

Abstract
Glycan microarrays have become a powerful platform to investigate the interactions of carbohydrates with a variety of biomolecules. However, the number and diversity of glycans available for use in such arrays represents a key bottleneck in glycan array fabrication. To address this challenge, we describe a novel glycan array platform based on surface patterning of engineered glycophages that display unique carbohydrate epitopes. Specifically, we show that glycophages are compatible with surface immobilization procedures and that phage-displayed oligosaccharides retain the ability to be recognized by different glycan-binding proteins (e.g., antibodies, lectins) after immobilization. A key advantage of glycophage arrays is that large quantities of glycophages can be produced biosynthetically from recombinant bacteria and isolated directly from bacterial supernatants without laborious purification steps. Taken together, the glycophage array technology described here should help to expand the diversity of glycan libraries and provide a complement to the existing toolkit for high-throughput analysis of glycan-protein interactions.
Keywords: filamentous phage, functional glycomics, glycan-binding protein, glycoarray, lectin
Introduction
Carbohydrate chains known as glycans are involved in myriad molecular interactions and are crucial for the development, growth, function, and survival of many organisms [1]. Their importance is derived in part from the incredible diversity of forms that have been observed in nature, either as free structures or as conjugates to other biomolecules (such as proteins, lipids, and nucleic acids). In fact, the spectrum of all glycan structures, known as the glycome, in some organisms is so large that it exceeds the number of proteins encoded in the genome by orders of magnitude [2].
To date, substantial knowledge regarding glycans has been gained from studies employing traditional analytical tools. However, there remains a pressing need for high-throughput techniques that allow systematic investigation of structurally and functionally important glycans, which often involve glycan-binding proteins (GBPs) that recognize specific glycan sequences and thereby ‘decode’ the complex structural information in glycans. A major technical breakthrough along these lines was the development of printed glycan microarrays, in which many defined glycan structures are immobilized on a solid support in a spatially defined arrangement and simultaneously presented to GBPs or microorganisms including viruses and bacteria (reviewed in [3-8]). Such microarrays enable multiplex analysis of glycan-protein interactions using only tiny amounts of material and thus have emerged as a powerful methodology for decoding the informational content of the glycome. One of the first reports of such a system was in 2002 by Wang et al. who described the fabrication of a carbohydrate array to identify specific microbial polysaccharides that interacted with human antibodies [9]. Since that time, numerous reports have demonstrated the use of glycan microarrays for determining the glycan-binding specificity of GBPs and shedding light on key mechanistic details underlying mammalian biology, host-pathogen interactions, and immune recognition of glycans [3, 5, 10, 11].
In general, the glycan substrates for most microarrays are either synthesized via chemical, enzymatic and/or chemo-enzymatic routes, which can be expensive; or isolated from natural sources such as cells, tissues, pathogens, milk or urine, which yields low amounts of glycan and requires several purification steps [4, 7]. As a result, most glycan microarray studies only use a small fraction of the total structural diversity found in nature. While this has been sufficient to obtain useful results in a variety of applications [9, 12-16], the full potential of defined glycan microarrays will only be realized by (i) increasing the quantity and diversity of glycan structures and (ii) developing reliable and reproducible chemistries for the immobilization of chemically and structurally diverse glycan probes onto a solid support with retention of functionality. Hence, the chemistries related to the synthesis, harvesting, and immobilization of glycans are paramount for microarray technology development.
Here, we describe a facile alternative for glycan array fabrication using engineered phages that display defined glycan epitopes, so-called “glycophages” [17, 18]. Since its inception in 1985 [19], phage display has been used primarily for library-based selection of ligand-binding proteins [20-23] as well as stabilized protein variants, regulatable enzymes and protein catalysts [23-25]. While far less common, recombinant phages can also be immobilized on a solid support in a spatially defined arrangement such that phage-displayed proteins or peptides are accessible for subsequent high-throughput protein-protein interaction screening. For example, microarrays have been patterned with recombinant phages displaying antibody clones for the detection of leukocyte surface antigens [26], small peptides for the diagnosis of the immune response in patients with cancer [27] or HIV [28], and even cDNA libraries for the diagnosis of non-small cell lung cancer [29]. In all of these cases, phage particles were entirely compatible with the microarray patterning process and the displayed molecules (e.g., proteins, peptides) were accessible for subsequent protein-protein interaction analysis using standard microarray scanners. Building on these ideas, we have combined the immobilization compatibility of phage particles with our recent demonstration of glycan display on filamentous phage [17] to create a novel glycan array technology for detecting GBP interactions. Specifically, we show that glycophages are compatible with surface immobilization procedures, can be modified with different glycan structures, and retain the ability following immobilization to be recognized by different GBPs (e.g., antibodies, lectins) that recognize the displayed oligosaccharides. The advantages of glycophage-based arrays include the low cost and scalability of phage/glycan production, which are biosynthetic processes involving the cultivation of recombinant Escherichia coli cells, and the ease with which glycophages can be recovered from the bacterial supernatant without laborious purification steps. For these reasons, we anticipate that glycophage arrays will become a useful tool for identifying and profiling GBPs, including mammalian lectins (C-type lectins, galectins, and siglecs), plant lectins, antibodies, viral and bacterial lectins, and intact viruses.
