

Abstract
The incidence of certain angiogenesis-dependent diseases is higher in Caucasians than in African Americans. Angiogenesis is amplified in wound healing and cornea models in albino C57 mice compared with black C57 mice. Moreover, mouse and human melanocytes with low pigmentation stimulate endothelial cell (EC) proliferation and migration in vitro more than melanocytes with high pigmentation. This effect is due, in part, to the secretion of an angiogenic protein called fibromodulin (FMOD) from lowly pigmented melanocytes. Herein, we expand upon the mechanism contributing to increased angiogenesis in lighter skin and report that monocyte chemotactic protein-1 (MCP-1) is secreted by nonpigmented mouse melanocytes by 5- to 10-fold more than pigmented melanocytes. MCP-1 protein stimulates EC proliferation and migration in vitro and angiogenesis in vivo. Mechanistic studies determine that FMOD is upstream of MCP-1 and promotes its secretion from both melanocytes and activated ECs via stimulation of NF-κB activity. Mice injected with FMOD-neutralizing antibodies show 2.3-fold decreased levels of circulating MCP-1. Human studies confirmed that, on average, Caucasians have 2-fold higher serum levels of MCP-1 than African Americans. Taken together, this study implicates the FMOD/MCP-1 pathway in the regulation of angiogenesis by local melanocytes and suggests that melanogenic activity may protect against aberrant angiogenic diseases.—Adini, I., Adini, A., Bazinet, L., Watnick, R. S., Bielenberg, D. R., and D’Amato, R. J. Melanocyte pigmentation inversely correlates with MCP-1 production and angiogenesis-inducing potential.
Keywords: vascular biology, endothelial cells, FMOD, NF-κB
Neovascularization, the formation of new blood vessels sprouting from preexisting capillaries, is regulated by the balance between endogenous stimulators and inhibitors in the cellular microenvironment. Certain angiogenesis-dependent skin and eye diseases have been shown to be more prevalent in Caucasians than in African Americans (1–3), suggesting that lower pigmentation is a risk factor. Our previous studies have shown that pathways involved in melanogenesis alter the local angiogenic balance (4). Melanogenesis is the process of melanin synthesis that is restricted to mature melanosomes in melanocytes (5). l-Tyrosine and l-dopa, besides serving as substrates and intermediates of melanogenesis, also act as inducers and positive regulators of the melanogenic pathway (6, 7). Melanocytes produce and secrete cytokines and extracellular matrix (ECM) components with reciprocal effects on surrounding cells and tissues when stimulated by UV light, cytokine mediators, and hormones (8–10). One possible explanation for these pigment-based epidemiologic observations may be due to melanocyte secretion of fibromodulin (FMOD), a small proteoglycan containing leucine-rich repeats with 4 keratan sulfate chains. Melanocytes with low pigment levels produce high levels of FMOD, significantly shifting the angiogenic balance toward the initiation of angiogenesis (4).
Angiogenesis can be stimulated by a range of factors secreted by stromal and inflammatory cells in the microenvironment (11, 12). These cells secrete proangiogenic factors including VEGF, TNF-α, IL-1β, and other cytokines that increase vascular permeability, promote endothelial cell (EC) migration, and support vessel invasion and sprouting (13–15). In this study, we focused on the cytokines secreted by human and mouse melanocytes with low vs. high pigment. Using a mouse angiogenesis antibody array, we determined that monocyte chemotactic protein-1 (MCP-1), also known as chemokine (C-C motif) ligand 2, was highly up-regulated in nonpigmented melanocytes compared to highly pigmented melanocytes. MCP-1 regulates the trafficking of monocytes to inflammatory sites, up-regulates VEGF expression, and stimulates EC proliferation and migration (16–18). MCP-1 induces the secretion of proteases such as matrix metalloproteinase-1 from ECs to degrade the ECM. This leads to the release of matrix-bound growth factors such as fibroblast growth factor, VEGF, and platelet-derived growth factor, which in turn promote neovascularization (19). MCP-1 can also be secreted by activated ECs, vascular smooth muscle cells, monocytes, and fibroblasts (20).
As a major regulator of neovascularization and inflammation, MCP-1 and other inflammatory mediators are known to contribute to the pathologic processes of diseases such as age-related macular degeneration (ARMD), a debilitating ocular condition involving invasion of choroidal blood vessels into the retina. Ambati et al. (20) have shown that choroidal endothelial production of MCP-1 mediates macrophage recruitment into aged choroids of wild-type mice. This macrophage infiltration is seen clinically in a multitude of retinal and choroidal angiogenic diseases (21). Likewise, Goede et al. (18) and Aplin et al. (22) have shown that MCP-1 is an indirect inducer of angiogenesis via macrophage recruitment.
Herein, we present data that lowly pigmented melanocytes secrete MCP-1, which stimulates angiogenesis. We show that MCP-1 is downstream of FMOD and NF-κB, and correlatively that darkly pigmented African Americans have lower circulating levels of MCP-1.
MATERIALS AND METHODS
Materials
Rabbit anti-mouse MCP-1 antibody and goat anti-human MCP-1 were obtained from Santa Cruz Biotechnology (Dallas, TX, USA) and R&D Systems (Minneapolis, MN, USA), respectively. Mouse anti-human FMOD antibody for neutralization was obtained from Alexis Biochemicals (San Diego, CA, USA) and Proteintech Group, Incorporated (Chicago, IL, USA). Antibodies for the canonical and noncanonical NF-κB pathway were obtained from Cell Signaling Technology (Danvers, MA, USA), and those for mouse anti-β-actin were from Sigma-Aldrich (St. Louis, MO, USA). Recombinant TNF-α and FMOD proteins were purchased from OriGene (Rockville, MD, USA), R&D Systems, and Proteintech Group, Incorporated. Recombinant VEGF-A and MCP-1 were purchased from R&D Systems, Cycloheximide was obtained from Sigma-Aldrich, and 6-amino-4-(4-phenoxyphenylethylamino) quinazoline (QNZ) was obtained from Millipore (Billerica, MA, USA). Human serum was purchased from Innovative Research (Novi, MI, USA).
