

Abstract
Secreted from adipose tissue, adiponectin is a vital endocrine hormone that acts in glucose metabolism, thereby establishing its crucial role in diabetes, obesity, and other metabolic disease states. Insulin exposure to primary adipocytes cultured in static conditions has been shown to stimulate adiponectin secretion. However, conventional, static methodology for culturing and stimulating adipocytes falls short of truly mimicking physiological environments. Along with decreases in experimental costs and sample volume, and increased temporal resolution, microfluidic platforms permit small-volume flowing cell culture systems, which more accurately represent the constant flow conditions through vasculature in vivo. Here, we have integrated a customized primary tissue culture reservoir into a passively operated microfluidic device made of polydimethylsiloxane (PDMS). Fabrication of the reservoir was accomplished through unique PDMS “landscaping” above sampling channels, with a design strategy targeted to primary adipocytes to overcome issues of positive cell buoyancy. This reservoir allowed three-dimensional culture of primary murine adipocytes, accurate control over stimulants via constant perfusion, and sampling of adipokine secretion during various treatments. As the first report of primary adipocyte culture and sampling within microfluidic systems, this work sets the stage for future studies in adipokine secretion dynamics.
Introduction
Due to the recent, overwhelming increases in the prevalence of diabetes, obesity, metabolic syndrome, and the many related disorders in the developed world, it is imperative to push the limits in understanding the fundamentals of hormone secretion that control the function of endocrine systems. Until recently, adipose tissue was only considered a fat storage system, but studies have found that adipose plays a significant endocrinological role in maintaining glucose homeostasis 1–6. Adipocytes, the main cell type of the adipose tissue, respond to extracellular stimuli and secrete hormones that function in glucose metabolism, hunger, and the immune system 3. Secreted from adipocytes, adiponectin is a 30 kDa protein hormone which exists in the bloodstream in three major multimeric forms – trimeric, low molecular weight (LMW), and high molecular weight (HMW). Although its ties with obesity and insulin resistance are part of ongoing research, adiponectin’s main function is thought to be in glucose homeostasis and improving insulin sensitivity 7–9.
To further probe the metabolic function of adipocytes, studies in acute secretion and secretory dynamics are imperative. However, conventional methods for sampling from primary adipocytes involve culturing the cells in a collagen matrix on relatively large volume well-plates under static conditions. Although this method has been proven useful in sampling from adipocytes for prolonged periods of time, studies on acute secretion are difficult to impossible, depending on the temporal resolution needed. Additionally, due to the volume or number of cells needed for well-plates and downstream assays, multiple animals (typically mice or rats) are required for even simple studies 10, 11. Offering alternatives to static sampling methodology, microfluidic systems not only allow for significant reductions in cell numbers, sample volumes, and reagents needed, but such constant-flow culture systems are arguably better representations of in vivo, biological conditions 12–14, 15, 16.
In order to properly integrate primary adipocyte culture and sampling onto a microfluidic device, certain challenges must be addressed. Adipocytes have inherent positive buoyancy due to lipid storage, causing them to float in aqueous media. Matrices such as collagen can be used to anchor the cells and negate cell buoyancy in the well-plate format; however, this culture matrix can easily clog microfluidic channels. Another hurdle, particularly with extracted primary adipocytes, is that cell density is inherently low from the sizeable lipid storage per cell. Device engineering is thus driven toward larger culturing regions that are closely linked with microfluidic sampling channels yet prevent channel clogging. In this work, we address these issues with methodology for “landscaping” of the polydimethylsiloxane (PDMS) device toward primary adipocyte culture and secretion sampling. Beginning with concepts outlined by Desai et al. 17, plastic inserts were fabricated as masters for customized PDMS reservoirs in passively operated microdevices 18. This novel microfluidic interface promotes 3D culture of small numbers of adipocytes, allows constant perfusion of the adipocytes cultured on the device, and facilitates small-volume sampling. As the first to report primary adipocyte culture and sampling on a microfluidic system, this work represents an important step toward high resolution adipokine secretion dynamics to help improve our understanding of the role of adipose tissue in the endocrine system.