Materials and Methods
Bacterial strains and growth conditions
E. coli strain DH5α (F- endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG ϕ80lacZΔM15 Δ(lacZYA-argF)U169, hsdR17(rK− mK+), λ−) was used for cloning of phagemids, while strain TG1 (supE hsdΔ5 thi Δ(lac-proAB) F’ [traD36 proABlacIlacZΔM15] was used for phage titering. To quantify phage titers, fresh TG1 cells were infected with the recovered phages and then selected on ampicillin to determine colony-forming units (CFUs). Strain TG1 lacking the waaL gene (TG1 ΔwaaL) was generated by P1vir phage transduction using strain TG1 as recipient and strain BW25113 waaL::Kan from the Keio collection [30] as donor. The resulting strain was used for all glycophage production. VCSM13 (Stratagene) was used as the helper phage and propagated in wild-type TG1 cells as described [31]. E. coli strains were grown in Luria-Bertani (LB) or 2xTY medium (during phage production) at 37°C or 30°C (induction phase). Culture medium was supplemented with 1% glucose (w/v) or 0.2% (w/v) L-arabinose (induction phase), as well as with the appropriate antibiotics at the following concentrations: 100 μg/ml ampicillin (Amp), 20 μg/ml chloroamphenicol (Cm), and 35 μg/ml kanamycin (Kan). M9 minimal medium was used to select for the presence of the F’ plasmid when needed.
Construction of phagemids
The phagemids constructed here were based on the phagemid pBAD-MBPDQNAT-g3p [17], encoding E. coli maltose-binding protein (MBP) modified with a C-terminal DQNAT motif, and cloned using standard protocols. To generate phagemid pBAD-MBP4xDQNAT-g3p, DNA encoding MBP4xDQNAT was PCR-amplified from plasmid pTrc-MBP-GT [32] as template. Next, pBAD-MBPDQNAT-g3p was digested with EcoRI-SalI to remove MBPDQNAT, followed by ligation of PCR-amplified MBP4xDQNAT into the same sites resulting in pBAD-MBP4xDQNAT-g3p. To generate the phagemid pBAD-MBP4xDQNAT-CT::PglB for bicistronic expression of MBP4xDQNAT fused to the C-terminal domain of g3p (CT; residues 230-406) [33] and PglB, the bicistronic phagemid pBAD-AcrA::PglB (laboratory stock) was digested with EcoRI and XmaI to remove the gene encoding AcrA. Next, DNA encoding MBP4xDQNAT-g3p was PCR-amplified from pBAD-MBP4xDQNAT-g3p as template and then ligated into EcoRI-XmaI digested pBAD-AcrA::PglB, resulting in phagemid pBAD-MBP4xDQNAT-g3p::PglB. The gene encoding full-length g3p (406 amino acids) was removed from pBAD-MBP4xDQNAT- g3p::PglB by digestion with SalI and XmaI and DNA encoding the C-terminal domain of g3p (residues 230-406) [33] was ligated into the same sites, resulting in phagemid pBAD-MBP4xDQNAT-CT::PglB. A nearly identical strategy was used to construct the negative control phagemids pBAD-MBP4xAQNAT-g3p::PglB and pBAD-MBP4xAQNATCT::PglB in which all four acceptor motifs were changed to AQNAT, a sequence that is inefficiently recognized by Campylobacter jejuni PglB [34, 35]. All phagemids were confirmed by DNA sequencing.
Plasmids used for glycan biosynthesis included: pMW07pglΔB for the C. jejuni heptasaccharide glycan (Cj-hepta) [35]; pACYCpgl4 for the C. lari hexasaccharide glycan (Cl-hexa) [36]; pYCG for the eukaryotic Man3GlcNAc2 core glycan (Man3) [37]; pMW07-O78 for E. coli O78 O-antigen (Ec-OAg (O78); laboratory stock); pMW07-O148 for E. coli O148 O-antigen (Ec-OAg (O148); laboratory stock); pGAB2 for Francisella tularensis SchuS4 O-antigen (Ft-OAg) [38], pLPS2 for Psuedomonas aeruginosa O11 O-antigen (Pa-OAg) [39], and pSS37 for Shigella dysenteriae serotype 1 O-antigen (Sd-OAg) [40].
