

Abstract
Background and Purpose
Rotigotine acts as a dopamine receptor agonist with high affinity for the dopamine D2, D3, D4 and D5 receptors but with a low affinity for the dopamine D1 receptor. We have investigated this further in radioligand binding and functional studies and compared the profile of rotigotine with that of other drugs used in the treatment of Parkinson's disease (PD).
Experimental Approach
The binding of rotigotine to human dopamine D1, D2, D3, D4 and D5 receptors was determined in radioligand binding studies using [3H]rotigotine and compared with that of standard antagonist radioligands. Functional interactions of rotigotine with human dopamine receptors was also determined.
Key Results
[3H]rotigotine can be used as an agonist radioligand to label all dopamine receptor subtypes and this can be important to derive agonist affinity estimates. Rotigotine maintains this high affinity in functional studies at all dopamine receptors especially D1, D2 and D3 receptors and, to a lesser extent, D4 and D5 receptors. Rotigotine, like apomorphine but unlike ropinirole and pramipexole, was a potent agonist at all dopamine receptors.
Conclusions and Implications
Rotigotine is a high-potency agonist at human dopamine D1, D2 and D3 receptors with a lower potency at D4 and D5 receptors. These studies differentiate rotigotine from conventional dopamine D2 agonists, used in the treatment of PD, such as ropinirole and pramipexole which lack activity at the D1 and D5 receptors, but resembles that of apomorphine which has greater efficacy in PD than other dopamine agonists but has suboptimal pharmacokinetic properties.
Tables of Links
| TARGETS |
|---|
| Dopamine D1 receptors |
| Dopamine D2 receptors |
| Dopamine D3 receptors |
| Dopamine D4 receptors |
| Dopamine D5 receptors |
| LIGANDS | |
|---|---|
| [3H]raclopride | Dopamine |
| [3H]SCH23390 | Lisuride |
| [3H]spiperone | Pramipexole |
| 7-OH-DPAT | Quinpirole |
| Apomorphine | Ropinirole |
| Chlorpromazine | Rotigotine |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Parkinson's disease (PD) is a chronic, degenerative neurological disease which is characterized by the progressive loss of nigrostriatal dopaminergic pathways giving rise to a range of motor symptoms including rigidity, tremor and bradykinesia. Currently available medications improve signs and symptoms for several years but do not slow the progression of the underlying disease. These medications focus on replacing, or compensating for, the central dopamine deficiency. Levodopa has been the gold standard therapy for the treatment of PD, but it is now clear that the initial control of motor symptoms is confounded after long-term treatment by the development of motor complications (dyskinesias; Jenner, 1995; 2008). Recent advances in medication have focused on the development of novel non-ergoline dopaminergic agonists, as ergoline agonists are associated with rare but severe side effects such as cardiac valve fibrosis (Tan, 2003). These novel dopamine agonists include rotigotine, ropinirole and pramipexole, all of which show high efficacy in reducing motor symptoms with a reduced propensity to induce dyskinesias compared with levodopa especially in younger patients (Clarke and Guttman, 2002; Needham and Worth, 2012). This has led to the use of dopamine agonists as monotherapy in early/mild PD to avoid or delay the need for levodopa (Wood, 2010). Dopamine agonists vary in their pharmacokinetic properties and mode of administration, with many of the drugs requiring repeat daily administration (see Rascol et al., 2007).
Dopamine receptors (see Schwartz et al., 1998; Beaulieu and Gainetdinov, 2011) are divided into two subclasses: the D1-like which consists of D1 and D5 receptors and the D2-like which includes D2, D3 and D4 receptors. The dopamine D2 receptor exists as a short and long splice variant (D2S and D2L with a 29 amino acid insert in the third intracellular loop), while the D4 receptor is highly polymorphic in humans with a repeat region in the third cytoplasmic loop. Agonist binding to the D2-like receptors results in activation of a variety of signalling pathways including inhibition of adenylyl cyclase, which is sensitive to the effects of Pertussis toxin, indicating that these responses are mediated by Gi/o proteins. All of the non-ergoline agonists currently in clinical use share the property of binding and activating the D2-like family of dopamine receptors, although they differ in their relative efficacy at these receptors (Millan et al., 2002; Newman-Tancredi et al., 2002). Thus, pramipexole preferentially stimulates dopamine D3 receptors compared with D2 receptors, whereas ropinirole stimulates both dopamine D2 and D3 receptors. Although the D4 receptor is also in the D2-like family, little is known concerning its role in relation to basal ganglia circuits and motor function. The D4 receptor is present within the prefrontal cortex, amygdala, hippocampus, thalamus, caudate putamen and cerebellum with lower levels in the mid-brain (see Lauzon and Laviolette, 2010). Selective antagonists (see Beaulieu and Gainetdinov, 2011) exist which preferentially block the D2, D3 and D4 receptors. The dopamine D1 and D5 receptors activate the Gαs/olf family of G-proteins to stimulate cAMP production. They are found exclusively postsynaptically, they lack introns in their coding regions and are 80% homologous in their transmembrane domains. The D1 subtype is the predominant form and is localized in the primary dopaminergic projection areas such as striatum, nucleus accumbens and cortical areas whereas the D5 subtype is expressed in cortical, subcortical and limbic areas (Kahn et al., 2000). There is currently no selective agent, either agonist or antagonist, which discriminates between D1 and D5 receptors.
Rotigotine ([-]2-(N-propyl-N-2-thienylethylamino)-5-hydroxytetralin) was launched as a non-ergoline dopamine receptor agonist for the treatment of idiopathic PD (Baldwin and Keating, 2007) and for the treatment of restless legs syndrome (Baldwin and Keating, 2008; Oertel et al., 2011). In vitro receptor binding studies (Scheller et al., 2009) have shown that rotigotine has a 10-fold selectivity for D3 (pKi 9.2) receptors compared with D2, D4 and D5 (pKi 8.5–8.0) and a 100-fold selectivity compared with D1 receptors (pKi 7.2). In functional studies, rotigotine behaved as full agonist at all dopamine receptors but notably the potency for stimulation of D1 receptors was similar to that for D2 and D3 receptors (pEC50 respectively: 9.0, 9.4–8.6, 9.7). Differences between binding affinities and functional potency can be the result of many factors such as receptor reserve and different affinity states. For other GPCRs where agonist and antagonist radioligands exist, marked differences in agonist and antagonist potency are often seen. For example, at the muscarinic M1 receptor, unlike antagonist ligands, agonist ligands displayed marked differences in their potency to inhibit the binding of the agonist radioligand [3H]oxotremorine-M and the antagonist radioligand [3H]N-methyl scopolamine (Freedman et al., 1988). These differences reflect the presence of high-and low-affinity agonist states of the GPCR (DeLean et al., 1980) and have been demonstrated for a variety of GPCRs (Watson et al., 2000).