Experimental
Reagents and Materials
Polydimethylsiloxane (PDMS) precursors, Sylgard® 184 elastomer base and curing agent, were obtained from Dow Corning (Midland, Maryland, USA). Silicon wafers were obtained from Silicon Inc. (Boise, ID, USA), and SU-8 photoresist and developer were purchased from Microchem (Newton, Massachusetts, USA). Tubing for interfacing syringes and devices was obtained from Small Parts (TGY-020-5C; 0.02 in. ID, 0.06 in. OD, 0.02 in. wall), as were blunt ended needles (NE-223PL-C 22G).
NaCl, CaCl2•2H2O, bovine serum albumin (BSA), NaOH, and fetal bovine serum (FBS) were purchased from VWR (West Chester, Pennsylvania, USA). KH2PO4, NaH2PO4, 4-2-hydroxyethyl-1-piperazineethanesulfonic acid (HEPES), nystatin, insulin, penicillin/streptomycin, and D-glucose were obtained from Sigma-Aldrich (St. Louis, Missouri, USA). MgSO4•7H2O was purchased from Fischer Scientific (Waltham, MA, USA). LipidTOX™, 4′,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI), Dulbecco’s Modified Eagle Medium (DMEM), Minimum Essential Media (MEM), and MEM non-essential amino acids were obtained from Gibco Invitrogen Life Technologies (Grand Island, NY, USA). PureCol collagen was purchased from Advanced Biomatrix (San Diego, CA, USA).
Microfluidic Interface Fabrication
In this section that mirrors the workflow depicted in Fig. 1, we describe the stepwise fabrication of the customized adipocyte culturing interface. A 1-cm diameter plastic insert was fabricated using a two-part epoxy resin (Smooth-Cast 310; Smooth-On plastics) 17, 18. As shown in Fig. 1A, molds for three support pegs were created using a 1-mm thickness PDMS slab as a template. Three holes (1 mm) were punched in the slab in a triangular pattern, and the slab was plasma oxidized and irreversibly bonded to a glass substrate. A cylindrical piece of PDMS (2.5-mm height, 2-mm diameter) was reversibly sealed by pressing to the PDMS slab in the center of the triangular pattern. A 7-mm reservoir hole was then punched into a thick (~2 cm) PDMS slab, which was centered on the triangular pattern and reversibly sealed by pressing to the thin PDMS slab to finalize the mold for Smooth-Cast 310 (Fig. 1A). Silicone oil was used to pretreat the PDMS mold as a release agent. Smooth-Cast 310 precursors were degassed for 10 min then mixed in a 1:1 ratio. It was found that curing a thin layer of Smooth-Cast (~200 μL) at the bottom of the mold and into the three support pegs allowed for efficient degassing of the featured regions. Once the thin layer was partially cured, more degassed mixture was poured into the large reservoir and allowed to cure overnight. After curing the Smooth-Cast insert, the thick PDMS slab was carefully peeled away (Fig. 1B), and PDMS debris was removed from the bottom of the insert using an X-Acto™ knife. Finally, the insert was used as a template for landscaping the PDMS fluidic interface above microfluidic channels defined by a SU-8/silicon wafer pattern. As shown in Fig. 1B, the three pegs were used to define the thickness of the PDMS under the large fluidic reservoir, and the Smooth-Cast insert structure facilitated creation of a “moat” region for 3D culture of primary adipocytes in a collagen matrix. Prior to final bonding of the PDMS to a glass channel floor, additional access holes were punched, including a 1-mm diameter access hole to the microfluidic channel, centred through the 2-mm raised central region of the PDMS moat.
Figure 1.

Fabrication and use of the Smooth-Cast 310 insert. A) The PDMS mold for the insert served as an initial proxy for the final device, including the raised island and moat region customized for culture of primary adipocytes. B) The resultant Smooth-Cast insert was used to define a customized macro-to-micro interface to microfluidic channels for secretion sampling.
