

Abstract
Background: Prenatal exposure to maternal cigarette smoking (prenatal smoke exposure) had been associated with altered DNA methylation (DNAm) at birth.
Objective: We examined whether such alterations are present from birth through adolescence.
Methods: We used the Infinium HumanMethylation450K BeadChip to search across 473,395 CpGs for differential DNAm associated with prenatal smoke exposure during adolescence in a discovery cohort (n = 132) and at birth, during childhood, and during adolescence in a replication cohort (n = 447).
Results: In the discovery cohort, we found five CpGs in MYO1G (top-ranking CpG: cg12803068, p = 3.3 × 10–11) and CNTNAP2 (cg25949550, p = 4.0 × 10–9) to be differentially methylated between exposed and nonexposed individuals during adolescence. The CpGs in MYO1G and CNTNAP2 were associated, respectively, with higher and lower DNAm in exposed versus nonexposed adolescents. The same CpGs were differentially methylated at birth, during childhood, and during adolescence in the replication cohort. In both cohorts and at all developmental time points, the differential DNAm was in the same direction and of a similar magnitude, and was not altered appreciably by adjustment for current smoking by the participants or their parents. In addition, four of the five EWAS (epigenome-wide association study)–significant CpGs in the adolescent discovery cohort were also among the top sites of differential methylation in a previous birth cohort, and differential methylation of CpGs in CYP1A1, AHRR, and GFI1 observed in that study was also evident in our discovery cohort.
Conclusions: Our findings suggest that modifications of DNAm associated with prenatal maternal smoking may persist in exposed offspring for many years—at least until adolescence.
Citation: Lee KW, Richmond R, Hu P, French L, Shin J, Bourdon C, Reischl E, Waldenberger M, Zeilinger S, Gaunt T, McArdle W, Ring S, Woodward G, Bouchard L, Gaudet D, Davey Smith G, Relton C, Paus T, Pausova Z. 2015. Prenatal exposure to maternal cigarette smoking and DNA methylation: epigenome-wide association in a discovery sample of adolescents and replication in an independent cohort at birth through 17 years of age. Environ Health Perspect 123:193–199; http://dx.doi.org/10.1289/ehp.1408614
Introduction
Maternal cigarette smoking during pregnancy was and still is common in the industrialized world. In the United States, for example, 30–40% of pregnant women smoked in the 1960s and 1970s, and 16% of pregnant women smoke today (Substance Abuse and Mental Health Services Administration 2012). Prenatal exposure to maternal cigarette smoking (prenatal smoke exposure) has been associated with a number of health problems in the exposed offspring (Oken et al. 2008; Power et al. 2010; Syme et al. 2010). The underlying mechanisms of these associations are not clear but may involve lasting modulations of DNA methylation (DNAm).
DNAm is a chemical modification constituted most commonly by the addition of a methyl group to cytosines in CpG dinucleotides (CpGs) (Relton and Davey Smith 2010). It is catalyzed by two main types of DNA methyltransferases (DNMTs): de novo DNMTs, which play a key role in establishing new DNAm patterns during cell differentiation and embryogenesis; and maintenance DNMTs, which are responsible for copying these patterns from cell to cell during successive mitotic divisions (Jeltsch 2006; Tang et al. 2009). The main function of DNAm is the regulation of gene expression and genomic architecture (Levin and Moran 2011; Tsukahara et al. 2009).
Current research suggests that cigarette smoke is a powerful environmental modifier of DNAm (reviewed by Lee and Pausova 2013). Cigarette smoke contains a large number of chemicals, such as carcinogens, nicotine, and carbon monoxide, that have been shown to modify DNAm in differentiating and dividing cells (Cuozzo et al. 2007; Di et al. 2012; Han et al. 2001; Huang et al. 2013; Mercer et al. 2009; Mortusewicz et al. 2005; Satta et al. 2008; Shahrzad et al. 2007). Consistent with these possible effects, reproducible differences in DNAm have been reported in peripheral blood cells of smokers versus nonsmokers (Breitling et al. 2011; Philibert et al. 2012; Shenker et al. 2013; Zeilinger et al. 2013).
Many chemicals contained in cigarette smoke can easily pass from a pregnant smoker to the developing embryo/fetus (Lambers and Clark 1996). Accordingly, differences in DNAm have been reported in cord blood cells of babies born to smoking versus nonsmoking mothers (Joubert et al. 2012). Whether these DNAm differences persist postnatally in peripheral blood cells of the exposed offspring has not been studied; this is the main subject of the present investigation. Prenatal exposure to cigarette smoke may induce lasting and soma-wide (present in all somatic cells) modifications of DNAm in the exposed offspring, particularly if occurring during early stages of embryogenesis when global erasure and reestablishment of DNAm patterns take place in yet-undifferentiated stem cells (Feng et al. 2010; Smith et al. 2012). These DNAm modifications are then propagated during embryonic development to all somatic cell lineages (including precursors of peripheral blood cells) and maintained throughout life by the action of maintenance DNMTs, which copy these modifications from cell to cell during successive cell divisions (Davies et al. 2012; Faulk and Dolinoy 2011; Jurkowska et al. 2011; Lee and Pausova 2013; Petronis 2010).
To investigate whether DNAm modifications induced by prenatal smoke exposure persist postnatally (at least until adolescence), we first conducted an epigenome-wide association study (EWAS) to search for CpGs methylated differentially between exposed and nonexposed adolescents in one cohort, the Saguenay Youth Study (SYS) (Pausova et al. 2007). Subsequently, we tested whether the identified CpGs were also methylated differentially between exposed and nonexposed newborns, children, and adolescents in another cohort, the Avon Longitudinal Study of Parents and Children (ALSPAC) (Boyd et al. 2013).
Methods
Discovery cohort: The Saguenay Youth Study (SYS). Recruitment and assessment of prenatal smoke exposure. The SYS (n = 1,028, 12–18 years of age) is a population-based cross-sectional study of cardiovascular, metabolic, brain and mental health in adolescents carried out in the genetic founder population of the Saguenay Lac St. Jean region of Quebec, Canada (Pausova et al. 2007). All participants were white Caucasians. Adolescents exposed (n = 490) and nonexposed (n = 538) to maternal cigarette smoking during pregnancy were matched at recruitment by maternal education and school attended, to reduce potential confounding related to socioeconomic status (Pausova et al. 2007). Participants were recruited via high schools. Exposure status was defined as having a mother who smoked at least one cigarette per day during the second trimester of pregnancy. Being nonexposed was defined as having a mother who did not smoke at least 1 year before and then throughout the pregnancy. The information on exposure was obtained from the mother via a structured telephone interview at the time of recruitment of her adolescent offspring, and was validated subsequently against medical records from the time of pregnancy for a subset of adolescents; kappa statistics with a mean (± SD) value of 0.69 ± 0.04 indicated a good strength of agreement in this subset (good agreement, 0.6–0.8) (Landis and Koch 1977). The main exclusion criteria were as follows: premature birth (< 35 weeks) or detached placenta; maternal alcohol abuse during pregnancy (> 210 mL of alcohol/week); positive medical history of type 1 diabetes, or of heart disease requiring surgery or sustained medication; and contraindications for magnetic resonance imaging. Additional recruitment details and selection criteria have been described previously (Pausova et al. 2007).
