

Abstract
New antibiotics are needed because as drug resistance is increasing, the introduction of new antibiotics is decreasing. Here, we discuss six possible approaches to develop ‘resistance-resistant’ antibiotics. First, multi-target inhibitors in which a single compound inhibits more than one target may be easier to develop than conventional combination therapies with two new drugs. Second, inhibiting multiple targets in the same metabolic pathway is expected to be an effective strategy due to synergy. Third, discovering multiple-target inhibitors should be possible by using sequential virtual screening. Fourth, re-purposing existing drugs can lead to combinations of multi-target therapeutics. Fifth, targets need not be proteins. Sixth, inhibiting virulence factor formation and boosting innate immunity may also lead to decreased susceptibility to resistance. Although it is not possible to eliminate resistance, the approaches reviewed here offer several possibilities for reducing the effects of mutations and in some cases suggest that sensitivity to existing antibiotics may be restored, in otherwise drug resistant organisms.
Keywords: antibiotics, resistance, multi-targeting, isoprenoids, molecular dynamics, innate immunity
The Rise of Antibiotic Resistance
With the recent issuance by the United States Centers for Disease Control and Prevention of its “Threat Report 2013” on antibiotic resistance[1] and the World Health Organization’s “Antimicrobial resistance: global report on surveillance 2014”[2], there can be little doubt that antibiotic resistance will be a major public health threat for the foreseeable future. This threat is recognized, for example, by the recent announcement of the (~$17 million) Longitude Prize 2014[3] on the 300th anniversary of the Longitude Act 1714 (the Prize was won a half-century later by John Harrison, for his chronometer), the current topic being: “How can we prevent the rise in resistance to antibiotics?” More recently, President Obama signed an Executive Order: Combating Antibiotic-Resistant Bacteria[4], offering a $20 million prize in the same general area as the Longitude Prize, focus in both cases being in the area of diagnostics. Here, we review some of the scientific possibilities for resistance-resistant antibiotics.
Antibiotics have been known for hundreds and in some cases, thousands of years-albeit not in pure form. For example, the “newest” anti-malarial in widespread use, artemisinin (1, Figure 1), was used in traditional Chinese medicine ~2000 years ago[5] but was only isolated in a pure form in the 1970s by Tu Youyou[6, 7, 8] apparently due to a request from Ho Chi Minh to Mao Zedong for an anti-malarial to replace chloroquine (2) (developed in WWII), where resistance had developed. The anti-malarial quinine (3), whose activity (in cinchona bark) was known in the 15th century by the Incas, was later brought to Europe and quinine was isolated, by French scientists, in 1820.[9] However, the mechanisms of action of all three drugs have been the topic of debate, suggesting, perhaps, that they might each have more than one target.
Figure 1.

Structures of some drugs, drugs leads and other compounds of interest. 1: artemisinin; 2: chloroquine; 3: quinine; 4: early proposed structure of Salvarsan; 5, 6: actual structures of Salvarsan; 7: Prontosil; 8: penicillin; 9: colistin; 10: ivermectin; 11: eflornithine; 12: miltefosine; 13: bisamidine, BPH-1358; 14: risedronate; 15: SQ109; 16: MBX-1066; 17: staphyloxanthin; 18: tuberculosinyl adenosine.
The first development of a synthetic antibiotic began about 100 years ago with the discovery of Salvarsan (compound 606; Arsphenamine) by Ehrlich[10], proposed in early work to be 4. However, the actual structures of Salvarsan 5, 6 were only discovered a decade ago[11] and the exact targets, presumably one or more proteins containing reactive thiols to which arsenic can bind, are still unknown. The next generation of antibiotics, sulfonamides such as Prontosil (7), were developed by IG Farben[12] and their target, dihydropteroate synthase, is known, but resistance occurred rapidly and the use of sulfonamides was largely replaced by the discovery and development of penicillin 8 (which does have multiple targets), shortly before and during WWII.
After the development of penicillin there was a “golden age” of antibiotic discovery during the 1950s and 1960s, however, after ~1985 there has been a sharp fall-off in new antibiotic drug discovery for reasons that are at least in part financial; an antibiotic that cures a patient in perhaps one week does not have a large profit potential. But perhaps more importantly, it does appear that many of the developments in high-throughput screening (HTS) of synthetic libraries, genomics, structure-based design, combichem and natural product screening have led to relatively few new progressable leads. While it is certain that Chemists need to improve their chemical libraries, this is in a sense a chicken/egg problem in that if it was clear how to do this, we would likely already have more antibiotics! Indeed, as discussed in two recent reviews[13, 14] most of the recently introduced antibiotics belong to existing antibiotic classes. Unfortunately, during this same time period, drug resistance with e.g. MRSA (methicillin resistant Staphylococcus aureus), multi-drug resistant as well as extensively drug resistant Mycobacterium tuberculosis, multiple cases of hospital acquired infections (Clostridium difficile, and infections with Gram-negatives such as Klebsiella pneumoniae, Pseudomonas aeruginosa and Acinetobacter baumannii) have occurred and are anti-correlated with the introduction of new anti-infectives [15, 16], although apparently there is now renewed interest in the development of new antibiotics by the pharmaceutical industry[17].
This lack of new antibiotics has been highlighted in the CDC and WHO reports, although such warnings are not new[18] and indeed were basically presaged by Fleming[19]. What is new is the discovery of organisms such as Klebsiella pneumoniae producing both NDM-1 carbapenemase and beta-lactamase, leading to the strong possibility of pan-resistance and in some cases, the re-introduction of an old drug, colistin (9; a polymyxin), a polycationic species that targets bacterial cell membranes, for treating such infections. In addition, there is now a report of the occurrence of vancomycin resistance in MRSA S. aureus.[20] Why these developments are so alarming is that patients undergoing any surgical procedure: for cancer, heart disease, joint replacements, for example, will be at greatly increased risk for acquiring an untreatable bacterial infection, not least because in many instances they will already have a compromised immune system.
In the less developed world, HIV/AIDS, malaria, tuberculosis, as well as the leishmaniases and Chagas disease, cause millions of deaths and hundreds of millions of DALYs (disability adjusted life years)[21], mostly in Africa, and once again, there have been few new drugs introduced because the profit motive is even less attractive for patients who have few (or no) resources. Some success stories have, however, been reported. For example, the drug ivermectin 10, developed to treat heartworm in dogs, is used to treat river blindness; the facial hair removal drug Vaniqua (eflornithine, 11) initially developed as an anti-cancer drug, has been used to treat sleeping sickness[22] and another anti-cancer drug lead, miltefosine 12 is now FDA-approved to treat the leishmaniases[23], the common theme here being that none of these drugs/drug leads were originally developed to treat the neglected tropical diseases but were later found to have activity against parasitic protozoa, and in some cases have been provided by pharmaceutical companies at low or no cost to patients. These developments clearly show an important role for drug re-purposing, a topic we discuss below.