Production and purification of glycophage particles
A single colony of E. coli TG1 ΔwaaL carrying a phagemid, either pBAD-MBP4xDQNAT-g3p::PglB or pBAD-MBP4xDQNATCT::PglB, and a plasmid for glycan biosynthesis was inoculated into 6 mL of M9 minimal medium supplemented with Amp and Cm and shaken at 37°C overnight. The following day, cells were subcultured into 40 mL of 2xTY medium supplemented with Amp, Cm, and glucose such that the initial OD600 was 0.06 and shaken at 37°C. The culture was superinfected with 3.2×1011 CFUs of VCSM13 helper phage when the OD600 reached 0.5-0.6 and incubated for 30 min at 37°C without shaking. For recovering cells, flasks were incubated for 10 min at 37°C with shaking and then centrifuged (3,300 × g, 10 min, 16°C). The resulting cell pellet was resuspended in 40 mL 2xTY medium supplemented with Amp, Cm and Kan. Resuspended cells were cultured for 30 min at 30°C with shaking, after which protein expression was induced with 0.2% arabinose for 16h. For glycophage recovery, the culture supernatant was separated from the cells by centrifugation (6,000 × g, 15 min, 4°C). Phage particles were precipitated from the supernatant by adding PEG/NaCl (1/5 volume of the supernatant) and incubated on ice for at least 2 h. Precipitated phage particles were collected by centrifugation (10,000 x g, 25 min, 4°C), after which the phage pellet was resuspended in 1 mL of 2% sarkosyl (Sigma) in phosphate buffered saline (PBS) to remove any unspecific glycan remnants on phage particles and reprecipitated twice with PEG/NaCl to wash off the detergent. The phage pellet was resuspended in PBS based on pellet size and the phage concentration was determined by OD268 [41]. Phage particles were stored for up to one week at 4°C or up to 4 months at −20°C.
Western blotting and glycophage ELISA
For Western blot analysis, glycophages (~6×1010 phage particles) were run on 7.5% SDS-polyacrylamide gels (BioRad), and Western blotting was performed as described previously [42]. For glycophage enzyme-linked immunosorbent analysis (ELISA), high-binding 96-well clear half-area microtiter plates (Corning) were coated with 1-20 × 1011 phage particles in 25 μL of PBS buffer in replicates, and incubated for 18-24 h at 4°C. The plate was blocked with blocking buffer (PBS + 2% nonfat milk) for 2 h at RT. The wells were washed 4 times with PBST (PBS with 0.1% Tween20) and probed with 50 μL of primary antibody and shaken for 1 h at RT, and if secondary antibody was required, the washed wells were probed for 1 hour at RT. After additional washings with PBS, o-phenylenediamine dihydrochloride (OPD) substrate (Sigma) was added to each well and after 40 min the reaction was stopped with 3 M H2SO4. The absorbance in each well was measured at 492 nm (Abs492). Blots or arrays were probed with one of the following: anti-MBP antibody conjugated with horseradish peroxidase (HRP) (New England Biolabs), hR6 rabbit serum that is specific for the C. jejuni heptasaccharide glycan and also recognizes the C. lari hexasaccharide glycan (kindly provided by Markus Aebi), monoclonal antibody 100-4G11-A that recognizes the Man3GlcNAc2 N-glycan [43], rabbit anti-E. coli O78 antibody (Abcam), rabbit anti-E. coli O148 antibody (Abcam), mouse anti-F. tularensis LPS antibody (Abcam), rabbit anti-P. aeruginosa O11 antibody (kindly provided by Markus Aebi), and Shigella Antiserum Poly Group A raised in rabbit (BD Diagnostic Systems). For non-HRP conjugated antibodies, anti-rabbit IgG-HRP or anti-mouse IgG-HRP (Promega) was used as the secondary antibody. HRP activity was detected using a chemiluminescent substrate (BioRad).