In the present study, we selected 66 exposed adolescents matched to 66 nonexposed by sex, age, and maternal education. The exposed adolescents were selected within each sex separately as those with the exposure being close to the sex-specific median (10 cigarettes/day in both sexes). This process resulted in a selection of 66 adolescents (33 males and 33 females) who were exposed to maternal cigarette smoking throughout pregnancy (all trimesters) at a level of 9 ± 4 cigarettes/day (range, 5–20 cigarettes/day). The studied adolescents were 12–18 years of age (Table 1). Written consent of the parents and assent of the adolescents were obtained. The research ethics committees of the Chicoutimi Hospital and the Hospital for Sick Children approved the study.
Table 1.
Basic characteristics of SYS adolescents (discovery sample).
| Characteristic | Nonexposed | Exposed | p-Value |
|---|---|---|---|
| Sex (males/females) | 33/33 | 33/33 | NAa |
| Age (years) | 15.6 ± 1.5 | 15.5 ± 1.4 | 0.95 |
| Maternal educationb | 5.4 ± 1.9 | 5.1 ± 2.0 | 0.47 |
| Family income ($CAD/year) | 60,384 ± 24,387 | 58,939 ± 25,548 | 0.74 |
| Current adolescent smoking (yes/no)c | 2/61 | 5/60 | 0.44 |
| Current paternal smoking (yes/no)d | 11/52 | 19/43 | 0.08 |
| Current maternal smoking (yes/no)e | 5/61 | 39/27 | < 0.0001 |
| Maternal smoking during pregnancy (cigarettes/day) | 0 | 8.9 ± 3.9 | NAa |
| Maternal age at delivery (years) | 27.0 ± 4.5 | 25.0 ± 4.3 | 0.01 |
| Length of gestation (weeks) | 39.6 ± 1.5 | 39.6 ± 1.6 | 0.83 |
| Birth weight (g) | 3,442 ± 406 | 3,275 ± 501 | 0.04 |
| NA, not applicable. Data are presented as mean ± SD or n. aDetermined by the selection of the EWAS-studied participants. bMaternal education was classified as the highest achieved level: primary school not completed, primary school completed, high school not completed, high school completed, college completed, or university completed. cCurrent adolescent smoking was defined as smoking of at least 1 cigarette in the preceding 30 days (data missing in 4 adolescents). dCurrent paternal smoking was defined as smoking of at least 1 cigarette in the preceding 30 days (data missing in 7 fathers). eCurrent maternal smoking was defined as smoking of at least 1 cigarette in the preceding 30 days. | |||
Adolescent and parent cigarette smoking. Adolescents self-reported their mental health and substance use (including cigarette smoking) by completing a questionnaire developed for the SYS by J. Séguin from the University of Montreal (Groupe de Recherche sur l’Inadaptation Psychosociale); this questionnaire is based on validated National Longitudinal Survey of Children and Youth (NLSCY) and Quebec Longitudinal Study of Child Development (QLSCD) protocols (Pausova et al. 2007). Blood collection and adolescent smoking assessment occurred on the same day. Parents self-reported their own mental health and substance use (including cigarette smoking) using a similar questionnaire, as adapted by colleagues at the Groupe de Recherche sur l’Inadaptation Psychosociale of the University of Montreal (Pausova et al. 2007).
Epityping with the Infinium HumanMethylation450K BeadChip. DNA samples were extracted from the buffy coat (peripheral blood cells) using the Qiagen Autopure LS (Qiagen, Venlo, the Netherlands). Bisulfite conversion was performed on 800 ng of DNA from each sample with the EZ-96 DNA Methylation Kit (Zymo Research, Irvine, CA, USA). DNA was subsequently used for hybridization on the Infinium HumanMethylation450K BeadChip (Illumina, San Diego, CA, USA). Bisulfite conversion and methylation array hybridization were performed at the Helmholtz Zentrum München German Research Center for Environmental Health (Neuherberg, Germany). The Infinium HumanMethylation450K BeadChip interrogates methylation at > 485,000 CpGs and provides coverage of > 99% of RefSeq genes (Sandoval et al. 2011). The DNAm score at each CpG, referred to as the DNAm β-value, is derived from the fluorescent intensity ratio [β = intensity of the methylated allele/(intensity of the unmethylated allele + intensity of the methylated allele + 100)] (Bibikova et al. 2011). All samples were randomly loaded onto 11 arrays (12 samples per array) and processed by the same technician at the same time to minimize batch effects. Methylation values were normalized using the pre-process Illumina algorithm implemented in Minfi R package (Aryee et al. 2014). Parameters were set to mimic the Illumina Genome Studio normalization procedure (Illumina). Quality control was performed by excluding CpGs with detection p ≥ 0.05 in > 20% of samples (764 CpGs). After excluding these probes, as well as control probes and probes on sex chromosomes, a total of 473,395 CpGs were analyzed. All 132 samples had > 98% sites with detection p < 0.05.
Statistical methods (EWAS). Methylation β-values at each of the 473,395 CpGs were transformed to obtain M-values, defined as log2 [β/(1–β)], which have greater statistical power than β-values to detect differential methylation at highly methylated and unmethylated CpGs (Du et al. 2010). Multivariable linear regression was used to perform association tests between the M-value at each CpG (n = 475,395) as the dependent variable and prenatal smoke exposure (exposed vs. nonexposed) as the independent variable. These analyses were adjusted for potential confounding by age, sex, batch, and blood cell fractions (model A). Blood cell fractions (granulocytes, B cells, CD8+ T cells, CD4+ T cells, NK cells, monocytes) were determined by the method of Houseman (Houseman et al. 2012); the fractions were analyzed with principal component analysis, and the first three components capturing 95% of variance were included in the regression model. The Manhattan and quantile–quantile plots for this model A are shown in Supplemental Material, Figure S1. None of the estimated blood cell fractions differed between exposed and nonexposed individuals (p = 0.31–0.89). In additional analyses, we also adjusted for current smoking by adolescents (model B) or for both current smoking by adolescents and secondhand smoking via parents (model C). Further, we used the above-described basic model A to analyze subsets of adolescents a) who did not smoke themselves (61 exposed and 64 nonexposed; model D) or b) who did not smoke themselves and their parents did not smoke at present (19 exposed and 49 nonexposed; model E). Finally, we ran an analysis that was not corrected for any potential confounders (model F). CpGs achieving Bonferroni-corrected statistical significance of p < 0.05 (uncorrected p < 1.1 × 10–7) were considered EWAS significant. False discovery rate (FDR)–corrected p-values were also determined according to the method of Benjamini and Hochberg (1995). These statistical analyses were performed using the R software (http://www.r-project.org/).