We have studied further the pharmacological interaction of rotigotine with all the dopamine receptors and compared this with other dopamine agonists used in the treatment of PD. We have performed these using two different approaches. First, we used radioligand binding comparing agonist and antagonist radioligands. We compared the receptor binding profile of the agonists used in the treatment of PD and of pharmacological standards on human recombinant dopamine D1, D2, D3, D4 and D5 receptors using both agonist [3H]rotigotine (Van der Weide et al., 1987) and [3H]antagonist radioligands to label these receptors. Second, we have studied the functional interaction of these compounds with the D1, D2, D3, D4 and D5 receptors. Functional studies in D1, D2 and D3 receptors were performed using the technique of cellular dielectric spectroscopy (CDS, so-called ‘label-free’) performed with the Molecular Devices (Wokingham, UK). CDS measures the changes in impedance of a cell layer that occur in response to receptor stimulation which reflects a morphological change in the cells and, as such, is independent of the signalling pathway utilized (Peters et al., 2010). Changes (both positive and negative) in cellular impedance are measured in real time in live cells and can be used to quantify various responses including full/partial agonism, antagonism and allosteric modulation (Rocheville and Jerman, 2009). The response measured represents an integrated response probably reflecting multiple cellular events downstream of the initial receptor event; for example, dopamine D1 and D2 receptors couple to different G-proteins. The advantage of using CDS is that we could study receptor activation at the three dopamine receptors using a single methodology. However, this method requires the use of adherent cells which attach to the electrode plate. For the human dopamine D4 receptor, the receptor was expressed in CHO cells grown in suspension and attempts to attach the cells to the plates using various coating did not give reproducible results. We therefore used agonist-induced G-protein activation as a functional measure at the D4 receptor. Our efforts to generate a human D5 receptor expressing cell line failed to yield sufficient expression for radioligand binding studies. The human D5 receptor cell line was therefore obtained from a commercial source which was coupled to measure changes in intracellular calcium.
Methods
Cell culture
All cells were cultured at 37°C in a humidified atmosphere of 5% CO2. Cells were grown in DMEM-F12 + GlutaMAX™-I medium (GIBCO ®, Invitrogen, Merelbeke, Belgium) containing 10% FBS (BioWhittaker ®, Lonza, Verviers, Belgium), 400 μg·mL−1 Geneticin (GIBCO), 100 IU·mL−1 penicillin and 100 IU·mL−1 streptomycin (Pen-Strep solution, BioWhittaker). LMtk (Ltk-) mouse fibroblast cells expressing the dopamine D1 receptor (BioSignal Inc, Montreal, Canada, now Perkin Elmer) were used as they have been shown to couple efficiently and give robust functional responses (Watts et al., 1995). CHO cells expressing the human dopamine D2 (long isoform which contains an additional 29 amino acids), D3 and D4 (long form with seven repeat polymorphism of exon III) receptors were developed in house. Cells stably expressing the human dopamine D5 receptor were obtained from Millipore (Merck-Millipore S.A/N.V., Overijse, Belgium) and cultured according to the manufacturer's instructions.
Membrane preparation
Adherent cells were cultured in 176 cm2 Petri dishes until confluent and the medium was removed. The cells were washed with 30 mL PBS at 25°C and detached by incubation with 30 mL 1 mM EDTA solution in PBS (pH 7.4) for 7 min at 37°C. After centrifugation (1500× g for 10 min at 4°C), the pellet was resuspended in 3 mL buffer [15 mM Tris-HCl buffer (pH 7.4) containing 1 mM EGTA, 0.3 mM EDTA, 2 mM MgCl2 with one tablet of Complete® Mini EDTA free (Roche, Vilvoorde, Belgium) per 20 mL buffer] per flask. The cells were homogenized (Potter) and the homogenates were frozen in liquid nitrogen and defrosted in a 25°C water bath. This step was repeated once more to complete the cell disruption. After equilibration at 25°C, DNAse (final concentration 10 IU·mL−1) was added to the membrane suspension and incubated for 10 min at 25°C followed by centrifugation (40 000× g for 25 min at 4°C). The pellet was resuspended in Tris-sucrose buffer (20 mM Tris-HCl buffer pH 7.4 containing 250 mM sucrose). The membrane preparation was frozen in aliquots in liquid nitrogen before storage at −140°C. Suspension cells were centrifuged and treated as described earlier.
Radioligand binding assays
Binding assays were performed in 96-well polypropylene tubes in a final volume of 2 mL for D1 and D4 membranes and 1 mL for D2, D3 and D5 membranes containing: 50 μL radioligand, 10 μL drug/buffer/non-specific binding, buffer (final concentration 50 mM Tris-HCl pH 7.4, MgCl2 2 mM) and membranes (5 μg protein for D2 and D3 and 25 μg protein for D1 and D5). Following 120 min of incubation at 25°C, bound radioligand was determined by rapid vacuum filtration through A/C glass fibre filters (Pall Corporation, Zaventem, Belgium) presoaked in 0.1% polyethylenimine. The filters were washed four times with 2 mL ice-cold washing buffer (Tris-HCl 50 mM, pH 7.4 at 4°C) and retained radioactivity was determined by liquid scintillation counting.
In kinetic studies, association and dissociation [induced by an excess (10 μM) of chlorpromazine for D2, D3 and D4 receptors and apomorphine for D1 and D5 receptors] were followed at different times up to 180 min.
For saturation studies, the concentration of radioligand used was typically 0.02–5 nM. For competition and kinetic studies, [3H]rotigotine was used at 0.8 nM for dopamine D1 and 0.2 nM for dopamine D2, D3, D4 and D5 receptors. For antagonist radioligands in competition studies, [3H]SCH23390 was used at 0.03 nM for D1 and D5 receptors, [3H]raclopride at 0.65 nM for D2 receptors and [3H]spiperone at 0.3 nM for D3 receptors and 0.05 nM for D4 receptors. These conditions were selected to give a robust signal window in the absence of ligand depletion (which was observed for [3H]spiperone in the CHO D2 cells). Competition curves were performed using 10 concentrations (half-log dilutions) in triplicate.
CDS
CDS measurements were performed with the Cellkey (MDS Sciex) at 37°C. Cells were seeded into the wells of a 96-well microplate in 200 μL medium and incubated overnight (37°C; 5% CO2). Growth medium was then exchanged to 135 μL incubation buffer [Hank's balanced salt solution (HBSS) containing 20 mM HEPES, pH 7.4]. Plates were placed onto the Cellkey system and baseline measurements were taken for 5 min. The test agonist (15 μL) at varying concentrations (ranging from 0.1 pM to 10 μM) was added and impedance measurements were collected for 30 min.
[35S]GTPγS binding
Membranes (CHO hD4, 20 μg per assay) were incubated with TRIS-MgCl2, drug/H2O/agonist, containing final concentrations: NaCl 50 mM; MgCl2 3 mM; GDP 1 μM; saponin 10 μg·mL−1 in 200 μL for 15 min at 25°C. Then, 20 μL of [35S]GTPγS (0.15–0.20 nM in 0.01N HCl) was added to each well and incubated for 60 min at 25°C. The plates were filtered, dried and retained radioactivity was determined.
Intracellular calcium
Changes in intracellular calcium were followed in cells stably expressing the human dopamine D5 receptor and G-protein Gα15 (see Smart and Wood, 2001). Cells were pre-incubated for 60 min with 4 μM Fluo-4 AM (Molecular Probes F-14202; Life Technologies, Gent, Belgium) with pluronic acid in the presence of probenecid (0.8 mM final). Cells (20 000 per well) were washed and incubated in HBSS with 20 mM HEPES (pH 7.4), basal readings taken, drugs added and changes in intracellular calcium were determined using a FlexStation (Molecular Devices, St. Grégoire Cédex, France).
Data analysis
Data were analysed using Excel and PRISM (GraphPad Software, La Jolla, CA, USA). Association and dissociation data were fitted to one-phase and two-phase models and the best fit was determined using the extra sum-of-squares F-test (PRISM). Equilibrium saturation curves were analysed in Microsoft Office Excel 2003 using one-site or two-site analysis, depending on the outcome of the sum-of-squares F-test performed with PRISM. Competition curves were fitted to the three-parameter logistic equation (slope fixed to unity as the slope was not significantly different from 1) and pIC50 corrected to pKi according to Cheng and Prusoff (1973). In functional studies, EC50 values were determined by non-linear regression analysis of the curves using the log(agonist) versus response model in PRISM (slope = 1 as it was not significantly different from unity).