Microarray fabrication
All chemicals were purchased from Sigma-Aldrich and used without further purification unless otherwise indicated. Non-contact printing was performed on N-hydroxysuccinimide (NHS)-activated slides (NEXTERION® Slide H, Schott North America) as previously described [16] as 16 subarrays per slide using a Perkin Elmer Piezorray Printer. For multi-panel experiments on a single slide, the array layout was designed using Piezorray software according to the dimension of a standard 16-chamber adaptor. For glycan printing, 100 μM of 2-amino-N-(2-aminoethyl)-benzamide (AEAB)-conjugated glycans, prepared as previously described [16], were used. All glycan-AEAB (GAEAB) structures are given in Supplementary Fig. S1. For glycophage printing, 10 μl of glycophage samples (containing ~1×1012 CFU/mL) were used. All phage-displayed glycan structures are given in Table 1. All samples were printed in phosphate buffer (100 mM sodium phosphate, pH 8.5) in replicates of 5, and the average volume delivered to the array was 0.3 nanoliter +/−10% (intra-tip). After printing, slides were placed in a high-moisture chamber at 55°C. After 1 h, the slides were blocked with 50 mM ethanolamine in 0.1 M Tris buffer (pH 9.0) for 1 h, washed, and subsequently dried by centrifugation and stored desiccated at −20°C. For analysis, the 16-chamber adaptor was applied on the slide to separate the single slide to 16 chambers sealed from each other during the assay. Prior to binding assays, the subarrays were rehydrated for 5 min in TSM buffer (20 mM Tris- HCL, 150 mM sodium chloride (NaCl), 0.2 mM calcium chloride (CaCl2) and 0.2 mM magnesium chloride (MgCl2).
Table 1.
Summary of phage-displayed glycan library construction
| Glycan | Plasmid | Structure | Titer* | Ref. |
|---|---|---|---|---|
| Cj-hepta N-glycan | pMW07pglAB | GalNAc-α1,4-GalNAc-α1,4-(Glcβ1,3)-GalNAc-α1,4-GalNAc-α1,4-GalNAc-α1,3-Bac-β1 | 1.5×1011 | [35, 54] |
| Cl-hexa N-glycan | pACYCpgl4 | GalNAc-α1,4-GalNAc-α1,4-GalNAc-α1,4-GalNAc-α1,4-GalNAc-α1,3-Bac-β1 | 6.4×1010 | [36] |
| Man3 N-glycan | pYCG | Manα1-3(Manα1-6)-Manβ1-4-GlcNAcβ1-4-GlcNAc | 3.2×109 | [37] |
| Ec-OAg (O78) O-antigen | pMW07-O78 | →3)-β-D-GlcNAc-(1→4)-β-D-GlcNAc-(1→4)-β-D-Man-(1→4)-α-D-Man-(1→ | 5.6×1010 | [55] |
| Ec-OAg (O148) O-antigen | pMW07-O148 | →3)-α-L-Rhap-(1→3)-α-L-Rhap-(1→2)-α-D-Glcp-(1→3)-α-D-GlcpNAc-(1→ | 1.6×1011 | [56] |
| Ft-OAg O-antigen | pGAB2 | →4)-α-D-GalNAcAN-(1→4)-α-D-GalNAcAN-(1→3)-β-D-QuiNAc-(1→2)-β-D-Qui4NFm-(1→ | 1.1×1011 | [38, 57] |
| Pa-OAg O-antigen | pLPS2 | →3)-α-L-FucNAc-(1→3)-β-D-FucNAc-(1→2)-β-D-Glc-(1→ | 1.9×1011 | [39, 58] |
| Sd-OAg O-antigen | pSS37 | →3)-α-L-rhap-(1→3)-α-L-rhap-(1→2)-α-D-Galp-(1→3)-α-D-GlcpNAc-(1→ | 6.0×1010 | [40, 59] |
CFU/mL culture supernatant
GBP binding assays using microarrays
For interrogation of the printed subarrays, GBPs were diluted in binding buffer (TSM buffer with 1% BSA and 0.05% Tween20). The array was interrogated with the following biotinylated lectins (Vector Labs): Helix pomatia agglutinin (HPA), Maakia amurensis lectin (MAL), Sambucus nigra agglutinin (SNA), and soybean agglutinin (SBA), which are specific for defined GAEAB or glycophage structures on the array. An aliquot (200 μl) of each GBP or antibody was applied to individual subarrays and incubated for 1 h at room temperature. After washing with TSM buffer to remove excess GBP, biotinylated lectins were detected by a second incubation with 0.5 μg/ml Cy5-streptavidin (Cy5-SA; Invitrogen Life Technologies), anti-glycan antibodies were detected by a secondary incubation with 5 μg/ml Cy5-conjugated secondary antibodies (Invitrogen Life Technologies) at room temperature for 1 h followed by a wash step to remove excess reagent and finally with water to remove residual salt.
Microarray scanning
The slides were scanned with a Perkin Elmer ProScanArray microarray scanner equipped with 4 lasers covering an excitation range from 488 nm to 637 nm. For Cy5 fluorescence, the excitation wavelength was 649 nm and the emission wavelength was 670 nm. The scanned images were analyzed with the ScanArray Express software. The results are reported as the average (n=6) relative fluorescence units (RFU) with the error reported as the standard deviation of the mean.