Biochemical methods
ELISA was performed with Quantikine Human or Mouse MCP-1 ELISA kits (R&D Systems). MCP-1 was quantified by ELISA and is expressed in the figures as picogram per milliliter per 1 million cells for conditioned medium (CM) experiments.
Cell culture
Adult dermal human microvascular endothelial cells (HMVECs) (Invitrogen, Life Technologies, Grand Island, NY, USA) and human melanocytes (ScienCell Research Laboratories, Carlsbad, CA, USA) were grown according to the manufacturers’ protocols. The nonpigmented (albino) mouse melanocytes were isolated from BALB/c mice and contain a point mutation (TGT→TCT) in the tyrosinase gene, resulting in an inactive enzyme and a lack of melanin production. BALB/c pigmented melanocytes were genetically repaired by editing this mutation using an RNA-DNA oligonucleotide (23). Mouse BALB/c melanocytes and C57 melan-a cells were originally from Dr. Dorothy Bennett (St. George’s, University of London) and the Wellcome Trust Functional Genomics Cell Bank and acquired from Dr. Vitali Alexeev (Thomas Jefferson University, Philadelphia, PA, USA) (23–25). Choroidal and iris melanocytes were obtained from choroidal and iris tissue of albino C57 and black C57 mice. Cells were isolated by mechanical disruption and established in RPMI 1640 medium containing 10% fetal bovine serum and β-mercaptoethanol (100 μM) for 2 wk, and then analyzed for MCP-1 secretion.
Melanocyte CM for ECs
Equal numbers of pigmented or nonpigmented melanocytes were plated on tissue culture plates overnight. One day later, melanocyte medium was replaced with EGM2-MV (EC medium). After 24–48 h, CM was collected, and the melanocytes were counted.
Angiogenesis antibody array
CMs from pigmented and nonpigmented melanocytes were analyzed for protein expression with the use of RayBio Mouse Angiogenesis Antibody Array (catalog #AAM-ANG-1; RayBiotech, Norcross, GA, USA) according to the manufacturer’s instructions. Blots were analyzed with ImageJ software (NIH, Bethesda, MD, USA). Results of interest in the angiogenesis antibody array were verified by ELISA (R&D Systems).
Cell migration assay
Cell migration was performed as previously described (26) with the following modifications: ECs were seeded (105 cells/100 μl) onto the upper chamber and allowed to migrate toward CM from melanocytes or antibodies present in the bottom of the Transwell. After 4 h, cells were stained with Giemsa and counted in 10 random fields (×400) across the membrane. Migrated cells were detached, lysed, labeled with a fluorescent dye, and measured with a fluorescence microplate reader (PerkinElmer Life and Analytical Sciences, Waltham, MA, USA). Experiments were repeated at least 3 times.
Cell proliferation assay
HMVECs were plated at 10,000 cells/ml onto 96-well culture plates in CM from nonpigmented melanocytes treated with IgG or anti-MCP-1 antibody. WST-1 was used to assess viable cells (Roche Diagnostics Corporation, Indianapolis, IN, USA). Colorimetric absorbance of the samples was read at 450 nm. Experiments were repeated at least 3 times.
Luciferase assay
HMVECs (106 cells) were transfected with MetLucNF-κB using Nucleofector and Nucleofector Kit (Lonza, Walkersville, MD, USA). Cells were incubated for 24 h, and FMOD was added for an additional 24 h. Medium was collected and luciferase detected using the Ready-To-Glow Secreted Luciferase Vector kit (Clontech Laboratories, Mountain View, CA, USA).
Mice
All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee of Boston Children’s Hospital. BALB/c, C57Bl/6, and B6(Cg)-Tyrc-2J/J mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA).
Corneal micropocket assay
The corneal micropocket angiogenesis assay was performed as previously described (27). Pellets containing MCP-1 (0.05 μg/pellet equivalent to 5.2 pmol/pellet), VEGF (positive control, 0.2 μg/pellet equivalent to 5.2 pmol/pellet), or sham pellet (vehicle only) were implanted into the corneas of C57BL/6J mice and analyzed 6 d postimplantation. Vessel growth was measured and neovascular area calculated using the following equation: vessel area (VA) (mm2) = [π × clock hour × vessel length (mm) × 0.2 mm]. The experiment was repeated 3 times; each trial included 10 eyes per group.
Matrigel plug assay
Matrigel (BD Biosciences, San Jose, CA, USA) was mixed with Caucasian melanocytes and goat anti-human MCP-1 or control goat IgG (R&D Systems) and injected subcutaneously on the dorsal flank of C57BL/6 mice. After 7 d, plugs were enzymatically digested, and fluorescence-activated cell sorting (FACS) analysis was performed. Quantification of ECs was defined as CD31+CD45−. Flow cytometry was performed using FACSCalibur and CellQuest software (BD Biosciences).
Statistics
Data are expressed as the mean ± SD. Statistical significance was assessed using Student’s unpaired 2-tailed t test. P < 0.05 was considered statistically significant.
