

Abstract
An R345W mutation in the N-glycoprotein, fibulin-3 (F3), results in inefficient F3 folding/secretion and higher intracellular F3 levels. Inheritance of this mutation causes the retinal dystrophy malattia leventinese. N-Linked glycosylation is a common cotranslational protein modification that can regulate protein folding efficiency and energetics. Therefore, we explored how N-glycosylation alters the protein homeostasis or proteostasis of wild-type (WT) and R345W F3 in ARPE-19 cells. Enzymatic and lectin binding assays confirmed that WT and R345W F3 are both primarily N-glycosylated at Asn249. Tunicamycin treatment selectively reduced R345W F3 secretion by 87% (vs. WT F3). Genetic elimination of F3 N-glycosylation (via an N249Q mutation) caused R345W F3 to aggregate intracellularly and adopt an altered secreted conformation. The endoplasmic reticulum (ER) chaperones GRP78 (glucose-regulated protein 78) and GRP94 (glucose-regulated protein 94), and the ER lectins calnexin and calreticulin were identified as F3 binding partners by immunoprecipitation. Significantly more N249Q and N249Q/R345W F3 interacted with GRP94, while substantially less N249Q and N249Q/R345W interacted with the ER lectins than their N-glycosylated counterparts. Inhibition of GRP94 ATPase activity reduced only N249Q/R345W F3 secretion (by 62%), demonstrating this variant’s unique reliance on GRP94 for secretion. These observations suggest that R345W F3, but not WT F3, requires N-glycosylation to acquire a stable, native-like structure.—Hulleman, J. D., Kelly, J. W. Genetic ablation of N-linked glycosylation reveals two key folding pathways for R345W fibulin-3, a secreted protein associated with retinal degeneration.
Keywords: EFEMP1, endoplasmic reticulum, malattia leventinese, unfolded protein response
Age-related macular degeneration (AMD) is the most common cause of irreversible blindness in the Western world (1, 2). AMD is characterized by the extracellular deposition of lipids and proteins (drusen), retinal pigment epithelium (RPE) apoptosis, and complement activation [reviewed in (3)]. Although AMD affects a significant (and growing) portion of the population, its pathogenesis is still poorly understood, primarily as a result of a variety of age-related cellular changes, environmental factors, and complex genetics. However, studying early-onset inherited retinal degenerative diseases with phenotypic similarities to AMD has provided mechanistic insights that potentially underlie AMD (4, 5). One such familial disease is malattia leventinese (ML), also known as Doyne honeycomb retinal dystrophy or familial dominant drusen. ML is a macular dystrophy caused by a single Arg345Trp (R345W) mutation in fibulin-3 (F3). Like those with AMD, ML patients develop sub-RPE drusen, and RPE atrophy and vacuolization (6, 7). Mice expressing R345W F3 display these same symptoms and also show signs of complement C3 activation (8–10). For these reasons, we decided to pursue a deeper understanding of how RPE cells handle wild-type (WT) and R345W F3, which should provide an experimental framework to better understand the etiology of ML and AMD.
F3 is a multidomain, disulfide-rich, secreted N-glycoprotein that is involved in the formation of the extracellular matrix [reviewed in (11)]. F3 is composed of a series of tandem calcium-binding epidermal growth factor (cbEGF) domains, a predicted N-linked glycosylation site at Asn249 (in cbEGF domain 3), and a C-terminal fibulin-type domain [Fig. 1A; reviewed in (12)]. The R345W mutation that disrupts F3 protein folding and causes ML is located within the sixth EGF domain [Fig. 1A (13, 14)]. As a result of this apparent misfolding, R345W F3 is inefficiently secreted from a variety of human cell lines and exhibits higher intracellular steady-state levels than WT F3 (13–17).
Figure 1.
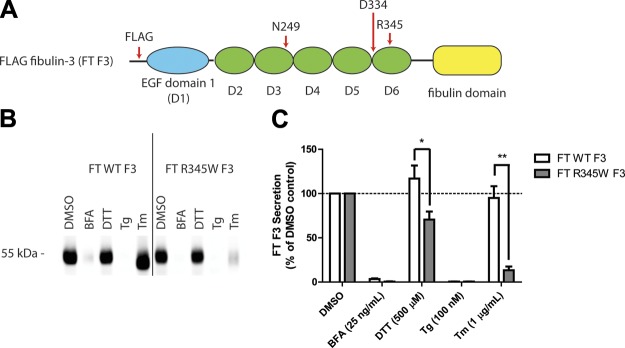
ER stressors differentially affect F3 secretion. A) Schematic of FT F3 used in the studies. FT F3 contains an N-terminal FT, 1 atypical EGF domain (blue oval) followed by 5 canonical cbEGF domains (green ovals), and a C-terminal fibulin domain (yellow rectangle). Specific residues of interest are highlighted. B) ARPE-19 cells expressing WT or R345W F3 were treated with the ER stressors: BFA (25 ng/ml), DTT (500 μM), Tg (100 nM), or Tm (1 μg/ml) for 24 h. F3 was IP’d from the conditioned media using M1 anti-FLAG agarose beads and analyzed by Western blot analysis with an anti-F3 antibody. Representative data are shown of ≥4 experiments. C) Quantification of conditioned media IP experiments (*P < 0.05, **P < 0.01, t test). Note migration changes in WT and R345W F3 after treatment with Tm, indicating effective inhibition of F3 N-linked glycosylation.
When misfolded proteins accumulate within the endoplasmic reticulum (ER) at levels that exceed the ER protein homeostasis network’s capacity to clear them, 1 or more protein folding sensors within the ER (IRE-1 [inositol requiring enzyme 1], activating transcription factor [ATF]-6, or PERK [protein kinase RNA-like ER kinase]) is activated. This triggers activation of one or more of the unfolded protein response [UPR; reviewed in (18)] stress-responsive signaling pathways. Global UPR activation leads to the generation of 3 transcription factors (XBP1 [X-box binding protein-1] spliced, cleaved ATF-6 and ATF-4), which in turn up-regulate profolding chaperones/enzymes, ER-associated degradation (ERAD) components, and can cause translational attenuation, which lowers the protein load processed by the ER protein homeostasis network [reviewed in (18)]. Many genetic ocular diseases are associated with UPR activation, including autosomal-dominant retinitis pigmentosa [rhodopsin mutations (19, 20)] and ocular anterior segment dysgenesis [COL4A1 mutations (21)]. Roybal et al. (17) have shown that overexpression of R345W F3 (and to a lesser extent, WT F3) in ARPE-19 cells causes activation of the IRE-1 and ATF-6 arms of the UPR. However, this is likely due to the supraphysiologic buildup of the recombinant F3 protein, because other cell culture (13) and in vivo studies on F3 (8) did not detect UPR activation. Nonetheless, because the UPR controls protein homeostasis in the secretory pathway, UPR activation will likely alter F3 folding and secretion vs. degradation decisions within the ER (22).
Consistent with this hypothesis, we have previously demonstrated that inefficient R345W F3 secretion can be partially rescued by a variety of perturbations, including selective activation of the PERK arm of the UPR, growth temperature reduction, and translational attenuation (13, 15). These protein homeostasis network adaption strategies may function by increasing the protein homeostasis network component to F3 stoichiometry (13, 15). Additionally, in an unbiased chemical screen, we identified phorbol myristate 12-acetate as a compound that preferentially reduced R345W F3 secretion, apparently acting through activation of the ATF-6 arm of the UPR (16).
