

Abstract
Triptolide is a key component of the traditional Chinese medicinal plant Thunder God Vine and has potent anticancer and immunosuppressive activities. It is an irreversible inhibitor of eukaryotic transcription through covalent modification of XPB, a subunit of the general transcription factor TFIIH. Cys342 of XPB was identified as the residue that undergoes covalent modification by the 12,13-epoxide group of triptolide. Mutation of Cys342 of XPB to threonine conferred resistance to triptolide on the mutant protein. Replacement of the endogenous wild-type XPB with the Cys342Thr mutant in a HEK293T cell line rendered it completely resistant to triptolide, thus validating XPB as the physiologically relevant target of triptolide. Together, these results deepen our understanding of the interaction between triptolide and XPB and have implications for the future development of new analogues of triptolide as leads for anticancer and immunosuppressive drugs.
Keywords: inhibitors, medicinal chemistry, natural products, target validation, transcription factors
Triptolide (1, TPL), a diterpene triepoxide (Figure 1), was isolated from Trypterygium Wilfordii Hook F (Lei Gong Teng or Thunder God Vine), which has been used in traditional Chinese medicine for centuries.[1] Numerous cellular activities have been found for triptolide, including inhibition of the activity of a number of unrelated transcription factors and global inhibition of mRNA synthesis.[2] A number of putative cellular targets of triptolide have been reported to date. Among them are the calcium channel polycystin-2, the membrane protease ADAM10, the dCTP pyrophosphatase (DCTPP1), and the kinase-regulating protein TAB1.[3] By using a systematic top-down approach with the inhibitory effect of triptolide on de novo RNA synthesis as the starting point, we recently identified the Xeroderma Pigmentosum B (XPB)/ERCC3 subunit of TFIIH as a new molecular target of triptolide.[4] We showed that triptolide forms a covalent complex with XPB and inhibits its DNA-dependent ATPase activity without affecting its DNA helicase activity.
Figure 1.
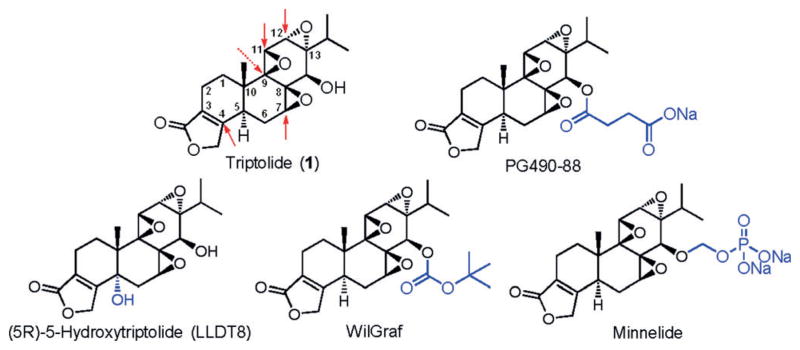
Structures of triptolide and triptolide analogues under clinical development. Potential sites of attack by a nucleophile from a protein are marked with red arrows. Sections for which the analogues differ in structure from triptolide are highlighted in blue.
Several analogues of triptolide have been developed as potential anticancer and immunosuppressive drug leads (Figure 1). They include PG490-88 and WilGraf for treating graft rejection after organ transplantation, LLDT8 for treating rheumatoid arthritis, and Minnelide for treating cancer.[5] Among these analogues, Minnelide is currently undergoing Phase I clinical trial for cancer.[6] It is noteworthy that all analogues of triptolide in clinical development contain the intact core structure of triptolide.
Triptolide is decorated with four potentially reactive chemical groups that may covalently react with XPB: the butenolide moiety in the five-membered lactone or one of the three epoxide groups (Figure 1). There have been disagreements in the literature as to which of the epoxide groups is the most reactive electrophile for thiols. One group reported that the 9,11-epoxide of triptolide is opened by propanethiol to form an adduct at C9 (2; Figure 2a).[7] Years later, another group reported that the same reaction led to the opening of the 12,13-epoxide of triptolide at the C12 position (3; Figure 2a).[8] To distinguish between those two alternative paths, we reacted N-acetyl-L-cysteine methyl ester (10 mM) with triptolide (0.1 mM) in a PBS buffer (pH 7.4, 1 mM MgCl2 and 1% DMSO) at room temperature for 72 h. Only one product was detected and isolated in 55% yield, with 45% unreacted triptolide recovered (Figure S1a in the Supporting Information). By means of LC–MS (Figure S1b) and 2D-NMR (H-H COSY, HMBC, NOE; Figure S2), the structure of the product was determined to be the C12 adduct of N-acetyl-L-cysteine methyl ester (4; Figure 2a). These results suggest that the 12,13-epoxide of triptolide is intrinsically the most reactive toward thiols.