Replication cohort: Avon Longitudinal Study of Parents and Children (ALSPAC). Recruitment and assessment of prenatal smoke exposure. ALSPAC is a population-based longitudinal birth cohort ascertained in the former Avon Health Authority in southwest England between 1 April 1991 and 31 December 1992; the initial cohort included 14,541 pregnancies and 13,971 children alive at 12 months of age (Boyd et al. 2013). From this cohort, approximately 500 children have been epityped with the Infinium HumanMethylation450K BeadChip (Illumina) as part of a larger methylation project, ARIES, which is generating epigenetic information for 1,000 mother–offspring pairs at multiple time points across the life course (ARIES 2014). The individuals included in the present study are a subset of the offspring participants in ARIES for whom 450K data were available at the time of submission of this manuscript. Of these individuals, 82% (430/526) had methylation data available for at least two of the following three time points: at birth, 7 years of age, and 17 years of age. DNA samples were extracted from cord blood cells (birth) and peripheral blood cells (at 7 and 17 years of age). From these, we selected exposed individuals as child participants whose mothers smoked ≥ 5 cigarettes/day throughout their gestation, and nonexposed individuals as child participants whose mothers did not smoke approximately 4/5 months before pregnancy and throughout all trimesters of their gestation (see Supplemental Material, Table S1). Prenatal exposure to maternal cigarette smoking was assessed during pregnancy. Methylation values were normalized using the approach of Touleimat and Tost (2012).
Statistical methods. Associations between prenatal exposure to maternal cigarette smoking and M-values at the five EWAS-significant CpGs identified in the SYS were tested using linear regression models adjusted for age, sex, maternal education, household social class, and batch. We included maternal education and household social class in the model, as unlike in the SYS exposed and nonexposed participants were not matched for any indices of socioeconomic status at recruitment. In additional analyses, we also adjusted for parent (secondhand) smoking at 7 years of age, and for both participant smoking and parent (secondhand) smoking at 17 years of age. Statistical analyses were performed using R software.
Results
EWAS search for differential DNAm associated with prenatal smoke exposure in the SYS adolescents. The EWAS search was carried out in the SYS, which is a population-based cross-sectional study of 12- to 18-year-old adolescents aimed at investigating long-term health outcomes associated with prenatal exposure to maternal cigarette smoking (Table 1). All participants are from the genetic founder population of the Saguenay Lac St. Jean region of Quebec, Canada (Pausova et al. 2007).
The EWAS search in the SYS identified 5 EWAS-significant (p < 1.1 × 10–7) CpGs (Figure 1A and Table 2, model A). Four of them, including the most significant one (cg12803068, p = 3.3 × 10–11), were located within a 1-kb segment of the myosin IG gene (MYO1G); they were all associated with higher DNAm in exposed than in nonexposed adolescents (Figure 1A and Table 2, model A). DNAm at all 4 CpGs was correlated (r = 0.83–0.92, p < 0.0001), and as such the 4 CpGs are not independent signals. The other significant CpG was found in the contactin associated protein-like 2 gene (CNTNAP2, cg25949550, p = 4.0 × 10–9). In contrast with the MYO1G CpGs, it was associated with lower DNAm in exposed than nonexposed adolescents (Figure 1A and Table 2, model A). None of the above CpGs were cross-reactive or polymorphic as listed in studies by Chen et al. (2013) or Price et al. (2013). These results were obtained while adjusting for potential confounders, namely age, sex, batch, and blood cell fractions (model A). They remained virtually unchanged when not adjusted for any of these potential confounders (model F; see also Supplemental Material, Table S2). In both adjusted and unadjusted analyses, differential DNAm (DNAm β difference between exposed and nonexposed) ranged from +0.04 to +0.09 in MYO1G, and it was –0.02 in CNTNAP2.
Figure 1.
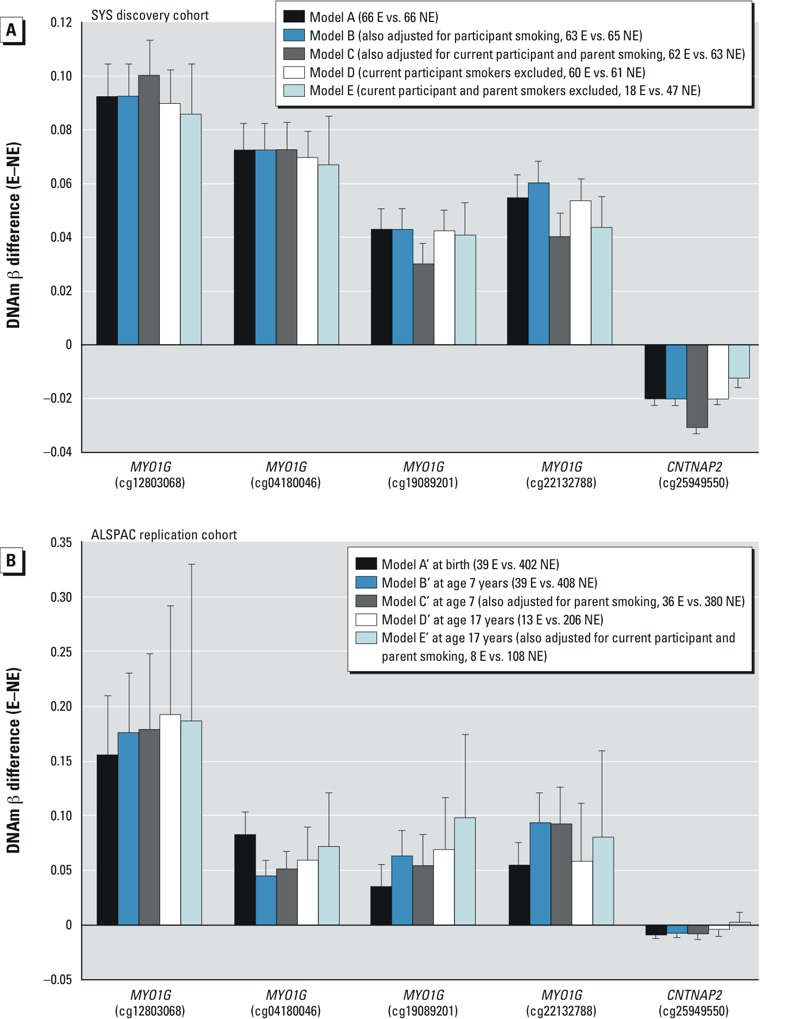
EWAS-significant CpGs in the discovery SYS (A) and replication ALSPAC (B) cohorts. Mean differences in DNAm β-values (and their 95% confidence intervals) between exposed (E) and nonexposed (NE) individuals during adolescence in the discovery SYS cohort and at birth, during childhood, and during adolescence in the replication ALSPAC cohort are presented. In all presented models, the differences in the SYS cohort were adjusted for age, sex, batch, and blood cell fractions, and the differences in the ALSPAC cohort were adjusted for age, sex, batch, maternal education, and parental social class. p-Values of the associations are presented in Tables 2 and 3.
Table 2.