In the following, we thus enumerate and expand on a series of six general propositions, with examples, that may help facilitate the development of new therapeutic strategies and leads. The focus is largely though not exclusively on new applications of multi-target inhibitors in infectious diseases (bacterial and protozoan), based in part on new concepts and discoveries as to how some drugs/drug leads function.
Multi-target inhibitors
For drugs with a single mechanism of action, combination therapies are needed to combat resistance. However, there are intrinsic difficulties associated with developing multiple new drugs for combination therapies: multi-targeting might be easier. In many cases, the root cause of antibiotic resistance is that a mutation occurs in the target, usually a protein, rendering an inhibitor ineffective. In some cases, larger inhibitors can circumvent resistance due to enhanced drug-target interactions (as with e.g. the anti-fungal posaconazole versus fluconazole), but this may not always be possible. In general, however, if the probability of resistance due to mutations developing in target A is PA and in target B, PB, the likelihood that resistance develops in both targets will be given by the conditional probability PAPB, a small number. Indeed, the anti-infective drugs that have been most successful in mono-therapy often have more than one target[24, 25], while all TB, malaria and HIV/AIDS drugs are only now effective in combination therapies, due to resistance[24]. It is, however, already very difficult (and expensive) to find just one new drug or one new drug lead, and the development of two drugs for use in such a combination will be twice as difficult. Nevertheless, the importance of this approach is recognized by e.g. the issuance in 2013[26] of the U. S. Food and Drug Administration’s “Guidance for Industry” for the development of anti-infective combination therapies, in particular for two drugs acting in the same metabolic pathway[26], where synergistic interactions are often found.
For an individual drug acting on a single target, there are at least 10 requirements or factors to be satisfied for success (Figure 2A). In addition to efficacy against a given target, ADME-Tox (adsorption, distribution, metabolism, excretion, toxicity), ease of synthesis, cost of synthesis, intellectual property position, and market size, are all important. Combination therapies involving two new drugs acting on two new targets (Figure 2B) would require at least 2×10=20 properties to be satisfied, a daunting challenge, but one that is of course met in all combination therapies that are in clinical use. However, if an individual drug or drug lead can also inhibit a second target, the multi-target approach, then there would be just 10+1=11 properties to be satisfied (Figure 2C). If it is argued that n is >10, then (n+1)/n will decrease, which is desirable.
Figure 2.

Therapeutic strategies for antibiotic development. A to D, Schematic illustration showing the approximate number of factors or requirements that need to be met for different types of therapies illustrating the advantage of multi-target inhibition. A, For an individual drug acting on a single target, there are at least 10 requirements or factors to be satisfied for success. B, Combination therapies involving two new drugs acting on two new targets double the requirements for success. C, Multi-target inhibitors retain the advantages of combination therapies but require fewer properties to be satisfied for success. D, Combination multi-targeting is expected to be highly active and resistance-resistant. E to G, There are three main classes of multi-target inhibitor: series, parallel and network. E, Series inhibitors work on sequential targets in the same metabolic pathway. F, parallel inhibitors work on unrelated pathways (e.g. DNA and membrane targets). G, Network inhibition is a combination of series and parallel inhibitors. H, Hybrid inhibitors contain overlapping or fused pharmacophores for 2 or more targets. E to G are reprinted with permission from reference [35]. Copyright 2014 American Chemical Society.
Activity against the second target is clearly essential, and there are good examples of such activity[27, 28, 29, 30, 31] and ongoing discovery programs on type II topisomerases, DNA Gyrase and Topo IV[32]. Moreover, computational methods such as QSAR (quantitative structure-activity relationships) do enable optimization against two or more targets, (e.g. in the manner that Khanna et al.[33] used the concept of the additivity of molecular fields to optimize dual activators of PPARα and PPARγ. It also follows that combinations of dual target inhibitors would be particularly active and again, arguably more readily developed than in the case of four new drugs affecting four new targets (~40 criteria to be met; Figure 2D). Examples of these different therapeutic approaches are shown in Figure 2. In addition, combinations of a new multi-target inhibitor with an existing drug (though not covered by the FDA proposals) should be more readily developed than would be two new drugs. Such combinations could also be used to restore drug sensitivity to otherwise resistant organisms[34]. The development of multi-targeted anti-bacterials is likely to be difficult, but since there are existing multi-targeted drugs, is possible. It will be important to have good activity against both targets, otherwise resistance will rapidly develop, and determinations of activity against two targets is clearly more difficult to determine experimentally with a single multi-target drug lead than with conventional combination therapies, and likely requires computational modeling[35].
Multi-targeting can account for the (early) successes found with anti-infectives such as penicillin, and probably with drugs such as quinine and artemisinin, although of course, resistance has now developed, as it always will, for purely thermodynamic reasons. In the following, we thus next discuss where multi-targeting is likely to be most effective; different types of multi-target inhibition; new routes to inhibitor discovery that do not involve solely enzyme targets; as well as anti-virulence/innate immunity based approaches that may be less prone to the development of resistance.
Targeting metabolic pathways
Metabolic pathways involving many coupled reactions should be good targets for multiple targeting, and this approach is one of those quoted by the FDA in its recent guidelines for the development of novel anti-infectives[26]. But which pathways should be targeted? Many metabolic pathways would not be good antibacterial targets because they would be remediable through import from the host. However, the enzymes involved in isoprenoid biosynthesis, together with later stages involved in sterol and bacterial cell wall biosynthesis, represent one of the largest biosynthetic networks and the enzymes that catalyze many of these reactions are the targets of currently used drugs such as statins, bisphosphonates, terbinafine, the azole antifungals, methicillin and vancomycin (Figure 3). There are two pathways involved in the earliest stages of isoprenoid biosynthesis: the mevalonate pathway (in humans as well as in yeasts, fungi, most protozoa and some bacteria, e.g. S. aureus), and the “non-mevalonate” (methylerythritol phosphate) pathway[36] (present in e.g. Mycobacterium tuberculosis and the malaria parasite, Plasmodium falciparum). The development of new inhibitors with novel targets and multiple-target mechanisms of action in these pathways is of great interest, as is the possibility of combining new inhibitors with existing drugs, restoring drug sensitivity to otherwise resistant species (e.g. MRSA[34]), as discussed below.
Figure 3.

Isoprenoid biosynthesis pathways and inhibitors, showing that there are multiple validated drug targets and corresponding inhibitors in these pathways. Unlike many biosynthetic pathways, isoprenoid biosynthesis produces end-products that are (in most cases) only found in pathogens and are not available from the host, making isoprenoid biosynthesis a good drug target. For clarity, the enzyme targets are omitted but the isoprenoid products are shown in blue, inhibitors in pink. Abbreviations used: G3P, glycerol-3-phosphate; DHAP, dihydroxyacetone phosphate; HMBPP, (E)-hydroxy-2-methyl-but-2-enyl-4-diphosphate; DMAPP, dimethylallyl diphosphate; IPP: isopentenyl diphosphate; HMGCoA: 3-hydroxy-3-methyl-glutaryl-coenzyme A; FPP, farnesyl diphosphate; GGPP, geranylgeranyl diphosphate; UPP, undecaprenyl diphosphate; STX, staphyloxanthin; DHS, dehydrosqualene; GGTI, protein geranylgeranyl transferase inhibitor; FTI, protein farnesyl transferase inhibitor; BPs, bisphosphonates; BPH-652, a biphenyl phosphonosulfonate.