Figure 2.

The C12,13-epoxide of triptolide (TPL) forms a covelant bond with XPB. A) The C12,13-epoxide of triptolide reacts with N-acetyl-L-cysteine methyl ester to form the adduct 4. B) Inhibition of cell proliferation and the ATPase activity of TFIIH by triptolide analogues. Mean values ± SEM from two independent experiments are shown. C) Covalent binding of triptolide analogues to XPB as determined by the lack of recovery of XPB ATPase activity upon dialysis of the analogue–XPB complex. Mean values ± SD from two independent experiments are shown. DMSO=dimethyl sulfoxide.
To assess which of the four potentially reactive groups in triptolide is involved in covalent modification of XPB, we synthesized four analogues of triptolide in which the bute-none unit and each of the three epoxide groups were eliminated either individually or in combination (Figure 2b). We then assessed the cellular activity as well as the ability of each of the four analogues to covalently bind to XPB by using a “binding-dialysis-activity recovery” sequence. All four analogues suffered from a significant loss in activity for inhibition of cell proliferation and the ATPase activity of TFIIH, albeit to different degrees (Figure 2b). Among the four analogues, only 5, which lacks the 12,13-epoxide, completely lost its activity at the highest soluble concentrations, while the remaining analogues retained partial anti-proliferative activity. To further determine the ability of the analogues to covalently bind XPB, we incubated each analogue with purified TFIIH for 2 h before subjecting the incubation mixture to extensive dialysis. Only if an analogue is covalently bound to XPB, would there be no recovery of XPB activity after dialysis. Significant recovery of the ATPase activity of XPB was observed with analogue 5, while the remaining analogues 6–8 retained the ability to covalently bind XPB and caused irreversible inhibition similar to that caused by triptolide (Figure 2c, and Figure S3). Together, these observations indicate that only the 12,13-epoxide of triptolide mediates covalent modification of XPB, while the other two epoxide groups, as well as the butenolide moiety, are dispensable for covalent binding of triptolide to XPB.
Having identified the epoxide group in triptolide that is involved in the covalent modification of XPB, we turned to the reciprocal question of which amino acid residue in XPB is covalently modified by triptolide. HeLa cell nuclear extract was incubated with triptolide to allow the formation of the triptolide–XPB covalent complex, which was then immunoprecipitated using an anti-XPB antibody and subsequently subjected to SDS-PAGE. The XPB protein band, as judged by its molecular mass, was excised from the gel and subjected to in-gel digestion with trypsin. The tryptic peptide mixture was extensively analyzed by using high-resolution mass spectrometry. With this approach, 82% coverage of the XPB protein sequence was achieved (Figure S4). All identified tryptic peptides gave the expected molecular masses with one exception. Further MS/MS sequencing of the peptide with a mismatch in molecular mass revealed that it spans residues 335–346 of XPB (335SGVIVLPCGAGK346) and there is a mass shift of +360.1573 Da for this peptide at Cys342, which corresponds to the exact mass of triptolide (Figure 3a). These results suggest that Cys342 is likely the residue in XPB that is covalently modified by triptolide.
Figure 3.

Triptolide binds covalently to C342 of XPB. A) MS/MS spectrum of a peptide from human XPB (TFIIH) treated with triptolide. Insets: MS/MS spectrum of m/z 730.8895, which led to the identification of a mass shift of +360.1573 Da at the Cys342 residue of 335SGVIVLPCGAGK346. The labels (b) and (y) designate N- and C-terminal fragments of the peptide, respectively. The label Δ designates (b) or (y) ions with water and/or ammonia loss. B) [3H]-triptolide does not bind covalently to recombinant C342A, C342S, or C342T XPB mutant proteins.