Materials
All cell culture reagents were obtained from Invitrogen unless otherwise stated.
Radioligands used were: [3H]rotigotine (54 Ci·mmol−1, Tritec AG, Teufen, Switzerland); [3H]SCH23390 (88 Ci·mmol−1, GE Healthcare, Diegem, Belgium), [3H]raclopride (83 Ci·mmol−1, Perkin Elmer, Zaventem, Belgium) and [3H]spiperone (15 Ci·mmol−1, Perkin Elmer). Rotigotine was synthesized at UCB (Braine-l'Alleud, Belgium), reagents and standards were obtained from Sigma-RBI (Diegem, Belgium).
Results
Kinetics of [3H]rotigotine binding
The binding of [3H]rotigotine to all dopamine receptor subtypes was reversible and showed time dependence. Although dissociation from the D2 and D3 receptors was slow, a partial inhibition model did not better describe the data than a complete inhibition model; therefore, data were analysed assuming complete reversibility would be reached. The association of [3H]rotigotine with the dopamine D2, D4 and D5 receptors was best described by a two-phase model, whereas the dopamine D1 and D3 receptor association data were best described by a one-phase model (Figure 1). The dissociation data at all receptors were best described by a one-phase model. Binding kinetic constants are shown in Table 1.
Figure 1.

Association and dissociation curves for [3H]rotigotine binding to human dopamine D1, D2 and D3 receptors. Data are representative of three independent experiments. The curves are the best fit as described with calculated constants shown in Table 1.
Table 1.
Radioligand binding kinetic constants of [3H]rotigotine binding to human dopamine D1, D2, D3 and D4 receptors
| Kinetic parameters | Receptor | ||||
|---|---|---|---|---|---|
| D1 | D2 | D3 | D4 | D5 | |
| Dissociation | |||||
| koff (min−1) | 0.08 ± 0.01 | 0.007 ± 0.002 | 0.008 ± 0.001 | 0.03 ± 0.01 | 0.16 ± 0.01 |
| t1/2 (min) | 8.6 ± 0.2 | 110 ± 30 | 87 ± 7 | 24 ± 3.5 | 4.5 ± 0.4 |
| Association | |||||
| kobs fast (min−1) | 1.2 ± 0.2 | 0.42 ± 0.09 | 0.11 ± 0.02 | 0.26 ± 0.04 | 0.25 ± 0.08 |
| kobs slow (min−1) | 0.06 ± 0.03 | 0.04 ± 0.01 | – | 0.035 ± 0.006 | – |
| %fast | 45 ± 2 | 70 ± 6 | – | 84 ± 9 | – |
| t1/2 fast (min) | 0.56 ± 0.07 | 1.7 ± 0.3 | 6.62 ± 1.6 | 2.7 ± 0.4 | 1.5 ± 0.7 |
| t1/2 slow (min) | 12 ± 5 | 19 ± 4 | – | 25 ± 4 | – |
Results are mean ± SD from three separate experiments. Kinetic constants were determined as described. koff is the dissociation rate constant and kobs = konL + koff where kon is the association rate constant and L is the concentration of radioligand. t1/2 is the half time for dissociation or association.
Comparison of saturation binding of agonist and antagonist binding at dopamine D1, D2, D3, D4 and D5 receptors
Equilibrium saturation assays were performed in triplicate with [3H]rotigotine and with the radiolabelled antagonist [3H]SCH23390 for D1 and D5, [3H]raclopride for D2 and [3H]spiperone for D3 and D4 receptors (Table 2 and Figure 2). Analysis of the saturation binding of [3H]rotigotine to dopamine D1 and D2 receptors was best described by a two-site model and by a one-site model for the D3, D4 and D5 receptors. For the two-site analysis, the Bmax of the low-affinity site was poorly defined because of the low signal at high radiolabel concentrations but the fit was well described assuming a similar Bmax value to that seen with the antagonist radioligand. At the dopamine D1 and D5 receptors, the number of sites (Bmax) identified by [3H]rotigotine was markedly lower than that labelled by [3H]SCH23390. At the D2, D3 and D4 receptors, the number of sites labelled by [3H]rotigotine was similar to that labelled by [3H]raclopride and [3H]spiperone respectively. At the dopamine D1 receptor, the presence of 5'-guanylyl imidodiphosphate (GppNHp; 10 μM) attenuated the high-affinity [3H]rotigotine binding site whereas GppNHp had no significant effect on [3H]rotigotine binding to the D2 and D3 receptors.
Table 2.
Equilibrium binding constants for [3H]rotigotine and [3H]antagonist radioligands at human recombinant dopamine receptors
| Receptor | Radioligand | KD (nM) | Bmax (fmol·mg−1 protein) | ||
|---|---|---|---|---|---|
| High affinity site | Low affinity site | High affinity site | Low affinity site | ||
| D1 | [3H]rotigotine | 0.19 ± 0.13 | 6.80 ± 3.23 | 440 ± 420 | 2 970 ± 400 |
| [3H]rotigotine +Gpp(NH)p | – | 10.0 ± 5.5 | – | 2 700 ± 1 300 | |
| [3H]SCH23390 | 0.69 ± 0.23 | – | 29 700 ± 5 500 | – | |
| D2 | [3H]rotigotine | 0.09 ± 0.02 | 4.57 ± 4.4 | 6 390 ± 3 600 | 20 530 ± 4 600 |
| [3H]rotigotine +Gpp(NH)p | 0.20 ± 0.14 | 4.08 ± 3.9 | 4 120 ± 2 300 | 23 300 ± 9 300 | |
| [3H]raclopride | 0.68 ± 0.07 | – | 17 600 ± 2 600 | – | |
| D3 | [3H]rotigotine | 0.21 ± 0.06 | – | 22 700 ± 5 600 | – |
| [3H]rotigotine +Gpp(NH)p | 0.20 ± 0.04 | – | 26 540 ± 1 545 | – | |
| [3H]spiperone | 0.12 ± 0.03 | – | 18 100 ± 950 | – | |
| D3 | [3H]rotigotine | 0.83 ± 0.06 | – | 1 550 ± 640 | – |
| [3H]spiperone | 0.055 ± 0.01 | – | 1 440 ± 670 | – | |
| D4 | [3H]rotigotine | 6.50 ± 1.85 | – | 1 300 ± 290 | – |
| [3H]SCH23390 | 1.08 ± 0.44 | – | 8 200 ± 1 200 | – | |
Data shown are the mean ± SD calculated parameters from three to four saturation experiments. Parameters were calculated in PRISM using one-site or two-site analysis, depending on the outcome of the sum-of-squares F-test.
Figure 2.