Results
Redesign of glycophage display system for array fabrication
We hypothesized that glycan arrays could be fabricated by patterning E. coli-derived glycophages on a solid support such as a nitrocellulose membrane, a microtiter plate, or a glass microscope slide (Fig. 1). Towards this goal, we previously developed a method for the display of asparagine-linked (N-linked) glycans on filamentous phage M13 [17]. At the heart of this system is a plasmid for expressing a fusion between a target N-linked glycoprotein and the minor phage coat protein g3p. The target glycoprotein in this plasmid is E. coli MBP, which is modified with a C-terminal glycosylation tag comprising a D/E-X1-N-X2-S/T acceptor motif that is known to be efficiently glycosylated by the C. jejuni oligosaccharyltransferase (OST) PglB [32, 34]. When this plasmid was transformed in E. coli cells that also carried the pACYCpgl plasmid encoding the protein glycosylation locus (pgl) from C. jejuni [44], the resulting MBPDQNAT-g3p fusion protein produced by these cells was efficiently glycosylated with the C. jejuni heptasaccharide glycan (GalNAc5(Glc)Bac where Bac is bacillosamine or 2,4-diacetamido-2,4,6-trideoxyglucose) [17]. Moreover, phage particles prepared from these cells displayed the C. jejuni glycan, which was readily detected by glycan-specific serum antibodies or captured by the lectin SBA that binds the terminal N-acetyl-galactosamine (GalNAc) of the C. jejuni oligosaccharide [17].
Figure 1. Schematic of glycophage array technology.
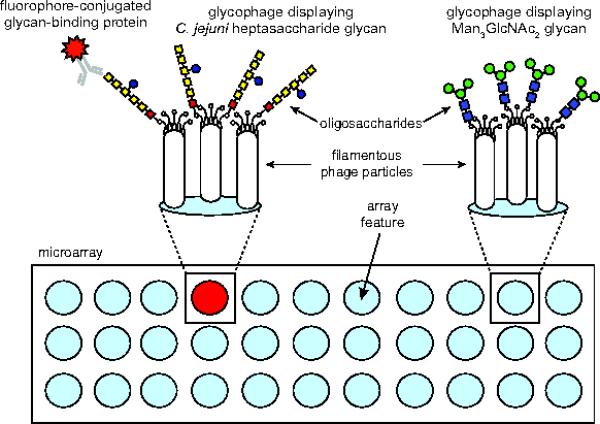
Detection of GBP binding activity enabled by covalent printing of diverse glycophages, each displaying a different glycan, onto an amino-reactive glass surface followed by image analysis using standard microarray technology. The displayed glycan is produced biosynthetically in engineered E. coli cells carrying a plasmid that encodes the glycan biosynthesis pathway and a phagemid that encodes the acceptor protein-phage coat protein fusion (e.g., MBP4xDQNAT-g3p). In the presence of the OST PglB from the bacterium C. jejuni, structurally diverse glycans can be covalently linked to MBP4xDQNAT-g3p and subsequently displayed on phage particles.
In this study, the above glycophage system was redesigned for use in glycan array fabrication in several ways. First, we introduced additional acceptor sites for glycan attachment to MBP. This involved replacing MBPDQNAT in pBAD-MBPDQNAT-g3p with a version of MBP that was C-terminally modified with four tandem repeats of the bacterial glycan acceptor motif DQNAT, resulting in phagemid pBAD-MBP4xDQNAT-g3p. Second, given the observation that the WaaL O-antigen ligase in E. coli transfers lipid-linked glycans onto lipid A and reduces protein glycosylation efficiency by depleting the pool of available substrates for PglB [45], we used P1vir phage transduction to knockout the waaL gene in E. coli strain TG1. Third, we generated the bicistronic phagemid pBAD-MBP4xDQNAT-g3p::PglB that encodes both the MBP-g3p fusion and the C. jejuni PglB enzyme, thereby affording inducible control over OST expression. Moreover, PglB is needed for transferring other recombinantly produced glycans (e.g., O-antigen polysaccharides) [45]; hence, this bicistronic phagemid provides a copy of the PglB enzyme for future glycan library building. Fourth, because display efficiency and fusion stability often decrease with protein size, possibly as a consequence of coat protein proteolysis, we decreased the overall size of the displayed fusion protein by truncating g3p. This strategy was motivated by the fact that fusion to the C-terminal domain of g3p can be more stable than fusion to full-length g3p [46] and also that shortened g3p enables better display of certain structures by relieving spatial constraints that occur with full-length g3p [47]. Importantly, following helper phage superinfection, the resulting packaged phagemid carries both native g3p of the helper phage, which is necessary for infection, and the encoded fusion comprised of the C-terminal domain of g3p, which is displayed for screening [48].