EWAS: prenatal smoke exposure–associated differential DNA methylation during adolescence in the discovery SYS sample.
| CpG ID | Gene | Model A | Model B | Model C | Model D | Model E | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| DNAm β difference (SE) | p‑Value | DNAm β difference (SE) | p‑Value | DNAm β difference (SE) | p‑Value | DNAm β difference (SE) | p‑Value | DNAm β difference (SE) | p‑Value | ||
| cg12803068 | MYO1G | 0.09 (0.01) | 3.3 × 10–11* | 0.09 (0.01) | 7.0 × 10–11* | 0.10 (0.01) | 2.2 × 10–8* | 0.09 (0.01) | 4.9 × 10–10* | 0.09 (0.02) | 2.2 × 10–5 |
| cg04180046 | MYO1G | 0.07 (0.01) | 3.9 × 10–11* | 0.07 (0.01) | 1.2 × 10–10* | 0.08 (0.01) | 1.2 × 10–8* | 0.07 (0.01) | 4.4 × 10–10* | 0.07 (0.02) | 4.0 × 10–5 |
| cg19089201 | MYO1G | 0.04 (0.01) | 1.6 × 10–9* | 0.04 (0.01) | 4.0 × 10–9* | 0.05 (0.01) | 3.5 × 10–7 | 0.04 (0.01) | 1.8 × 10–8* | 0.04 (0.01) | 7.9 × 10–5 |
| cg25949550 | CNTNAP2 | –0.02 (0.00) | 4.0 × 10–9* | –0.02 (0.00) | 8.2 × 10–10* | –0.02 (0.00) | 9.4 × 10–7 | –0.02 (0.00) | 1.2 × 10–8* | –0.01 (0.01) | 1.6 × 10–2 |
| cg22132788 | MYO1G | 0.06 (0.01) | 8.4 × 10–9* | 0.06 (0.01) | 1.5 × 10–8* | 0.06 (0.01) | 1.5 × 10–6 | 0.05 (0.01) | 4.6 × 10–8* | 0.04 (0.01) | 1.3 × 10–3 |
| cg23222488 | NA | 0.03 (0.01) | 1.2 × 10–6 | 0.03 (0.01) | 3.9 × 10–6 | 0.03 (0.01) | 3.7 × 10–5 | 0.02 (0.01) | 1.2 × 10–5 | 0.01 (0.01) | 8.6 × 10–3 |
| cg27214774 | FAM65C | 0.01 (0.00) | 1.9 × 10–6 | 0.01 (0.00) | 3.3 × 10–6 | 0.01 (0.00) | 9.3 × 10–5 | 0.01 (0.00) | 1.9 × 10–5 | 0.01 (0.00) | 7.6 × 10–2 |
| cg00253658 | NA | 0.07 (0.01) | 2.0 × 10–6 | 0.07 (0.01) | 3.3 × 10–6 | 0.07 (0.01) | 8.8 × 10–5 | 0.07 (0.01) | 5.9 × 10–6 | 0.05 (0.03) | 1.3 × 10–3 |
| cg27641820 | ANKRD11 | –0.01 (0.00) | 5.3 × 10–6 | 0.00 (0.00) | 8.2 × 10–6 | –0.00 (0.00) | 2.1 × 10–4 | –0.01 (0.00) | 5.4 × 10–6 | –0.00 (0.00) | 1.3 × 10–1 |
| cg13323902 | VTRNA1-1 | –0.01 (0.00) | 6.6 × 10–6 | –0.01 (0.01) | 3.8 × 10–6 | –0.01 (0.01) | 2.4 × 10–4 | –0.02 (0.01) | 7.9 × 10–7 | –0.03 (0.01) | 1.2 × 10–5 |
| cg05176844 | HCG27 | –0.02 (0.00) | 7.2 × 10–6 | –0.02 (0.00) | 4.1 × 10–6 | –0.02 (0.00) | 1.3 × 10–5 | –0.02 (0.01) | 2.1 × 10–6 | –0.02 (0.01) | 1.0 × 10–3 |
| cg05549655 | CYP1A1 | 0.03 (0.01) | 7.6 × 10–6 | 0.03 (0.01) | 4.5 × 10–6 | 0.03 (0.01) | 4.6 × 10–4 | 0.03 (0.01) | 6.5 × 10–6 | 0.02 (0.02) | 1.9 × 10–2 |
| cg01586011 | NA | 0.02 (0.01) | 8.3 × 10–6 | 0.01 (0.01) | 4.0 × 10–5 | 0.01 (0.01) | 6.3 × 10–3 | 0.01 (0.01) | 1.0 × 10–4 | –0.01 (0.01) | 2.1 × 10–1 |
| cg17293195 | KRTAP5-7 | –0.02 (0.01) | 1.2 × 10–5 | –0.02 (0.01) | 8.8 × 10–6 | –0.02 (0.01) | 1.0 × 10–3 | –0.02 (0.01) | 4.2 × 10–5 | –0.02 (0.01) | 4.5 × 10–2 |
| cg17924476 | AHRR | 0.03 (0.01) | 1.2 × 10–5 | 0.03 (0.01) | 5.0 × 10–5 | 0.03 (0.01) | 6.6 × 10–4 | 0.03 (0.01) | 1.8 × 10–4 | 0.03 (0.01) | 1.2 × 10–1 |
| cg13570656 | CYP1A1 | 0.04 (0.01) | 1.3 × 10–5 | 0.04 (0.01) | 5.3 × 10–6 | 0.04 (0.01) | 2.9 × 10–4 | 0.04 (0.01) | 1.1 × 10–5 | 0.00 (0.02) | 1.2 × 10–2 |
| cg15982595 | C13orf35 | –0.01 (0.00) | 1.5 × 10–5 | –0.01 (0.00) | 3.0 × 10–5 | –0.01 (0.00) | 1.7 × 10–3 | –0.01 (0.00) | 3.3 × 10–5 | –0.01 (0.00) | 6.1 × 10–2 |
| cg17101494 | NA | –0.04 (0.02) | 1.6 × 10–5 | –0.04 (0.02) | 1.6 × 10–5 | –0.04 (0.02) | 2.8 × 10–4 | –0.04 (0.02) | 1.9 × 10–5 | –0.04 (0.02) | 6.0 × 10–4 |
| cg11936410 | TPM1 | –0.01 (0.00) | 1.7 × 10–5 | –0.01 (0.00) | 9.2 × 10–6 | –0.01 (0.00) | 1.5 × 10–3 | –0.01 (0.00) | 1.7 × 10–5 | –0.01 (0.00) | 3.1 × 10–4 |
| cg00213123 | CYP1A1 | 0.02 (0.01) | 1.9 × 10–5 | 0.02 (0.01) | 1.2 × 10–5 | 0.02 (0.01) | 8.1 × 10–4 | 0.02 (0.01) | 1.5 × 10–5 | 0.00 (0.01) | 2.3 × 10–2 |
| cg13418795 | GUCY2D | –0.01 (0.00) | 1.9 × 10–5 | –0.01 (0.00) | 8.8 × 10–5 | –0.01 (0.00) | 9.9 × 10–5 | –0.01 (0.00) | 2.9 × 10–5 | –0.01 (0.01) | 2.7 × 10–3 |
| cg05552796 | NA | 0.00 (0.00) | 2.0 × 10–5 | 0.00 (0.00) | 2.4 × 10–5 | 0.01 (0.00) | 1.2 × 10–4 | 0.00 (0.00) | 1.4 × 10–4 | –0.00 (0.00) | 1.9 × 10–2 |
| cg04515311 | NA | –0.01 (0.00) | 2.1 × 10–5 | –0.01 (0.00) | 1.0 × 10–4 | –0.01 (0.00) | 4.5 × 10–3 | –0.01 (0.00) | 9.4 × 10–5 | –0.01 (0.01) | 7.4 × 10–2 |
| cg12245321 | NA | –0.01 (0.00) | 2.1 × 10–5 | –0.01 (0.00) | 2.8 × 10–5 | –0.01 (0.00) | 2.1 × 10–3 | –0.01 (0.00) | 3.3 × 10–5 | –0.01 (0.01) | 9.3 × 10–4 |
| cg07719772 | NA | 0.02 (0.01) | 2.7 × 10–5 | 0.02 (0.01) | 1.4 × 10–5 | 0.02 (0.01) | 1.3 × 10–3 | 0.02 (0.01) | 4.9 × 10–6 | 0.01 (0.01) | 1.1 × 10–1 |
| Model A: adjusted for age, sex, batch, and blood cell fractions (66 exposed vs. 66 nonexposed). Model B: adjusted for current participant smoking in addition to age, sex, batch, and blood cell fractions (63 exposed vs. 65 nonexposed). Model C: adjusted for current participant and parent smoking in addition to age, sex, batch, and blood cell fractions (62 exposed vs. 63 nonexposed). Model D: current participant smokers excluded; adjusted for age, sex, batch, and blood cell fractions (60 exposed vs. 61 nonexposed). Model E: current participant and/or parent smokers excluded; adjusted for age, sex, batch, and blood cell fractions (18 exposed vs. 47 nonexposed). Differential DNAm between adolescents exposed and nonexposed prenatally to maternal cigarette smoking was assessed with linear regression using M-values at 473,395 CpGs. The results are presented as adjusted mean differences in DNAm β-values between exposed and nonexposed individuals (DNAm β difference) and their respective SEs and uncorrected (nominal) p-values from the linear regression described above. Chromosome position (indicated by chromosome number followed by position) is based on the NCBI Human Reference Genome Assembly Build 37.p10 (http://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.22/). *Indicates FDR-significance (p < 0.05). | |||||||||||