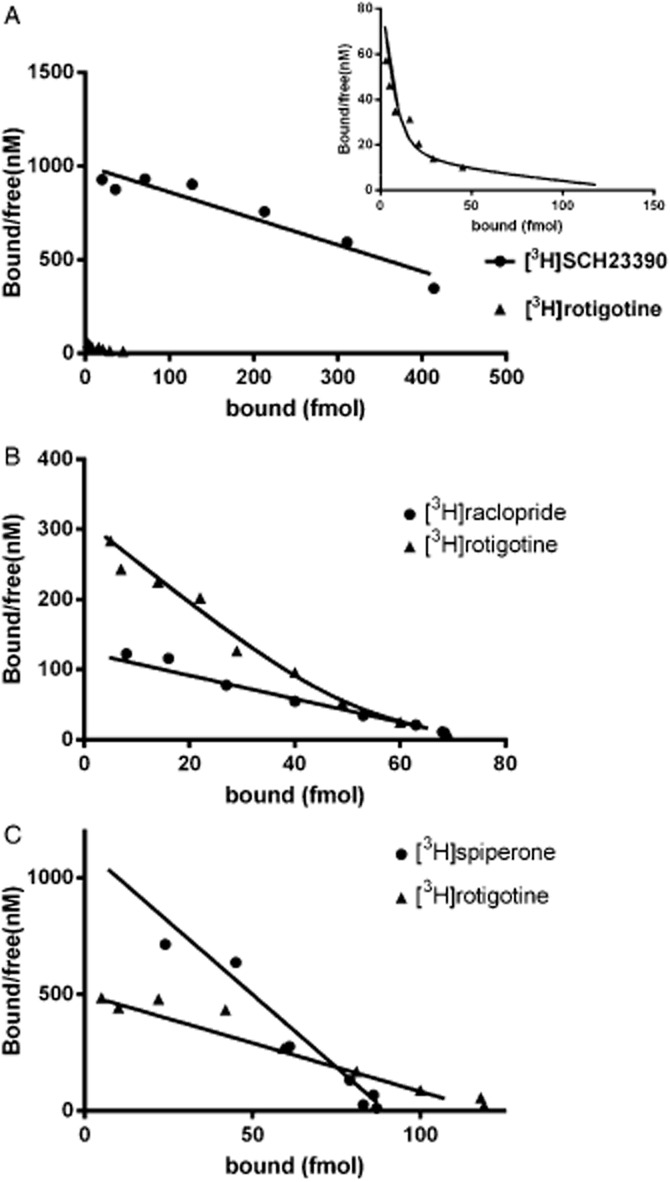
Representative Scatchard plots from radioligand saturation binding studies. (A) Shows the specific binding of [3H]SCH23390 and [3H]rotigotine to the human dopamine D1 receptor expressed in LMtk cells with inset showing enlargement to show two-site fit for [3H]rotigotine. (B) Shows the specific binding of [3H]raclopride and [3H]rotigotine to the human dopamine D2 receptor expressed in CHO cells. (C) Shows the specific binding of [3H]spiperone and [3H]rotigotine to the human dopamine D3 receptor expressed in CHO cells.
Competition profile at human dopamine D1, D2, D3, D4 and D5 receptors
To compare the pharmacological profile of the site labelled by the agonist radioligand [3H]rotigotine with that labelled by antagonist radioligands, competition binding assays were performed. The assays were carried out using both [3H]rotigotine as the agonist radioligand and using [3H]SCH23390, [3H]raclopride and [3H]spiperone as the antagonist radioligand for D1 and D5, D2, D3 and D4 receptors respectively (Table 3). Rotigotine itself displaced [3H]rotigotine binding at the human dopamine receptors with pKi values in agreement with the measured pKD. At the D1 and D2 receptors, the observed pKi was consistent with the high-affinity site observed in saturation experiments (pKD 9.7 and 10.0 respectively).
Table 3.
Affinity of selected compounds for human recombinant dopamine D2, D3 and D4 receptors identified using both [3H]rotigotine and [3H]standard antagonist in radioligand binding studies
| Compounds | pKi D2 | pKi D3 | pKi D4 | |||
|---|---|---|---|---|---|---|
| [3H]Rotig | [3H]Spip | [3H]Rotig | [3H]Rac | [3H]Rotig | [3H]Spip | |
| Agonists | ||||||
| Rotigotine | 10.2 ± 0.2 | 8.7 ± 0.1 | 9.9 ± 0.1 | 10.0 ± 0.2 | 8.5 ± 0.1 | 7.7 ± 0.5 |
| Lisuride | 9.3 ± 0.4 | 8.3 ± 0.2 | 8.5 ± 0.1 | 8.9 ± 0.1 | 6.8 ± 0.1 | 6.8 ± 0.1 |
| Pramipexole | 8.6 ± 0.1 | 6.7 ± 0.1 | 9.4 ± 0.1 | 9.6 ± 0.3 | 7.9 ± 0.1 | 7.2 ± 0.1 |
| Ropinirole | 7.8 ± 0.1 | 6.4 ± 0.1 | 8.2 ± 0.1 | 8.0 ± 0.1 | 6.9 ± 0.1 | 6.6 ± 0.1 |
| Dopamine | 8.5 ± 0.1 | 6.7 ± 0.2 | 8.7 ± 0.2 | 8.5 ± 0.1 | 8.2 ± 0.2 | 7.1 ± 0.1 |
| (-)3-PPP | 7.2 ± 0.2 | 5.5 ± 0.2 | 7.3 ± 0.1 | 7.0 ± 0.1 | 7.0 ± 0.1 | 6.2 ± 0.2 |
| Quinpirole | 8.2 ± 0.2 | 6.5 ± 0.1 | 8.7 ± 0.1 | 8.7 ± 0.1 | 8.1 ± 0.1 | 7.6 ± 0.1 |
| Apomorphine | 9.3 ± 0.1 | 8.0 ± 0.1 | 8.3 ± 0.1 | 8.4 ± 0.2 | 8.9 ± 0.1 | 8.4 ± 0.2 |
| 7-OH-DPAT | 8.4 ± 0.3 | 7.1 ± 0.2 | 9.1 ± 0.1 | 9.1 ± 0.1 | – | – |
| Antagonists | ||||||
| SCH23390 | 7.2 ± 0.1 | 6.9 ± 0.4 | 6.3 ± 0.1 | 6.2 ± 0.1 | 6.7 ± 0.5 | 6.6 ± 0.1 |
| Raclopride | 8.7 ± 0.1 | 9.1 ± 0.3 | 7.0 ± 0.1 | 7.1 ± 0.1 | 5.7 ± 0.5 | 5.8 ± 0.3 |
| Haloperidol | 10.5 ± 0.1 | 9.9 ± 0.5 | 8.4 ± 0.2 | 8.7 ± 0.1 | – | – |
| Spiperone | 10.3 ± 0.1 | 10.4 ± 0.2 | 9.6 ± 0.2 | 9.9 ± 0.1 | 9.8 ± 0.2 | 10.1 ± 0.2 |
| SB277011A | 6.3 ± 0.2 | 6.2 ± 0.1 | 7.8 ± 0.1 | 7.7 ± 0.1 | 5.4 ± 0.2 | 5.6 ± 0.2 |
Data are mean ± SD from three separate experiments. Shown are the inhibitory affinity constants (pKi) to inhibit the binding of [3H]rotigotine (Rotig), [3H]spiperone (Spip) and [3H]raclopride (Rac).
For the antagonist compounds, SB277011A (Reavill et al., 2000) displayed some selectivity for the dopamine D3 receptor; SCH23390 displayed marked selectivity for the dopamine D1 and D5 receptors and raclopride, spiperone and haloperidol exhibited high affinity for the dopamine D2, D3 and D4 receptors. There was a marked similarity in binding affinities for all of these antagonists at each dopamine receptor labelled with either [3H]rotigotine or the respective antagonist radioligand.