UPR activation is only one mechanism that the cell uses to promote proper protein quality control [i.e., to make the folding and secretion vs. degradation decision in the early secretory pathway; reviewed in (22)] during stress. Another pathway that the cell utilizes to achieve protein homeostasis is to glycosylate difficult-to-fold proteins. Most secreted or trafficked proteins are co- or posttranslationally modified through the addition of oligosaccharide chains on either specific Ser/Thr residues (O-linked glycosylation) or on specific Asn residues (N-linked glycosylation) within the Asn-Xxx-Thr/Ser recognition sequence [or sequon, where Xxx is any amino acid but Pro; reviewed in (23)]. Although the role of O-linked glycosylation in proteostasis remains unclear, N-linked glycosylation, specifically the en bloc transfer of a triantennary Glc3Man9(GlcNAc)2 oligosaccharide to the Asn within the sequon, is known to provide an extrinsic mechanism by which ER oligosaccharide binding chaperone pathways (i.e., the calnexin [CNX] and calreticulin [CRT] pathways) engage nascent polypeptide chains to assist in their folding [including disulfide bond formation (24)] and trafficking [reviewed in (25)]. It is noteworthy that the N-linked glycan also provides a means to achieve protein quality control through mannose trimming, which leads to ERAD [reviewed in (25)]. N-Glycosylation can also intrinsically accelerate N-glycoprotein folding (26) and reduce protein aggregation (26–28).
Given the extrinsic importance of N-linked glycosylation to enable N-glycoprotein clients to engage the dedicated N-glycoprotein proteostasis network folding and degradation pathways, as well as the intrinsic effects of N-glycosylation on folding energetics, we explored whether F3 protein homeostasis was altered by the pharmacologic inhibition or genetic ablation of N-linked glycosylation. Herein we demonstrate that Asn249-linked N-glycosylation of R345W F3 is an important modification that strongly affects F3’s folding fitness and intracellular steady-state levels, as well as dictates F3’s ER protein homeostasis network interactome. Collectively, our observations reveal that R345W F3, and to a lesser extent WT F3, requires N-glycosylation to acquire and maintain a stable, native-like structure.
MATERIALS AND METHODS
Construct generation
An N-terminal FLAG-tagged version of F3 was constructed by replacing the native signal sequence of F3 with a preprotrypsin signal sequence (amino acids: MSALLILALVGAAVA), followed by a FLAG tag sequence (amino acids: DYKDDDDK), followed by a leucine and amino acids 28–493 of F3, creating FLAG-tagged (FT) F3 (Fig. 1A). This FT construct was then introduced into the pENTR1A entry vector (Life Technologies, Carlsbad, CA, USA). Site-directed mutagenesis was performed using the QuikChange method (Agilent, Santa Clara, CA, USA). The sequences of all constructs were verified.
Cell culture
Human retinal pigment epithelium cells (ARPE-19; American Type Culture Collection CRL-2302; ATCC, Manassas, VA, USA) were routinely maintained at 37°C, 5% CO2 in DMEM/F-12 media (Corning, Corning, NY, USA) supplemented with 10% FBS (Omega Scientific, Tarzana, CA, USA), and 1% penicillin/streptomycin and glutamine (Corning) (denoted as complete media). Human embryonic kidney cells that stably express the tet-repressor (TREx-293, Life Technologies) were routinely maintained at 37°C, 5% CO2 in DMEM (Corning) supplemented with 10% FBS (Omega Scientific), and 1% penicillin/streptomycin and glutamine (Corning). Passaging was accomplished using 0.25% trypsin/EDTA (Corning).
Adenovirus production
Constructs used in these studies were shuttled from the pENTR1A entry vector into the pAD-CMV-V5-DEST vector by an LR Clonase II reaction (Life Technologies). Adenovirus encoding for each FT construct was produced as described previously (16). Virus DNA was quantified by the AdenoX qPCR titer kit (Clontech, Mountain View, CA, USA), and levels of FT F3 expression after infection were verified by quantitative PCR (qPCR). The relative amounts of virus used in experiments were normalized according to transcript levels as determined by qPCR.
Quantitative PCR
Changes in transcript levels of stress-responsive genes or F3 were assessed by qPCR as described previously (16). In brief, RNA was extracted from ARPE-19 cells using the RNeasy kit (Qiagen, Germantown, MD, USA) according to the manufacturer’s protocol, and then 500 ng RNA was reverse transcribed using the Quantitect Reverse Transcription Kit (Qiagen). The resulting cDNA was diluted with DNase/RNase-free water before being used for amplification. qPCR primers [the same as those listed elsewhere (16)] were selected using Primer3 software (http://frodo.wi.mit.edu/). cDNA was amplified using Sybr Green Fast Master Mix (Roche, Piscataway, NJ, USA) and quantified using an Applied Biosystems 7900 qPCR machine (Life Technologies). The data were then analyzed using Data Assist software (Life Technologies).
Immunoprecipitation
ARPE-19 cells were plated at a density of 100,000 cells/cm2, allowed to attach to tissue culture dishes for 48 h, and were then infected for an additional 48 h with adenovirus encoding for FT F3 at a multiplicity of infection (MOI) of 5. It is noteworthy that this level of infection (∼2.5 to 5 times endogenous F3 levels) does not activate the UPR in FT WT F3 vs. FT R345W F3-expressng cells as assessed by qPCR of candidate genes (Supplemental Fig. 1A–D). After 48 h of infection, media were changed with fresh, complete media or serum-free Opti-MEM (Life Technologies). After an additional 24 h, media were removed and FT F3 was immunoprecipitated (IP’d) overnight at 4°C using anti-FLAG M1 antibody conjugated to agarose beads (Sigma-Aldrich, St. Louis, MO, USA). Soluble intracellular FT F3 was IP’d by first lysing cells in RIPA buffer (50 mM Tris, pH 7.4, 150 mM NaCl2, 1% Triton X-100 [v/v], 0.1% SDS [w/v], 0.5% sodium deoxycolate [w/v]), followed by centrifugation. The soluble protein fraction was quantified using a bicinchoninic acid assay (Thermo Fisher Scientific, Waltham, MA, USA), and equal amounts of total protein were used for the IP. The beads were then washed 3 times with RIPA buffer supplemented with 1–3 mM CaCl2. Samples were eluted in 1× SDS-PAGE buffer by boiling for 5 min without reductant. A portion of the eluted sample (20 μl of a 30 μl elution) was transferred to a new tube containing β-mercaptoethanol. Samples were boiled again for 5 min before SDS-PAGE followed by Western blot analysis. Equal amounts of total transferred protein were confirmed by Ponceau S and/or housekeeping protein levels.
Denaturing Western blot analysis
Denatured protein samples were separated on a 10% Tris-Gly SDS-PAGE gel (Life Technologies) and transferred to a nitrocellulose membrane. Membranes were probed with a rabbit anti-F3 antibody (1:500; ProSci, Poway, CA, USA), mouse anti–β-actin (1:10000; Sigma-Aldrich), rabbit anti-GRP94 (glucose-regulated protein 94; 1:1500; GeneTex, Irvine, CA, USA), rabbit anti-CRT (1:500; GeneTex), rabbit anti-CNX (1:1000; Enzo Life Sciences, Farmingdale, NY, USA), or mouse anti-GRP78 (glucose-regulated protein 78; 1:500; Santa Cruz, Santa Cruz, CA, USA). Blots were then imaged with a LI-COR Odyssey infrared imager (LI-COR, Lincoln, NE, USA) using a goat anti-mouse 680 nm or goat anti-rabbit 800 nm secondary antibody (1:10,000; LI-COR). All Western blot quantification was performed using LI-COR software.