Next, we examined whether current cigarette smoking by the adolescent participants or by their parents confounded these associations. In our sample, only 5 of 66 exposed and 2 of 66 nonexposed (Table 1) adolescents reported “smoking at least 1 cigarette in the last 30 days.” Associations of prenatal maternal smoking and differential DNAm were comparable in models that also adjusted for participant smoking (Figure 1A, Model B) or that excluded current participant smokers (Figure 1A, model D). Adjusting for current smoking by parents as well as by the participants had little influence on point estimates, though p-values were increased somewhat: 2 of the 5 CpGs remained EWAS-significant, whereas the remaining 3 showed p < 1.5 × 10–6 (Figure 1A and Table 2, model C). Excluding adolescents who were current smokers or whose parents were current smokers reduced the sample size from 66 exposed and 66 nonexposed to 18 exposed and 47 nonexposed. Nevertheless, all 5 CpGs remained significant (at p < 0.05) even in this smaller subsample (Figure 1A and Table 2, model E).
Replication and persistence of differential DNAm associated with prenatal smoke exposure in the ALSPAC newborns, children, and adolescents. The replication and persistence studies were carried out in an independent sample, the ALSPAC cohort (Boyd et al. 2013) (see Supplemental Material, Table S1). Specifically, we tested whether the 5 EWAS-significant CpGs identified in the discovery SYS sample of adolescents were also differentially methylated in exposed versus nonexposed newborns (39 vs. 402), children (7 years; 39 vs. 408), and adolescents (17 years; 13 vs. 206) from the ALSPAC cohort. We found that all 5 CpGs were associated with prenatal smoke exposure at birth and during childhood (Table 3, models A´ and B´, respectively)—and 4 of the 5 CpGs were associated with prenatal smoke exposure during adolescence (Table 3 model D´). The direction and magnitude of differential DNAm at all three developmental time points for all 5 CpGs (Figure 1B) was consistent with the findings in the discovery cohort (Figure 1A). These results remained significant (p < 0.05) after adjusting for exposure to secondhand smoke (i.e., current smoking by parents) during childhood (Table 3, model C´) and for current participant smoking and parent smoking during adolescence (Table 3, model E´), despite substantially reduced sample sizes (Figure 1B).
Table 3.
Prenatal smoke exposure–associated differential DNAm at EWAS-significant CpGs from the discovery SYS sample in newborns, children, and adolescents in the replication ALSPAC sample.
| CpG ID | Gene | Model A´ | Model B´ | Model C´ | Model D´ | Model E´ | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| DNAm β difference (SE) | p-Value | DNAm β difference (SE) | p-Value | DNAm β difference (SE) | p-Value | DNAm β difference (SE) | p-Value | DNAm β difference (SE) | p-Value | ||
| cg12803068 | MYO1G | 0.15 (0.03) | 5.4 × 10–10 | 0.17 (0.03) | 4.8 × 10–11 | 0.18 (0.04) | 1.6 × 10–7 | 0.19 (0.05) | 1.7 × 10–5 | 0.18 (0.07) | 3.4 × 10–3 |
| cg04180046 | MYO1G | 0.08 (0.01) | 7.7 × 10–12 | 0.04 (0.01) | 2.1 × 10–8 | 0.05 (0.01) | 4.9 × 10–7 | 0.06 (0.02) | 6.8 × 10–4 | 0.07 (0.03) | 1.1 × 10–2 |
| cg19089201 | MYO1G | 0.03 (0.01) | 2.6 × 10–4 | 0.06 (0.01) | 2.2 × 10–7 | 0.05 (0.02) | 6.8 × 10–4 | 0.07 (0.03) | 3.8 × 10–3 | 0.10 (0.04) | 1.2 × 10–2 |
| cg25949550 | CNTNAP2 | –0.01 (0.00) | 6.3 × 10–7 | –0.01 (0.00) | 4.0 × 10–4 | –0.01 (0.00) | 2.6 × 10–3 | –0.00 (0.00) | 1.6 × 10–1 | 0.00 (0.01) | 8.7 × 10–1 |
| cg22132788 | MYO1G | 0.05 (0.01) | 4.0 × 10–10 | 0.09 (0.02) | 5.3 × 10–13 | 0.09 (0.02) | 3.9 × 10–8 | 0.06 (0.03) | 1.9 × 10–3 | 0.08 0.04 | 2.8 × 10–3 |
| Model A´: at birth (39 exposed vs. 402 nonexposed). Model B´: at age 7 years (39 exposed vs. 408 nonexposed). Model C´: at age 7 years (36 exposed vs. 380 nonexposed); additionally adjusted for current parent smoking. Model D´: at age 17 years (13 exposed vs. 206 nonexposed). Model E´: at age 17 years (8 exposed vs. 108 nonexposed); additionally adjusted for current parent and adolescent smoking differential DNAm between individuals exposed and nonexposed prenatally to maternal cigarette smoking was assessed with linear regression using M-values at 5 CpGs that were EWAS-significant in the SYS discovery cohort. All models (A´–E´) were adjusted for age, sex, maternal education, household social class, and batch. The results are presented as adjusted mean differences in DNAm β-values between exposed and nonexposed individuals (DNAm β difference) and their respective SEs and uncorrected (nominal) p-values from the linear regression described above. Chromosome position (indicated by chromosome number followed by position) is based on the NCBI Human Reference Genome Assembly Build 37.p10 (http://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.22/). | |||||||||||
Discussion
The results of the present study suggest that prenatal smoke exposure is associated with modifications of DNAm that persist in the exposed offspring for years—from birth until at least adolescence—and that these associations remain after adjusting for blood-cell fractions, current smoking by adolescent participant, and current smoking by parents.