There are also four main types of multi-target inhibitor that, conceptually, can guide inhibitor design (Figure 2E to Figure 2H). A series inhibitor (Figure 2E) targets two enzymes that are adjacent to one another in a metabolic pathway. The product (“A”) of the first enzyme is the substrate for the second enzyme, suggesting the possibility that an inhibitor that resembles “A” might inhibit both enzymes. Parallel inhibition (Figure 2F) involves mimicking a substrate “B” which is the substrate for more than one enzyme (that is not in series). For example, some analogs of farnesyl diphosphate[37] can inhibit formation of both the staphyloxanthin virulence factor in S. aureus (inhibiting dehydrosqualene synthase, CrtM), as well as bacterial cell wall biosynthesis (inhibiting undecaprenyl diphosphate synthase), as discussed below. Network inhibition (Figure 2G) involves both series and parallel targeting and is expected to be particularly effective, since many targets are involved. Hybrid inhibitors (Figure 2H) are molecules that contain overlapping or fused pharmacophores for two (or more) targets and, logically, should be the most straightforward to design. Moreover, proteins need not be the only targets, as discussed below.
The use of molecular dynamics (MD) simulations can lead to the discovery of new leads
Finding new inhibitors, potential drug leads, is an expensive process and often requires chemical libraries of ~500,000 compounds, typically found only in pharmaceutical companies or large screening centers. However, the results from GlaxoSmithKline in which 70 high throughput screening campaigns were investigated, were disappointing[38]. Reasons for this are uncertain but could be related to the diversity of the libraries that were screened. An alternative approach that can in principle be used to screen even larger libraries is to use computational “in silico” or virtual screening (VS). Plus, sequential virtual screening against multiple targets is theoretically possible. The VS approach is, however, often limited by the number of X-ray crystallographic structures available, and these structures represent only a small fraction of the conformational space accessible to the target. One approach to help circumvent this problem is to use molecular dynamics (MD) simulations to sample large conformational spaces, then to select a diverse library of MD-derived protein structures for use in VS. Hits obtained from such VS can then be used as targets for similarity searches.
As an example, we recently reported the use of MD simulations to investigate farnesyl diphosphate synthase (FPPS)[39] and undecaprenyl diphosphate synthase (UPPS), two enzymes involved in isoprenoid biosynthesis (Figure 3). The MD result on UPPS[40] is shown in Figure 4A and indicates that for most of the time the active site is “closed” (volume V~400 Å3) but at ~12 nsec it opens up to V~900Å3. The frequency of occurrence of a given pocket volume is shown in Figure 4B, with the largest MD pocket having a volume very similar to that seen in an inhibitor-bound structure (PDB ID code 2E98) containing 4 inhibitor molecules[41]. The closed and open UPPS structures are shown in Figure 4C and Figure 4D. The more open structure enabled an excellent correlation between IC50 and docking scores[40]. We used a series of FPPS MD structures in VS finding several leads that inhibited both FPPS as well as UPPS, then used similarity searches to find additional UPPS inhibitor leads, resulting in the bisamidine 13 (IC50 = 100 nM) that was active in cells (a MRSA MIC ~ 250 ng/mL) and in vivo[34] (Figure 4E). The compound also synergized with methicillin against a MRSA strain of S. aureus (USA300; FICI = 0.25; Figure 4F), a potential route to restoring sensitivity to MRSA strains. The use of MD-based structures thus opens up new possibilities for in silico screening-there are very many structures in an MD trajectory--and even allosteric sites (outside the active site) can be targeted. The drawback of the approach is that some of the better programs used in computational docking are expensive, and while large amounts of computer time are generally available, corresponding commercial software may be less so.
Figure 4.

Molecular dynamics as a route to drug lead discovery. A–D, MD results for E. coli UPPS. A, volume of the binding pocket along the MD trajectory of E. coli undecaprenyl diphosphate synthase (UPPS). The black line shows data taken every 10 ps, the over-layed gray line is the average over every 100 ps; B, frequency at which different volumes of the pocket are sampled; C, the apo crystal structure with 1 Å spheres filling the active site pocket; D, a bisphosphonate-bound crystal structure with 1 Å spheres filling the active site pocket. Note the significantly larger pocket size in the bisphosphonate-bound structure when compared to the apo crystal structure. The MD-based structures provide the best correlation between experimental IC50 values and docking scores. E, activity of 13 (bisamidine, BPH-1358) in a mouse model of S. aureus (USA200) infection; F, in vitro synergy showing isobologram for BPH-1358 + methicillin inhibition of S. aureus (USA300) cell growth, FICI = 0.25. A to D are reprinted with permission from reference [40]. E and F are reprinted with permission from reference [34].
Finding multi-target inhibitor leads by repurposing existing drugs
One attractive route to finding new multi-target leads is to find alternative uses for existing drugs, basically following the aphorism frequently attributed to Sir James Black (inventor of propranolol and cimetidine) that “The most fruitful basis for discovery of a new drug is to start with an old drug”. This obviously leads to the idea of drug “re-purposing”, but which compounds should be re-purposed? And is it possible to develop re-purposed multi-target leads? For the most common diseases (certainly of the developed world) it seems likely that most FDA-approved drugs will already have been screened in both cell-based assays as well as against many putative enzyme targets. However, this may not be the case for the neglected tropical diseases.
In our group we proposed [42] a “knowledge-based” approach to help find potential (in some cases multi-target) leads. Knowledge-based means that a broad range of literature (including the “popular” literature such as Science, Nature Reviews Drug Discovery and Microbes magazine) is used to come up with new ideas for drug leads. For example, the discovery[43] that bisphosphonates kill trypanosomes arose from our observation[44] that trypanosomes (T. cruzi) contain large amounts of diphosphate and the bisphosphonate risedronate (14), containing a diphosphate “isostere”, had reported activity against the protozoan Entamoeba histolytica[45]. This led to the discovery that bisphosphonates inhibited farnesyl diphosphate synthase[46] and geranylgeranyl diphosphate synthase[47], and to the development of bisphosphonates active against malaria parasites[48]. These compounds also activate gamma delta T-cells (containing the Vγ2Vδ2 T-cell receptor)[49] which can then kill bacteria[50].