Detergent insoluble F3 was analyzed by infection of ARPE-19 cells at an MOI of 25 for 48 h. Cells were collected and lysed in mammalian lysis buffer supplemented with a complete protease inhibitor cocktail (Roche). After lysis, the insoluble debris was isolated by centrifugation (21,000 × g, 10 min, 4°C). The insoluble material was then resuspended in 6× Laemmli buffer containing 12% SDS and boiled for 20 min. A portion of the resuspended insoluble material was loaded onto a TrisGly SDS-PAGE gel followed by Western blot analysis.
Glycosylation analysis
To test whether FT F3 was glycosylated via asparagines (N-linked) or serines/threonines (O-linked), media aliquots from cells expressing FT F3 were treated with PNGaseF (New England Biolabs, Ipswich, MA, USA) or neuraminidase/O-glycosidase, respectively. In brief, serum-free media samples containing FT F3 were denatured for 10 min at 100°C in 1× denaturing buffer, after which Nonidet P-40 (1% [v/v] final concentration) and G7 buffer (1×) were added. Samples were digested with PNGaseF (1 μl), neuraminidase (1 μl), or neuraminidase and O-glycosidase (1 μl each) for 1 h at 37°C, followed by boiling in reducing SDS-PAGE buffer for 5 min. Samples were then analyzed by Western blot analysis.
Concanavalin A binding
Concanavalin A (ConA)–conjugated agarose beads (Vector Laboratories, Burlingame, CA, USA) were used to enrich for glycosylated F3. In brief, ConA agarose beads were washed 3 times with 1 ml of wash buffer (10 mM HEPES, pH 7.4, 250 mM NaCl, 1 mM CaCl2, 1 mM MgCl2) before applying conditioned media to the beads, followed by rotation at 4°C overnight. ConA beads were washed 3 times with wash buffer and eluted in 1× SDS-PAGE buffer by boiling for 5 min without reductant. A portion of the eluted sample (20 μl of a 30 μl elution) was transferred to a new tube containing 140 mM β-mercaptoethanol (final concentration). Samples were boiled again for 5 min before SDS-PAGE followed by Western blot analysis.
Blue native PAGE
To assess the size of secreted and intracellular FT F3 variants under native, nondenaturing conditions, blue native (BN) PAGE was used (29). ARPE-19 cells were infected with FT F3 for 48 h, after which the media was replaced with Opti-MEM. After 24 h, conditioned media aliquots were taken, and cells were washed once in HBSS (Corning), scraped from the plate, and lysed by freeze/thaw in HBSS supplemented with protease inhibitors (Complete, mini, EDTA-free; Roche). Protein concentrations were measured by a bicinchoninic acid assay, and samples were diluted with BN PAGE sample buffer (5% [v/v] glycerol, 0.25% [w/v] Coomassie Blue G-250, 0.01% [w/v] Ponceau S). A total of ∼50 μg was loaded per lane on a NativePAGE 4% to 16% gradient Bis-Tris gel (Life Technologies). Gels were run for 30 min at 100 V at room temperature and then run at 200 V for 3 h at 4°C using a cathode buffer lacking Coomassie Blue. Proteins were then blotted onto PVDF and probed with either a mouse anti-FLAG M2 antibody (1:2000; Agilent) or a rabbit anti-F3 antibody (1:500; ProSci), followed by a horseradish peroxidase–conjugated secondary antibody (1:15,000; Thermo Fisher Scientific) and substrate addition (Luminata Forte; Millipore, Billerica, MA, USA). F3 molecular weight was estimated on the basis of NativeMark protein standards (Life Technologies).
Cross-linking
To determine F3 binding partners, we cross-linked proteins in the cell using the cell-permeable, thiol-cleavable primary amine cross-linker dithiobis(succinimidyl) propionate (DSP; G-Biosciences, St. Louis, MO, USA). Cells were treated with 500 μM DSP for 30 min at room temperature, followed by quenching of the cross-linker with an excess of Tris-HCl, pH 8.8, for 15 min at room temperature. After cross-linking, cells were lysed in RIPA buffer supplemented with protease inhibitors, and FT F3 was IP’d as described above.
RESULTS
Tunicamycin treatment significantly reduces R345W F3 secretion in comparison to other ER stressors
Given the established importance of N-linked glycosylation in regulating disulfide bond formation, protein folding/secretion, degradation, and protein stability (26–28), we tested whether pharmacologic inhibition of this cotranslational modification would perturb F3 proteostasis. ARPE-19 cells were infected with adenovirus encoding a FT WT or R345W F3. We titered the virus such that the levels of R345W F3 used did not elicit differential activation of the UPR compared with WT F3 (Supplemental Fig. 1A–C). Infected cells were then treated with ER stressors for 24 h. Pharmacologic inhibition of N-linked glycosylation, achieved through administration of tunicamycin (Tm) (30), caused a significant reduction in R345W F3 secretion (down to 13% of DMSO treated R345W F3 levels; Fig. 1B, C) without affecting WT F3 secretion (maintained 95% of DMSO treated WT F3 levels; Fig. 1B, C). These relative secretion differences after Tm treatment were not simply due to differential toxicity because viability measurements showed no significant differences between WT vs. R345W F3-expressing cells treated with Tm for 24 h (Supplemental Fig. 1E).
Because Tm inhibits the N-linked glycosylation of all newly synthesized glycoproteins (not just F3), it causes substantial protein misfolding within the ER and global activation of the UPR (Supplemental Fig. 1A–C). To determine whether the reduction in R345W F3 secretion after Tm treatment was due to UPR activation, inhibition of N-linked glycosylation, or both, we treated F3-expressing cells with a series of distinct ER stressors which disrupt protein trafficking (brefeldin A [BFA]), disulfide bond formation (DTT), and calcium influx into the ER (thapsigargin [Tg]). Treatment with BFA and Tg, similar to Tm, resulted in substantial activation of the UPR in both WT and R345W F3-expressing cells (Supplemental Fig. 1A–C). In contrast, DTT did not sustainably activate the UPR over 24 h, most likely as a result of the ability of the ER to rapidly reoxidize itself (31). However, DTT did cause a weak, transient activation of the UPR after a 1 h treatment (Supplemental Fig. 2A). Surprisingly, prolonged treatment (24 h) with these chemical ER stressors did not substantially reduce cellular viability in 3 out of 4 cases, the exception being BFA treatment (Supplemental Fig. 1E).
Intriguingly, treatment with low levels of DTT (500 μM) over the 24 h period had a slight enhancing effect on WT F3 (117% of the DMSO control), while modestly, but significantly, reducing R345W F3 secretion (71% of DMSO control; Fig. 1B, C). Treatment with BFA and Tg all but prevented WT and R345W secretion from ARPE-19 cells (Fig. 1B, C) and elicited a similar activating effect on the distinct arms of the UPR (Supplemental Fig. 1A–C). Thus, it is apparent that the Tm-mediated reduction in R345W F3 secretion is not simply due to global UPR activation and the consequences thereof. Instead, it is possible that the Tm-mediated selective reduction in R345W F3 secretion is due either to the inability of glycosylation-deficient R345W F3 to effectively use the CNX and/or CRT N-glycoprotein folding/degradation pathways or due to the effects of Tm on other glycoproteins that F3 relies on for secretion.