To our knowledge, our study is the first to report that differential DNAm associated with prenatal smoke exposure may persist in the exposed offspring for years after their birth. The MoBa study (Norwegian Mother and Child Cohort Study) reported that prenatal smoke exposure was associated with differential DNAm at birth (Joubert et al. 2012). Importantly, all 5 CpGs we found to be differentially methylated in the present study at birth and during childhood (in the ALSPAC replication cohort) and during adolescence (in the SYS discovery sample and in the ALSPAC cohort) were also differentially methylated in the MoBa study at birth (Joubert et al. 2012), with the differences between exposed and nonexposed being of the same direction and similar magnitude (Table 4). Other CpGs with prenatal smoke exposure–associated differential DNAm in the MoBa study were also significant at the strict look-up level (26 CpGs, p < 1.92 × 10–3) in the SYS adolescents, including CpGs in CYP1A1, AHRR, and GFI1, and the gene-unmapped CpG 04598670 (Table 4). Differential DNAm across the 26 MoBa CpGs was correlated between the SYS and MoBa samples (r2 = 0.53, p < 0.0001) (see Supplemental Material, Figure S2). Taken together, these studies indicate that prenatal smoke exposure is associated with specific DNAm modifications observed at birth (MoBa, ALSPAC at birth) and that some of these modifications may persist in the exposed offspring until at least adolescence (SYS, ALSPAC).
Table 4.
Look-up in the SYS discovery sample of adolescents of the 26 CpGs identified previously at p < 1.10–7 in the MoBa sample of newborns.
| CpG ID | Chromosome | Gene | MoBa newbornsa | SYS adolescents | ||
|---|---|---|---|---|---|---|
| DNAm β difference (%) | p-Value | DNAm β difference (%) | p-Value | |||
| cg12803068 | 7 | MYO1Gb | 8.3 | 8.51 × 10–19 | 9.2 | 3.34 × 10–11b |
| cg22549041 | 15 | CYP1A1b | 7.2 | 4.52 × 10–9 | 4.3 | 9.70 × 10–5b |
| cg18092474 | 15 | CYP1A1b | 5.9 | 1.10 × 10–8 | 2.9 | 3.60 × 10–4b |
| cg04180046 | 7 | MYO1Gb | 5.3 | 8.76 × 10–20 | 7.2 | 3.90 × 10–11b |
| cg12477880 | 21 | RUNX1 | 4.6 | 1.02 × 10–9 | 6.2 | 3.46 × 10–3 |
| cg05549655 | 15 | CYP1A1b | 3.5 | 2.96 × 10–10 | 2.9 | 7.57 × 10–6b |
| cg11924019 | 15 | CYP1A1b | 3.2 | 2.62 × 10–8 | 2.8 | 3.35 × 10–5b |
| cg23067299 | 5 | AHRRb | 3.2 | 4.21 × 10–10 | 1.1 | 5.74 × 10–4b |
| cg22132788 | 7 | MYO1Gb | 2.8 | 1.98 × 10–18 | 5.5 | 8.41 × 10–9b |
| cg19089201 | 7 | MYO1Gb | 1.4 | 3.22 × 10–10 | 4.3 | 1.58 × 10–9b |
| cg03346806 | 8 | NA | –1.5 | 3.08 × 10–8 | –0.3 | 3.44 × 10–1 |
| cg25949550 | 7 | CNTNAP2b | –1.8 | 4.15 × 10–30 | –2.0 | 3.97 × 10–9b |
| cg18655025 | 14 | TTC7B | –1.8 | 2.07 × 10–8 | –0.7 | 1.32 × 10–1 |
| cg03991871 | 5 | AHRR | –2.2 | 2.04 × 10–11 | –2.6 | 7.17 × 10–2 |
| cg21161138 | 5 | AHRR | –2.3 | 1.52 × 10–11 | –1.1 | 6.17 × 10–2 |
| cg04598670 | 7 | NAb | –3 | 1.29 × 10–11 | –3.4 | 1.27 × 10–4b |
| cg10399789 | 1 | GFI1 | –3.7 | 4.08 × 10–13 | 0.6 | 2.09 × 10–1 |
| cg06338710 | 1 | GFI1 | –5.8 | 1.34 × 10–18 | 0.5 | 5.94 × 10–1 |
| cg09662411 | 1 | GFI1 | –6.6 | 2.26 × 10–20 | 0.0 | 8.37 × 10–1 |
| cg18316974 | 1 | GFI1 | –7.1 | 6.43 × 10–24 | 0.5 | 8.90 × 10–1 |
| cg05575921 | 5 | AHRR | –7.5 | 2.85 × 10–39 | –1.7 | 1.68 × 10–2 |
| cg11715943 | 6 | HLA-DPB2 | –7.5 | 1.00 × 10–8 | –0.7 | 7.10 × 10–1 |
| cg14179389 | 1 | GFI1b | –8.6 | 5.38 × 10–28 | –4.7 | 5.13 × 10–5b |
| cg12876356 | 1 | GFI1 | –11.9 | 2.29 × 10–30 | –0.5 | 6.63 × 10–1 |
| cg18146737 | 1 | GFI1 | –12.3 | 2.42 × 10–30 | 0.6 | 8.30 × 10–1 |
| cg09935388 | 1 | GFI1 | –13.7 | 1.05 × 10–38 | –1.5 | 1.31 × 10–1 |
| DNA methylation (DNAm) β-value differences between exposed and nonexposed individuals are shown. The SYS model A (adjusted for age, sex, batch, and blood cell fractions) is presented. aJoubert et al. (2012). bCpGs replicated in the discovery SYS sample at strict look-up–level significance (26 tests, p < 1.92 × 10–3). | ||||||
The EWAS-significant CpGs were located in MYO1G and CNTNAP2. MYO1G encodes a plasma membrane–associated class I myosin that is expressed abundantly in hematopoietic cells; it regulates cell elasticity and migration (Olety et al. 2010; Patino-Lopez et al. 2010). CNTNAP2 encodes a member of the neurexin family of cell-adhesion molecules that plays a critical role in brain development (Anderson et al. 2012), and has been implicated in a number of neurodevelopmental disorders, including autism (Anney et al. 2012). One of the main functions of DNAm is the regulation of gene expression. The 4 EWAS-significant CpGs in MYO1G (but not the one in CNTNAP2) were located in a region containing binding sites for a number of transcription factors and enhancers (Encyclopedia of DNA Elements at UCSC, 2003–2012; http://genome.ucsc.edu/ENCODE) and thus might alter mRNA expression of the gene. This possibility remains to be studied, however.