Microfluidic Channel Design and Fabrication
All devices were fabricated using PDMS as the channelled substrate and No. 1 glass coverslips as the floor substrate 19. An SU-8 patterned silicon master was fabricated using standard photolithography. The microfluidic channel layout (Fig. 2A) was designed in Adobe Illustrator software and sent to Fineline Imaging (Colorado Springs, CO) for photomask printing at 50 800 dpi resolution (negative image). Deeper channel patterns were 156 μm deep, 600 μm wide, and 1.5 mm in length; and shallow sampling channels were 15 μm deep, 60 μm wide, and 10.8 mm long. PDMS precursors were mixed in a 10:1 ratio (elastomer to curing agent) and degassed prior to curing. A few grams of uncured PDMS was poured onto the SU-8/silicon wafer, and a small amount was injected into the hole of the insert followed by removal of air bubbles. The insert was then carefully aligned around the channels and set directly onto the wafer into the layer of uncured PDMS. The remainder of uncured PDMS was slowly poured onto the wafer to ensure the inserts were not disturbed, and the entire assembly was placed into an oven for curing overnight at 65 °C. Periodically, insert positioning was monitored to ensure alignment during PDMS curing. Following curing, the insert was removed, and the resulting device contained a primary adipocyte culture region, with a fluidic “moat,” patterned into the PDMS above the microfluidic channels. A cross-section of this PDMS landscaping is shown in Fig. 2B.
Figure 2.
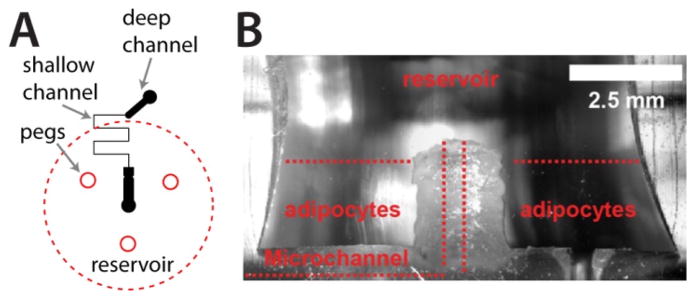
Macro-to-micro interface for primary tissue culture and sampling. A) The microfluidic channel layout is depicted in black, with deep (156 μm) and shallow (15 μm) channels included. Insert peg locations and reservoir placement are shown in red. B) A cross section of a typical PDMS interface, highlighting the “moat” region for culture of primary adipocytes that addresses cell buoyancy issues and allows passive secretion sampling into the microchannel.
Primary Adipocyte Isolation and Culture
Epididymal fat pads were obtained by surgical extraction from 18–20 week old male C57BL/6J mice (Jackson Laboratories, Bar Harbor, Maine). The adipose tissue was placed in a 60 mm plate to obtain the wet weight of the tissue. Fat pads were minced and digested with type I collagenase at 37 °C with shaking for 30 min 10, 11, 20. After washing with HEPES buffer, the suspension was then filtered through a 210 μm filter (spectra mesh). The solution was centrifuged at 900 rpm for 6 min at 4 °C, and the infranatant was removed. Adipocytes were re-suspended in HEPES buffer, centrifuged, and the infranatant was removed for a second wash. Cells were subsequently suspended and incubated in a serum media mix consisting of DMEM containing 1.2 % of nystatin, penicillin/streptomycin, fetal bovine serum, and MEM nonessential amino acids for 30 min at 37 °C in 5% CO2.