All the ER stressors used above elevated the intracellular steady-state concentrations of F3, ranging from an average of 1.4-fold (WT F3, DTT treatment) to 5.9-fold (R345W F3, Tm treatment) (Fig. 2A, B). Only after Tm treatment did WT and R345W F3 intracellular levels diverge significantly relative to each other (Fig. 2A, B). Given that BFA and Tg all but prevent F3 secretion, it is intriguing that so much R345W F3 is retained intracellularly only after Tm treatment. This suggests that BFA and Tg treatment not only prevent F3 secretion but also enhance its degradation, potentially through ERAD and/or through suppressing F3 transcription (Supplemental Fig. 1D).
Figure 2.

ER stressors cause higher intracellular steady-state F3 levels. A) ARPE-19 cells expressing WT or R345W F3 were treated with BFA (25 ng/ml), DTT (500 μM), Tg (100 nM), or Tm (1 μg/ml) for 24 h. Cells were then collected and lysed, and F3 was then IP’d from the cell lysates using anti-FLAG M1 agarose beads and analyzed by Western blot analysis with an anti-F3 antibody. Representative data are shown of ≥4 experiments. B) Quantification of lysate IP (*P < 0.05, t test).
The major secreted F3 species is N- but not O-glycosylated
Given that Tm treatment reduces R345W F3 secretion (Fig. 1B, C) and increases R345W F3 intracellular steady-state levels (Fig. 2A, B), we decided to biochemically analyze F3 glycosylation using a series of glycosidases and glycoprotein binding assays. We focused on the 55 kDa secreted form of F3 because it is the predominant species secreted from ARPE-19 cells (Supplemental Fig. 2B). Treatment of conditioned media from ARPE-19 cells expressing WT or R345W F3 with PNGaseF resulted in a faster migrating species (Fig. 3A), indicating the presence of N-linked glycosylation, most likely at N249, as suggested elsewhere (32), and by the UniProtKB database (UniProtKB ID: Q12805). Treatment of the same conditioned media with neuraminidase (alone) or with neuraminidase and O-glycosidase did not alter the migration of the primary, 55 kDa F3 species, indicating that this isoform is not O-glycosylated (Fig. 3A). Additionally, we biochemically analyzed the glycosylation pattern of conditioned media from Tm-treated ARPE-19 cells expressing WT or R345W F3. F3, which was secreted while N-linked glycosylation was inhibited by Tm, was resistant to all 3 glycosidases used (Supplemental Fig. 2D), corroborating the hypothesis that the migration change in Fig. 3A after PNGaseF digestion is due to N-linked glycosylation.
Figure 3.

The major F3 isoform secreted from ARPE-19 cells is N-glycosylated but not O-glycosylated. A) Conditioned media aliquots from ARPE-19 cells were denatured and treated with PNGaseF (1 μl), neuraminidase (1 μl), or neuraminidase (1 μl) and O-glycosidase (1 μl), followed by Western blot analysis. B) Conditioned media aliquots from ARPE-19 cells expressing F3 were either IP’d with anti-FLAG M1 agarose beads or with ConA agarose beads (25 μl slurry). Representative data are shown of ≥3 experiments.
As a confirmatory assay for the presence of glycosylated F3, we also tested the ability of F3 to bind to agarose-conjugated ConA. ConA is a lectin that binds to terminal mannosyl and glucosyl residues of N- and O-linked glycoproteins. Both WT and R345W F3 originating from vehicle-treated ARPE-19 cells bound to ConA, but neither WT nor R345W F3 was able to bind to ConA beads when N-linked glycosylation was inhibited with Tm (Fig. 3B). These results, in combination with those comprising Fig. 3A, indicate that N-linked glycosylation, most likely at N249, is the primary glycosylation modification of the predominant 55 kDa secreted F3 species.
Recently, Djokic et al. (32) biochemically characterized the proteolysis and glycosylation of a series of secreted short fibulin proteins, including WT F3 (but not R345W F3), which were purified from the conditioned media of human embryonic kidney (HEK)-293 cells. Although this was an important study, HEK cells do not endogenously express F3, and thus it is quite possible that F3 is differentially glycosylated in HEK-based vs. ARPE-19-based cells, as has been noted in the past with other proteins of interest (33). Indeed, we have observed that the relative abundance of F3 isoforms is apparently different in conditioned media originating from ARPE-19 vs. HEK cells expressing WT F3 (Supplemental Fig. 2B, C).
Genetic elimination of F3 N-linked glycosylation does not reduce the extent of R345W F3 secretion from ARPE-19 cells
To follow up on the general pharmacologic inhibition of F3 N-linked glycosylation by Tm, we generated variants of WT and R345W F3 that cannot be N-glycosylated, i.e., N249Q and N249Q/R345W F3, respectively. Additionally, for comparative purposes, we generated a folding-defective mutant of F3 in which the first Asp of a single cbEGF domain (a highly conserved residue within all cbEGF domains) was mutated to a Gly (D334G; Fig. 1A). Elimination of this conserved Asp mitigates Ca+2 binding, which prevents domain folding (34, 35). This variant enables us to assess and compare the characteristics of F3 when it is unable to fold in the ER.
We hypothesized that elimination of F3 N-linked glycosylation would affect WT and R345W F3 secretion analogously to Tm treatment; namely that N249Q F3 would fold and be secreted as efficiently as WT F3 and that N249Q/R345W F3 would be inefficiently secreted and accumulate intracellularly at higher steady-state levels. The N249Q mutation caused a significant molecular weight migration change in conditioned media and lysates, indicating a lack of an N-linked glycan at Asn249 (Fig. 4A, C). However, surprisingly, N249Q F3 was more efficiently secreted than WT F3 (191% of WT F3 levels; Fig. 4A, B). Even more surprising was that N249Q/R345W F3, while still inefficiently secreted compared to WT F3 or N249Q F3, was secreted as efficiently as R345W F3 (100% of R345W F3 levels; Fig. 4A, B). In contrast, the D334G mutation in either the WT or R345W F3 background completely prevented any detectible secretion of F3 (Fig. 4A, B).
Figure 4.

Genetic elimination of N-linked glycosylation at Asn249 does not hinder F3 secretion. A) Conditioned media aliquots from ARPE-19 cells expressing WT, N249Q, D334G, R345W, N249Q/R345W, or R345W/D334G F3 were IP’d for F3, followed by Western blot analysis. B) Quantification of secreted F3 protein in the Western blot tests in (A), n ≥ 3 (C) Corresponding cell lysates from (A) were IP’d for intracellular F3. D) Quantification of intracellular F3 in the Western blot tests in (C), n ≥ 5. E) Low levels of N249Q/R345W F3 slightly activate the UPR. UPR-responsive transcripts were measured in ARPE-19 cells after 72 h of F3 expression. Data shown are a combination of 3 independent experiments.
While having minimal detrimental effects on WT F3 secretion, the N249Q mutation did elevate intracellular steady-state F3 levels in both the WT and R345W F3 background (211% and 349% of WT and R345W F3 levels, respectively; Fig. 4C, D). However, these levels are much lower than the intracellular levels of the D334G mutants, which accumulate at levels >11-fold over WT and R345W F3 levels (Fig. 4C, D). The elevated intracellular levels of N249Q/R345W F3 correlate with an increased likelihood of causing F3-dependent UPR activation (Fig. 4E). Although these levels did not reach statistical significance, asparagine synthetase transcripts (PERK activation) were elevated 1.7-fold, ERDJ4 (DnaJ homolog, subfamily B, member 9; IRE1 activation) was elevated 1.7-fold, and GRP78 (ATF-6 activation) was elevated 1.8-fold (Fig. 4E), consistent with higher N249Q/R345W F3 steady-state levels (Fig. 4C, D). These N249Q-dependent alterations of R345W F3 protein homeostasis were not simply due to differences in expression levels (Supplemental Fig. 3A) or cell viability (Supplemental Fig. 3B, C).