In addition to prenatal exposure to cigarette smoke, active smoking has been associated with differential DNAm in peripheral blood cells (Breitling et al. 2011; Philibert et al. 2012; Shenker et al. 2013; Zeilinger et al. 2013). In our discovery sample, only 5% of adolescents reported current cigarette smoking, and excluding these participants or adjusting for their current smoking had little influence on model estimates. Further, we considered that postnatal exposure to secondhand smoke might be a confounder of the associations between prenatal smoke exposure and DNAm during childhood and adolescence. Although secondhand smoke exposure has not been reported to affect DNAm in peripheral blood cells, some evidence exists that it is associated with differential DNAm in cancer tissues (though to a lesser degree than variations observed in active smokers) (Scesnaite et al. 2012; Wilhelm-Benartzi et al. 2011). Our results showed, however, that the differential DNAm associated with prenatal smoke exposure seen in the SYS adolescents was present (albeit somewhat attenuated) even after additional adjusting for secondhand smoke exposure to parental smoking. Importantly, the prenatal smoke exposure–associated differential DNAm we observed in the ALSPAC replication sample also remained significant after additional adjusting for adolescents’ smoking and their exposure to secondhand smoke generated by the parents. Across both SYS (adolescence) and ALSPAC (birth, childhood, adolescence) samples and all statistical models we tested (with varying numbers of available participants), the magnitude of DNAm differences between exposed and nonexposed individuals was generally consistent. Collectively, these results support the postnatal persistence of prenatal smoke exposure–associated DNAm modifications in the exposed offspring.
Conclusion
Our results suggest that prenatal smoke exposure may induce reproducible alterations of DNAm in the offspring, and that some of these alterations may persist for many years—well into adolescence. The potential implications of such epigenetic modifications for the “programming” of health require further study. Likewise, their possible utility as blood biomarkers of prenatal environmental exposures, such as maternal cigarette smoking, needs further exploration. Promotion of smoking cessation remains imperative (Ng et al. 2014); gaining knowledge about molecular mechanisms capable of influencing health trajectories in the exposed population is critical for the prevention and treatment of associated disorders.
Supplemental Material
Footnotes
We thank the following individuals for their contributions in acquiring data for the Saguenay Youth Study (SYS): M. Bernard (database architect, The Hospital for Sick Children) and H. Simard, and her team of research assistants (Cégep de Jonquière). We also thank M. Lemire (Ontario Institute for Cancer Research) for his assistance with data analysis. We also thank The Centre for Applied Genomics, The Hospital for Sick Children for assistance with EWAS (epigenome-wide association study) statistical analyses. In addition, we thank the entire Avon Longitudinal Study of Parents and Children (ALSPAC) team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists, and nurses. Finally, we thank T. Hudson (Ontario Institute for Cancer Research) and A. Patterson (The Hospital for Sick Children) for critical review of the manuscript.
The Canadian Institutes of Health Research and the Heart and Stroke Foundation of Canada provide funding for the SYS. The McLaughlin Centre at the University of Toronto provided supplementary funds for the DNA methylation studies in the SYS. The UK Medical Research Council and the Wellcome Trust (grant 092731), and the University of Bristol provide core support for ALSPAC. L.B. is a junior research scholar from the Fonds de Recherche du Quebec (FRQS) and a member of the FRQS-funded Centre de recherche clinique Étienne-Le Bel.
The authors declare they have no actual or potential competing financial interests.
References
- Anderson GR, Galfin T, Xu W, Aoto J, Malenka RC, Südhof TC. Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development. Proc Natl Acad Sci USA. 2012;109:18120–18125. doi: 10.1073/pnas.1216398109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anney R, Klei L, Pinto D, Almeida J, Bacchelli E, Baird G, et al. Individual common variants exert weak effects on the risk for autism spectrum disorders. Hum Mol Genet. 2012;21:4781–4792. doi: 10.1093/hmg/dds301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARIES. ARIES: Accessible Resource for Integrated Epigenomic Studies. Homepage. 2014. Available: http://www.ariesepigenomics.org.uk/ [accessed 24 June 2013] [DOI] [PMC free article] [PubMed]
- Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA Methylation microarrays. Bioinformatics. 2014;30:1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Boyd A, Golding J, Macleod J, Lawlor DA, Fraser A, Henderson J, et al. Cohort profile: the ‘children of the 90s’—the index offspring of the Avon Longitudinal Study of Parents and Children. Int J Epidemiol. 2013;42:111–127. doi: 10.1093/ije/dys064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet. 2011;88:450–457. doi: 10.1016/j.ajhg.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8:203–209. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A, et al. 2007DNA damage, homology-directed repair, and DNA methylation. PLoS Genet 3e110; 10.1371/journal.pgen.0030110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S, et al. 2012Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol 13R43; 10.1186/gb-2012-13-6-r43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di YP, Zhao J, Harper R. Cigarette smoke induces MUC5AC protein expression through the activation of Sp1. J Biol Chem. 2012;287:27948–27958. doi: 10.1074/jbc.M111.334375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, et al. 2010Comparison of beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 11587; 10.1186/1471-2105-11-587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulk C, Dolinoy DC. Timing is everything: The when and how of environmentally induced changes in the epigenome of animals. Epigenetics. 2011;6:791–797. doi: 10.4161/epi.6.7.16209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Jacobsen SE, Reik W. Epigenetic reprogramming in plant and animal development. Science. 2010;330:622–627. doi: 10.1126/science.1190614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Lin IG, Hsieh CL. Protein binding protects sites on stable episomes and in the chromosome from de novo methylation. Mol Cell Biol. 2001;21:3416–3424. doi: 10.1128/MCB.21.10.3416-3424.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. 2012DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 1386; 10.1186/1471-2105-13-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Okuka M, Lu W, Tsibris JC, McLean MP, Keefe DL, et al. Telomere shortening and DNA damage of embryonic stem cells induced by cigarette smoke. Reprod Toxicol. 2013;35:89–95. doi: 10.1016/j.reprotox.2012.07.003. [DOI] [PubMed] [Google Scholar]