Phylogenetic sequence alignment revealed that Cys342 is conserved among eukaryotes but is changed to either a threonine or a valine in various archaeal species (Figure S5).[9] To verify that Cys342 mediates the covalent binding of triptolide to XPB, we mutated it to Ser, Thr, and Ala. Each of the mutants, as well as the wild-type XPB protein, was produced through baculovirus-driven overexpression in insect cells, followed by purification. Although wild-type XPB bound covalently to [3H]-triptolide, none of the Cys342 mutants were capable of forming a covalent complex with [3H]-triptolide (Figure 3b). In comparison to wild-type XPB, all three mutants have lower intrinsic ATPase activity (Figure S6). When the mutant proteins were assayed in the presence of triptolide, none were inhibited by up to 100 μM of triptolide. These results support the hypothesis that Cys342 is the residue that is covalently modified by triptolide and that this covalent modification is essential for the inhibition of XPB by triptolide.
In addition to assessing the intrinsic ATPase activity of the Cys342 mutant XPB proteins, we also purified their corresponding TFIIH complexes (Figure S7). Of the three mutants, the C342T mutant TFIIH complex possessed the highest enzymatic activity and remained resistant to 100 μM triptolide (Figure 4a, and Figure S8). The retention of significant enzymatic activity of the C342T mutant in the context of TFIIH and its insensitivity to triptolide offered a precious opportunity to assess the importance of XPB as a mediator of the effect of triptolide on cell proliferation by replacing endogenous wild-type XPB with this mutant. Since the complete knock-out of XPB is lethal, we used a combination of CRISPR/Cas9 and piggyBac transposase methods to generate a single-allele knock-in mutant line.[10] We introduced a point mutation (C342T) to one XPB allele with a piggyBac transposon based homologous recombination donor that facilitates footprint-free gene editing, followed by knock-out of the remaining wild type allele of XPB by using CRISPR/Cas9, thereby generating a HEK293T cell line (T7115) that only expresses the XPB C342T mutant (Figure S13). The knock-out of one XPB allele and mutation of the other (C342T) were verified by PCR/restriction digestion with AflII and sequencing (Figures S9,S10). As expected, the resultant T7115 line had a slower growth rate in comparison with wild-type HEK293T cells as judged by cell density, which likely reflects the lower enzymatic activity of the XPB C342T mutant (Figure S11). Importantly, the XPB-C342T-expressing T7115 line was completely resistant to triptolide at concentrations up to 30 μM, while the proliferation of wild-type HEK293T cells was completely inhibited at 100 nM (Figure 4b). It is noteworthy that the T7115 cells were still sensitive to two other inhibitors of transcription, namely actinomycin D and α-amanitin,[11] thus suggesting that the resistance conferred by the C342T mutant of XPB is highly specific to triptolide (Figure S12).
Figure 4.
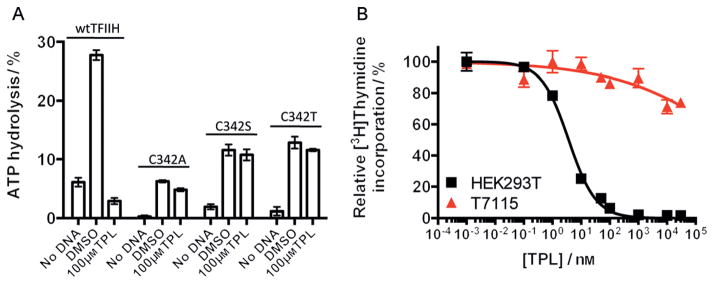
The C342T XPB mutant is resistant to triptolide in vitro and confers high-level triptolide resistance to HET293T cells. A) C342A, C342S, and C342T XPB mutant TFIIH complexes were resistant to triptolide at concentrations up to 100 μM and C342T-TFIIH has the highest ATPase activity. Mean values ± SD from two independent experiments are shown. B) A mutant knock-in cell line (T7115) expressing only the C342T XPB mutant is selectively resistant to triptolide in cell proliferation assay. Mean values ± SEM from four independent experiments are shown.