An example of a combination of two re-purposed multi-target drugs is that of amiodarone (or dronedarone) with an azole such as posaconazole, stimulated by a report in Nature Reviews Drug Discovery[51]. Amiodarone is an anti-arrhythmia drug that was serendipitously found[52] to kill S. cerevisiae, an effect that was potentiated by an azole drug[53] (an anti-fungal that blocks ergosterol biosynthesis). We found[54] that amiodarone killed T. cruzi, blocking ergosterol biosynthesis at the oxidosqualene cyclase level, in addition to causing an increase in [Ca2+]i (as in yeast) and it acted synergistically with posaconazole (which inhibits ergosterol biosynthesis at the lanosterol 14α-demethylase level). In addition, we found that posaconazole also increased [Ca2+]i, that is, it unexpectedly affected ion channels. More recently, it has been shown that amiodarone collapses the proton motive force (PMF), acting as an uncoupler[55]. The two FDA approved drugs thus both act as multi-target inhibitors of T. cruzi (but not host) cell growth affecting ΔpH, Δψ, [Ca2+]i and ergosterol biosynthesis: a potent combination multi-target approach to therapy, with amiodarone now having been used clinically[55, 56, 57] as an anti-parasitic (against T. cruzi and Leishmania mexicana.) A potential problem with repurposing (and with combination therapies) is that of possible toxicity, although this possibility exists with any new drug/drug lead and in principle, seems more likely when two drugs (rather than a single multi-target lead) are involved, since there would be more possibilities for the production of toxic breakdown products. Moreover, it would be easier to get approval for a single new chemical entity, rather than two NCEs, but if combinations of multi-target inhibitors are safe, they are expected to be very effective and resistance-resistant.
Proteins need not be the only targets in multi-target inhibition
In recent work[35] we found that the TB drug/drug lead SQ109[58], thought to target the trehalose monomycolate transporter MmpL3 (Mycobacterial membrane protein Large 3[59]), also inhibited two enzymes involved in menaquinone biosynthesis (MenA and MenG) and additionally, was an uncoupler, collapsing ΔpH and Δψ (the pH gradient and the membrane potential): multi-target inhibition[35]. In this case there is series inhibition of two (sequential) enzymes in a metabolic pathway, together with effects on a non-protein target (the membrane lipid bilayer), as well as involvement of a third protein, MmpL3, a transporter presumably powered by the PMF: network inhibition (Figure 2G). SQ109 (15) is an ethylene diamine, an analog of ethambutol, and acts as uncoupler, in addition to collapsing the membrane potential, resulting in a decrease in ATP synthesis, as observed in E. coli inverted membrane vesicles and M. smegmatis cells[35]. These effects on the proton motive force (PMF = ΔpH + Δψ) are expected to block PMF/ATP-powered transporters and when combined with enzyme inhibition (MenA, MenG and MmpL3), result in potent M. tuberculosis cell growth inhibition, as well as the lack of resistance under serial passage, and a proposed multi-target network inhibition scheme is shown in Figure 5[35]. Notably, other bacteria (Clostridium difficile, Helicobacter pylori) as well as the malaria parasite P. falciparum, are also killed by SQ109 (which is in clinical trials for tuberculosis), additional examples of drug (or drug lead) re-purposing.
Figure 5.
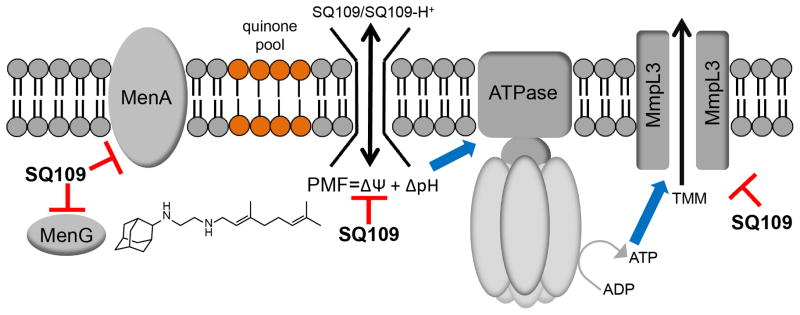
Membrane and protein targeting of SQ109 and its analogues. MenA, MenG targeting can affect respiration/electron transfer; PMF (ΔpH, Δψ) collapse leads to decreased ATP biosynthesis, reduction inPMF/ATP-powered transporters (e.g., MmpL3), increased TMM accumulation, and decreased cell wall biosynthesis. Reprinted with permission from reference [35]. Copyright 2014 American Chemical Society.
The concept of membrane targeting is certainly not new - the antifungal amphotericin binds to ergosterol[60] in fungal cell membranes (and in some protozoa[61]), and it has a relatively low rate of resistance. There are, however, resistant strains[62] which correlate with changes in sterol composition, and it is well-known that bacteria can modulate their lipid composition[63]. Thus, it is likely that purely membrane-targeting antibiotics will eventually lose efficacy due to changes in cell lipid membrane composition, hence the need for inhibitors that can target both a lipid membrane, and a protein (or other) target.
Multi-targeting need not be restricted to proteins or membranes: protein+DNA targeting may be possible. rRNA inhibitors are certainly very well-known[64] and in most cases, single target mutations do not lead to high level resistance (rRNA has multiple copies, except in M. tuberculosis[65], where there is high level resistance). But what about DNA targeting for anti-infectives? This sounds a risky, counter-intuitive proposition but there may be instances where pathogen DNA is a good target. For example, compounds such as the bisamidines (e.g. pentamidine[66]) bind to DNA[67] and have been used clinically as anti-parasitics and against Pneumocystis jirovecii pneumonia, for many years. What is interesting about this class of compound is that several of them also inhibit bacterial cell wall biosynthesis and in our lab we found[34] that some bisamidines inhibit isoprenoid (undecaprenyl diphosphate synthase and farnesyl diphosphate synthase) biosynthesis, act synergistically with methicillin (in the MRSA300 strain of S. aureus) and were active in vivo in a mouse model of infection[34], suggesting multi-site targeting. Interestingly, small structural changes in bisamidine structure can switch a molecule from targeting cell wall biosynthesis to inhibiting DNA biosynthesis (as deduced from macro-molecular labeling experiments).[68] Specifically, the Microbiotix compound MBX-1066 (16) with a 5-membered ring inhibits (primarily) DNA biosynthesis, the 6-membered ring homolog (MBX-1162) inhibits cell wall biosynthesis.
A concern with compounds that target DNA is that they might have genotoxicity. Interestingly, however, it has been found in trypanosomes that the bisamidines only affect replicating kinetoplast DNA (k-DNA) and not nuclear DNA, with k-DNA not being present in host cells, opening up possibilities, perhaps, for more selective activity against, in this case, pathogenic protozoa. In addition, pentamidine can act as an uncoupler[69], as with SQ109. Moreover, DNA minor groove binders such as MGB-BP-3 (from MGB Biopharma) apparently function by targeting specific motifs in bacterial DNA, affecting transcription factor function but not cell growth[70, 71].
Targeting virulence and the innate immune system are under-exploited areas of drug discovery for infectious diseases
Current antibiotics target cell killing or cell growth, but alternate approaches, such as those targeting virulence and the innate immune system, are also of interest [72]. Targeting virulence may place an organism under less selective pressure for the development of resistance[72, 73] and even if it does not, it is still an interesting new approach, since logically, very narrow spectrum inhibitors might be developed (including multi-target inhibitors). This would be of considerable importance because disruption of our normal commensal micro-flora often results in overgrowth with other pathogens, such as C. difficile.