RESULTS
Pigmentation alters the secretion of MCP-1 by melanocytes
Following our recent discovery that pigmentation levels in melanocytes regulate angiogenesis, in part, through the expression of FMOD (4), we sought to determine whether other cytokines contribute to this effect. Primary cultures of melanocytes isolated from albino BALB/c mice that lack tyrosinase activity, an enzyme that catalyzes tyrosine to melanin by oxidation, were compared to melanocytes in which pigmentation was restored by repairing the tyrosinase gene (23). The CMs from these 2 melanocyte lines, which differ only in 1 base pair, were compared using a mouse angiogenesis antibody array to detect differential secretion of proangiogenic cytokines. Several cytokines were variably expressed, and MCP-1 showed the greatest difference between nonpigmented and pigmented melanocytes (Fig. 1A, box). MCP-1 was secreted at a 2.5-fold higher level by mouse nonpigmented melanocytes than by mouse pigmented melanocytes (comparing protein dot blot and equalizing to the control dots) (Fig. 1A). These results were confirmed and expanded by ELISA and showed a 4-fold reduction in MCP-1 secretion in genetically repaired BALB/c pigmented melanocytes compared to wild-type, nonpigmented BALB/c melanocytes (Fig. 1B, right bars). In addition, endogenously pigmented C57 immortalized mouse melanocyte cells called melan-a secreted greater than 10-fold lower concentrations of MCP-1 than BALB/c melanocytes (Fig. 1B, left bars).
Figure 1.

Effect of melanocytes on MCP-1. A) Mouse angiogenesis antibody array expression profiles of multiple cytokines induced by CM from nonpigmented melanocytes (left) and CM from pigmented melanocytes (right). B) Quantification of MCP-1 level secreted by various melanocytes. C) MCP-1 in melanocytes in tyrosine-depleted media. D) MCP-1 in the iris and choroid of Alb-C57 and C57 mice. E) MCP-1 in the serum from Alb-C57 and C57 mice. F) Quantification of MCP-1 level secreted by ex vivo monocytes of Alb-C57 and C57. G) Quantification of MCP-1 level secreted by ECs after incubation in CM from pigmented and nonpigmented melanocytes. H) MCP-1 in the serum from Caucasians and African Americans. *P < 0.01; **P < 0.001; ***P < 0.0001.
Melanogenesis and the activity of tyrosinase can be regulated by their substrate, tyrosine (28, 29). Therefore, we next explored whether tyrosinase activity or melanin production could directly regulate MCP-1 secretion. Accordingly, we cultured pigmented melanocytes in normal media that contain tyrosine (tyrosine depletion −) and in media depleted of tyrosine (tyrosine depletion +) and measured the concentration of secreted mouse MCP-1. Nonpigmented melanocytes served as a control (Fig. 1C, left bar). In the absence of exogenous tyrosine, melanocytes produce a lower level of pigment or intermediate pigment products (30). Depletion of tyrosine from the media resulted in a doubling of MCP-1 secretion (65–123 pg/ml) in pigmented melanocytes (Fig. 1C, compare middle and right bars). MCP-1 was secreted 3.2-fold higher by mouse nonpigmented melanocytes than by mouse pigmented melanocytes (Fig. 1C, compare left and middle bars). Levels of MCP-1 in pigmented cells depleted of tyrosine never reached that of nonpigmented cells, possibly because of the fact that cells can convert phenylalanine to tyrosine. These findings indicate that MCP-1 production in melanocytes is inhibited by pigment production.
To expand upon our in vitro data, we examined whether the levels of MCP-1 differ in various pigmented tissues in white vs. black mice. BALB/c (albino) and C57 (black) mice are different strains of mice and are, therefore, genetically different. In order to impartially compare between tissues, we used wild-type (tyrosinase-expressing) “black” C57BL/6J (referred to hereafter as C57) and mutant (G→T mutation at nucleotide 291 in the tyrosinase gene) “white/albino” B6(Cg)-Tyrc-2J/J mice (referred to hereafter as Alb-C57), as described previously (4). Two “pigmented” tissues, iris and choroid, were isolated from Alb-C57 and C57 mice, and the cells were cultured in vitro (without passage). Secreted MCP-1 levels were higher in Alb-C57 mice than C57 mice in both tissues (15-fold in iris; 2.5-fold in choroid) (Fig. 1D). Next, we compared circulating mouse MCP-1 levels in serum from Alb-C57 and C57 mice (n = 10 mice/group) and found 2.2-fold higher levels of MCP-1 in albino mice serum (Fig. 1E).
Because it has been demonstrated that MCP-1 is also produced by other cell types including monocytes and ECs (20), we examined the production of MCP-1 from monocytes isolated from Alb-C57 and C57 mice. Monocytes (cultured from spleen) from nonpigmented mice secrete more than twice the amount of MCP-1 than those from pigmented mice (Fig. 1F). We next measured the level of MCP-1 secreted by ECs treated with the CM of pigmented and nonpigmented melanocytes. In order to distinguish between EC-secreted and melanocyte-secreted MCP-1, we treated human dermal ECs (HMVECs) with CM from nonpigmented or pigmented BALB/c mouse melanocytes and measured MCP-1 with a human-specific ELISA. We found that ECs treated with CM from nonpigmented cells displayed a 4.5-fold increase in MCP-1 secretion (Fig. 1G).
Given these results, we subsequently measured human MCP-1 levels in human serum from Caucasian Americans (34 samples) and African Americans (44 samples). We observed that serum from Caucasians contained 2-fold higher levels of MCP-1 than African Americans (Fig. 1H).
Collectively, these findings indicate that MCP-1 protein is regulated by pigment in both humans and mice. White mice or lightly pigmented people have higher systemic levels of MCP-1. This difference correlates on the tissue and cellular level. The secretion of MCP-1 is proportional to the activity of the tyrosinase enzyme and its substrate tyrosine.
