible that VSC2 might concurrently act on other pathways that influence the system.
VSC2 appeared to directly interact with the Keap1 protein. Keap1 is known to contain multiple cysteine residues, some of which have been identified as the thiol whose modification causes a conformational change in the protein (Kaspar et al., 2009). VSC2 has an activated vinyl group, which is a Michael-type addition acceptor expected to be attacked by cellular nucleophiles such as the thiol group of cysteine. Previous studies have shown that Michael reaction acceptors activate the Nrf2 pathway (Dinkova-Kostova et al., 2001) and that the α,β-unsaturated carbonyl group acceptor interferes with Keap1-Nrf2 binding (Wu et al., 2010). It is therefore possible that VSC2 acts on the cysteine thiol(s) of Keap1 causing a conformational change and liberation of Nrf2.
Natural compounds such as sulforaphane, curcumin and chalcones exhibit antioxidant and anti-inflammatory properties. However, many of the natural compounds have limited metabolic stability and low brain penetration rate, resulting in low efficacy especially when aiming at disorders of the CNS. We found that our vinyl sulfone compounds have stability against microsomal and plasma enzymes in an acceptable range (data not shown). This may have contributed to the relatively potent microglia-suppressing and neuroprotective effects of VSC2 in our animal model. VSC2 was effective at a moderately low dose of 10 mg·kg−1, given by oral gavage. In comparison, a higher dose seems to be required for sulforaphane: Clarke et al. (2011) showed that after oral gavage of 35 or 140 mg·kg−1, the amount of sulforaphane detected in the brain was 1/1000 of that in the small intestine and that no free sulforaphane was detected after 2 h. Pharmacokinetic and toxicity studies are being carried out to assess the utility of VSC2 as a drug for the CNS.
Use of drugs that activate the Nrf-2 signalling pathway should be especially relevant for treatment of PD, among age-related chronic diseases. The nigral system is particularly prone to ROS generation due to the presence of dopamine itself and the related enzymes. Degenerating nigral dopaminergic neurons release a number of cellular factors such as neuromelanin, α-synuclein and matrix metalloproteinase-3 that trigger microglial activation (see Hwang, 2013). Cytotoxic and inflammatory mediators released from the microglia in turn exert damage to nearby dopaminergic neurons, completing the vicious cycle that is thought to underlie the progressive nature of the neurodegeneration (see Blandini, 2013). As such, VSC2 should be able to block this chain of events at both ends. In the dopaminergic neurons, it can prevent oxidative damage and release of immunogenic molecules and in microglia it can preclude accumulation of ROS and proinflammatory molecules. Therefore, the neuroprotection by VSC2 shown in the PD animal model is likely to come from the combined effects of these mechanisms.
In conclusion, we have synthesized a novel compound VSC2 that, in cultured microglia and in vivo, (i) prevents NF-κB activation and production of iNOS, NO, COX-2, TNF-α and IL-1β; and (ii) induces activation of Nrf2 signalling and expression of antioxidant enzymes HO-1, NQO1, GCLC and GCLM. This is accompanied by protection of the SN dopaminergic neurons. Together with our previous finding that VSC2 can directly protect dopaminergic neurons and alleviate the PD-associated motor dysfunction, VSC2 should be useful towards the development of a therapy for PD.
Acknowledgments
This study was supported by the National Agenda Project from Korea Research Council of Fundamental Science and Technology (O. H.), the Korea Institute of Science and Technology (2E23870 & 2E22520, K. D. P.), the Korea Health Technology R&D Project of the Ministry of Health and Welfare (HI12C1022, K. D. P.) and the National Research Foundation of Korea (NRF-2013-R1A1A2059669, H. J. S.; NRF-2009-0081674, D. J. K.; NRF-2009-0081675, O. H.).
Glossary
- ARE
antioxidant response element
- GCLC
glutamate-cysteine ligase catalytic subunit
- GCLM
glutamate-cysteine ligase modulatory subunit
- HO-1
haem oxygenase-1
- iNOS
inducible NOS
- NQO1
NAD(P)H: quinone oxidoreductase 1
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- PD
Parkinson's disease
- ROS
reactive oxygen species
- RT
reverse transcription
- SN
substantia nigra
- TH
tyrosine hydroxylase
- VSC2
(E)-1-(2-((2-methoxyphenyl)sulfonyl)vinyl)-2-chlorobenzene
Author contributions
J. A. L., J. H. K. and S. Y. W. made equal contributions. J. A. L., J. H. K., H. J. S. and S. H. H. performed all the biological and Surface plasmon resonance experiments. S. Y. W., B. K. J. and J. W. C. synthesized the compounds. O. H., K. D. P. and D. J. K. designed the study. O. H. and K. D. P. analysed the data and wrote the manuscript.