One example of the anti-virulence factor therapeutic approach is found in S. aureus, which makes the carotenoid virulence factor staphyloxanthin (17; Figure 1) to protect itself from reactive oxygen species (ROS) from the innate immune system (neutrophils and macrophages).
The first committed step in staphyloxanthin biosynthesis (Figure 6A) involves the condensation of two molecules of farnesyl diphosphate to form presqualene diphosphate and thence, dehydrosqualene, reactions catalyzed by the enzyme CrtM (dehydrosqualene synthase). CrtM knockout mutants do not produce the carotenoid virulence factor, are white, and are non-infective[74]. We noticed a report[75] of this work in ASM News (now Microbes magazine) which led to the idea that CrtM[74] must be structurally very similar to squalene synthase[76], used by humans to make presqualene diphosphate and thence, squalene, in cholesterol biosynthesis. This in turn led to the discovery that cholesterol-lowering drug leads developed by Bristol-Myers Squibb[77] were also anti-virulence drug leads for S. aureus infections (Figure 6B to D), boosting innate immunity by preventing formation of the virulence factor, staphyloxanthin, by inhibiting CrtM[76]. In this system, there is reduced drug pressure for the development of resistance due to “environmental specificity” [73, 76]. Susceptible S. aureus (no carotenoid pigment) are killed at the site of infection (by reactive oxygen species of the innate immune system) and there would indeed be selection for resistance at this site. However, populations at commensal sites (e.g. the nose) would not be under selection for resistance (since the drug had no effect on cell growth/nasal colonization). Such environmental specificity would not be expected with conventional antibiotics. [73]
Figure 6.

Targeting virulence factor formation in S. aureus. A, Initial step in the metabolic pathways to the virulence factor staphyloxanthin and cholesterol/ergosterol are the same. B, The cholesterol-lowing drug lead BPH-652 inhibits CrtM and inhibits formation of the orange carotenoid virulence factor. C, Bacteria treated with BPH-652 are killed by whole blood (ROS from neutrophils, macrophages). D. Mice treated with BPH-652 control a S. aureus infection (bacteria CFUs reduced by 98%). Reprinted with permission from reference [76].
There are undoubtedly many other isoprenoid virulence factors that can be targeted, e.g. the tuberculosinyl-adenosine (18) virulence factor in M. tuberculosis (Rv3378c)[78]. Tuberculosinyl adenosine synthase (Rv3378c) has some structural resemblance[78, 79] to cis-decaprenyl diphosphate synthase (involved in M. tuberculosis cell wall biosynthesis), and both should be (and are) amenable to multi-target inhibition.
A second (and potentially multi-target) approach to boosting innate immunity derives from other work by Nizet et al.[80] Specifically, the observation that in several studies, patients on statin therapies had decreased occurrence of death from sepsis led to the discovery that statins induced formation of anti-bacterial neutrophil and macrophage extra-cellular traps[80], NETs and METs, an effect that was replicated by use of a squalene synthase (SQS) inhibitor[80]. This discovery then led to the idea that it might be possible to develop inhibitors that target SQS as well as CrtM (since they have very similar 3D structures), inhibiting both virulence factor formation as well as inducing NET formation: multi-targeting of innate immunity. [81]
A third intriguing possibility involves combining direct pathogen killing with boosting innate immunity. The molecule HMBPP (E-1-hydroxy-2-methyl-but-2-enyl 4-diphosphate) is used by the enzyme IspH to make the building blocks of isoprenoid biosynthesis, isopentenyl diphosphate and dimethylallyl diphosphate (Figure 3). IspH is used by malaria parasites and by bacteria containing the non-mevalonate pathway, and is a drug target.[82] Inhibiting IspH will inhibit bacterial cell wall biosynthesis (as well as diverse other processes) and will result in accumulation of high levels of the HMBPP substrate. The interesting point here is that HMBPP and related compounds[83] are exceptionally potent “phosphoantigens” that stimulate the expansion of γδ T cells (containing the Vγ2Vδ2 T cell receptor) which can then differentiate into multi-functional effector sub-populations capable of producing cytokines, INFγ and perforin/granulysin[83]. Thus, an IspH inhibitor will kill bacteria in a direct fashion, in addition to boosting innate immunity via γδ T cell expansion. As one proof-of-concept, a Salmonella ispH− mutant (complemented with genes from the mevalonate pathway) stimulated a profound expansion of Vγ2Vδ2 T cells in monkeys without side-effects or anergy induction[84]. IspH inhibitors should have the same effect, in addition to inhibiting bacterial cell wall biosynthesis.
Concluding remarks
In the above, we argue that multi-target inhibition is one logical route to the development of new drugs/drug leads that, by their nature, should be more resistant to the development of resistance: “resistance-resistant” antibiotics. The basic concept is not new since it is known that several antibiotics do have multiple targets, but concepts such as using MD methods in virtual screening; targeting both proteins as well as e.g. the PMF; ways to get at new leads by using knowledge-based approaches (amiodarone as an anti-parasitic; SQ109 as an uncoupler; targeting virulence in S. aureus using a cholesterol-lowering drug lead) are all promising novel routes to anti-infective drug development. When combined with HTS, structure-based design, and combinational chemistry to expand upon new leads, these approaches will hopefully lead to new therapeutics that are less prone to the development of resistance that arises from spontaneous mutations, though obviously problems due to e.g. drug modifications and efflux pumps will still occur. Nevertheless, reducing the effects of target mutations would be a good start to help answer the question: “How can we prevent the rise in resistance to antibiotics?”
Unfortunately, as new antibiotics are discovered, they will fail if there continues to be massive antibiotic misuse. However, to misquote Wilde: “To lose one antibiotic… may be regarded as a misfortune; to lose all antibiotics looks like carelessness”[85].
Six approaches to developing resistance-resistant antibiotics are reviewed
Multi-targeting and inhibiting isoprenoid biosynthesis pathways are very attractive
Molecular dynamics simulations can lead to new routes to multi-targeting
Targeting innate immunity and virulence factors offer new opportunities
Acknowledgments
This work was supported in part by the United States Public Health Service (National Institutes of Health grants AI074233, GM065307 and CA158191), a Harriet A. Harlin Professorship (E.O.), the University of Illinois Foundation/Oldfield Research Fund, and a Pre-doctoral Fellowship from the American Heart Association, Midwest Affiliate (13PRE14510056 to X.F.).
Footnotes
Conflict of Interest
The authors declare they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Threat Report 2013: Antibiotic/Antimicrobial Resistance. Centers for Disease Control and Prevention; http://www.cdc.gov/drugresistance/threat-report-2013/ [Google Scholar]
- 2.Antimicrobial resistance: global report on surveillance 2014. World Health Organization; http://www.who.int/drugresistance/documents/surveillancereport/en/ [Google Scholar]
- 3.http://www.longitudeprize.org/.