Melanocyte-derived MCP-1 stimulates angiogenesis
MCP-1 is reported to stimulate angiogenesis (19, 31, 32). We tested the ability of MCP-1 protein to elicit neovascularization in the corneal micropocket assay (27). Pellets containing MCP-1 (0.05 μg/pellet equivalent to 5.2 pmol/pellet), VEGF (positive control, 0.2 μg/pellet equivalent to 5.2 pmol/pellet), or sham pellet (vehicle only) were implanted into the corneas and analyzed 6 d postimplantation. Upon analysis, we found that pellets releasing MCP-1 protein stimulated corneal neovascularization with a VA significantly greater than sham pellets (0.22 vs. 0.00 mm2) but less than the VEGF-positive control (Fig. 2A).
Figure 2.

MCP-1 stimulates angiogenesis. A) Corneal micropocket assay with pellets containing MCP-1, VEGF-A (positive control), or sham (negative control). Significant difference was seen between MCP-1 and control (n = 10 eyes/group). B) Caucasian melanocytes were suspended in Matrigel, and anti-MCP-1 or IgG (control) was added. After 7 d, cells were stained for CD31 and CD45 and analyzed by FACS (n = 10/group). C) HMVEC viability (WST-1 O.D.450) in response to melanocyte CM treated with anti-MCP-1. D) HMVEC migration in response to melanocyte CM treated with anti-MCP-1. **P < 0.001; ***P < 0.0001.
Once we established that MCP-1 protein itself could induce neovascularization, we next sought to determine whether melanocyte-associated angiogenesis was MCP-1 dependent. We utilized a modified Matrigel plug assay, in which the CMs from human nonpigmented (Caucasian) melanocytes were mixed with Matrigel in the ratio of 1:5, in the presence of anti-human MCP-1 antibody or control IgG, and injected s.c. into the flanks of C57 mice. After 7 d, the plugs were removed, digested to yield single cells, and stained for the pan-endothelial marker, CD31 (33). Because MCP-1 also recruits macrophages, which express CD31, the isolated cells were costained with CD45. Quantification by FACS analysis revealed that MCP-1 neutralization reduced the number of invading CD31+CD45− cells by 35% (Fig. 2B). These data suggest that melanocytes recruit ECs and promote angiogenesis via MCP-1.
To decipher the effects of MCP-1 on angiogenesis and determine the mechanism of MCP-1, we moved to an in vitro system. ECs were grown in CM from nonpigmented mouse melanocytes treated with anti-MCP-1 antibody or control IgG and analyzed for their ability to proliferate or migrate. Cell viability was evaluated after 2 d by using WST-1 reagent and showed that anti-MCP-1 antibody inhibited HMVEC viability by 20% compared to IgG (Fig. 2C). EC migration was examined using a Transwell migration assay with CM from melanocytes in the lower wells. Nonpigmented melanocytes promoted HMVEC migration 3-fold more than pigmented melanocytes (Fig. 2D). In addition, neutralizing MCP-1 resulted in ∼50% inhibition of EC migration induced by nonpigmented melanocyte CM (Fig. 2D). Taken together, these studies suggest that nonpigmented melanocytes can stimulate proliferation and migration of ECs and that MCP-1 enhances both of these processes. Relatively speaking, MCP-1 increased migration more than proliferation.
FMOD induces MCP-1 expression
Our laboratory recently reported the identification of a new angiogenic factor secreted by nonpigmented melanocytes, namely FMOD (4). Here, we examined whether FMOD could directly regulate MCP-1 expression in ECs. HMVECs were treated with FMOD or control for 24 h, and MCP-1 protein was measured in the CM by ELISA. FMOD stimulated nearly double the amount of MCP-1 from ECs as compared to control (Fig. 3A). A dose response of recombinant FMOD on ECs revealed that FMOD in the picomolar range was most stimulatory toward MCP-1 (Fig. 3B). Because nonpigmented melanocytes express MCP-1, we treated these cells with a neutralizing FMOD antibody for 24 h, and the resulting CM was analyzed by ELISA for MCP-1. Nonpigmented mouse melanocytes secreted only one-third the amount of MCP-1 in the presence of anti-FMOD (Fig. 3C). MCP-1 levels were constitutively higher in serum from Alb-C57 mice (Fig. 1E); therefore, we treated these mice with anti-FMOD (i.p.) or control IgG in vivo. Five days postinjection of antibody, the serum was analyzed, and the results revealed that mice treated with anti-FMOD contained 2.3-fold lower levels of circulating MCP-1 compared with mice injected with IgG (Fig. 3D). Based on these findings, we conclude that nonpigmented melanocytes secrete both MCP-1 and FMOD and that FMOD stimulates the secretion of MCP-1 from melanocytes and ECs.
Figure 3.
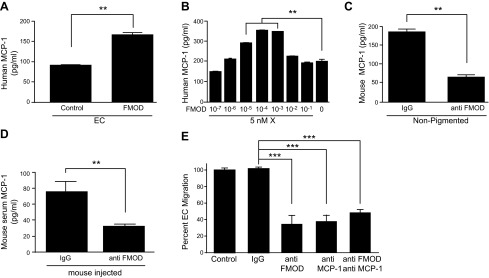
FMOD induced secretion of MCP-1. A) MCP-1 ELISA on ECs treated with FMOD or control. B) MCP-1 ELISA on ECs treated with dose response of FMOD. C) MCP-1 ELISA on nonpigmented melanocytes treated with anti-FMOD. D) MCP-1 ELISA on serum from C57 mice treated daily for 5 d with 8 μg/kg per day anti-FMOD intraperitoneally or IgG control. E) HMVEC migration in response to nonpigmented melanocyte CM treated with anti-FMOD, anti-MCP-1, or combination of anti-FMOD and anti-MCP-1. **P < 0.001; ***P < 0.0001.