Conflict of interest
There is no conflict of interest to disclose.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blandini F. Neural and immune mechanisms in the pathogenesis of Parkinson's disease. J Neuroimmune Pharmacol. 2013;8:189–201. doi: 10.1007/s11481-013-9435-y. [DOI] [PubMed] [Google Scholar]
- Botta D, White CC, Vliet-Gregg P, Mohar I, Shi S, McGrath MB, et al. Modulating GSH synthesis using glutamate cysteine ligase transgenic and gene-targeted mice. Drug Metab Rev. 2008;40:465–477. doi: 10.1080/03602530802186587. [DOI] [PubMed] [Google Scholar]
- Bukhari SN, Jantan I, Jasamai M. Anti-inflammatory trends of 1, 3-diphenyl-2-propen-1-one derivatives. Mini Rev Med Chem. 2013;13:87–94. [PubMed] [Google Scholar]
- Cho Y, Son HJ, Kim EM, Choi JH, Kim ST, Ji IJ, et al. Doxycycline is neuroprotective against nigral dopaminergic degeneration by a dual mechanism involving MMP-3. Neurotox Res. 2009;16:361–371. doi: 10.1007/s12640-009-9078-1. [DOI] [PubMed] [Google Scholar]
- Clarke JD, Hsu A, Williams DE, Dashwood RH, Stevens JF, Yamamoto M, et al. Metabolism and tissue distribution of sulforaphane in Nrf2 knockout and wild-type mice. Pharm Res. 2011;28:3171–3179. doi: 10.1007/s11095-011-0500-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Massiah MA, Bozak RE, Hicks RJ, Talalay P. Potency of Michael reaction acceptors as inducers of enzymes that protect against carcinogenesis depends on their reactivity with sulfhydryl groups. Proc Natl Acad Sci U S A. 2001;98:3404–3409. doi: 10.1073/pnas.051632198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourquet S, Guerois R, Biard D, Toledano MB. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J Biol Chem. 2010;285:8463–8471. doi: 10.1074/jbc.M109.051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. San Diego: Academic Press; 1997. pp. 59–67. [Google Scholar]
- Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72:1493–1505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Hara H, Ohta M, Adachi T. Apomorphine protects against 6-hydroxydopamine-induced neuronal cell death through activation of the Nrf2-ARE pathway. J Neurosci Res. 2006;84:860–866. doi: 10.1002/jnr.20974. [DOI] [PubMed] [Google Scholar]
- Hwang O. Role of oxidative stress in Parkinson's disease. Exp Neurobiol. 2013;22:11–17. doi: 10.5607/en.2013.22.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innamorato NG, Rojo AI, García-Yagüe AJ, Yamamoto M, de Ceballos ML, Cuadrado A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J Immunol. 2008;181:680–689. doi: 10.4049/jimmunol.181.1.680. [DOI] [PubMed] [Google Scholar]
- Innamorato NG, Jazwa A, Rojo AI, García C, Fernández-Ruiz J, Grochot-Przeczek A, et al. Different susceptibility to the Parkinson's toxin MPTP in mice lacking the redox master regulator Nrf2 or its target gene heme oxygenase-1. PLoS ONE. 2010;5:e11838. doi: 10.1371/journal.pone.0011838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazwa A, Rojo AI, Innamorato NG, Hesse M, Fernández-Ruiz J, Cuadrado A. Pharmacological targeting of the transcription factor Nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxid Redox Signal. 2011;14:2347–2360. doi: 10.1089/ars.2010.3731. [DOI] [PubMed] [Google Scholar]
- Jeong WS, Kim IW, Hu R, Kong AN. Modulatory properties of various natural chemopreventive agents on the activation of NF-kappaB signaling pathway. Pharm Res. 2004;21:661–670. doi: 10.1023/b:pham.0000022413.43212.cf. [DOI] [PubMed] [Google Scholar]
- Kaidery NA, Banerjee R, Yang L, Smirnova NA, Hushpulian DM, Liby KT, et al. Targeting Nrf2-mediated gene transcription by extremely potent synthetic triterpenoids attenuate dopaminergic neurotoxicity in the MPTP mouse model of Parkinson's disease. Antioxid Redox Signal. 2013;18:139–157. doi: 10.1089/ars.2011.4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang CH, Kim MJ, Seo MJ, Choi YH, Jo WS, Lee KT, et al. 5-Hydroxy-3,6,7,8,3′4′-hexamethoxyflavone inhibits nitric oxide production in lipopolysaccharide-stimulated BV2 microglia via NF-κB suppression and Nrf-2-dependent heme oxygenase-1 induction. Food Chem Toxicol. 2013;57:119–125. doi: 10.1016/j.fct.2013.03.019. [DOI] [PubMed] [Google Scholar]
- Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. 2009;47:1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SS, Lim J, Bang Y, Gal J, Lee SU, Cho YC, et al. Licochalcone E activates Nrf2/antioxidant response element signaling pathway in both neuronal and microglial cells: therapeutic relevance to neurodegenerative disease. J Nutr Biochem. 2012;23:1314–1323. doi: 10.1016/j.jnutbio.2011.07.012. [DOI] [PubMed] [Google Scholar]