- 4.Executive Order -- Combating Antibiotic-Resistant Bacteria. The White House; http://www.whitehouse.gov/the-press-office/2014/09/18/executive-order-combating-antibiotic-resistant-bacteria. [Google Scholar]
- 5.Tu Y. The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nat Med. 2011;17:1217–1220. doi: 10.1038/nm.2471. [DOI] [PubMed] [Google Scholar]
- 6.Cui W, Zhou Y. Ancient Chinese anti-fever cure becomes panacea for malaria. Bull World Health Organ. 2009;87:743–744. doi: 10.2471/BLT.09.051009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J. A detailed chronological record of project 523 and the discovery and development of qinghaosu (artemisinin) Strategic Book Publishing; 2013. [Google Scholar]
- 8.Honigsbaum M. The fever trail: In search of the cure for malaria. Farrar, Straus and Giroux; 2002. [Google Scholar]
- 9.Pelletier PJ, Caventou JB. Suite: Des researches chimiques sur les quinquinas. Annales de Chimie et de Physique. 1820;15:337–365. [Google Scholar]
- 10.Hüntelmann AC. Making Salvarsan: Experimental therapy and the development and marketing of Salvarsan at the interface between science, clinic, industry and public health. In: Gaudillière J-P, Hess V, editors. Ways of Regulating Drugs in the 19th and 20th Centuries. Palgrave Macmillan; 2012. [Google Scholar]
- 11.Lloyd NC, Morgan HW, Nicholson BK, Ronimus RS. The composition of Ehrlich’s salvarsan: Resolution of a century-old debate. Angew Chem Int Ed Engl. 2005;44:941–944. doi: 10.1002/anie.200461471. [DOI] [PubMed] [Google Scholar]
- 12.Domagk G. Nobel lecture: Further progress in chemotherapy of bacterial infections. Nobel Media AB; 2013. http://www.nobelprize.org/nobel_prizes/medicine/laureates/1939/domagk-lecture.pdf. [Google Scholar]
- 13.Hede K. Antibiotic resistance: An infectious arms race. Nature. 2014;509:S2–S3. doi: 10.1038/509S2a. [DOI] [PubMed] [Google Scholar]
- 14.Laxminarayan R. Antibiotic effectiveness: Balancing conservation against innovation. Science. 2014;345:1299–1301. doi: 10.1126/science.1254163. [DOI] [PubMed] [Google Scholar]
- 15.Leeb M. Antibiotics: A shot in the arm. Nature. 2004;431:892–893. doi: 10.1038/431892a. [DOI] [PubMed] [Google Scholar]
- 16.Nathan C. Antibiotics at the crossroads. Nature. 2004;431:899–902. doi: 10.1038/431899a. [DOI] [PubMed] [Google Scholar]
- 17.Reed J, Mullard A. An audience with John Reed. Nat Rev Drug Discov. 2014;13:170–171. doi: 10.1038/nrd4262. [DOI] [PubMed] [Google Scholar]
- 18.Davies S, Grant J, Catchpole M. The drugs don’t work: A global threat. Penguin Books Ltd; 2013. [Google Scholar]
- 19.Fleming A. Nobel Lectures, Physiology or Medicine 1942–1962. Elsevier Publishing Company; 1945. Nobel Lecture: Penicillin. http://www.nobelprize.org/nobel_prizes/medicine/laureates/1945/fleming-lecture.pdf. [Google Scholar]
- 20.Rossi F, Diaz L, Wollam A, Panesso D, Zhou Y, Rincon S, Arias CA. Transferable vancomycin resistance in a community-associated MRSA lineage. N Engl J Med. 2014;370:1524–1531. doi: 10.1056/NEJMoa1303359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murray CJL, Vos T, Lozano R, Naghavi M, Flaxman AD, Michaud C, Lopez AD. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2197–2223. doi: 10.1016/S0140-6736(12)61689-4. [DOI] [PubMed] [Google Scholar]
- 22.Lutje VSJ, Kennedy A. Chemotherapy for second-stage human African trypanosomiasis. Cochrane Database of Systematic Reviews 2013. 2013 doi: 10.1002/14651858.CD006201.pub3. http://onlinelibrary.wiley.com/doi/10.1002/14651858.CD006201.pub3/abstract. [DOI] [PMC free article] [PubMed]
- 23.FDA approves Impavido to treat tropical disease leishmaniasis. U.S. Food and Drug Administration; 2014. http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm389671.htm. [Google Scholar]
- 24.Silver LL. Multi-targeting by monotherapeutic antibacterials. Nat Rev Drug Discov. 2007;6:41–55. doi: 10.1038/nrd2202. [DOI] [PubMed] [Google Scholar]
- 25.Silver LL. Viable screening targets related to the bacterial cell wall. Antimicrobial Therapeutics Reviews: The Bacterial Cell Wall as an Antimicrobial Target. 2013;1277:29–53. doi: 10.1111/nyas.12006. [DOI] [PubMed] [Google Scholar]
- 26.Guidance for industry - Codevelopment of two or more new investigational drugs for use in combination. U.S. Food and Drug Administration; 2013. http://www.fda.gov/ForConsumers/ByAudience/ForPatientAdvocates/ucm237264.htm. [Google Scholar]
- 27.Morphy JR, Harris CJ. Designing multi-target drugs. RSC Publishing; 2012. [Google Scholar]
- 28.Medina-Franco JL, Giulianotti MA, Welmaker GS, Houghten RA. Shifting from the single to the multitarget paradigm in drug discovery. Drug Discov Today. 2013;18:495–501. doi: 10.1016/j.drudis.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prati F, Uliassi E, Bolognesi ML. Two diseases, one approach: Multitarget drug discovery in Alzheimer’s and neglected tropical diseases. MedChemComm. 2014;5:853–861. [Google Scholar]
- 30.East SP, Silver LL. Multitarget ligands in antibacterial research: Progress and opportunities. Expert Opin Drug Discov. 2013;8:143–156. doi: 10.1517/17460441.2013.743991. [DOI] [PubMed] [Google Scholar]
- 31.Silver LL. Challenges of antibacterial discovery. Clin Microbiol Rev. 2011;24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pucci MJ, Wiles JA. Bacterial topoisomerase inhibitors: Quinolones and beyond. In: Marinelli F, Genilloud O, editors. Antimicrobials: New and old molecules in the fight against multi-resistant bacteria. Springer; Berlin Heidelberg: 2014. [Google Scholar]
- 33.Khanna S, Sobhia ME, Bharatam PV. Additivity of molecular fields: CoMFA study on dual activators of PPAR alpha and PPAR gamma. J Med Chem. 2005;48:3015–3025. doi: 10.1021/jm049383s. [DOI] [PubMed] [Google Scholar]