We again performed EC migration assays in the presence of nonpigmented melanocyte CM (which contains both MCP-1 protein and FMOD protein) and examined the migration after incubation with anti-MCP-1, anti-FMOD, or a combination of both antibodies. All antibody treatments resulted in an approximate two-third decrease in EC migration (Fig. 3E). Interestingly, the combination of anti-MCP-1 and anti-FMOD together did not increase the inhibition of migration, indicating that these 2 proteins are likely in the same pathway.
We next sought to determine whether the increase in MCP-1 expression was due to the secretion of existing (stored) MCP-1 or new protein synthesis of MCP-1. To test this, we inhibited protein translation with cycloheximide, with and without FMOD, in HMVECs. We observed an increase in MCP-1 secretion with the addition of FMOD, as expected; but in the presence of cycloheximide, there was no MCP-1 production, with or without FMOD (Fig. 4A). In fact, cycloheximide alone reduced MCP-1 levels 10-fold below baseline. These findings indicate that the FMOD-mediated increase in MCP-1 levels in EC CM is the result of new protein synthesis.
Figure 4.
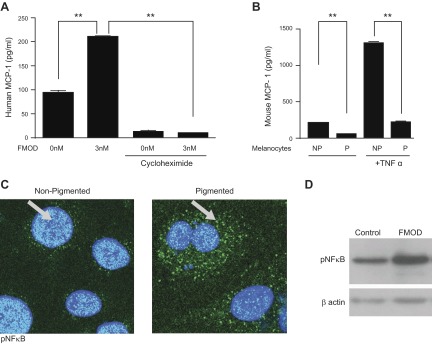
FMOD affects MCP-1 expression through activation of NF-κB. A) MCP-1 ELISA on ECs treated with cycloheximide (20 μg/ml) with or without FMOD (3 nM). B) MCP-1 ELISA on ECs treated with CM from pigmented (P) and nonpigmented (NP) melanocytes with or without TNF-α. C) Nonpigmented (left) and pigmented (right) melanocytes were stained with pNF-κB (green), and nuclei were counterstained with DAPI (blue). D) Western blot of pNF-κB in ECs treated with FMOD (3 nM) for 30 min. β-Actin was used as a loading control. All experiments were repeated at least 3 times. **P < 0.001.
FMOD activates the NF-κB pathway
TNF-α has been shown to stimulate the expression of MCP-1 in monocytes (34). Therefore, we investigated whether TNF-α could also regulate the expression of MCP-1 in melanocytes. Specifically, we treated pigmented and nonpigmented melanocytes with TNF-α and measured MCP-1 levels by ELISA. We observed that TNF-α increased MCP-1 secretion by 6.4-fold in nonpigmented melanocytes. In contrast, TNF-α only stimulated MCP-1 secretion in pigmented melanocytes by ∼2-fold (Fig. 4B).
The up-regulation of MCP-1 in monocytes has been shown to be mediated by NF-κB, and the MCP-1 promoter contains consensus NF-κB binding sites (34, 35). Therefore, we examined the localization of phosphorylated NF-κB (pNF-κB) in pigmented and nonpigmented mouse melanocytes. We observed that nonpigmented melanocytes induced the translocation of NF-κB (phosphorylated p65 S468 subunit) to the nucleus, whereas pigmented melanocytes did not (Fig. 4C).
NF-κB exists in a complex with IκB in the cytoplasm. Upon activation of upstream inflammatory pathways, IκB is phosphorylated by the IKK complex, ubiquitinated, and degraded, allowing NF-κB to translocate into the nucleus and bind the promoters of its target genes (36). We examined the signaling cascade in ECs following treatment with FMOD. Western blot verified that FMOD induced the pNF-κB in HMVECs (Fig. 4D). To elucidate the activation of NF-κB by FMOD, we next assessed the expression of associated kinases. Treatment of ECs with FMOD resulted in increased IKK1 and IKK2 (Fig. 5A). Additionally, TNF receptor-associated factor 2 (TRAF2), which promotes the prolonged IKK activity, was elevated after FMOD (37). Treatment of ECs with FMOD resulted in decreased IKBα, an inhibitor of ubiquitination kinase (Fig. 5B). Taken together, these data suggest that NF-κB activation is downstream of FMOD.
Figure 5.
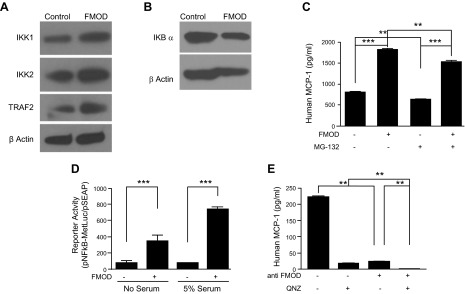
FMOD affects the NF-κB pathway. A) Western blot for IKK1, IKK2, TRAF2, and β-actin in HMVECs treated with FMOD (3 nM). B) Western blot for IKBα and β-actin in HMVECs treated with FMOD (3 nM). C) MCP-1 ELISA on HMVECs treated with 1 μM NF-κB inhibitor (MG-132), with or without FMOD (3 nM). D) Luminescence analyzed luciferase secretion after HMVECs were transfected with MetLucNF-κB and treated with FMOD (3 nM). E) Quantification of MCP-1 level secreted by ECs after treatment with QNZ (10 ng/ml) or anti-FMOD (0.2 mg/ml). All experiments were repeated at least 3 times. **P < 0.001; ***P < 0.0001.