Culture and Sampling of Adipocytes
To culture the cells onto a 6-well plate, 450 μL of collagen matrix (Pure-Col collagen tablets and 1 % MEM) and 90 μL of cells were pipetted directly into the center of the well. The plate was shaken to mix and distribute the collagen cell mixture throughout the well 10, 11, 20. After a 50-min incubation for collagen coagulation, the cells were placed in 2 mL of serum media, and allowed to incubate overnight at 37 °C with 5% CO2 (conditions for all incubations to follow). Cells were then washed with 1 mL of fresh serum media and allowed to incubate again for 1 h. Next, the 1 mL of serum-containing media was replaced with 1 mL of serum free media (SFM; DMEM with FBS replaced by 0.2 % BSA), and cells were incubated for 3 h. The SFM was replaced after 3 h with fresh SFM, and cells were treated with 100 nM insulin; untreated cells were considered the controls. Samples were collected after 1 h and stored at −20 °C for subsequent adiponectin quantitation.
For the microfluidic systems, 30 μL of collagen matrix was combined with 10 μL of cells in PCR tubes. The cell/collagen mixture was gently pipetted up and down to mix, then loaded into the moat region of the microfluidic device. After a 50 min incubation at 37 °C with 5% CO2, cells were placed in serum media and incubated overnight, followed by one wash with serum media and another 1 h incubation. Media was replaced with SFM and incubated for 3 h. 200 μL of fresh SFM was then added for initial priming and interfacing of the tubing (Tygon), manifold, and vacuum syringe. Solution was drawn through the channels and tubing using vacuum driven flow by a 100-mL glass syringe. Once solution entered the tubing, the vacuum was released, meniscuses were marked on each tube, and the cells were allowed to incubate for 10 min before sampling. Stimulation of the cells with 100 nM insulin or 10 μM niacin was immediately followed by vacuum-based flow, settling at a flow rate of 40 μL h−1 18. Control cells were left untreated. Perfusion of the cells continued for 1 h before the vacuum was released. Distances were recorded between the starting and ending meniscuses to confirm individual sample volumes, and secretory samples (~40 μL each) were collected into PCR tubes and stored at −20 °C for subsequent adiponectin quantitation.
Adiponectin Quantitation
Total secreted adiponectin was quantified in collected plate and microdevice samples using Mouse Adiponectin ELISA Kits (EZMADP-60K; Millipore), according to the manufacturer’s instructions. An 8-channel pipettor (Eppendorf) and an automated microplate shaker (VWR) and well washer (Wellwash Versa; Thermo Scientific) were used to minimize measurement error. Enzymatic product absorbance was measured at both 450 and 590 nm with a DTX 880 multimode microplate reader (Beckman Coulter). Population means of these measurements were compared using two-tailed Student’s t tests with equal variances using Microsoft Excel. Values for the probability, p, that the means are equal are listed in the text and figures. Data sets with p ≤ 0.05 were considered statistically different.
Adipocyte Imaging
Cells were first loaded into surrogate PDMS reservoirs (to facilitate high magnification imaging) then fixed in a 3.7 % solution of formaldehyde for 30 min at 25 °C and then incubated with LipidTOX™ (diluted per manufacturer’s instructions) for 30 min at 25° C with gentle rocking. After washing the cells with PBS buffer, a 300 nM solution of DAPI was added, and cells were incubated for 5 min at 25° C with gentle rocking. Cells were then washed 3 times with PBS buffer prior to imaging with a Nikon Ti-E wide-field inverted fluorescence microscope. LipidTOX™ emission was imaged at 525 ± 25 nm after excitation at 470 ± 20 nm, and DAPI emission was imaged at 460 ± 25 nm after excitation at 350 ± 25 nm. Collected images were overlaid using ImageJ (NIH) software21, and false coloring was applied for visualization (green for LipidTOX, blue for DAPI).
Results and Discussion
Microfluidic Interface Design
Interfacing strategies for microfluidic devices, particularly those made from PDMS, have been largely limited to straight, punched reservoirs or tubing connectors that intersect with the microfluidic channels near the bottom of the device. However, since microchannels (~50 μm depths) account for <1% of the typical device thickness (~7 mm in our laboratory), the bulk of the material is often underutilized. In this work, we exploit millimetre-scale patterning in the bulk material, referred to as PDMS landscaping, giving us exquisite control over the macro-to-micro interface. This way, an easily accessible 3D culture region in close proximity with microfluidic sampling channels could be designed specifically for primary adipocytes, allowing positive cell buoyancy issues to be overcome.