For the agonist compounds, some marked differences were seen. The agonists pramipexole, ropinirole, (-)3-(3-hydroxyphenyl)-N-n-piperidine ((-)3-PPP) and quinpirole lacked affinity for the D1 and D5 receptors. All of the other agonists displayed a higher affinity for the D1 receptor labelled with [3H]rotigotine compared with the antagonist ligand, [3H]SCH23390. At the dopamine D2 receptor, all of the agonists inhibited [3H]rotigotine binding with a higher affinity than required to inhibit binding of the antagonist [3H]raclopride. This difference in inhibitory affinity was greater at the dopamine D1 receptor. At the dopamine D3 receptor, there was no difference in inhibitory affinity for these agonists using either agonist ([3H]rotigotine) or antagonist ([3H]spiperone) radioligands. The agonists apomorphine, dopamine and rotigotine all displayed similar affinities for the [3H]rotigotine binding site on the dopamine D1, D2 and D3 receptors. The agonists lisuride, pramipexole, ropinirole, (-)3-PPP, quinpirole and 7-OH-DPAT all showed selectivity for the dopamine D2 and D3 receptor compared with the D1 receptor. Further, pramipexole and 7-OH-DPAT showed some selectivity for the dopamine D3 receptor which was more apparent when using [3H]antagonist ligands than [3H]rotigotine.
At the dopamine D4 receptor, most of the agonists tested displayed a lower affinity for this receptor (on both [3H]rotigotine and [3H]spiperone/antagonist binding) than for the D2 and D3 receptors. There was also a tendency for the potency to be higher on [3H]rotigotine binding than on [3H]spiperone which was most marked with dopamine. Dopamine itself exhibited similar affinity for the D2, D3 and D4 receptors.
For the dopamine D5 receptor, although the pharmacological profile was similar to that seen at the D1 receptor, the difference in inhibitory potency of agonist ligands at the agonist ([3H]rotigotine) and antagonist ([3H]SCH23390) radioligand binding was not as marked as that seen at the D1 receptor.
Functional profile at human dopamine D1, D2, D3, D4 and D5 receptors
The optimal cell density was determined using 10 μM dopamine and 10 μM ATP: 50 000 cells per well was chosen for LMtk D1, 30 000 cells per well for CHO D2 and 35 000 cells per well for CHO D3 cells to perform the functional assays.
In LMtk D1 cells, dopamine produced a concentration-dependent decrease in cellular impedance [maximal response 25–30 dZiec (Ohms)]. In D2 CHO and D3 CHO cells, dopamine produced a concentration-dependent increase in cellular impedance with a maximal response around 80 dZiec (Ohms) for D2 and 40 dZiec (Ohms) for D3.
Almost all of the compounds tested appeared as full agonists at the dopamine D1, D2 and D3 receptors in that they all produced maximal responses similar to that of the endogenous agonist dopamine (data shown for D1 receptor Figure 3). The exception was (-)3-PPP which gave a submaximal response at the dopamine D2 receptor (88% compared with dopamine 100%, P < 0.05 Student's paired t-test). There was a good correlation between functional potency of the agonists examined (Table 4) and their binding affinity versus [3H]rotigotine (Table 5) at the three dopamine receptors (Figure 4). At the dopamine D2 receptor, there is a good correlation between functional potency and binding affinity when using [3H]rotigotine as an agonist radioligand and when using [3H]spiperone as an antagonist radioligand (Figure 4), but there is a 100-fold lower binding affinity. At the dopamine D3 receptor, although there was a good correlation, the functional potency of compounds tended to be lower than their binding affinity at the D3 receptor and this was most notable for rotigotine, lisuride and pramipexole. Rotigotine, lisuride, dopamine and apomorphine displayed agonist properties at all three dopamine receptors, whereas pramipexole, ropinirole, (-)3-PPP and quinpirole only displayed agonist activity at dopamine D2 and D3 receptors.
Figure 3.
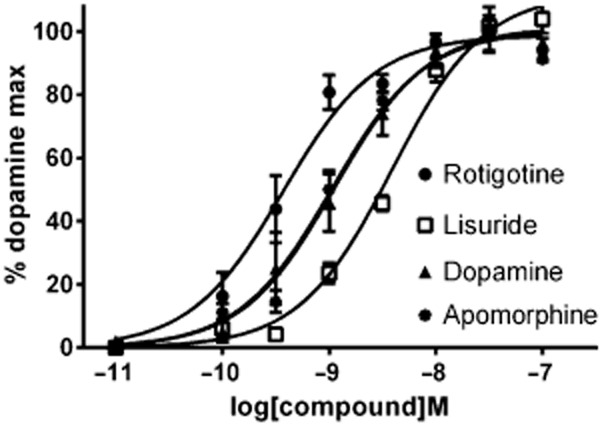
The effect of rotigotine and other dopamine agonists on the human dopamine D1 receptor in functional studies on LMtk cells. Results are mean changes in cellular dielectric spectroscopy (with SEM) expressed as a % of the maximum fitted response to the dopamine concentration-response curve from three separate determinations.
Table 4.
Functional profile of dopaminergic agonists at human recombinant dopamine receptor subtypes
| Compound | Receptor | ||||||
|---|---|---|---|---|---|---|---|
| D1 | D2 | D3 | D4 | D5 | |||
| pEC50 | pEC50 | pEC50 | pEC50 | Emax (%) | pEC50 | Emax (%) | |
| Rotigotine | 9.6 ± 0.1 | 10.4 ± 0.1 | 8.2 ± 0.2 | 7.7 ± 0.1 | 62 ± 2 | 7.7 ± 0.2 | 64 ± 5 |
| Lisuride | 8.5 ± 0.1 | 8.4 ± 0.1 | 6.8 ± 0.2 | – | – | – | – |
| Pramipexole | <5 | 9.1 ± 0.2 | 8.3 ± 0.1 | 7.4 ± 0.1 | 62 ± 4 | <5 | – |
| Ropinirole | <5 | 8.5 ± 0.1 | 8.1 ± 0.1 | 6.3 ± 0.1 | 67 ± 6 | <5 | – |
| Dopamine | 9.0 ± 0.4 | 8.6 ± 0.3 | 8.3 ± 0.1 | 7.5 ± 0.1 | 100 | 8.2 ± 0.3 | 100 |
| (-)3-PPP | <5 | 7.5 ± 0.1 | 7.5 ± 0.1 | 6.4 ± 0.2 | 2 ± 3 | – | – |
| Quinpirole | <5 | 8.9 ± 0.1 | 8.0 ± 0.2 | 7.1 ± 0.5 | 81 ± 9 | <5 | – |
| Apomorphine | 9.1 ± 0.1 | 9.5 ± 0.8 | 8.1 ± 0.1 | 8.3 ± 0.5 | 63 ± 1 | 7.9 ± 0.2 | 80 ± 5 |
Data are mean ± SD from three to four separate experiments.
Table 5.
Affinity of selected compounds for human recombinant dopamine D1 and D5 receptors identified using both [3H]rotigotine and [3H]standard antagonist in radioligand binding studies
| Compounds | pKi D1 | pKi D5 | ||
|---|---|---|---|---|
| [3H]Rotig | [3H]SCH | [3H]Rotig | [3H]SCH | |
| Agonists | ||||
| Rotigotine | 9.2 ± 0.1 | 6.6 ± 0.1 | 7.8 ± 0.2 | 7.4 ± 0.1 |
| Lisuride | 7.5 ± 0.1 | 6.0 ± 0.6 | – | – |
| Pramipexole | <5 | <5 | <5 | <5 |
| Ropinirole | <5 | <5 | <5 | <5 |
| Dopamine | 9.0 ± 0.2 | 5.8 ± 0.1 | 8.2 ± 0.1 | 7.5 ± 0.1 |
| (-)3-PPP | <5 | <5 | <5.5 | <5.5 |
| Quinpirole | <5 | <5 | <5 | <5 |
| Apomorphine | 8.8 ± 0.4 | 6.5 ±.0.1 | 7.9 ± 0.1 | 7.4 ± 0.1 |
| 7-OH-DPAT | 6.7 ± 0.1 | 4.7 ± 0.1 | 6.0 ± 0.1 | 5.2 ± 0.1 |
| Antagonists | ||||
| SCH23390 | 9.4 ± 0.2 | 9.3 ± 0.2 | 9.2 ± 0.1 | 9.5 ± 0.1 |
| Raclopride | <5 | <5 | <5 | <5 |
| Haloperidol | 8.3 ± 0.2 | 8.1 ± 0.2 | 9.1 ± 0.1 | 9.3 ± 0.6 |
| Spiperone | 6.8 ± 0.1 | 6.6 ± 0.1 | 6.0 ± 0.2 | 6.1 ± 0.1 |
| SB277011A | <5 | <5 | <5 | <5 |
Data are mean ± SD from three separate experiments. Shown are the inhibitory affinity constants (pKi) to inhibit the binding of [3H]rotigotine (Rotig) and [3H]SCH23390 (SCH).