The intrinsic electrophilicity of epoxide groups led to an earlier hypothesis that triptolide likely reacts with thiol groups in enzymes to elicit its antitumor activity.[7] Indeed, several epoxide-containing natural products, such as fumagillin, trapoxin B, and E-64, are known to work through covalent modification of key residues, including cysteine, of target proteins.[12] We showed that the α, β-unsaturated lactone, 7,8-epoxide, and 9,11-epoxide groups are all dispensable for the covalent binding of triptolide to XPB. A 9,11-deoxytriptolide analogue has been previously shown to be slightly more potent than triptolide for the inhibition of cancer cell proliferation,[13] which is consistent with our finding. The identification of the 12,13-epoxide of triptolide as the sole functional group required for the covalent modification of XPB suggests that the reaction of the cysteine thiol from XPB takes advantage of the intrinsic chemical reactivity of the 12,13-epoxide of triptolide.[7]
The identification of Cys342 as the sole residue of XPB to mediate the formation of a covalent complex with triptolide is in agreement with the earlier hypothesis that triptolide works by modifying cysteine thiols in enzymes.[7] It is interesting to note that the same cysteine residue was also detected by using a clickable iodoacetamide probe in conjunction with mass spectrometry to profile active cysteine residues in proteins.[14] That the same cysteine is not absolutely conserved in all organisms suggests that it is not essential for the intrinsic enzymatic activity of XPB.[9] Indeed, we found that the mutation of Cys342 to alanine, serine, or threonine did not completely abolish its ATPase activity (Figure 4a, and Figures S6,S8). The inability of any of the three cysteine mutants to form a covalent complex with [3H]-triptolide indicates that Cys342 is the only residue that undergoes covalent modification by triptolide.
To date, several putative target proteins of triptolide have been reported, including polycystin-2, ADAM10, DCTPP1, TAB1, and XPB, thus raising the question of which targets are physiologically relevant for its antitumor activity.[3] In a recent study to establish an elegant new target validation method involving a combination of in vitro selection of drug-resistant cancer cell lines followed by next-generation sequencing to identify responsible mutations, Smurnyy et al. found multiple triptolide-resistant mutations in XPB (ERCC3) and its partner protein GTF2H4.[15] Importantly, no triptolide-resistant mutations were found in polycystin-2, ADAM10, DCTPP1, or TAB1. As described above, we took advantage of the triptolide-resistant C342T mutant and generated a mutant HEK293T cell line (T7115) that is dependent on a single C342T mutant allele of XPB for growth and survival. The XPB C342T mutant renders the T7115 cell line nearly completely resistant to triptolide. It is noteworthy that the level of resistance conferred by the C342T mutation is approximately 100-fold higher than the most triptolide-resistant mutants previously identified.[15] Together, these results validate XPB as the key target that is responsible for the antiprolifeative activity of triptolide.
One of the major hurdles for triptolide to becoming a clinically useful drug is its toxicity, which has been attributed in part to the presence of multiple reactive electrophiles, including epoxide groups and the butenolide moiety.[16] Most, if not all, analogues of triptolide in preclinical and clinical development to date have all three epoxide and the butenolide groups intact (Figure 1), which likely contributes to their toxicity. The identification and verification of the 12,13-epoxide group as the only group involved in the covalent modification of XPB suggests that the other epoxide groups and the butenolide are likely to be dispensable for the anticancer activity of triptolide through inhibition of transcriptional initiation. It would thus be logical to design and synthesize novel triptolide analogues devoid of the other epoxide and the butenolide groups as the next generation of triptolide analogues to determine whether they have reduced toxicity while maintaining their anticancer activity.
Supplementary Material
Footnotes
This work was supported in part by a discretionary fund and FAMRI (J.O.L.), NIGMS training grant (D.T), Robert A. Welch Foundation (A-1280 to D.R.) and NIH (R37 GM 069874 to D.R.), NIH GM105933 and CA160036 (Y.Z.) and Maryland Stem Cell Research Fund (2011-MSCRFE-0087 to Z.Y.). Support of the Natural Products LINCHPIN Laboratory from the Vice President for Research, College of Science, and the Department of Chemistry at Texas A&M University is gratefully acknowledged.