For macro-to-micro interfacing, a Smooth-Cast 310 plastic insert was fabricated as shown in Fig. 1A (details in Experimental section). The insert was designed with three pegged feet to define the thickness of the PDMS remaining above the channels, and it was cylindrical, with a small, recessed cylindrical region at the bottom. Using the insert as a template during PDMS device fabrication, this architecture defined a central, raised island of PDMS in the final device, surrounded by a PDMS “moat” for 3D adipocyte culture around the fluidic entrance to the microchannels (device cross section shown in Fig. 2B). Access to a single sampling microchannel was punched through the raised island region. When vacuum was applied at the outlet, solution flowed over the cells, from the moat region through the raised island, and into the channel. This allowed us to apply previously-developed passive microfluidic sampling methods 18 to a more challenging cell type, primary murine adipocytes. As shown herein, the raised island region prevented collagen or cells from entering and blocking the microchannel, which rendered previous generations of the devices unusable. The design allowed ample cell numbers to be cultured in collagen on the chip and eliminated buoyancy issues as well as spatial confinements that could limit cell density.
Microfluidic 3D Culture of Primary Adipocytes
Conventional static containers, such as Petri dishes or well-plates, fall short of mimicking genuine physiological systems, which have constant flow and intricate vasculature to supply nutrients and remove waste products. As such, microfluidic systems still have untapped potential that could significantly impact biological sciences, as they hold the promise of reproducing in vivo nutrient supply, blood flow, etc. 14, 22. Recent breakthroughs in organ-on-a-chip or human-on-a-chip platforms have provided evidence to this point and have further heightened interest within the scientific community 12, 13. However, there have been relatively few reports in which microfluidic technology has been employed for analysis of adipocytes 2, 23–25, and to our knowledge, no reports to date have integrated primary adipocyte culture into a microfluidic system—perhaps a result of the inherent difficulties noted here.
Using the custom plastic inserts shown in Fig. 1, as well as a modified approach for collagen-based 3D cell culture, we have successfully transplanted primary adipocytes from animals (C57BL/6J mice) to microfluidic reservoirs for small-volume sampling and secretion quantitation (Fig. 3). Standard well plate-based culture techniques 11 are incompatible with smaller-scale fluidic systems due to the higher collagen viscosity and the possibility of obstructing flow in microfluidic channels. Ultimately, for our small-volume system, we reduced the volume of cells, increased the cell-to-collagen ratio, and used gentle, repeated pipetting to mix within the on-chip reservoir during collagen polymerization (Fig. 3, left). As shown by the inset image in Fig. 3, this protocol allowed 3D culture of the extracted adipocytes within PDMS reservoirs. Fluorescence microscopy, with DAPI stained nuclei (blue) and LipidTOX stained lipid storage (green), confirmed that collagenase digestion during extraction did not disrupt the cells’ physical integrity. Adipocyte size in the 3D matrix varied, but some individual cells were found to be in the 100 μm diameter range, resulting in much lower cell density compared to other tissue types such as pancreatic islets, which can contain thousands of cells in a diameter of 100 μm. To counter this effect and ensure adequate levels of hormone secretion, PDMS reservoirs were enlarged to accommodate more adipocytes per device.
Figure 3.

Cartoon of the modified procedure for preparing 3D culture of primary adipocytes using a collagen matrix, followed by loading into the custom microfluidic interface. The inset image from fluorescence microscopy shows a false-color composite image of adipocytes stained with lipid-staining LipidTOX (green) and nuclear stain DAPI (blue). The presence of a nucleus (blue circle) distinguishes an intact cell from a lipid droplet.