Figure 4.
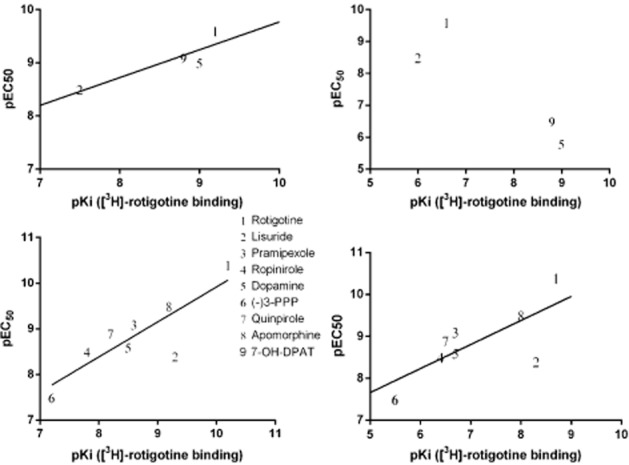
Correlation between functional potency (pEC50) and binding affinity (pKi) at the dopamine D1 receptor (top panels) and D2 receptor (bottom panels) using agonist ([3H]rotigotine) and antagonist ([3H]SCH23390 and [3H]spiperone for D1 and D2 respectively).
At the dopamine D4 receptor, most compounds (except apomorphine) displayed a lower potency compared with the D2 and D3 receptors. Further, all of the agonists appeared as partial agonists compared with dopamine, with efficacy of 60–80% with respect to dopamine maximal response. Similarly, at the D5 receptor, those agonists tested displayed a lower potency than that seen at the dopamine D1 receptor and they appeared as partial agonists.
Discussion and conclusions
In preclinical studies, rotigotine has been shown to act as a dopamine D2 receptor agonist (Van der Weide et al., 1987; Tan, 2003; Rascol et al., 2007). In radioligand binding studies (Scheller et al., 2009), rotigotine exhibited the highest affinity for the dopamine D3 receptor (Ki 0.71 nM), high affinity for D2, D5 and D4 receptors (Ki 4–15 nM) and low affinity for the dopamine D1 receptor (Ki 83 nM). However, in functional studies in the same paper, it was found that rotigotine acts as an agonist at all dopamine receptors with a moderate selectivity for the dopamine D2 receptor-like family. In the present study, we show that rotigotine is a high-affinity agonist at several dopamine receptors, notably D1, D2 and D3.
Using [3H]rotigotine as an agonist radioligand, we show that it can be used to label all the dopamine receptors and that it displays a high affinity for dopamine D1, D2 and D3 receptors with a lower affinity for dopamine D4 and D5 receptors. Further, there is evidence that [3H]rotigotine labels an agonist state at some of these receptors. In kinetic studies, there was evidence for the presence of two states of the D1, D2, D4 and D5 receptors whereas only one state was evident for the D3 receptor. Interestingly, this was seen in association studies but was not evident in dissociation studies. There was also evidence in saturation studies where two affinity states were observed for the D1 and D2 receptors. This profile of multiple affinity states, sensitivity to guanyl nucleotides and reduced binding capacity compared with antagonist radioligands, is consistent with [3H]rotigotine being an agonist at these GPCRs. The lack of this profile at the dopamine D3 receptor may reflect the poor functional coupling of this receptor to G-proteins and its selective coupling to Go (Zaworski et al., 1999; Lane et al., 2008). For the dopamine D2 receptor, where two affinity states for [3H]rotigotine were observed which were insensitive to the effects of GppNHp, these states may represent different conformations of the D2 receptor bound to different G-proteins present in the cell. The D2 receptor couples promiscuously to several members of the Gαi/o family for which it displays different potencies (Lane et al., 2008) and this may be apparent in cell lines where the D2 receptor is overexpressed giving rise to apparent multiple binding states. In competition binding studies, the pharmacological profile of [3H]rotigotine binding was again consistent with labelling an agonist state. Thus, in general, agonists, and notably dopamine, were more potent displacers of [3H]rotigotine binding than of the [3H]antagonist binding at all dopamine receptors except the D3 receptor. This may reflect the poor functional coupling seen with the D3 receptor as noted earlier.
Further, we show that rotigotine is a high potency agonist at all dopamine receptors, notably D1, D2 and D3. Indeed, rotigotine exhibited subnanomolar potency for the dopamine D1 and D2 receptors, low nanomolar potency for the D3 receptor and high nanomolar potency for the D4 and D5 receptors in functional studies. Although this is the first study which has compared functional interactions across all dopamine receptors, the results are in agreement with the limited published studies. The potency values obtained in the present study with ropinirole, pramipexole and dopamine agree with those reported by Coldwell et al. (1999) and Perachon et al. (1999) who also found these compounds to be full agonists. There was also good agreement on functional potencies at D3 receptors, with the results of Newman-Tancredi et al. (2002) with the exception of lisuride which was reported to be more potent and a partial agonist. This difference may reflect the slow kinetics of lisuride such that equilibrium may not be reached in functional studies (Coldwell et al., 1999). Recently, the concept of biased signalling or functional selectivity has emerged which may explain some of the differences seen in agonist pharmacology (Urban et al., 2007). However, one mechanism for such functional selectivity involves signalling through multiple G-proteins and, although this has been demonstrated for the dopamine D2 receptor (Beaulieu et al., 2005), the D3 receptor has been shown to selectively couple to GαO1 (Lane et al., 2008). It would be interesting to study the effects of these agonists on G-protein activation (e.g. [35S]GTPγS binding) and on other signalling pathways, such as cAMP, ERK, Akt. Potency and efficacy generally tended to be lower at the D4 receptor compared with the closely related D2 and D3 receptors, such that agonists display partial agonist properties consistent with other studies on the D4 receptor (Newman-Tancredi et al., 2002; Heusler et al., 2009). Similarly, functional potency at the D5 receptor tended to be lower than that seen at the D1 receptor. As discussed earlier, functional potency and efficacy can be affected by levels of receptor expression/receptor reserve and by stimulus-response coupling. However, the lower potency at the D4 and D5 receptors for the agonists examined was also seen in radioligand binding studies suggesting that this may be a function of the receptor.