To determine the overall effectiveness of these modifications, E. coli TG1 ΔwaaL cells were co-transformed with plasmid pMW07pglΔB [35] that encodes the entire C. jejuni pgl pathway except for PglB and a bicistronic phagemid which encodes PglB and the glycoprotein MBP4xDQNAT fused to the N-terminus of either full-length g3p (406 amino acids) or truncated g3p (residues 230-406) [33]. Phage production by these cells was initiated by superinfecting cultures with VCSM13 helper phage followed by induction of fusion protein expression. At the end of the induction phase, the culture densities were virtually identical (data not shown), indicating that the shortened g3p did not adversely affect growth of the host strain. Next, recombinant phage particles were recovered from these cells. The resulting phage titers from TG1 ΔwaaL cells expressing MBP4xDQNAT-g3p or MBP4xDQNAT-CT were indistinguishable (data not shown), indicating that the shortened g3p did not adversely affect phage production.
Immunoblot analysis of phage preparations confirmed that the MBP4xDQNAT-g3p and MBP4xDQNAT-CT fusions were incorporated into phage particles at nearly identical levels (Fig. 2A). If the VCSM13 helper phage was omitted, no bands were observed indicating the lack of phage production. In addition, both phage-displayed fusions were glycosylated with the C. jejuni glycan as determined by immunoblotting with hR6 serum (Fig. 2A), which is specific for the C. jejuni heptasaccharide glycan [36]. When the acceptor motifs were all changed to AQNAT, a sequence that is inefficiently recognized by C. jejuni PglB [34, 35], no bands were observed with the hR6 serum indicating that neither of the fusions was glycosylated (Fig. 2A). It is noteworthy that several prominent glycosylated degradation products were observed for the larger MBP4xDQNAT-g3p fusion but not the MBP4xDQNAT-CT fusion (Fig. 2A), suggesting that the fusion with shortened g3p was more stable under the conditions tested here. For these reasons, we opted to use the pBAD-MBP4xDQNAT-CT::PglB phagemid for all glycan display hereafter. Using this phagemid under optimal growth and protein expression conditions, we regularly achieved phage titers of ~1×1011 CFUs per mL of culture supernatant (Table 1), indicating that very high phage titers could be obtained with our redesigned glycophage system. Importantly, glycosylated phage production using shortened g3p was found to be dependent on helper phage addition, the phagemid encoding MBP4xDQNAT-CT, and the glycan biosynthesis pathway (Supplementary Fig. S2).
Figure 2. Generation of phage-displayed glycan library members.
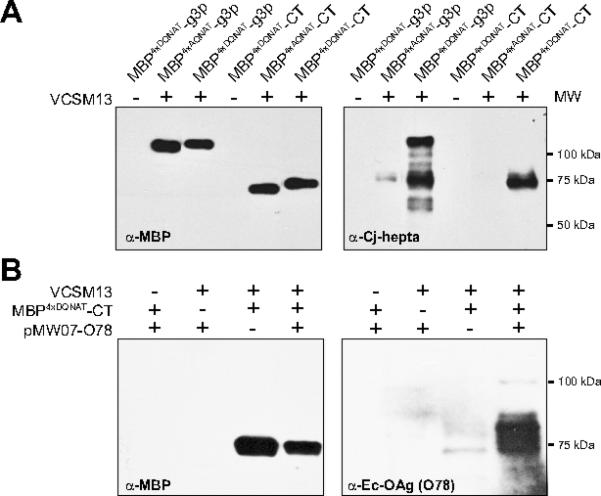
(A) Western blot analysis of phage preparations (containing 4×1010 glycophage particles) derived from E. coli TG1 ΔwaaL cells co-transformed with pMW07pglΔB and one of the following phagemids: pBAD-MBP4xDQNAT-g3p::PglB, pBAD-MBP4xAQNAT-g3p::PglB, pBADMBP4xDQNAT-CT::PglB or pBAD-MBP4xAQNAT-CT::PglB. (B) Western blot analysis of phage preparations (containing 6×1010 glycophage particles) derived from E. coli TG1 ΔwaaL cells co-transformed with phagemid pBAD-MBP4xDQNAT-CT::PglB and pMW07-O78. Phage preparation involved addition (+) or omission (-) of VCSM13 helper phage. Additional controls involved omitting either the phagemid or the glycan biosynthesis pathway. Blots were probed with anti-MBP, hR6 serum (anti-Cj-hepta), or anti-Ec-OAg (O78) as indicated. Molecular weight (MW) markers are indicated at right.