- 34.Zhu W, Zhang Y, Sinko W, Hensler ME, Olson J, Molohon KJ, Oldfield E. Antibacterial drug leads targeting isoprenoid biosynthesis. Proc Natl Acad Sci USA. 2013;110:123–128. doi: 10.1073/pnas.1219899110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li K, Schurig-Briccio LA, Feng X, Upadhyay A, Pujari V, Lechartier B, Oldfield E. Multitarget drug discovery for tuberculosis and other infectious diseases. J Med Chem. 2014;57:3126–3139. doi: 10.1021/jm500131s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rohmer M. From molecular fossils of bacterial hopanoids to the formation of isoprene units: Discovery and elucidation of the methylerythritol phosphate pathway. Lipids. 2008;43:1095–1107. doi: 10.1007/s11745-008-3261-7. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, Lin F-Y, Li K, Zhu W, Liu Y-L, Cao R, Oldfield E. HIV-1 integrase inhibitor-inspired antibacterials targeting isoprenoid biosynthesis. ACS Med Chem Lett. 2012;3:402–406. doi: 10.1021/ml300038t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 39.Lindert S, Zhu W, Liu Y-L, Pang R, Oldfield E, McCammon JA. Farnesyl diphosphate synthase inhibitors from in silico screening. Chem Biol Drug Des. 2013;81:742–748. doi: 10.1111/cbdd.12121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sinko W, de Oliveira C, Williams S, Van Wynsberghe A, Durrant JD, Cao R, McCammon JA. Applying molecular dynamics simulations to identify rarely sampled ligand-bound conformational states of undecaprenyl pyrophosphate synthase, an antibacterial target. Chem Biol Drug Des. 2011;77:412–420. doi: 10.1111/j.1747-0285.2011.01101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo RT, Cao R, Liang PH, Ko TP, Chang TH, Hudock MP, Wang AH. Bisphosphonates target multiple sites in both cis- and trans-prenyltransferases. Proc Natl Acad Sci USA. 2007;104:10022–10027. doi: 10.1073/pnas.0702254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oldfield E. Targeting isoprenoid biosynthesis for drug discovery: Bench to bedside. Acc Chem Res. 2010;43:1216–1226. doi: 10.1021/ar100026v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin MB, Grimley JS, Lewis JC, Heath HT, Bailey BN, Kendrick H, Oldfield E. Bisphosphonates inhibit the growth of Trypanosoma brucei, Trypanosoma cruzi, Leishmania donovani, Toxoplasma gondii, and Plasmodium falciparum: A potential route to chemotherapy. J Med Chem. 2001;44:909–916. doi: 10.1021/jm0002578. [DOI] [PubMed] [Google Scholar]
- 44.Urbina JA, Moreno B, Vierkotter S, Oldfield E, Payares G, Sanoja C, Docampo R. Trypanosoma cruzi contains major pyrophosphate stores, and its growth in vitro and in vivo is blocked by pyrophosphate analogs. J Biol Chem. 1999;274:33609–33615. doi: 10.1074/jbc.274.47.33609. [DOI] [PubMed] [Google Scholar]
- 45.Bruchhaus I, Jacobs T, Denart M, Tannich E. Pyrophosphate-dependent phosphofructokinase of Entamoeba histolytica: Molecular cloning, recombinant expression and inhibition by pyrophosphate analogues. Biochem J. 1996;316:57–63. doi: 10.1042/bj3160057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin MB, Arnold W, Heath IIIHT, Urbina JA, Oldfield E. Nitrogen-containing bisphosphonates as carbocation transition state analogs for isoprenoid biosynthesis. Biochem Biophys Res Commun. 1999;263:754–758. doi: 10.1006/bbrc.1999.1404. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Y, Cao R, Yin F, Hudock MP, Guo RT, Krysiak K, Oldfield E. Lipophilic bisphosphonates as dual farnesyl/geranylgeranyl diphosphate synthase inhibitors: An X-ray and NMR investigation. J Am Chem Soc. 2009;131:5153–5162. doi: 10.1021/ja808285e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.No JH, de Macedo Dossin F, Zhang Y, Liu YL, Zhu W, Feng X, Oldfield E. Lipophilic analogs of zoledronate and risedronate inhibit Plasmodium geranylgeranyl diphosphate synthase (GGPPS) and exhibit potent antimalarial activity. Proc Natl Acad Sci USA. 2012;109:4058–4063. doi: 10.1073/pnas.1118215109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, Cao R, Yin F, Lin FY, Wang H, Krysiak K, Oldfield E. Lipophilic pyridinium bisphosphonates: Potent gammadelta T cell stimulators. Angew Chem Int Ed Engl. 2010;49:1136–1138. doi: 10.1002/anie.200905933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang L, Kamath A, Das H, Li L, Bukowski JF. Antibacterial effect of human Vγ2Vδ2 T cells in vivo. J Clin Invest. 2001;108:1349–1357. doi: 10.1172/JCI13584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ellis C. Doing the two step. Nat Rev Drug Discov. 2003;2:605–605. [Google Scholar]
- 52.Courchesne WE. Characterization of a novel, broad-based fungicidal activity for the antiarrhythmic drug amiodarone. J Pharm Exp Ther. 2002;300:195–199. doi: 10.1124/jpet.300.1.195. [DOI] [PubMed] [Google Scholar]
- 53.Gupta SS, Ton V-K, Beaudry V, Rulli S, Cunningham K, Rao R. Antifungal activity of amiodarone is mediated by disruption of calcium homeostasis. J Biol Chem. 2003;278:28831–28839. doi: 10.1074/jbc.M303300200. [DOI] [PubMed] [Google Scholar]
- 54.Benaim G, Sanders JM, Garcia-Marchán Y, Colina C, Lira R, Caldera AR, Urbina JA. Amiodarone has intrinsic anti-Trypanosoma cruzi activity and acts synergistically with posaconazole. J Med Chem. 2006;49:892–899. doi: 10.1021/jm050691f. [DOI] [PubMed] [Google Scholar]
- 55.Benaim G, Paniz Mondolfi AE. The emerging role of amiodarone and dronedarone in Chagas disease. Nat Rev Cardiol. 2012;9:605–609. doi: 10.1038/nrcardio.2012.108. [DOI] [PubMed] [Google Scholar]
- 56.Paniz-Mondolfi AE, Perez-Alvarez AM, Reyes-Jaimes O, Socorro G, Zerpa O, Slova D, Concepcion JL. Concurrent Chagas’ disease and borderline disseminated cutaneous leishmaniasis: The role of amiodarone as an antitrypanosomatidae drug. Ther Clin Risk Manag. 2008;4:659–663. doi: 10.2147/tcrm.s2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paniz-Mondolfi AE, Perez-Alvarez AM, Lanza G, Marquez E, Concepcion JL. Amiodarone and itraconazole: A rational therapeutic approach for the treatment of chronic Chagas’ disease. Chemotherapy. 2009;55:228–233. doi: 10.1159/000219436. [DOI] [PubMed] [Google Scholar]
- 58.Protopopova M, Hanrahan C, Nikonenko B, Samala R, Chen P, Gearhart J, Nacy CA. Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J Antimicrob Chemother. 2005;56:968–974. doi: 10.1093/jac/dki319. [DOI] [PubMed] [Google Scholar]
- 59.Tahlan K, Wilson R, Kastrinsky DB, Arora K, Nair V, Fischer E, Boshoff HI. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2012;56:1797–1809. doi: 10.1128/AAC.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ellis D. Amphotericin B: Spectrum and resistance. J Antimicrob Chemother. 2002;49:7–10. doi: 10.1093/jac/49.suppl_1.7. [DOI] [PubMed] [Google Scholar]