On the basis of radioligand binding and functional studies, there therefore appears to be two classes of dopamine agonists. First, apomorphine and rotigotine which display a high affinity and potency for dopamine D1, D2 and D3 receptors and a lower affinity and potency for dopamine D4 and D5 receptors. Second, ropinirole, pramipexole and lisuride which display high affinity and potency for the dopamine D2 and D3 receptors, lower affinity and potency for dopamine D4 receptors and with negligible activity at the dopamine D1 and D5 receptors. This may have clinical implications in the treatment of PD as, despite the recent advances with the use of dopamine agonists, levodopa remains the gold standard of symptomatic efficacy in treating the motor deficits in PD (Poewe, 2009). It is generally accepted that levodopa (after conversion to dopamine) acts via stimulation of all dopamine receptors. Because there are similar numbers of dopamine D1 and dopamine D2 receptors in the striatum, simultaneous stimulation of both receptor types may be one of the explanations for the greater efficacy of levodopa. In support of this, intermittent s.c. injections of apomorphine, which is a full agonist at dopamine D1 receptors (inter alia; Tiberi and Caron, 1994) and which shows high efficacy at dopamine D2 receptors (inter alia; Newman-Tancredi et al., 2002), have been shown to produce anti-Parkinsonian efficacy similar to that of levodopa (Poewe and Wenning, 2000). Unfortunately, the use of apomorphine in the clinic is limited by its high first-pass metabolism and its side effects such as emesis (Poewe and Wenning, 2000). It was therefore proposed that a synergistic activation of both dopamine D1 and D2 receptors may be important for full reversal of Parkinsonian motor deficits (Robertson, 1992; Vermeulen et al., 1994; 1999,). The dopamine D4 receptor has been suggested to play a role in emotional processing and plasticity and in novelty/impulsive behavioural phenomena (see Lauzon and Laviolette, 2010). As such, it has been suggested that the D4 receptor may play a role in attention deficit hyperactivity disorder and in the mechanism of action of drugs used in the treatment of such disorders. Impulse-control disorders are a common side effect associated with dopamine agonists (Perez-Lloret and Rascol, 2010). They are seen in 5 to 15% of patients and may affect patients severely, as in the case of pathological gambling). It is tempting to speculate that an action at the D4 receptor may contribute to these adverse effects. Little is known regarding the role of the dopamine D5 receptor in the mechanism of action of the dopamine agonists in PD. There are no selective pharmacological agents which discriminate between D1 and D5 receptors. D5 receptors exhibit much lower concentrations than D1 receptors and while D1 receptors are expressed widely throughout the brain, D5 receptors are restricted in expression patterns, particularly in cortical and limbic regions (see Homes et al., 2001). Evidence from D5 receptor knockout mice suggest that the D5 receptor does not play a major role in many dopamine-mediated behaviours, but may contribute to exploratory locomotion, startle and pre-pulse inhibition. As well as a role in locomotor activity, the D1 receptor plays a role in memory and learning so an agonist action at this receptor may be beneficial in elderly patients where cognitive impairment is often seen.
In conclusion, rotigotine acts as a high-affinity dopamine receptor agonist at D1, D2, D3 and, to a lesser extent, D4 and D5 receptor subtypes and, as such, is differentiated from compounds such as ropinirole and pramipexole which act mainly as dopamine D2, D3 and, to a lesser extent, D4 receptor agonists. Indeed, the profile of rotigotine more closely resembles that of apomorphine. The present studies were performed in vitro using recombinant expression systems. It will therefore be of interest to study these effects in native tissue systems. It also remains to determine whether this D1 receptor activation by rotigotine contributes to its in vivo profile and whether this translates to its clinical profile in the treatment of PD.
Acknowledgments
We thank Mr David Urbain, Mme Murielle Martini and Mr Michel Famelart for excellent technical assistance and support in this study.
Glossary
- (-)3-PPP
(-)3-(3-hydroxyphenyl)-N-n-piperidine
- CDS
cellular dielectric spectroscopy
- GppNHp
5'-guanylyl imidodiphosphate
- HBSS
Hank's balanced salt solution
- PD
Parkinson's disease
Author contributions
M. W, M. G. and D. S. conceived and designed the experiments. V. D. performed the experiments. M. W. and M. G. wrote the paper.
Conflict of interest
Authors declare that they have not any conflict of interest.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The concise guide to PHARMACOLOGY 2013/2014: G protein-coupled receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin CM, Keating GM. Rotigotine transdermal patch. A review of its use in the management of Parkinson's disease. CNS Drugs. 2007;21:1039–1055. doi: 10.2165/00023210-200721120-00007. [DOI] [PubMed] [Google Scholar]
- Baldwin CM, Keating GM. Rotigotine transdermal patch in restless leg syndrome. CNS Drugs. 2008;22:797–806. doi: 10.2165/00023210-200822100-00001. [DOI] [PubMed] [Google Scholar]
- Beaulieu J-M, Gainetdinov PR. The physiology, signalling and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- Beaulieu J-M, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov PR, Caron MG. An Akt/β-arrestin 2/PP2A signalling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Clarke CE, Guttman M. Dopamine agonist monotherapy in Parkinson's disease. Lancet. 2002;360:1767–1769. doi: 10.1016/s0140-6736(02)11668-0. [DOI] [PubMed] [Google Scholar]
- Coldwell MC, Boyfield I, Brown AM, Hagan JJ, Middelmiss DN. Comparison of the functional potencies of ropinirole and other dopamine agonist at human D2(long), D3 and D4.4 receptors expressed in Chinese hamster ovary cells. Br J Pharmacol. 1999;127:1696–1702. doi: 10.1038/sj.bjp.0702673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLean A, Stadel JM, Lekkowitz RJ. A ternary complex model explains the agonist specific binding properties of the adenylyl cyclase-coupled beta-adrenergic receptor. J Biol Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- Freedman SB, Harley EA, Iversen LL. Relative affinities of drugs acting at cholinoceptors in displacing agonist and antagonist radioligands: the NMS/Oxo-M ratio as an index of efficacy at cortical muscarinic receptors. Br J Pharmacol. 1988;93:437–445. doi: 10.1111/j.1476-5381.1988.tb11451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusler P, Bruins Slot L, Rauly-Lestienne I, Palmier C, Tardiff S, Tourette A, et al. Activation of G proteins and extracellular signal-regulated kinase1/2 phosphorylation via human dopamine D4.4 receptors: differential pathway-dependent potencies of receptor agonists. Naunyn Schmeidebergs Arch Pharmacol. 2009;379:87–99. doi: 10.1007/s00210-008-0333-4. [DOI] [PubMed] [Google Scholar]
- Homes A, Hollon TR, Gleason TC, Liu Z, Dreiling Z, Sibley DR, et al. Behavioral characterization of dopamine D5 receptor null mutant mice. Behav Neurosci. 2001;115:1129–1144. [PubMed] [Google Scholar]
- Jenner P. The rationale for the use of dopamine agonists in Parkinson's disease. Neurol. 1995;45(Suppl. 3):S6–S12. doi: 10.1212/wnl.45.3_suppl_3.s6. [DOI] [PubMed] [Google Scholar]