Adiponectin Secretion Quantitation with Microfluidics
In standard 6-well microplates, a significant difference in adiponectin secretion was observed between the untreated and insulin treated cells (Fig. 4, left bars), as anticipated. Levels increased from 7.58 ± 2.25 pg min−1 μL−1 (untreated) to 17.45 ± 1.47 pg min−1 μL−1 (treated; p<0.02). However, to fill one 6-well plate required pooled adipose tissue from at least two mice 10, 11, 20. Attempts to scale down to smaller well-plates failed, with adiponectin secretion amounts after all treatments falling below the detection limit of the ELISA, even after varying the cell density and minimizing media volumes. We believe these results highlight some deficiencies in the static, plate-based methodology for adipocyte secretion sampling. The large well format demands high amounts of tissue (high animal numbers) and does not allow for high temporal resolution. Secreted adipokines are thus diluted significantly prior to sampling and measurement.
Figure 4.
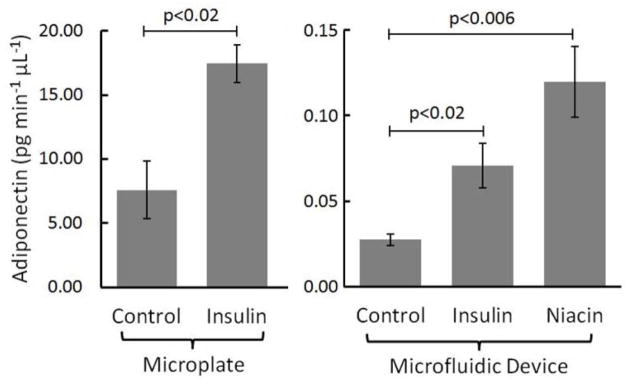
Primary adipocytes were cultured on standard microplates and in microfluidic devices with customized culture interfaces. 100 nM insulin treatments induced similar changes in adiponectin secretion between culture systems. Differences in secretion rate magnitudes suggest the possibility of autocrine effects of secreted agents. Additionally, on-chip stimulation with 10 μM niacin confirmed earlier findings by Judd et al. 11, 26
In contrast, constant-flow microfluidic systems have the potential to minimize dilution, even from very small numbers of cells, and permit downstream analysis of secreted hormones for high temporal resolution 18, 25, 27, 28. The continuous replacement of media in microfluidic systems removes waste effectively and provides excellent control over paracrine or autocrine feedback on secretion rates, while simultaneously providing a more in vivo-like environment for cell culture and sampling 12–14. Our microfluidic device is passively operated, such that flow rate is controlled by the channel’s fluidic resistance 18, 29. A flow rate of 40 μL h−1 was generated with a hand-held syringe, as optimized in previous work using these channel dimensions 18. Adipocytes were cultured in collagen using the custom microfluidic interface then incubated in serum free media for 3 h at 37 °C. Media was replaced, tubing was interfaced from the syringe to the device, and a vacuum was applied at the outlet. This way, cell media and purfusates were drawn into the channels. During this final media replacement, adipocytes were either stimulated with unaltered media (control), 100 nM insulin, or 10 μM niacin. Secretory samples were collected for 1 h, then stored at −20 °C prior to adiponectin quantitation using ELISA. As shown in Fig. 4 (right bars), significant increases in adiponectin secretion were observed after both insulin and niacin treatments compared to the control, as expected 11, 26, 30. Adipose tissue from 5 different C57BL/6J mice was used for this experiment, with 6 sets of cells for the control and 13 sets of cells each for the insulin and niacin treatments. Rather than simply reporting relative secretion, data is reported in units pg min−1 μL−1, representing the amount of adiponectin secreted over a 60 min time period per μL of cell volume loaded onto the device. Levels increased from 0.027 ± 0.003 pg min−1 μL−1 (untreated) to 0.071 ± 0.013 pg min−1 μL−1 (insulin treated; p<0.02) or to 0.120 ± 0.021 pg min−1 μL−1 (niacin treated; p<0.006). Even with large decreases in overall cell number, minimized dilution permitted quantitative analysis of secreted adiponectin. Interestingly, the amount of hormone secreted per volume of cells was >200-fold lower in the flowing, microfluidic system (from both treated and untreated cells), suggesting that autocrine effects7, 31–33 may account for much of the secreted hormones in static systems. However, further biological studies are necessary for confirmation.