Phage-displayed glycan library construction
Next, we sought to create an array of diverse phage-displayed glycan structures using our redesigned glycophage system. Our strategy was to transform TG1 ΔwaaL cells carrying the pBAD-MBP4xDQNAT-CT phagemid with a second plasmid encoding a glycan biosynthesis pathway (Table 1). For example, the plasmids pACYCpgl4 and pYCG enabled glycosylation of MBP4xDQNATCT with the C. lari hexasaccharide N-glycan [36] and the eukaryotic Man3GlcNAc2 N-glycan [37], respectively (data not shown). In addition to N-glycans, we also generated phages displaying O-antigen polysaccharides from different Gram-negative bacteria. This was accomplished by leveraging the fact that recombinant O-antigens can be assembled on a lipid carrier (bactoprenyl pyrophosphate) at the cytoplasmic side of the inner membrane of E. coli, flipped into the periplasm by the O-antigen translocating flippase Wzx, and transferred to asparagine residues of target proteins by PglB [45]. PglB is the only protein of the bacterial N-glycosylation machinery required for this transfer and its relaxed specificity toward the glycan structure enables transfer of numerous structurally distinct O-antigens [38, 45, 49]. To extend this concept to glycophage display, TG1 ΔwaaL cells carrying pBAD-MBP4xDQNAT-CT were transformed with plasmid pMW07-O78, which encodes enzymes for the biosynthesis of Ec-OAg (O78) composed of tetrasaccharide repeating-units whose structure is given in Table 1. High recombinant phage titers were recovered from these cells, similar to the titers achieved with the different N-glycan biosynthesis pathways (Table 1). When these phage preparations were analyzed by immunoblotting, a ladder of higher molecular weight bands were detected by the anti-Ec-OAg (O78) antiserum (Fig. 2B). This laddering of O78 polysaccharide-linked phages was consistent with the typical variability of the chain length of the O-polysaccharides generated by the Wzy polymerase [45]. The high-molecular-weight bands were absent in the samples obtained from cells that were not infected with helper phage, lacked the phagemid, or lacked the O-antigen biosynthesis pathway (Fig. 2B). Altogether, these results suggest that O78 polysaccharide was covalently linked to MBP4xDQNAT-CT. Nearly identical phage glycosylation was obtained for four additional O-antigens including: Ec-OAg (O148) produced from pMW07-O148; Ft-OAg produced from pGAB2 [38]; Pa-OAg produced from pLPS2 [39]; and Sd-OAg produced from pSS37 [40]. The structures of these antigens and their production levels are summarized in Table 1.
Detection of glycan-protein interactions by glycophage ELISA
We next determined whether glycans displayed on the surface of phages could still be detected by GBPs following immobilization to a solid surface. Various methods have been employed to immobilize phages on surfaces for array development including noncovalent adsorption to polystyrene or nitrocellulose [28, 29]. Indeed, when glycophages prepared from TG1 ΔwaaL cells carrying the pBAD-MBP4xDQNAT-CT::PglB phagemid and plasmid pMW07pglΔB were used to coat the surface of microtiter plates, a strong binding signal was observed following incubation with hR6 serum antibodies (Fig. 3A). In contrast, only background signals were measured for the helper phage control or the glycophage sample prepared from TG1 ΔwaaL cells carrying pMW07pglΔB along with the pBADMBP4xAQNAT-CT::PglB phagemid, in which the acceptor motifs have been mutated (Fig. 3A). As expected, ELISA signals for all of the other immobilized glycophages were significantly lower than that of the C. jejuni N-glycan, except for the C. lari hexasaccharide glycan which is structurally similar to the C. jejuni N-glycan and reacts with hR6 serum [36]. Collectively, these data indicate that glycophages are compatible with surface immobilization procedures and that phage-displayed glycans retain the ability to be recognized by a cognate GBP after immobilization. Identical glyophage ELISAs were performed for six additional GBPs that recognized one of the glycans in the library. The resulting dataset was used to generate an orthogonality matrix of glycan binding for each of the GBPs (Fig. 3B). Importantly, all of the GBPs bound most efficiently to their target glycan structures as evidenced by the strong blue signal along the diagonal of the matrix. The signal-to-noise ratios (S:N) generated using this assay were significant, with values of 3.0, 3.5, 6.7, and 10 for the anti-Cj hepta, anti-Cl hexa, anti-FtOAg, anti-PaOAg antibodies, respectively (where S:N was determined by normalizing the activity measured for GBP binding of cognate glycan (signal) by the highest activity measured for GBP binding of non-cognate glycan (noise). For the poorer S:N cases, such as the anti-Ec-OAg antibodies (S:N = 1.9 for O78 and 1.8 for O148) and the anti-SD-OAg antibody (S:N = 1.5), it is unclear whether this cross reactivity was due to unexpectedly low specificity of the GBPs themselves or the need for further optimization of assay conditions to reduce background caused by non-specific cross-reaction (e.g., reduce possible phage cross-contamination which arises more readily than bacterial contaminations, but can be prevented using filtered tips).