- Jenner P. Preventing and controlling dyskinesia in Parkinson's disease – a view of current knowledge and future opportunities. Mov Disord. 2008;23:S585–S598. doi: 10.1002/mds.22022. [DOI] [PubMed] [Google Scholar]
- Kahn ZU, Gutierrez A, Martin R, Penafiel A, Rivera A, De La Calle A. Dopmaine D5 receptors in rat and human brain. Neurosci. 2000;100:689–699. doi: 10.1016/s0306-4522(00)00274-8. [DOI] [PubMed] [Google Scholar]
- Lane JR, Powney B, Wise A, Rees S, Milligan G. G protein coupling and ligand selectivity of the D2L and D3 dopamine receptors. J Pharmacol Exp Ther. 2008;325:319–330. doi: 10.1124/jpet.107.134296. [DOI] [PubMed] [Google Scholar]
- Lauzon NM, Laviolette SR. Dopamine D4-recpetor modulation of cortical neuronal network activity and emotional processing: implications for neuropsychiatric disorders. Behav Brain Res. 2010;208:12–22. doi: 10.1016/j.bbr.2009.11.037. [DOI] [PubMed] [Google Scholar]
- Millan MJ, Maifoss L, Cussac D, Audinot V, Boutin J-A, Newman-Tancredi A. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. I. A multivariate analysis of the binding profile of 14 drugs at 21 native and cloned human receptor subtypes. J Pharmacol Exp Ther. 2002;303:791–804. doi: 10.1124/jpet.102.039867. [DOI] [PubMed] [Google Scholar]
- Needham E, Worth P. Parkinson's disease: a guide to pharmacological management. Prescriber. 2012;23:21–31. [Google Scholar]
- Newman-Tancredi A, Cussac D, Audinot V, Nicolas J-P, De Ceuninck F, Boutin J-A, et al. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. II. Agonist and antagonist properties at subtypes of dopamine D2-like receptor and α1/α2-adrenoceptor. J Pharmacol Exp Ther. 2002;303:805–814. doi: 10.1124/jpet.102.039875. [DOI] [PubMed] [Google Scholar]
- Oertel W, Trenkwalder C, Benes H, Ferini-Strambi L, Hogl B, Poewe W, et al. Long-term safety and efficacy of rotigotine transdermal patch for moderate to severe idiopathic restless legs syndrome: a 5-year open-label extension. Lancet Neurol. 2011;10:710–720. doi: 10.1016/S1474-4422(11)70127-2. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42:D1098–D1106. doi: 10.1093/nar/gkt1143. (Database Issue): [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perachon S, Schwartz J-C, Sokoloff P. Functional potencies of new antiparkinsonian drugs at recombinant human dopamine D1, D2 and D3 receptors. Eur J Pharmacol. 1999;366:293–300. doi: 10.1016/s0014-2999(98)00896-6. [DOI] [PubMed] [Google Scholar]
- Perez-Lloret S, Rascol O. Dopamine receptor agonists for the treatment of early or advanced Parkinson's disease. CNS Drugs. 2010;24:941–968. doi: 10.2165/11537810-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Peters MF, Vaillancourt F, Heroux M, Valiquette M, Scott CW. Comparing label-free biosensors for pharmacological screening with cell-based functional assays. Assay Drug Dev Technol. 2010;8:219–227. doi: 10.1089/adt.2009.0232. [DOI] [PubMed] [Google Scholar]
- Poewe W. Treatments for Parkinson disease–past achievements and current clinical needs. Neurology. 2009;72:S65–S73. doi: 10.1212/WNL.0b013e31819908ce. [DOI] [PubMed] [Google Scholar]
- Poewe W, Wenning GK. Apomorphine: an underutilised therapy for Parkinson's disease. Mov Disord. 2000;15:789–794. doi: 10.1002/1531-8257(200009)15:5<789::aid-mds1005>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Rascol O, Slaoui T, Regragui W, Ory-Magne F, Brefel-Courbon C, Montastruc J-L. Dopamine agonists. In: Koller WC, Melamed E, et al., editors. Handbook of Clinical Neurology Vol 83 (3rd Series) Parkinson's Disease and Related Disorders, Part II. Amsterdam: Elsevier B.V; 2007. pp. 73–92. [DOI] [PubMed] [Google Scholar]
- Reavill C, Taylor SG, Wood MD, Ashmeade T, Austin NE, Avenell KY, et al. Pharmacological actions of a novel, high-affinity, and selective human dopamine D(3) receptor antagonist, SB-277011-A. J Pharmacol Exp Ther. 2000;294:1154–1165. [PubMed] [Google Scholar]
- Robertson HA. Dopamine receptor interactions: some implications for the treatment of Parkinson's disease. Trends Neurosci. 1992;15:201–206. doi: 10.1016/0166-2236(92)90034-6. [DOI] [PubMed] [Google Scholar]
- Rocheville M, Jerman J. 7TM pharmacology measured by label-free: a holistic approach to cell signalling. Curr Opin Pharmacol. 2009;9:643–649. doi: 10.1016/j.coph.2009.06.015. [DOI] [PubMed] [Google Scholar]
- Scheller D, Ullmer C, Berkels R, Gwarek M, Lubbert H. The in vitro receptor profile of rotigotine: a new agent for the treatment of Parkinson's disease. Naunyn Schmeidebergs Arch Pharmacol. 2009;379:73–86. doi: 10.1007/s00210-008-0341-4. [DOI] [PubMed] [Google Scholar]
- Schwartz J-C, Carlsson A, Caron M, Scatton B, Civelli O, Kebabian JW. Dopamine receptors. In: Girdlestone D, et al., editors. The IUPHAR Compendium of Receptor Characterization and Classification. London: IUPHAR Media; 1998. pp. 141–151. [Google Scholar]
- Smart D, Wood M. Real time receptor function in vitro: microphysiometry and the fluorometric imaging plate reader (FLIPR) In: Stanford C, Horton R, et al., editors. Receptors: Structure and Function. Oxford, UK: Oxford University Press; 2001. pp. 175–191. [Google Scholar]
- Tan E-K. Dopamine agonists and their role in Parkinson's disease treatment. Expert Rev Neurother. 2003;3:805–810. doi: 10.1586/14737175.3.6.805. [DOI] [PubMed] [Google Scholar]
- Tiberi M, Caron MG. High agonist-independent activity is a distinguishing feature of the dopamine D1B receptor subtype. J Biol Chem. 1994;269:27925–27931. [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastro M, Nichols DE, Koblika B, Weinstein H, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Van der Weide J, De Vries JB, Tepper PG, Horn AS. In vitro binding of the very potent and selective D-2 dopamine agonist, [3H]N-0437 to calf caudate membranes. Eur J Pharmacol. 1987;134:211–219. doi: 10.1016/0014-2999(87)90167-1. [DOI] [PubMed] [Google Scholar]
- Vermeulen RJ, Drukarch B, Sahadat MC, Goosen C, Wolters EC, Stoof JC. The dopamine D1 agonist SKF 81297 and the dopamine D2 agonist LY 171555 act synergistically to stimulate motor behavior of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned parkinsonian rhesus monkeys. Mov Disord. 1994;9:664–672. doi: 10.1002/mds.870090613. [DOI] [PubMed] [Google Scholar]
- Vermeulen RJ, Drukarch B, Wolters EC, Stoof JC. Dopamine D1 receptor agonists: the way forward for the treatment of Parkinson's disease? CNS Drugs. 1999;11:83–91. [Google Scholar]
- Watson J, Collin L, Ho M, Riley G, Scott C, Selkirk JV, et al. 5-HT1A receptor agonist-antagonist binding affinity difference as a measure of intrinsic activity in recombinant and native tissue systems. Br J Pharmacol. 2000;130:1108–1114. doi: 10.1038/sj.bjp.0703394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts VJ, Lawler CP, Gonzales AJ, Zhou Q-Y, Civelli O, Nichols DE, et al. Spare receptors and intrinsic activity: studies with D1 dopamine receptor agonists. Synapse. 1995;21:177–187. doi: 10.1002/syn.890210211. [DOI] [PubMed] [Google Scholar]
- Wood LD. Clinical review and treatment of select adverse effects of dopamine receptor agonists in Parkinson's disease. Drugs Aging. 2010;27:295–310. doi: 10.2165/11318330-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Zaworski PG, Alberts GL, Pregenzer JF, Im WB, Slightom JL, Gill GS. Efficient functional coupling of the human D3 dopamine receptor to G(o) subtype of G proteins in SH-SY5Y cells. Br J Pharmacol. 1999;128:1181–1188. doi: 10.1038/sj.bjp.0702905. [DOI] [PMC free article] [PubMed] [Google Scholar]