Given our surprise that genetic elimination of N-linked glycosylation did not reduce R345W F3 secretion levels from ARPE-19 cells (Fig. 4A, B), we decided to explore how cells of different origin handled the same F3 variants. Introduction of the N249Q F3 variants into TREx-293 cells (an HEK-based cell line) yielded similar levels of secreted N249Q F3 vs. WT F3 to what we observed in ARPE-19 cells (on average 130% of WT F3 levels; Supplemental Fig. 4A, C). However, secreted N249Q/R345W F3 levels originating from TREx-293 cells were substantially reduced when compared to R345W F3 (on average 26% of R345W F3 levels, Supplemental Fig. 4A, C). Intracellular levels were similar in ARPE-19 and TREx-293 cells, with the N249Q mutation increasing steady-state levels of both WT and R345W F3 (Supplemental Fig. 4B, C). The D334G F3 mutants acted similarly in TREx-293 and ARPE-19 cells, with no detectible protein secretion and a large accumulation of intracellular F3 (Supplemental Fig. 4A, B). Thus, the only key difference between how ARPE-19 and TREx-293 cells handle F3 is that N249Q/R345W F3 is poorly secreted in TREx-293 cells (Supplemental Fig. 4A).
N249Q/R345W F3 adopts an altered structure in conditioned media and intracellularly under native conditions
Because the absence of N-linked glycosylation can affect protein aggregation (26–28) and disulfide bond formation (24), we assessed how genetic ablation of F3 N-linked glycosylation affected these 2 characteristics in conditioned media and intracellular lysates, respectively. Under denaturing conditions, we did not observe any difference in the migration of any of the intracellular or secreted F3 variants which would indicate protein aggregation (Fig. 4A, C). We therefore decided to use BN PAGE as a nondenaturing approach to assess for native F3 protein aggregates (29).
BN PAGE analysis of conditioned media from cells expressing WT, N249Q, or R345W F3 showed the presence of a single band that migrated at an approximate molecular weight of 110 kDa, the predicted size of a F3 dimer (Fig. 5A). There were no obvious differences in the migration pattern of these 3 variants, indicating that they are likely folded similar to one another (Fig. 5A). In contrast, we were unable to detect secreted N249Q/R345W F3 under identical conditions using 2 different antibodies (Fig. 5A and Supplemental Fig. 4D). On the basis of denaturing SDS-PAGE results, we know that N249Q/R345W F3 is secreted as efficiently as R345W F3, but it appears that N249Q/R345W F3 adopts an alternate conformation under native conditions, one that is distinct from WT, N249Q, or R345W F3.
Figure 5.

FT N249Q/R345W F3 adopts an altered conformation extracellularly and intracellularly under native conditions. A) Conditioned media from ARPE-19 cells expressing F3 variants were run on a BN PAGE gel under nondenaturing, nonreducing conditions, followed by Western blot analysis. B) ARPE-19 cells expressing WT, N249Q, D334G, R345W, N249Q/R345W, or D334G/R345W F3 were lysed, and proteins were separated by BN PAGE (under nondenaturing and nonreducing conditions), followed by Western blot analysis. C) N249Q/R345W F3 has a high propensity to form large, insoluble aggregates. ARPE-19 cells expressing F3 variants were lysed and the insoluble material was resuspended in 6× Laemmli buffer and run under nonreducing conditions. Bands shown originate from the SDS-PAGE stacking gel. Representative data of 2 independent experiments are shown. D) Conditioned media aliquots from ARPE-19 cells expressing F3 variants were IP’d and run under nonreducing SDS-PAGE conditions. Representative data of 3 independent experiments.
A similar nondenaturing analysis of cell lysates from ARPE-19 cells expressing WT, R345W or the N249Q and D334G variants demonstrated diffuse staining across a molecular weight range of ∼242 to 1048 kDa, indicating that intracellular N249Q/R345W, D334G, and D334G/R345W F3 misfold and aggregate under native conditions (Fig. 5B). Such aggregates were not detected in lysates from WT, N249Q, or R345W F3-expressing cells (Fig. 5B). Furthermore, we assayed for classic detergent-insoluble aggregates in F3-expressing cells by resuspending the insoluble portion of the cell lysates with 6× Laemmli buffer and running them under denaturing conditions. Under these conditions, N249Q/R345W F3 had the highest likelihood of forming large aggregates that were incapable of entering the SDS-PAGE gel (Fig. 5C).
We next assessed whether the secreted glycosylation deficient F3 showed signs of disulfide bonding problems to determine if that could be the reason for our inability to detect the protein under native conditions. Thus, we analyzed secreted F3 variants by SDS-PAGE under nonreducing conditions. We did not observe any differences in WT vs. R345W F3 migration under nonreducing conditions, as we have noted previously (13) (Fig. 5D); nor did we observe any difference in the apparent molecular weight of N249Q vs. N249Q/R345W F3 under identical conditions (Fig. 5D). These results suggest that the altered N249Q/R345W F3 native conformation is likely not due to aberrant disulfide bonding.
Glycosylation-deficient F3 interacts strongly with the major ER chaperone GRP94
Given our result that genetic ablation of F3 N-linked glycosylation has little effect on the efficiency of F3 secretion (but exhibits an apparently large effect on N249Q/R345W F3 stability based on its aggregation propensity), we hypothesized that F3 can utilize certain folding pathways in the ER that are dependent on the presence or absence of F3 N-glycosylation. To this end, we used cross-linking, IP, and Western blot analysis to identify F3 interacting partners.
We identified 3 new F3 interacting proteins in ARPE-19 cells: the ER chaperone GRP94, and the ER lectin chaperones CNX and CRT. GRP94 interacted weakly with WT F3, had a stronger interaction with R345W F3, and interacted more strongly with both N249Q and N249Q/R345W F3 than their respective WT and R345W F3 counterparts (Fig. 6A, B). Concomitantly, the association of N249Q and N249Q/R345W F3 with CNX and CRT decreased. Interestingly, the interaction of F3 with CRT is largely dependent on N-glycosylation, whereas the F3 interaction with CNX is less dependent on N-glycosylation (Fig. 6A). Thus, the N-glycosylation–deficient variants, especially N249Q/R345W F3, appear to rely more strongly on a GRP94-dependent folding pathway in the absence or reduction of lectin binding. These results reaffirm that N249Q/R345W F3 adopts an alternate conformation that is distinct from WT, N249Q, and R345W F3.
Figure 6.

Nonglycosylated F3 associates more strongly with GRP94 and less strongly with CNX and CRT. A) ARPE-19 cells expressing F3 variants were cross-linked and lysed, and F3 was IP’d, followed by Western blot analysis. In order to robustly observe the F3 interaction with the lectins, an MOI of 25 was necessary. Inputs represent 2% of the IP’d material. Similar relative amounts of F3 were observed in the MOI 5 and 25 inputs. Representative data of 3 independent experiments. B) Quantification of GRP94 binding to F3 by LI-COR. Data are presented as the ratio of GRP94/F3 and are normalized to the GRP94/F3 ratio of WT F3, n = 3 (*P < 0.05, t test).