These results not only confirmed the viability of the primary adipocytes cultured in our custom microfluidic interface, but they also highlight a major advantage in the low numbers of cells that can be interrogated with microfluidic systems. With only 10 μL of wet cell suspension for each experiment, 5 mice were used to generate 32 independent measurements in our system, made feasible due to minimized dilution with microscale sampling. This can be compared to the two mice that were needed just to fill the space in the 6-well plate system. With a typical mouse giving 0.5–1.0 mL of usable adipose tissue, the 6-well plate system permits only 5–10 treatments per mouse. At only 10 μL tissue needed per assay, our customized microfluidic system should allow 50–100 treatments per mouse, facilitating a large reduction in animal usage along with the concomitant statistical improvements.
Conclusions
A customized microfluidic interface has been designed and validated as a capable platform for culturing and sampling primary adipocytes. Novel methods for PDMS landscaping were presented, with the design targeted to 3D culture and sampling of adipose tissue. Cells were shown functional by comparison to the standard well-plate system, and significant advantages were noted with the microfluidic system, such as a 10-fold decrease in the amount of required tissue and the more in vivo-like, constant perfusion with nutrients. To our knowledge, this represents the first report of primary adipocyte culture integrated with sampling in a microfluidic system. As is, this system should be useful to reduce animal numbers needed for hormone secretion assays—particularly for expensive knock-out mice. With further improvements, the device should also be capable of probing adipokine secretion dynamics at the minute or sub-minute time scale, which would help improve our understanding of the endocrine function of adipose tissue.
Acknowledgments
Support for the work was provided by the National Institutes of Health (R01 DK093810) as well as the Department of Chemistry and Biochemistry and the College of Science and Mathematics at Auburn University. The authors would like to thank Charles Ellis and the Electrical Engineering Department of Auburn University for use of their clean room facilities, as well as Dr. Kennon Deal for help with wafer fabrication.
References
- 1.Ahima RS. Obesity (Silver Spring) 2006;14(Suppl 5):242S–249S. doi: 10.1038/oby.2006.317. [DOI] [PubMed] [Google Scholar]
- 2.Clark AM, Sousa KM, Chisolm CN, MacDougald OA, Kennedy RT. Anal Bioanal Chem. 2010;397:2939–2947. doi: 10.1007/s00216-010-3897-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davis KE, Scherer PE. Biochem J. 2008;416:e7–9. doi: 10.1042/BJ20082033. [DOI] [PubMed] [Google Scholar]
- 4.Desruisseaux MS, Nagajyothi, Trujillo ME, Tanowitz HB, Scherer PE. Infect Immun. 2007;75:1066–1078. doi: 10.1128/IAI.01455-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oh DK, Ciaraldi T, Henry RR. Diabetes Obes Metab. 2007;9:282–289. doi: 10.1111/j.1463-1326.2006.00610.x. [DOI] [PubMed] [Google Scholar]
- 6.Sun K, Kusminski CM, Scherer PE. J Clin Invest. 2011;121:2094–2101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. J Clin Invest. 2006;116:1784–1792. doi: 10.1172/JCI29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schraw T, Wang ZV, Halberg N, Hawkins M, Scherer PE. Endocrinology. 2008;149:2270–2282. doi: 10.1210/en.2007-1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shetty S, Kusminski CM, Scherer PE. Trends Pharmacol Sci. 2009;30:234–239. doi: 10.1016/j.tips.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 10.Plaisance EP, Grandjean PW, Brunson BL, Judd RL. Metabolism. 2008;57:404–409. doi: 10.1016/j.metabol.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 11.Plaisance EP, Lukasova M, Offermanns S, Zhang Y, Cao G, Judd RL. Am J Physiol Endocrinol Metab. 2009;296:E549–558. doi: 10.1152/ajpendo.91004.2008. [DOI] [PubMed] [Google Scholar]