We treated ECs with a proteasome inhibitor (MG-132) that blocks degradation of IκB and therefore blocks NF-κB from binding target genes (38, 39). We observed that treatment of ECs with MG-132 (1 μM) resulted in a 25% decrease in MCP-1 secretion as measured by ELISA (Fig. 5C, compare bar 1 and 3). In addition, MG-132 could significantly inhibit FMOD-induced MCP-1 secretion (Fig. 5C, compare bar 2 and 4). These results are in agreement with previous reports demonstrating that MCP-1 secretion is proteasome dependent (40).
We then examined the relative ability of FMOD to activate NF-κB in ECs using an NF-κB luciferase reporter assay. Specifically, ECs were transfected with MetLucNF-κB and then treated with FMOD in medium lacking or containing (5%) serum. FMOD resulted in a 4.6-fold increase in luciferase activity in no serum and a 9.5-fold increase in luciferase activity in 5% serum (Fig. 5D). Finally, we examined the effects of 6-amino-4-(phenoxyphenylethylamino) QNZ, a specific NF-κB inhibitor (41), on MCP-1 regulation and compared it to inhibiting MCP-1 with anti-FMOD. HMVECs treated with QNZ (1 ng/ml) reduced MCP-1 levels by 12-fold, whereas anti-FMOD reduced MCP-1 levels 8-fold (Fig. 5E). When given in combination, MCP-1 levels were barely detectable in ECs. Therefore, blocking FMOD upstream with antibodies or downstream with specific inhibitors led to the same result, namely the lack of MCP-1 expression. Taken together, these results strongly suggest that FMOD promotes the secretion of MCP-1 via stimulation of NF-κB activity.
DISCUSSION
Epidemiologic evidence of the association between pigmentation and the incidence of angiogenic diseases prompted us to investigate the possible mechanisms underlying these observations. We postulated that at least one explanation for these differences could be the differential expression of angiogenic factors by pigmented and nonpigmented melanocytes. Following this line of inquiry led us to discover that melanocyte-secreted FMOD was implicated in this process (4). The purpose of the current study was to determine if the FMOD-driven angiogenic activity of melanocytes is mediated in part by MCP-1, which has been previously reported to stimulate angiogenesis (42). Our earlier findings demonstrated that nonpigmented melanocytes promoted angiogenesis in vitro and in vivo and that FMOD is responsible for a portion of this effect (4). Herein, we report a correlation between low pigmentation, FMOD production and MCP-1 production. We demonstrate that both ECs and melanocytes secrete MCP-1 in an FMOD-dependent manner. We further demonstrate that MCP-1 is a significant contributor to proangiogenic activity in tissues containing melanocytes and can stimulate neovascularization in mouse models. Based upon the results in this report and our previous manuscript (4), FMOD and MCP-1 appear to be major contributing factors responsible for the angiogenic activity of melanocytes.
MCP-1 has been shown to be expressed by multiple cell types, including monocytes/macrophages, lymphocytes, vascular endothelium, and fibroblasts (43). However, the finding reported here that MCP-1 is produced by nonpigmented melanocytes is novel to the field. These findings were translated to humans by analysis of serum, which revealed that MCP-1 is present at higher levels in Caucasians compared to African Americans. In addition, our data demonstrating that FMOD significantly stimulated MCP-1 levels in mouse serum served as functional validation of the analyses of human samples.
Lastly, to delineate the mechanism of FMOD-MCP-1-mediated angiogenesis, we turned our attention to NF-κB, a transcription factor that plays an important role in inflammation. Herein, we show that NF-κB activity (translocation to nucleus) was preferentially induced in nonpigmented melanocytes vs. pigmented melanocytes. The mechanism of this differential activation was due to increased FMOD secretion by nonpigmented melanocytes. Recombinant FMOD recapitulated the activity present in the CM of nonpigmented melanocytes and accelerated NF-κB activity (via stimulation of IKK and depletion of IKBα), and thereby stimulated MCP-1 production. As such, the activation of NF-κB by FMOD may serve to potentiate both the immune response and angiogenesis observed in ARMD and other angiogenesis-dependent diseases associated with pigmentation. The ramifications of this study could provide new insight into the biochemical mechanism behind the epidemiologic correlation between pigmentation and angiogenesis-related diseases and potentially lead to novel treatment modalities.
Acknowledgments
The authors thank Alexeev Y. Vitali and the Wellcome Trust Functional Genomics Cell Bank for primary murine melanocyte cells and Pouya Pakneshan for helping with ELISAs. I.A. performed the experiments, analyzed the data, and wrote the manuscript. A.A. and L.B. assisted with the experiments. D.R.B. and R.S.W. edited the manuscript. R.J.D. supervised the analysis and edited the manuscript. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the U.S. National Institutes of Health (NIH). Research reported in this publication was supported, in part, by the NIH National Eye Institute under Award Number R01EY012726 (to R.J.D.). The authors declare no conflicts of interest.