Chemical inhibition of GRP94 ATPase activity selectively prevents N249Q/R345W F3 secretion
It appears that F3 can use distinct folding pathways that depend on F3’s N-glycosylation state. To determine how dependent N-glycosylation deficient F3 is on the GRP94 pathway, we used a chemical approach to reduce GRP94 activity. We treated F3-expressing cells with 17-N-allylamino-17-demethoxygeldanamycin (17-AAG, 1 μM), an inhibitor of HSP90 and GRP94 ATPase activity (36). 17-AAG had no detrimental effect on WT, N249Q, or R345W F3 secretion (104%, 120%, and 128% of DMSO-treated N249Q and R345W F3 levels, respectively; Fig. 7A, B) but greatly reduced the secretion of N249Q/R345W F3 (38% of DMSO-treated N249Q/R345W F3 levels; Fig. 7A, B), indicating a unique reliance of N249Q/R345W F3 on GRP94 for folding and secretion.
Figure 7.

N249Q/R345W F3 requires GRP94 ATPase activity for efficient secretion. A) ARPE-19 cells expressing WT, N249Q, R345W, or N249Q/R345W F3 were treated with DMSO or 17-AAG (1 μM) for 24 h and F3 was IP’d from the conditioned media, followed by Western blot analysis. Representative data of 3 independent experiments. B) Quantification of F3 secreted protein levels after treatment, n = 3 (**P ≤ 0.01, t test).
In addition to monitoring N249Q/R345W F3 secretion after 17-AAG treatment, we also used IP to determine how 17-AAG altered the N249Q/R345W F3 ER interactome. Treatment with 17-AAG significantly reduced the N249Q/R345W F3-GRP94 interaction while having no impact on its interaction with the prominent ER master chaperone GRP78, another newly identified F3 interacting partner (Fig. 8A, B). These results reaffirm the reliance of N249Q/R345W F3 on GRP94 for efficient secretion from ARPE-19 cells.
Figure 8.

17-AAG alters chaperone binding to N249Q/R345W F3. A) ARPE-19 cells were infected with FT N249Q/R345W F3, followed by treatment with DMSO or 17-AAG (1 μM) for 24 h. Cells were then cross-linked and F3 was IP’d. The IP’d proteins and corresponding input (2% of the IP) were separated on an SDS-PAGE, followed by Western blot analysis. B) Quantification of chaperone binding to F3. Data are presented as the ratio of chaperone to F3 in the IP’d material, n = 3 (*P ≤ 0.05, t test).
DISCUSSION
In the present study, we investigated the role that N-linked glycosylation plays in regulating the folding, secretion, and intracellular levels of F3, an important extracellular matrix protein. Tm treatment significantly reduced the secretion of the pathogenic ML variant R345W F3 and significantly elevated its intracellular steady-state levels. The main secreted species of WT and R345W F3 appears to be solely N-glycosylated at Asn249. Mutation of Asn249 prevented N-linked glycosylation of F3 but surprisingly had no detrimental effect on the amount of secreted N249Q/R345W F3. However, the quality of the intracellular and secreted N249Q/R345W F3 protein appears to be significantly diminished under nondenaturing, native conditions. The ER folding pathway used by this variant is unique, as the protein binds strongly to GRP94 and requires GRP94 activity for efficient secretion, unlike the other F3 variants. Together, these results support the importance of N-linked glycosylation of F3 at Asn249 and suggest that N-linked glycosylation makes the disease-causing R345W F3 protein less susceptible to aggregation and less sensitive to changes in the ER protein homeostasis machinery.
A series of independent groups have identified F3 binding partners that may be responsible for regulating F3 folding and/or function. Before the present study, there were 5 proteins known to interact with F3 [complement factor H (37), tissue inhibitor of matrix metalloproteinase 3 (38), extracellular matrix protein 1 (39), ER resident protein 57 (40), and ERdj5 (41)]. Given the multidomain structure of F3, it is not surprising that we were able to identify 4 more interacting partners of F3, the ER chaperones: GRP78 and GRP94, and the lectin chaperones CNX and CRT. Although all the F3 variants that we analyzed interacted with GRP94, N249Q/R345W F3 interacted most strongly. As validation of this interaction, GRP94 has been shown to bind to other EGF-rich, fibulin-like proteins, such as fibrillin-1 (42). Interestingly, it appears that the GRP94 interaction with WT, N249Q, and R345W F3 was not required for the secretion of each protein because targeting GRP94 ATPase activity with 17-AAG did not reduce their secretion. In contrast, 17-AAG significantly reduced N249Q/R345W F3 secretion (by over 60%), demonstrating a unique dependence of this variant on GRP94. Given these results, it is clear that there is a need to further understand the role of GRP94 in F3 protein homeostasis and to identify and characterize additional F3 interacting partners. Determining such partners will likely aid in advancing our understanding of F3 cellular protein homeostasis. Accordingly, we are currently performing a more in-depth and quantitative analysis of F3 intracellular and extracellular binding partners, which we hope to report in due course.
In the ER, there is a competition between protein folding and secretion vs. degradation, which determines the intracellular steady-state levels of a protein of interest [reviewed in (43)]. In general, N-glycosylated proteins such as F3 enter the lectin chaperone folding vs. degradation pathway immediately after they are cotranslationally imported into the ER and are covalently modified with a 14-member Glc3Man9(GlcNAc)2 oligosaccharide, which is subsequently trimmed by glucosidase I and II to yield Glc1Man9(GlcNAc)2. This trimmed glycan can then engage the lectin-assisted folding cycle composed of CNX and CRT [reviewed in (43)]. It is less likely that nonglycosylated candidates enter the lectin folding cycle; however, some groups have detected lectin-independent folding capabilities of both CNX and CRT with certain client proteins (44, 45). If the glycosylated client protein is unable to fold, it is trimmed by ER mannosidase I, recognized by ERAD components, retrotranslocated from the ER, and degraded by the proteasome [reviewed in (43, 46, 47)]. Our observation that genetic ablation of the glycosylation sequon does not reduce secreted levels of N249Q or N249Q/R345W F3, combined with the fact that intracellular levels of both proteins were higher, indicates that elimination of the N-linked glycan at Asn249 slows ERAD of both WT and R345W F3. Although the mechanism behind elevated intracellular F3 levels is not clear, we discovered that absence of N-linked glycosylation prevents F3 from fully engaging the CNX/CRT lectin-assisted folding and quality-control pathway. Although it seems that N249Q F3 can adopt a fold similar to WT F3 (as determined by BN PAGE), and thus can fold largely independent of CNX/CRT or GRP94, in contrast, N249Q/R345W F3 aggregates intracellularly and adopts an altered conformation extracellularly, indicating a dependence on CNX and CRT for proper folding. Thus, at least for R345W F3, it appears that N-linked glycosylation at Asn249 is critical for it to acquire and maintain its proper structure intracellularly and possibly extracellularly.
On the basis of our observations, secreted WT and R345W F3 are quantitatively N-glycosylated at Asn249. The N-glycosylation efficiency of intracellular F3 needs to be studied more carefully. Given that N-linked glycosylation appears to be necessary for effectively targeting intracellular F3 for degradation, it is conceivable that alteration of the N-linked glycosylation seqon could change the efficiency of F3 N-linked glycosylation and/or alter glycan elaboration. Thus, the use of a strategy such as the so-called enhanced aromatic sequon approach (48) could optimize F3 glycosylation, alter R345W F3 degradation, and provide more time for the protein to fold and be secreted. Furthermore, alteration of glycan occupancy or glycan elaboration likely influences the timing and extent of binding to F3 interacting partners and the ultimate fate of F3. As such, an in-depth quantitative analysis of glycan-dependent and -independent interacting partners of WT and R345W F3 warrants further study.