- Jeltsch A. Molecular enzymology of mammalian DNA methyltransferases. Curr Top Microbiol Immunol. 2006;301:203–225. doi: 10.1007/3-540-31390-7_7. [DOI] [PubMed] [Google Scholar]
- Joubert BR, Håberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, et al. 2012450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect 1201425–1431.; 10.1289/ehp.1205412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkowska RZ, Jurkowski TP, Jeltsch A. Structure and function of mammalian DNA methyltransferases. Chembiochem. 2011;12:206–222. doi: 10.1002/cbic.201000195. [DOI] [PubMed] [Google Scholar]
- Lambers DS, Clark KE. The maternal and fetal physiologic effects of nicotine. Semin Perinatol. 1996;20:115–126. doi: 10.1016/s0146-0005(96)80079-6. [DOI] [PubMed] [Google Scholar]
- Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics. 1977;33:159–174. [PubMed] [Google Scholar]
- Lee KW, Pausova Z.2013Cigarette smoking and DNA methylation. Front Genet 4132; 10.3389/fgene.2013.00132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin HL, Moran JV. Dynamic interactions between transposable elements and their hosts. Nat Rev Genet. 2011;12:615–627. doi: 10.1038/nrg3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer BA, Wallace AM, Brinckerhoff CE, D’Armiento JM. Identification of a cigarette smoke-responsive region in the distal MMP-1 promoter. Am J Respir Cell Mol Biol. 2009;40:4–12. doi: 10.1165/rcmb.2007-0310OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortusewicz O, Schermelleh L, Walter J, Cardoso MC, Leonhardt H. Recruitment of DNA methyltransferase 1 to DNA repair sites. Proc Natl Acad Sci USA. 2005;102:8905–8909. doi: 10.1073/pnas.0501034102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng M, Freeman MK, Fleming TD, Robinson M, Dwyer-Lindgren L, Thomson B, et al. Smoking prevalence and cigarette consumption in 187 countries, 1980–2012. JAMA. 2014;311:183–192. doi: 10.1001/jama.2013.284692. [DOI] [PubMed] [Google Scholar]
- Oken E, Levitan EB, Gillman MW. Maternal smoking during pregnancy and child overweight: systematic review and meta-analysis. Int J Obes (Lond) 2008;32:201–210. doi: 10.1038/sj.ijo.0803760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olety B, Wälte M, Honnert U, Schillers H, Bähler M. Myosin 1G (Myo1G) is a haematopoietic specific myosin that localises to the plasma membrane and regulates cell elasticity. FEBS Lett. 2010;584:493–499. doi: 10.1016/j.febslet.2009.11.096. [DOI] [PubMed] [Google Scholar]
- Patino-Lopez G, Aravind L, Dong X, Kruhlak MJ, Ostap EM, Shaw S. Myosin 1G is an abundant class I myosin in lymphocytes whose localization at the plasma membrane depends on its ancient divergent pleckstrin homology (PH) domain (Myo1PH). J Biol Chem. 2010;285:8675–8686. doi: 10.1074/jbc.M109.086959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pausova Z, Paus T, Abrahamowicz M, Almerigi J, Arbour N, Bernard M, et al. Genes, maternal smoking, and the offspring brain and body during adolescence: design of the Saguenay Youth Study. Hum Brain Mapp. 2007;28:502–518. doi: 10.1002/hbm.20402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465:721–727. doi: 10.1038/nature09230. [DOI] [PubMed] [Google Scholar]
- Philibert RA, Beach SR, Brody GH. Demethylation of the aryl hydrocarbon receptor repressor as a biomarker for nascent smokers. Epigenetics. 2012;7:1331–1338. doi: 10.4161/epi.22520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power C, Atherton K, Thomas C. Maternal smoking in pregnancy, adult adiposity and other risk factors for cardiovascular disease. Atherosclerosis. 2010;211:643–648. doi: 10.1016/j.atherosclerosis.2010.03.015. [DOI] [PubMed] [Google Scholar]
- Price ME, Cotton AM, Lam LL, Farré P, Emberly E, Brown CJ, et al. 2013Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin 64; 10.1186/1756-8935-6-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relton CL, Davey Smith G.2010Epigenetic epidemiology of common complex disease: prospects for prediction, prevention, and treatment. PLoS Med 7e1000356; 10.1371/journal.pmed.1000356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, et al. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics. 2011;6:692–702. doi: 10.4161/epi.6.6.16196. [DOI] [PubMed] [Google Scholar]
- Satta R, Maloku E, Zhubi A, Pibiri F, Hajos M, Costa E, et al. Nicotine decreases DNA methyltransferase 1 expression and glutamic acid decarboxylase 67 promoter methylation in GABAergic interneurons. Proc Natl Acad Sci USA. 2008;105:16356–16361. doi: 10.1073/pnas.0808699105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scesnaite A, Jarmalaite S, Mutanen P, Anttila S, Nyberg F, Benhamou S, et al. Similar DNA methylation pattern in lung tumours from smokers and never-smokers with second-hand tobacco smoke exposure. Mutagenesis. 2012;27:423–429. doi: 10.1093/mutage/ger092. [DOI] [PubMed] [Google Scholar]
- Shahrzad S, Bertrand K, Minhas K, Coomber BL. Induction of DNA hypomethylation by tumor hypoxia. Epigenetics. 2007;2:119–125. doi: 10.4161/epi.2.2.4613. [DOI] [PubMed] [Google Scholar]
- Shenker NS, Polidoro S, van Veldhoven K, Sacerdote C, Ricceri F, Birrell MA, et al. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet. 2013;22:843–851. doi: 10.1093/hmg/dds488. [DOI] [PubMed] [Google Scholar]
- Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, et al. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature. 2012;484:339–344. doi: 10.1038/nature10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Substance Abuse and Mental Health Services Administration. National Survey on Drug Use and Health, 2012 (ICPSR 34933). Washington, DC:U.S. Department of Health and Human Services. 2012. Available: http://www.icpsr.umich.edu/icpsrweb/SAMHDA/studies/34933 [accessed 24 June 2013]
- Syme C, Abrahamowicz M, Mahboubi A, Leonard GT, Perron M, Richer L, et al. Prenatal exposure to maternal cigarette smoking and accumulation of intra-abdominal fat during adolescence. Obesity (Silver Spring) 2010;18:1021–1025. doi: 10.1038/oby.2009.354. [DOI] [PubMed] [Google Scholar]
- Tang M, Xu W, Wang Q, Xiao W, Xu R. Potential of DNMT and its epigenetic regulation for lung cancer therapy. Curr Genomics. 2009;10:336–352. doi: 10.2174/138920209788920994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touleimat N, Tost J. Complete pipeline for Infinium® Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics. 2012;4:325–341. doi: 10.2217/epi.12.21. [DOI] [PubMed] [Google Scholar]
- Tsukahara S, Kobayashi A, Kawabe A, Mathieu O, Miura A, Kakutani T. Bursts of retrotransposition reproduced in Arabidopsis. Nature. 2009;461:423–426. doi: 10.1038/nature08351. [DOI] [PubMed] [Google Scholar]
- Wilhelm-Benartzi CS, Christensen BC, Koestler DC, Houseman EA, Schned AR, Karagas MR, et al. Association of secondhand smoke exposures with DNA methylation in bladder carcinomas. Cancer Causes Control. 2011;22:1205–1213. doi: 10.1007/s10552-011-9788-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilinger S, Kühnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, et al. 2013Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One 8e63812; 10.1371/journal.pone.0063812 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.