Glossary
- ARMD
age-related macular degeneration
- CM
conditioned medium
- EC
endothelial cell
- ECM
extracellular matrix
- FACS
fluorescence-activated cell sorting
- FMOD
fibromodulin
- HMVEC
human microvascular endothelial cell
- MCP-1
monocyte chemotactic protein-1, also called CCL-2
- pNF-κB
phosphorylated NF-κB
- QNZ
quinazoline
- TRAF
TNF receptor-associated factor
- VA
vessel area
REFERENCES
- 1.Vanderbeek B. L., Zacks D. N., Talwar N., Nan B., Musch D. C., and Stein J. D. (2011) Racial differences in age-related macular degeneration rates in the United States: a longitudinal analysis of a managed care network. Am. J. Ophthalmol. 152, 273–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Congdon N., O’Colmain B., Klaver C. C., Klein R., Muñoz B., Friedman D. S., Kempen J., Taylor H. R., Mitchell P.; Eye Diseases Prevalence Research Group (2004) Causes and prevalence of visual impairment among adults in the United States. Arch. Ophthalmol. 122, 477–485 [DOI] [PubMed] [Google Scholar]
- 3.Gottlieb J. L. (2002) Age-related macular degeneration. JAMA 288, 2233–2236 [DOI] [PubMed] [Google Scholar]
- 4.Adini I., Ghosh K., Adini A., Chi Z. L., Yoshimura T., Benny O., Connor K. M., Rogers M. S., Bazinet L., Birsner A. E., Bielenberg D. R., D’Amato R. J. (2014) Melanocyte-secreted fibromodulin promotes an angiogenic microenvironment. J. Clin. Invest. 124, 425–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slominski A., Paus R. (1990) Are l-tyrosine and l-dopa hormone-like bioregulators? J. Theor. Biol. 143, 123–138 [DOI] [PubMed] [Google Scholar]
- 6.Slominski A., Paus R., Schadendorf D. (1993) Melanocytes as “sensory” and regulatory cells in the epidermis. J. Theor. Biol. 164, 103–120 [DOI] [PubMed] [Google Scholar]
- 7.Slominski A., Zmijewski M. A., Pawelek J. (2012) l-Tyrosine and l-dihydroxyphenylalanine as hormone-like regulators of melanocyte functions. Pigment Cell Melanoma Res. 25, 14–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slominski A., Kim T. K., Brożyna A. A., Janjetovic Z., Brooks D. L., Schwab L. P., Skobowiat C., Jóźwicki W., Seagroves T. N. (2014) The role of melanogenesis in regulation of melanoma behavior: melanogenesis leads to stimulation of HIF-1α expression and HIF-dependent attendant pathways. [E-pub ahead of print]Arch. Biochem. Biophys. 563, 79–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slominski A., Tobin D. J., Shibahara S., Wortsman J. (2004) Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 84, 1155–1228 [DOI] [PubMed] [Google Scholar]
- 10.Slominski A. (2009) Neuroendocrine activity of the melanocyte. Exp. Dermatol. 18, 760–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim Y. W., West X. Z., Byzova T. V. (2013) Inflammation and oxidative stress in angiogenesis and vascular disease. J. Mol. Med. (Berl.) 91, 323–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coussens L. M., Raymond W. W., Bergers G., Laig-Webster M., Behrendtsen O., Werb Z., Caughey G. H., Hanahan D. (1999) Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 13, 1382–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noonan D. M., De Lerma Barbaro A., Vannini N., Mortara L., Albini A. (2008) Inflammation, inflammatory cells and angiogenesis: decisions and indecisions. Cancer Metastasis Rev. 27, 31–40 [DOI] [PubMed] [Google Scholar]
- 14.Costa C., Incio J., Soares R. (2007) Angiogenesis and chronic inflammation: cause or consequence? Angiogenesis 10, 149–166 [DOI] [PubMed] [Google Scholar]
- 15.Mantovani A., Sozzani S., Locati M., Allavena P., Sica A. (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23, 549–555 [DOI] [PubMed] [Google Scholar]
- 16.Charo I. F., Taubman M. B. (2004) Chemokines in the pathogenesis of vascular disease. Circ. Res. 95, 858–866 [DOI] [PubMed] [Google Scholar]
- 17.Salcedo R., Ponce M. L., Young H. A., Wasserman K., Ward J. M., Kleinman H. K., Oppenheim J. J., Murphy W. J. (2000) Human endothelial cells express CCR2 and respond to MCP-1: direct role of MCP-1 in angiogenesis and tumor progression. Blood 96, 34–40 [PubMed] [Google Scholar]
- 18.Goede V., Brogelli L., Ziche M., Augustin H. G. (1999) Induction of inflammatory angiogenesis by monocyte chemoattractant protein-1. Int. J. Cancer 82, 765–770 [DOI] [PubMed] [Google Scholar]
- 19.Gálvez B. G., Genís L., Matías-Román S., Oblander S. A., Tryggvason K., Apte S. S., Arroyo A. G. (2005) Membrane type 1-matrix metalloproteinase is regulated by chemokines monocyte-chemoattractant protein-1/ccl2 and interleukin-8/CXCL8 in endothelial cells during angiogenesis. J. Biol. Chem. 280, 1292–1298 [DOI] [PubMed] [Google Scholar]
- 20.Ambati J., Anand A., Fernandez S., Sakurai E., Lynn B. C., Kuziel W. A., Rollins B. J., Ambati B. K. (2003) An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat. Med. 9, 1390–1397 [DOI] [PubMed] [Google Scholar]
- 21.Elner S. G., Strieter R. M., Elner V. M., Rollins B. J., Del Monte M. A., Kunkel S. L. (1991) Monocyte chemotactic protein gene expression by cytokine-treated human retinal pigment epithelial cells. Lab. Invest. 64, 819–825 [PubMed] [Google Scholar]