In summary, our results provide evidence for the importance of N-linked glycosylation of R345W F3 for its cellular folding and function. R345W F3 depends strongly on interaction with CNX/CRT for regulating its folding and secretion vs. degradation, and when it cannot interact with the lectin-assisted folding cycle, nonglycosylated R345W F3 is strongly dependent on GRP94 for mediating its secretion, apparently in an altered conformation. Thus, the absence of R345W F3 N-glycosylation at Asn249 unmasks an apparent inherent destabilization in R345W F3 that is not present in nonglycosylated WT F3. It is currently unclear whether this destabilization is a result of intrinsic or extrinsic effects. This destabilization, although nearly undetectable under conditions that favor N-glycosylation, may ultimately contribute to causing ML as a consequence of aging-associated cellular changes.
Supplementary Material
Acknowledgments
The authors thank R. L. Wiseman for useful discussions. This work was supported by the Lita Annenberg Hazen Foundation; the Skaggs Institute for Chemical Biology; the Scripps Translational Science Institute (Grant UL1 RR025774 to J.W.K.); the U.S. National Institutes of Health (NIH) National Institute on Aging (Grant AG018917 to J.W.K.); the Roger and Dorothy Hirl Research Fund (to J.D.H.); the NIH National Eye Institute (Core Grant EY020799); and an unrestricted grant from Research to Prevent Blindness.
Glossary
- 17-AAG
17-N-allylamino-17-demethoxygeldanamycin
- AMD
age-related macular degeneration
- ATF
activating transcription factor
- BFA
brefeldin A
- BN, blue native; cbEGF
calcium-binding epidermal growth factor
- CNX
calnexin
- ConA
concanavalin A
- CRT
calreticulin
- ER
endoplasmic reticulum
- ERAD
endoplasmic reticulum–associated degradation
- ERDJ4
DnaJ homolog, subfamily B, member 9
- F3
fibulin-3
- FT
FLAG tagged
- GRP78
glucose-regulated protein 78
- GRP94
glucose-regulated protein 94
- IP
immunoprecipitation
- IRE-1
inositol requiring enzyme 1
- ML
malattia leventinese
- MOI
multiplicity of infection
- PERK
protein kinase RNA-like ER kinase
- qPCR
quantitative polymerase chain reaction
- RPE
retinal pigment epithelium
- Tg
thapsigargin
- Tm
tunicamycin
- UPR
unfolded protein response
- WT
wild type
- XBP1
X-box binding protein-1
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Tielsch J. M., Javitt J. C., Coleman A., Katz J., Sommer A. (1995) The prevalence of blindness and visual impairment among nursing home residents in Baltimore. N. Engl. J. Med. 332, 1205–1209 [DOI] [PubMed] [Google Scholar]
- 2.Attebo K., Mitchell P., Smith W. (1996) Visual acuity and the causes of visual loss in Australia. The Blue Mountains Eye Study. Ophthalmology 103, 357–364 [DOI] [PubMed] [Google Scholar]
- 3.de Jong P. T. (2006) Age-related macular degeneration. N. Engl. J. Med. 355, 1474–1485 [DOI] [PubMed] [Google Scholar]
- 4.Stone E. M., Sheffield V. C., Hageman G. S. (2001) Molecular genetics of age-related macular degeneration. Hum. Mol. Genet. 10, 2285–2292 [DOI] [PubMed] [Google Scholar]
- 5.Lin J. H., Lavail M. M. (2010) Misfolded proteins and retinal dystrophies. Adv. Exp. Med. Biol. 664, 115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stone E. M., Lotery A. J., Munier F. L., Héon E., Piguet B., Guymer R. H., Vandenburgh K., Cousin P., Nishimura D., Swiderski R. E., Silvestri G., Mackey D. A., Hageman G. S., Bird A. C., Sheffield V. C., Schorderet D. F. (1999) A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat. Genet. 22, 199–202 [DOI] [PubMed] [Google Scholar]
- 7.Piguet B., Haimovici R., Bird A. C. (1995) Dominantly inherited drusen represent more than one disorder: a historical review. Eye (Lond.) 9, 34–41 [DOI] [PubMed] [Google Scholar]
- 8.Fu L., Garland D., Yang Z., Shukla D., Rajendran A., Pearson E., Stone E. M., Zhang K., Pierce E. A. (2007) The R345W mutation in EFEMP1 is pathogenic and causes AMD-like deposits in mice. Hum. Mol. Genet. 16, 2411–2422 [DOI] [PubMed] [Google Scholar]
- 9.Garland D. L., Fernandez-Godino R., Kaur I., Speicher K. D., Harnly J. M., Lambris J. D., Speicher D. W., Pierce E. A. (2014) Mouse genetics and proteomic analyses demonstrate a critical role for complement in a model of DHRD/ML, an inherited macular degeneration. Hum. Mol. Genet. 23, 52–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marmorstein L. Y., McLaughlin P. J., Peachey N. S., Sasaki T., Marmorstein A. D. (2007) Formation and progression of sub-retinal pigment epithelium deposits in Efemp1 mutation knock-in mice: a model for the early pathogenic course of macular degeneration. Hum. Mol. Genet. 16, 2423–2432 [DOI] [PubMed] [Google Scholar]
- 11.Timpl R., Sasaki T., Kostka G., Chu M. L. (2003) Fibulins: a versatile family of extracellular matrix proteins. Nat. Rev. Mol. Cell Biol. 4, 479–489 [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y., Marmorstein L. Y. (2010) Focus on molecules: fibulin-3 (EFEMP1). Exp. Eye Res. 90, 374–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hulleman J. D., Kaushal S., Balch W. E., Kelly J. W. (2011) Compromised mutant EFEMP1 secretion associated with macular dystrophy remedied by proteostasis network alteration. Mol. Biol. Cell 22, 4765–4775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marmorstein L. Y., Munier F. L., Arsenijevic Y., Schorderet D. F., McLaughlin P. J., Chung D., Traboulsi E., Marmorstein A. D. (2002) Aberrant accumulation of EFEMP1 underlies drusen formation in Malattia Leventinese and age-related macular degeneration. Proc. Natl. Acad. Sci. USA 99, 13067–13072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hulleman J. D., Balch W. E., Kelly J. W. (2012) Translational attenuation differentially alters the fate of disease-associated fibulin proteins. FASEB J. 26, 4548–4560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hulleman J. D., Brown S. J., Rosen H., Kelly J. W. (2013) A high-throughput cell-based Gaussia luciferase reporter assay for identifying modulators of fibulin-3 secretion. J. Biomol. Screen. 18, 647–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roybal C. N., Marmorstein L. Y., Vander Jagt D. L., Abcouwer S. F. (2005) Aberrant accumulation of fibulin-3 in the endoplasmic reticulum leads to activation of the unfolded protein response and VEGF expression. Invest. Ophthalmol. Vis. Sci. 46, 3973–3979 [DOI] [PubMed] [Google Scholar]
- 18.Ron D., Walter P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 [DOI] [PubMed] [Google Scholar]