- 61.Cencig S, Coltel N, Truyens C, Carlier Y. Parasitic loads in tissues of mice infected with Trypanosoma cruzi and treated with ambisome. PLoS Negl Trop Dis. 2011;5:e1216. doi: 10.1371/journal.pntd.0001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Woods RA, Bard M, Jackson IE, Drutz DJ. Resistance to polyene antibiotics and correlated sterol changes in 2 isolates of Candida tropicalis from a patient with an amphotericin b-resistant funguria. J Infect Dis. 1974;129:53–58. doi: 10.1093/infdis/129.1.53. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Y-M, Rock CO. Membrane lipid homeostasis in bacteria. Nature Rev Microbiol. 2008;6:222–233. doi: 10.1038/nrmicro1839. [DOI] [PubMed] [Google Scholar]
- 64.Wilson DN. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nature Rev Microbiol. 2014;12:35–48. doi: 10.1038/nrmicro3155. [DOI] [PubMed] [Google Scholar]
- 65.Bercovier H, Kafri O, Sela S. Mycobacteria possess a surprisingly small number of ribosomal-RNA genes in relation to the size of their genome. Biochem Biophys Res Commun. 1986;136:1136–1141. doi: 10.1016/0006-291x(86)90452-3. [DOI] [PubMed] [Google Scholar]
- 66.Munday JC, Eze AA, Baker N, Glover L, Clucas C, Andres DA, De Koning HP. Trypanosoma brucei aquaglyceroporin 2 is a high-affinity transporter for pentamidine and melaminophenyl arsenic drugs and the main genetic determinant of resistance to these drugs. J Antimicrob Chemother. 2014;69:651–663. doi: 10.1093/jac/dkt442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Edwards KJ, Jenkins TC, Neidle S. Crystal structure of a pentamidine-oligonucleotide complex: Implications for DNA-binding properties. Biochemistry. 1992;31:7104–7109. doi: 10.1021/bi00146a011. [DOI] [PubMed] [Google Scholar]
- 68.Opperman TJ, Houseweart C, Aiello D, Williams JD, Peet NP, Moir DT, Bowlin TL. The mechanism of antibacterial action of novel bis-indole antibiotics. Microbiotix, Inc; http://www.microbiotix.com/posters/ICAAC-2010/F1-1631%20Opperman%20et%20al%20ICAAC2010.pdf. [Google Scholar]
- 69.Moreno SNJ. Pentamidine is an uncoupler of oxidative phosphorylation in rat liver mitochondria. Arch Biochem Biophys. 1996;326:15–20. doi: 10.1006/abbi.1996.0041. [DOI] [PubMed] [Google Scholar]
- 70.Tsutsumi LS, Owusu YB, Hurdle JG, Sun D. Progress in the discovery of treatments for C. difficile infection: A clinical and medicinal chemistry review. Curr Top Med Chem. 2014;14:152–175. doi: 10.2174/1568026613666131113154753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ravic M, Suckling C, Hunter I, Gemmell C, Warn P, Sattar A, Saunders M. Disposition of MGB-BP-3, a new class of antibacterial agent, after oral administration in a hamster model of severe Clostridium difficile associated diarrhoea (CDAD). 52nd Interscience Conference on Antimicrobial Agents and Chemotherapy.2012. [Google Scholar]
- 72.Antibiotic Resistance: Implications for Global Health and Novel Intervention Strategies: Workshop Summary. Institute of Medicine (US) Forum on Microbial Threats. National Academies Press (US); 2010. [PubMed] [Google Scholar]
- 73.Allen RC, Popat R, Diggle SP, Brown SP. Targeting virulence: Can we make evolution-proof drugs? Nat Rev Micro. 2014;12:300–308. doi: 10.1038/nrmicro3232. [DOI] [PubMed] [Google Scholar]
- 74.Liu GY, Essex A, Buchanan JT, Datta V, Hoffman HM, Bastian JF, Nizet V. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J Exp Med. 2005;202:209–215. doi: 10.1084/jem.20050846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Potera C. Pigments of pathogens might provide golden antimicrobial opportunities. ASM News. 2005;71:450–451. [Google Scholar]
- 76.Liu CI, Liu GY, Song Y, Yin F, Hensler ME, Jeng WY, Oldfield E. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science. 2008;319:1391–1394. doi: 10.1126/science.1153018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dickson JK, Biller SA, Magnin DR, Petrillo EW, Hillyer JW, Hsieh DC, Ciosek CP. Orally active squalene synthase inhibitors: Bis((acyloxy)alkyl) prodrugs of the α-phosphonosulfonic acid moiety. J Med Chem. 1996;39:661–664. doi: 10.1021/jm950735s. [DOI] [PubMed] [Google Scholar]
- 78.Layre E, Lee HJ, Young DC, Martinot AJ, Buter J, Minnaard AJ, Moody DB. Molecular profiling of Mycobacterium tuberculosis identifies tuberculosinyl nucleoside products of the virulence-associated enzyme Rv3378c. Proc Natl Acad Sci USA. 2014;111:2978–2983. doi: 10.1073/pnas.1315883111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chan HC, Feng X, Ko TP, Huang CH, Hu Y, Zheng Y, Guo RT. Structure and inhibition of tuberculosinol synthase and decaprenyl diphosphate synthase from Mycobacterium tuberculosis. J Am Chem Soc. 2014;136:2892–2896. doi: 10.1021/ja413127v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chow OA, von Kockritz-Blickwede M, Bright AT, Hensler ME, Zinkernagel AS, Cogen AL, Nizet V. Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe. 2010;8:445–454. doi: 10.1016/j.chom.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lin FY, Zhang Y, Hensler M, Liu YL, Chow OA, Zhu W, Oldfield E. Dual dehydrosqualene/squalene synthase inhibitors: Leads for innate immune system-based therapeutics. Chem Med Chem. 2012;7:561–564. doi: 10.1002/cmdc.201100589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang W, Oldfield E. Bioorganometallic chemistry with IspG and IspH: Structure, function, and inhibition of the [Fe4S4] proteins involved in isoprenoid biosynthesis. Angew Chem Int Edit. 2014;53:4294–4310. doi: 10.1002/anie.201306712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen CY, Yao S, Huang D, Wei H, Sicard H, Zeng G, Chen ZW. Phosphoantigen/IL2 expansion and differentiation of Vγ2Vδ2 T cells increase resistance to tuberculosis in nonhuman primates. PLoS Pathog. 2013;9:e1003501. doi: 10.1371/journal.ppat.1003501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Workalemahu G, Wang H, Puan K-J, Nada MH, Kuzuyama T, Jones BD, Morita CT. Metabolic engineering of Salmonella vaccine bacteria to boost human Vγ2Vδ2 T cell immunity. J Immunol. 2014;193:708–721. doi: 10.4049/jimmunol.1302746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wilde O, Frankau G. The importance of being earnest: a trivial comedy for serious people. London, England, UK: 1899. [Google Scholar]