- 12.Bhatia SN, Ingber DE. Nat Biotechnol. 2014;32:760–772. doi: 10.1038/nbt.2989. [DOI] [PubMed] [Google Scholar]
- 13.Bhise NS, Ribas J, Manoharan V, Zhang YS, Polini A, Massa S, Dokmeci MR, Khademhosseini A. J Control Release. 2014;190:82–93. doi: 10.1016/j.jconrel.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim S, Lee H, Chung M, Jeon NL. Lab Chip. 2013;13:1489–1500. doi: 10.1039/c3lc41320a. [DOI] [PubMed] [Google Scholar]
- 15.Giebink AW, Vogel PA, Medawala W, Spence DM. Diabetes Metab Res Rev. 2013;29:44–52. doi: 10.1002/dmrr.2359. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Giebink A, Spence DM. Integr Biol. 2014;6:65–75. doi: 10.1039/c3ib40187a. [DOI] [PubMed] [Google Scholar]
- 17.Desai SP, Freeman DM, Voldman J. Lab Chip. 2009;9:1631–1637. doi: 10.1039/b822081f. [DOI] [PubMed] [Google Scholar]
- 18.Godwin LA, Pilkerton ME, Deal KS, Wanders D, Judd RL, Easley CJ. Anal Chem. 2011;83:7166–7172. doi: 10.1021/ac201598b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duffy DC, McDonald JC, Schueller OJ, Whitesides GM. Anal Chem. 1998;70:4974–4984. doi: 10.1021/ac980656z. [DOI] [PubMed] [Google Scholar]
- 20.Digirolamo M, Thacker SV, Fried SK. Am J Physiol. 1993;264:E361–366. doi: 10.1152/ajpendo.1993.264.3.E361. [DOI] [PubMed] [Google Scholar]
- 21.Rasband WS. ImageJ. http://imagej.nih.gov/ij/
- 22.Whitesides GM. Nature. 2006;442:368–373. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 23.Clark AM, Sousa KM, Jennings C, MacDougald OA, Kennedy RT. Anal Chem. 2009;81:2350–2356. doi: 10.1021/ac8026965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dugan CE, Cawthorn WP, MacDougald OA, Kennedy RT. Anal Bioanal Chem. 2014;406:4851–4859. doi: 10.1007/s00216-014-7894-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dugan CE, Kennedy RT. Methods Enzymol. 2014;538:195–209. doi: 10.1016/B978-0-12-800280-3.00011-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wanders D, Graff EC, White BD, Judd RL. PLoS One. 2013;8:e71285. doi: 10.1371/journal.pone.0071285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dishinger JF, Reid KR, Kennedy RT. Anal Chem. 2009;81:3119–3127. doi: 10.1021/ac900109t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Easley CJ, Rocheleau JV, Head WS, Piston DW. Anal Chem. 2009;81:9086–9095. doi: 10.1021/ac9017692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Godwin LA, Deal KS, Hoepfner LD, Jackson LA, Easley CJ. Anal Chim Acta. 2013;758:101–107. doi: 10.1016/j.aca.2012.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. J Biol Chem. 1995;270:26746–26749. doi: 10.1074/jbc.270.45.26746. [DOI] [PubMed] [Google Scholar]
- 31.Lin Z, Tian H, Lam KS, Lin S, Hoo RC, Konishi M, Itoh N, Wang Y, Bornstein SR, Xu A, Li X. Cell Metab. 2013;17:779–789. doi: 10.1016/j.cmet.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 32.Vienberg SG, Brons C, Nilsson E, Astrup A, Vaag A, Andersen B. Eur J Endocrinol. 2012;167:49–57. doi: 10.1530/EJE-12-0039. [DOI] [PubMed] [Google Scholar]
- 33.Rasmussen MS, Lihn AS, Pedersen SB, Bruun JM, Rasmussen M, Richelsen B. Obesity (Silver Spring) 2006;14:28–35. doi: 10.1038/oby.2006.5. [DOI] [PubMed] [Google Scholar]