- 19.Kroeger H., Messah C., Ahern K., Gee J., Joseph V., Matthes M. T., Yasumura D., Gorbatyuk M. S., Chiang W. C., LaVail M. M., Lin J. H. (2012) Induction of endoplasmic reticulum stress genes, BiP and chop, in genetic and environmental models of retinal degeneration. Invest. Ophthalmol. Vis. Sci. 53, 7590–7599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kroeger H., Chiang W. C., Lin J. H. (2012) Endoplasmic reticulum–associated degradation (ERAD) of misfolded glycoproteins and mutant P23H rhodopsin in photoreceptor cells. Adv. Exp. Med. Biol. 723, 559–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gould D. B., Marchant J. K., Savinova O. V., Smith R. S., John S. W. (2007) Col4a1 mutation causes endoplasmic reticulum stress and genetically modifiable ocular dysgenesis. Hum. Mol. Genet. 16, 798–807 [DOI] [PubMed] [Google Scholar]
- 22.Guerriero C. J., Brodsky J. L. (2012) The delicate balance between secreted protein folding and endoplasmic reticulum–associated degradation in human physiology. Physiol. Rev. 92, 537–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kornfeld R., Kornfeld S. (1985) Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 54, 631–664 [DOI] [PubMed] [Google Scholar]
- 24.O’Connor S. E., Pohlmann J., Imperiali B., Saskiawan I., Yamamoto K. (2001) Probing the effect of the outer saccharide residues of N-linked glycans on peptide conformation. J. Am. Chem. Soc. 123, 6187–6188 [DOI] [PubMed] [Google Scholar]
- 25.Helenius A., Aebi M. (2001) Intracellular functions of N-linked glycans. Science 291, 2364–2369 [DOI] [PubMed] [Google Scholar]
- 26.Hanson S. R., Culyba E. K., Hsu T. L., Wong C. H., Kelly J. W., Powers E. T. (2009) The core trisaccharide of an N-linked glycoprotein intrinsically accelerates folding and enhances stability. Proc. Natl. Acad. Sci. USA 106, 3131–3136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitra N., Sinha S., Ramya T. N., Surolia A. (2006) N-linked oligosaccharides as outfitters for glycoprotein folding, form and function. Trends Biochem. Sci. 31, 156–163 [DOI] [PubMed] [Google Scholar]
- 28.Narhi L. O., Arakawa T., Aoki K. H., Elmore R., Rohde M. F., Boone T., Strickland T. W. (1991) The effect of carbohydrate on the structure and stability of erythropoietin. J. Biol. Chem. 266, 23022–23026 [PubMed] [Google Scholar]
- 29.Wittig I., Braun H. P., Schägger H. (2006) Blue native PAGE. Nat. Protoc. 1, 418–428 [DOI] [PubMed] [Google Scholar]
- 30.Heifetz A., Keenan R. W., Elbein A. D. (1979) Mechanism of action of tunicamycin on the UDP-GlcNAc:dolichyl-phosphate Glc-NAc-1-phosphate transferase. Biochemistry 18, 2186–2192 [DOI] [PubMed] [Google Scholar]
- 31.Appenzeller-Herzog C., Riemer J., Zito E., Chin K. T., Ron D., Spiess M., Ellgaard L. (2010) Disulphide production by Ero1α-PDI relay is rapid and effectively regulated. EMBO J. 29, 3318–3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Djokic J., Fagotto-Kaufmann C., Bartels R., Nelea V., Reinhardt D. P. (2013) Fibulin-3, -4, and -5 are highly susceptible to proteolysis, interact with cells and heparin, and form multimers. J. Biol. Chem. 288, 22821–22835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.May L. T., Shaw J. E., Khanna A. K., Zabriskie J. B., Sehgal P. B. (1991) Marked cell-type-specific differences in glycosylation of human interleukin-6. Cytokine 3, 204–211 [DOI] [PubMed] [Google Scholar]
- 34.Putnam E. A., Cho M., Zinn A. B., Towbin J. A., Byers P. H., Milewicz D. M. (1996) Delineation of the Marfan phenotype associated with mutations in exons 23–32 of the FBN1 gene. Am. J. Med. Genet. 62, 233–242 [DOI] [PubMed] [Google Scholar]
- 35.Baetens M., Van Laer L., De Leeneer K., Hellemans J., De Schrijver J., Van De Voorde H., Renard M., Dietz H., Lacro R. V., Menten B., Van, Criekinge W., De Backer J., De Paepe A., Loeys B., Coucke P. J. (2011) Applying massive parallel sequencing to molecular diagnosis of Marfan and Loeys-Dietz syndromes. Hum. Mutat. 32, 1053–1062 [DOI] [PubMed] [Google Scholar]
- 36.Schulte T. W., Neckers L. M. (1998) The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother. Pharmacol. 42, 273–279 [DOI] [PubMed] [Google Scholar]
- 37.Wyatt M. K., Tsai J. Y., Mishra S., Campos M., Jaworski C., Fariss R. N, Bernstein S. L., Wistow G. (2013) Interaction of complement factor h and fibulin3 in age-related macular degeneration. PLoS One. 8, e68088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klenotic P. A., Munier F. L., Marmorstein L. Y., Anand-Apte B. (2004) Tissue inhibitor of metalloproteinases-3 (TIMP-3) is a binding partner of epithelial growth factor–containing fibulin-like extracellular matrix protein 1 (EFEMP1). Implications for macular degenerations. J. Biol. Chem. 279, 30469–30473 [DOI] [PubMed] [Google Scholar]
- 39.Sercu S., Lambeir A. M., Steenackers E., El Ghalbzouri A., Geentjens K., Sasaki T., Oyama N., Merregaert J. (2009) ECM1 interacts with fibulin-3 and the beta 3 chain of laminin 332 through its serum albumin subdomain-like 2 domain. Matrix Biol. 28, 160–169 [DOI] [PubMed] [Google Scholar]
- 40.Jessop C. E., Chakravarthi S., Garbi N., Hämmerling G. J., Lovell S., Bulleid N. J. (2007) ERp57 is essential for efficient folding of glycoproteins sharing common structural domains. EMBO J. 26, 28–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oka O. B., Pringle M. A., Schopp I. M., Braakman I., Bulleid N. J. (2013) ERdj5 is the ER reductase that catalyzes the removal of non-native disulfides and correct folding of the LDL receptor. Mol. Cell 50, 793–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wallis D. D., Putnam E. A., Cretoiu J. S., Carmical S. G., Cao S. N., Thomas G., Milewicz D. M. (2003) Profibrillin-1 maturation by human dermal fibroblasts: proteolytic processing and molecular chaperones. J. Cell. Biochem. 90, 641–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vembar S. S., Brodsky J. L. (2008) One step at a time: endoplasmic reticulum–associated degradation. Nat. Rev. Mol. Cell Biol. 9, 944–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brockmeier A., Williams D. B. (2006) Potent lectin-independent chaperone function of calnexin under conditions prevalent within the lumen of the endoplasmic reticulum. Biochemistry 45, 12906–12916 [DOI] [PubMed] [Google Scholar]
- 45.Ireland B. S., Brockmeier U., Howe C. M., Elliott T., Williams D. B. (2008) Lectin-deficient calreticulin retains full functionality as a chaperone for class I histocompatibility molecules. Mol. Biol. Cell 19, 2413–2423 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.Merulla J., Fasana E., Soldà T., Molinari M. (2013) Specificity and regulation of the endoplasmic reticulum–associated degradation machinery. Traffic 14, 767–777 [DOI] [PubMed] [Google Scholar]
- 47.Ruggiano A., Foresti O., Carvalho P. (2014) Quality control: ER-associated degradation: protein quality control and beyond. J. Cell Biol. 204, 869–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Culyba E. K., Price J. L., Hanson S. R., Dhar A., Wong C. H., Gruebele M., Powers E. T., Kelly J. W. (2011) Protein native-state stabilization by placing aromatic side chains in N-glycosylated reverse turns. Science 331, 571–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.