- 22.Aplin A. C., Ligresti G., Fogel E., Zorzi P., Smith K., Nicosia R. F. (2014) Regulation of angiogenesis, mural cell recruitment and adventitial macrophage behavior by Toll-like receptors. Angiogenesis 17, 147–161 [DOI] [PubMed] [Google Scholar]
- 23.Alexeev V., Yoon K. (2000) Gene correction by RNA-DNA oligonucleotides. Pigment Cell Res. 13, 72–79 [DOI] [PubMed] [Google Scholar]
- 24.Bennett D. C., Cooper P. J., Hart I. R. (1987) A line of non-tumorigenic mouse melanocytes, syngeneic with the B16 melanoma and requiring a tumour promoter for growth. Int. J. Cancer 39, 414–418 [DOI] [PubMed] [Google Scholar]
- 25.Sviderskaya E. V., Kallenberg D. M., Bennett D. C. (2010) Resource. The Wellcome Trust Functional Genomics Cell Bank: holdings. Pigment Cell Melanoma Res. 23, 147–150 [Google Scholar]
- 26.Satchi-Fainaro R., Mamluk R., Wang L., Short S. M., Nagy J. A., Feng D., Dvorak A. M., Dvorak H. F., Puder M., Mukhopadhyay D., Folkman J. (2005) Inhibition of vessel permeability by TNP-470 and its polymer conjugate, caplostatin. Cancer Cell 7, 251–261 [DOI] [PubMed] [Google Scholar]
- 27.Rogers M. S., Birsner A. E., D’Amato R. J. (2007) The mouse cornea micropocket angiogenesis assay. Nat. Protoc. 2, 2545–2550 [DOI] [PubMed] [Google Scholar]
- 28.Słominski A., Moellmann G., Kuklinska E., Bomirski A., Pawelek J. (1988) Positive regulation of melanin pigmentation by two key substrates of the melanogenic pathway, L-tyrosine and L-dopa. J. Cell Sci. 89, 287–296 [DOI] [PubMed] [Google Scholar]
- 29.Slominski A., Moellmann G., Kuklinska E. (1989) L-tyrosine, L-dopa, and tyrosinase as positive regulators of the subcellular apparatus of melanogenesis in Bomirski Ab amelanotic melanoma cells. Pigment Cell Res. 2, 109–116 [DOI] [PubMed] [Google Scholar]
- 30.Rohan R. M., Fernandez A., Udagawa T., Yuan J., D’Amato R. J. (2000) Genetic heterogeneity of angiogenesis in mice. FASEB J. 14, 871–876 [DOI] [PubMed] [Google Scholar]
- 31.Shyy Y. J., Hsieh H. J., Usami S., Chien S. (1994) Fluid shear stress induces a biphasic response of human monocyte chemotactic protein 1 gene expression in vascular endothelium. Proc. Natl. Acad. Sci. USA 91, 4678–4682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shyy Y. J., Wickham L. L., Hagan J. P., Hsieh H. J., Hu Y. L., Telian S. H., Valente A. J., Sung K. L., Chien S. (1993) Human monocyte colony-stimulating factor stimulates the gene expression of monocyte chemotactic protein-1 and increases the adhesion of monocytes to endothelial monolayers. J. Clin. Invest. 92, 1745–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adini A., Fainaru O., Udagawa T., Connor K. M., Folkman J., D’Amato R. J. (2009) Matrigel cytometry: a novel method for quantifying angiogenesis in vivo. J. Immunol. Methods 342, 78–81 [DOI] [PubMed] [Google Scholar]
- 34.Rao P., Hayden M. S., Long M., Scott M. L., West A. P., Zhang D., Oeckinghaus A., Lynch C., Hoffmann A., Baltimore D., Ghosh S. (2010) IkappaBbeta acts to inhibit and activate gene expression during the inflammatory response. Nature 466, 1115–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ueda A., Ishigatsubo Y., Okubo T., Yoshimura T. (1997) Transcriptional regulation of the human monocyte chemoattractant protein-1 gene. Cooperation of two NF-kappaB sites and NF-kappaB/Rel subunit specificity. J. Biol. Chem. 272, 31092–31099 [DOI] [PubMed] [Google Scholar]
- 36.Lee J. I., Burckart G. J. (1998) Nuclear factor kappa B: important transcription factor and therapeutic target. J. Clin. Pharmacol. 38, 981–993 [DOI] [PubMed] [Google Scholar]
- 37.Zhang L., Blackwell K., Altaeva A., Shi Z., Habelhah H. (2011) TRAF2 phosphorylation promotes NF-κB-dependent gene expression and inhibits oxidative stress-induced cell death. Mol. Biol. Cell 22, 128–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jobin C., Panja A., Hellerbrand C., Iimuro Y., Didonato J., Brenner D. A., Sartor R. B. (1998) Inhibition of proinflammatory molecule production by adenovirus-mediated expression of a nuclear factor kappaB super-repressor in human intestinal epithelial cells. J. Immunol. 160, 410–418 [PubMed] [Google Scholar]
- 39.Parry G. C., Martin T., Felts K. A., Cobb R. R. (1998) IL-1beta-induced monocyte chemoattractant protein-1 gene expression in endothelial cells is blocked by proteasome inhibitors. Arterioscler. Thromb. Vasc. Biol. 18, 934–940 [DOI] [PubMed] [Google Scholar]
- 40.Wang X. C., Jobin C., Allen J. B., Roberts W. L., Jaffe G. J. (1999) Suppression of NF-kappaB-dependent proinflammatory gene expression in human RPE cells by a proteasome inhibitor. Invest. Ophthalmol. Vis. Sci. 40, 477–486 [PubMed] [Google Scholar]
- 41.Tobe M., Isobe Y., Tomizawa H., Nagasaki T., Takahashi H., Hayashi H. (2003) A novel structural class of potent inhibitors of NF-kappa B activation: structure-activity relationships and biological effects of 6-aminoquinazoline derivatives. Bioorg. Med. Chem. 11, 3869–3878 [DOI] [PubMed] [Google Scholar]
- 42.Hong K. H., Ryu J., Han K. H. (2005) Monocyte chemoattractant protein-1-induced angiogenesis is mediated by vascular endothelial growth factor-A. Blood 105, 1405–1407 [DOI] [PubMed] [Google Scholar]
- 43.Leonard E. J., Yoshimura T. (1990) Human monocyte chemoattractant protein-1 (MCP-1). Immunol. Today 11, 97–101 [DOI] [PubMed] [Google Scholar]