- Koh K, Kim J, Jang YJ, Yoon K, Cha Y, Lee HJ, et al. Transcription factor Nrf2 suppresses LPS-induced hyperactivation of BV-2 microglial cells. J Neuroimmunol. 2011;233:160–167. doi: 10.1016/j.jneuroim.2011.01.004. [DOI] [PubMed] [Google Scholar]
- Lee IS, Lim J, Gal J, Kang JC, Kim HJ, Kang BY, et al. Anti-inflammatory activity of xanthohumol involves heme oxygenase-1 induction via NRF2-ARE signaling in microglial BV2 cells. Neurochem Int. 2011;58:153–160. doi: 10.1016/j.neuint.2010.11.008. [DOI] [PubMed] [Google Scholar]
- Lin W, Wu RT, Wu T, Khor TO, Wang H, Kong AN. Sulforaphane suppressed LPS-induced inflammation in mouse peritoneal macrophages through Nrf2 dependent pathway. Biochem Pharmacol. 2008;76:967–973. doi: 10.1016/j.bcp.2008.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurother. 2010;7:366–377. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009;30:42–59. doi: 10.1016/j.mam.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morroni F, Tarozzi A, Sita G, Bolondi C, Zolezzi Moraga JM, Cantelli-Forti G, et al. Neuroprotective effect of sulforaphane in 6-hydroxydopamine-lesioned mouse model of Parkinson's disease. Neurotoxicology. 2013;36:63–71. doi: 10.1016/j.neuro.2013.03.004. [DOI] [PubMed] [Google Scholar]
- Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem Pharmacol. 2010;80:1895–1903. doi: 10.1016/j.bcp.2010.07.014. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo AI, Innamorato NG, Martín-Moreno AM, De Ceballos ML, Yamamoto M, Cuadrado A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson's disease. Glia. 2010;58:88–98. doi: 10.1002/glia.20947. [DOI] [PubMed] [Google Scholar]
- Rushworth SA, MacEwan DJ, O'Connell MA. Lipopolysaccharide-induced expression of NAD(P)H: quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J Immunol. 2008;181:6730–6737. doi: 10.4049/jimmunol.181.10.6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Gottfried-Blackmore AC, McEwen BS, Bulloch K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia. 2007;55:412–424. doi: 10.1002/glia.20468. [DOI] [PubMed] [Google Scholar]
- Son HJ, Lee JA, Shin N, Choi JH, Seo JW, Chi DY, et al. A novel compound PTIQ protects the nigral dopaminergic neurones in an animal model of Parkinson's disease induced by MPTP. Br J Pharmacol. 2012;165:2213–2227. doi: 10.1111/j.1476-5381.2011.01692.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuckenholz V, Bacher M, Balzer-Geldsetzer M, Alvarez-Fischer D, Oertel WH, Dodel RC, et al. The α7 nAChR agonist PNU-282987 reduces inflammation and MPTP-induced nigral dopaminergic cell loss in mice. J Parkinsons Dis. 2013;3:161–172. doi: 10.3233/JPD-120157. [DOI] [PubMed] [Google Scholar]
- Tansey MG, Goldberg MS. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teismann P, Ferger B. Inhibition of the cyclooxygenase isoenzymes COX-1 and COX-2 provide neuroprotection in the MPTP-mouse model of Parkinson's disease. Synapse. 2001;39:167–174. doi: 10.1002/1098-2396(200102)39:2<167::AID-SYN8>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve NF, Lau A, Zhang DD. Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: an insight into cullin-ring ubiquitin ligases. Antioxid Redox Signal. 2010;13:1699–1712. doi: 10.1089/ars.2010.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi N, Slocum SL, Skoko JJ, Shin S, Kensler TW. When NRF2 talks, who's listening? Antioxid Redox Signal. 2010;13:1649–1663. doi: 10.1089/ars.2010.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SY, Kim JH, Moon MK, Han SH, Yeon SK, Choi JW, et al. Discovery of vinyl sulfones as a novel class of neuroprotective agents toward Parkinson's disease therapy. J Med Chem. 2014;57:1473–1487. doi: 10.1021/jm401788m. [DOI] [PubMed] [Google Scholar]
- Wu RP, Hayashi T, Cottam HB, Jin G, Yao S, Wu CC, et al. Nrf2 responses and the therapeutic selectivity of electrophilic compounds in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2010;107:7479–7484. doi: 10.1073/pnas.1002890107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HF, Liu XY, Niu DB, Li FQ, He QH, Wang XM. Triptolide protects dopaminergic neurons from inflammation-mediated damage induced by lipopolysaccharide intranigral injection. Neurobiol Dis. 2005;18:441–449. doi: 10.1016/j.nbd.2004.12.005. [DOI] [PubMed] [Google Scholar]