Supporting information for this article (including experimental details) is available on the WWW under http://dx.doi.org/10.1002/anie.201408817.
Contributor Information
Dr. Qing-Li He, Department of Pharmacology, Johns Hopkins School of Medicine, 725 North Wolfe Street, Hunterian Building, Room 516, Baltimore, MD 21205 (USA).
Dr. Denis V. Titov, Department of Pharmacology, Johns Hopkins School of Medicine, 725 North Wolfe Street, Hunterian Building, Room 516, Baltimore, MD 21205 (USA).
Dr. Jing Li, Natural Products LINCHPIN Laboratory and Dept. of Chemistry, Texas A&M University, P.O. Box 30012, College Station, TX 77843 (USA)
Prof. Minjia Tan, Ben May Department for Cancer Research, University of Chicago, Chicago, IL 60637 (USA)
Prof. Zhaohui Ye, Institute for Cell Engineering, Johns Hopkins University, School of Medicine, Baltimore, MD 21205 (USA)
Prof. Yingming Zhao, Ben May Department for Cancer Research, University of Chicago, Chicago, IL 60637 (USA)
Prof. Daniel Romo, Natural Products LINCHPIN Laboratory and Dept. of Chemistry, Texas A&M University, P.O. Box 30012, College Station, TX 77843 (USA)
Prof. Jun O. Liu, Email: joliu@jhu.edu, Department of Pharmacology, Johns Hopkins School of Medicine, 725 North Wolfe Street, Hunterian Building, Room 516, Baltimore, MD 21205 (USA)
References
- 1.a) Kupchan SM, Court WA, Dailey RG, Jr, Gilmore CJ, Bryan RF. J Am Chem Soc. 1972;94:7194–7195. doi: 10.1021/ja00775a078. [DOI] [PubMed] [Google Scholar]; b) Zhao X-M. In: Supplement to Materia Medica. Xing Zhang’s Jie., editor. Tang Pulishing House; Qian Tang: 1765. [Google Scholar]
- 2.a) Qiu D, Zhao G, Aoki Y, Shi L, Uyei A, Nazarian S, Ng JC, Kao PN. J Biol Chem. 1999;274:13443–13450. doi: 10.1074/jbc.274.19.13443. [DOI] [PubMed] [Google Scholar]; b) Westerheide SD, Kawahara TL, Orton K, Morimoto RI. J Biol Chem. 2006;281:9616–9622. doi: 10.1074/jbc.M512044200. [DOI] [PubMed] [Google Scholar]; c) Lee KY, Chang W, Qiu D, Kao PN, Rosen GD. J Biol Chem. 1999;274:13451–13455. doi: 10.1074/jbc.274.19.13451. [DOI] [PubMed] [Google Scholar]; d) Leuenroth SJ, Crews CM. Cancer Res. 2008;68:5257–5266. doi: 10.1158/0008-5472.CAN-07-6207. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) McCallum C, Kwon S, Leavitt P, Shoop W, Michael B, Felcetto T, Zaller D, O’Neill E, Frantz-Wattley B, Thompson C, Forrest G, Carballo-Jane E, Gurnett A. Therapy. 2005;2:261–273. [Google Scholar]; f) Vispe S, DeVries L, Creancier L, Besse J, Breand S, Hobson DJ, Svejstrup JQ, Annereau JP, Cussac D, Dumontet C, Guilbaud N, Barret JM, Bailly C. Mol Cancer Ther. 2009;8:2780–2790. doi: 10.1158/1535-7163.MCT-09-0549. [DOI] [PubMed] [Google Scholar]
- 3.a) Leuenroth SJ, Okuhara D, Shotwell JD, Markowitz GS, Yu Z, Somlo S, Crews CM. Proc Natl Acad Sci USA. 2007;104:4389–4394. doi: 10.1073/pnas.0700499104. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Soundararajan R, Sayat R, Robertson GS, Marignani PA. Cancer Biol Ther. 2009;8:2054–2062. doi: 10.4161/cbt.8.21.9803. [DOI] [PubMed] [Google Scholar]; c) Corson TW, Cavga H, Aberle N, Crews CM. Chem Bio Chem. 2011;12:1767–1773. doi: 10.1002/cbic.201100007. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lu Y, Zhang Y, Li L, Feng X, Ding S, Zheng W, Li J, Shen P. Chem Biol. 2014;21:246–256. doi: 10.1016/j.chembiol.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 4.Titov DV, Gilman B, He QL, Bhat S, Low WK, Dang Y, Smeaton M, Demain AL, Miller PS, Kugel JF, Goodrich JA, Liu JO. Nat Chem Biol. 2011;7:182–188. doi: 10.1038/nchembio.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou ZL, Yang YX, Ding J, Li YC, Miao ZH. Nat Prod Rep. 2012;29:457–475. doi: 10.1039/c2np00088a. [DOI] [PubMed] [Google Scholar]
- 6.Chugh R, Sangwan V, Patil SP, Dudeja V, Dawra RK, Banerjee S, Schumacher RJ, Blazar BR, Georg GI, Vickers SM, Saluja AK. Sci Transl Med. 2012;4:156ra139. doi: 10.1126/scitranslmed.3004334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kupchan SM, Schubert RM. Science. 1974;185:791–793. doi: 10.1126/science.185.4153.791. [DOI] [PubMed] [Google Scholar]
- 8.Yu DQ, Zhang DM, Wang HB, Liang XT. Acta Pharmacol Sin. 1992;27:830–836. [PubMed] [Google Scholar]
- 9.Fan L, Arvai AS, Cooper PK, Iwai S, Hanaoka F, Tainer JA. Mol Cell. 2006;22:27–37. doi: 10.1016/j.molcel.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 10.a) Yusa K, Zhou L, Li MA, Bradley A, Craig NL. Proc Natl Acad Sci USA. 2011;108:1531–1536. doi: 10.1073/pnas.1008322108. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Choi SM, Kim Y, Shim JS, Park JT, Wang RH, Leach SD, Liu JO, Deng C, Ye Z, Jang YY. Hepatology. 2013;57:2458–2468. doi: 10.1002/hep.26237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Lindell TJ, Weinberg F, Morris PW, Roeder RG, Rutter WJ. Science. 1970;170:447–449. doi: 10.1126/science.170.3956.447. [DOI] [PubMed] [Google Scholar]; b) Reich E, Franklin RM, Shatkin AJ, Tatum EL. Science. 1961;134:556–557. doi: 10.1126/science.134.3478.556. [DOI] [PubMed] [Google Scholar]
- 12.a) Griffith EC, Su Z, Turk BE, Chen S, Chang YW, Wu Z, Biemann K, Liu JO. Chem Biol. 1997;4:461–471. doi: 10.1016/s1074-5521(97)90198-8. [DOI] [PubMed] [Google Scholar]; b) Sin N, Meng L, Wang MQW, Wen JJ, Bornmann WG, Crews CM. Proc Natl Acad Sci USA. 1997;94:6099–6103. doi: 10.1073/pnas.94.12.6099. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kijima M, Yoshida M, Sugita K, Horinouchi S, Beppu T. J Biol Chem. 1993;268:22429–22435. [PubMed] [Google Scholar]; d) Taunton J, Hassig CA, Schreiber SL. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]; e) Barrett AJ, Kembhavi AA, Hanada K. Acta Biol Med Ger. 1981;40:1513–1517. [PubMed] [Google Scholar]
- 13.Zhou B, Miao Z, Deng G, Ding J, Yang Y, Feng H, Li Y. Bioorg Med Chem Lett. 2010;20:6217–6221. doi: 10.1016/j.bmcl.2010.08.106. [DOI] [PubMed] [Google Scholar]
- 14.Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, Cravatt BF. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smurnyy Y, Cai M, Wu H, McWhinnie E, Tallarico JA, Yang Y, Feng Y. Nat Chem Biol. 2014;10:623–625. doi: 10.1038/nchembio.1550. [DOI] [PubMed] [Google Scholar]
- 16.a) Chen BJ. Leuk Lymphoma. 2001;42:253–265. doi: 10.3109/10428190109064582. [DOI] [PubMed] [Google Scholar]; b) Du F, Liu Z, Li X, Xing J. J Appl Toxicol. 2014;34:878–884. doi: 10.1002/jat.2906. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.