Figure 3. Glycan-protein interactions detected by glycophage ELISA.
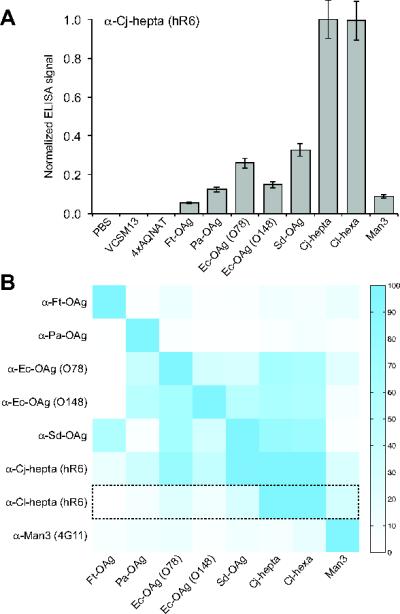
(A) Representative ELISA results obtained by immobilizing glycophage preparations on microtiter plate surfaces. Glycophage preparations were prepared from E. coli TG1 ΔwaaL cells co-transformed with the phagemid pBAD-MBP4xDQNAT-CT::PglB and one of the 8 different glycan encoding plasmids. Controls included PBS, helper phage only (VCSM13), and phages prepared from E. coli TG1 ΔwaaL cells co-transformed with pBAD-MBP4xAQNAT-CT::PglB and pMW07pglΔB (4xAQNAT). All samples were immobilized in triplicate and probed with hR6 serum antibodies (α-Cj-hepta). ELISA signals (Abs492) for each sample were normalized to the signal measured for the Cjhepta N-glycan sample. The results are reported as the average normalized ELISA signal (n=3) and error bars represent the standard deviation of the mean. (B) Orthogonality matrix depicting glycan specificity profiles for different GBPs. Glycophage ELISA was performed whereby each GBP (y-axis) was probed against the glycan library (x-axis) identically as described in (A) for hR6 serum antibodies. Each row depicts the normalized ELISA signals measured for the indicated GBP against each glycan sample, where data was independently normalized to the maximum signal obtained for that GBP. Color intensity scale is shown at right (highest signal = blue; lowest signal = white). Dashed box corresponds to data from (A) for hR6 serum antibodies.
Fabrication of glycophage-patterned microarrays
Encouraged by these ELISA results, we sought to prepare glycan arrays by directly spotting recombinant glycophages onto the surface of glass slides. Previous studies have shown that phages displaying peptides and proteins are compatible with robotic microarray spotters [26-28] and covalent chemical conjugation to glass slides through amino acids (e.g., lysine, cysteine, or any N- or C-terminal acids) that provide a suitable functional capacity for immobilization and modification chemistries [26]. Here, a similar strategy was adopted for covalently attaching glycophages to microglass slides by using standard microarray printing technology [16, 50]. Specifically, we used commercially available amine-reactive NHS-activated glass slides, which allowed rapid covalent coupling of terminal amines in glycophage coat proteins via formation of an amide bond under aqueous conditions at room temperature. In addition to spotting glycophage samples, we also printed PBS, biotin, and 10 different amine-functionalized GAEABs from the repository of the Consortium for Functional Glycomics (CFG) [16] (Supplementary Fig. S1), which served as controls for array fabrication and GBP binding.
To validate glycophage printing, we probed our microarrays with GBPs having specificity for the different glycans that were printed on the slides. For example, probing with serum antibodies against the P. aeruginosa O11 O-antigen followed by a Cy5-labeled secondary antibody resulted in strong binding to glycophages displaying the O11 O-antigen but not to other glycophages or any of the GAEAB control glycans (Fig. 4A and Supplementary Figs. S3 and S4). When the same array was probed with biotinylated lectins, HPA, MAL, and SNA and detected using Cy5-SA [16], only binding to cognate GAEABs was observed (Fig. 4A and Supplementary Fig. S3 and S4), consistent with their reported specificities. Importantly, none of these lectins bound to any of the glycophages. Nearly identical results were obtained with a second array that varied only in the identity and number of some of the GAEABs and glycophages that were printed. In particular, we observed that biotinylated SBA, which recognizes terminal GalNAc residues such as those in the C. jejuni and C. lari N-glycans [17], bound strongly and selectively to glycophages displaying the C. jejuni N-glycan structure (Fig. 4B). Collectively, these results signify that glycophages can be printed on NHS-derivatized microglass slides using standard robotic printing technology and that covalently linked phages retain their ability to be recognized by GBPs.
Figure 4. Detection of phage-displayed O-antigens using glycan microarray.
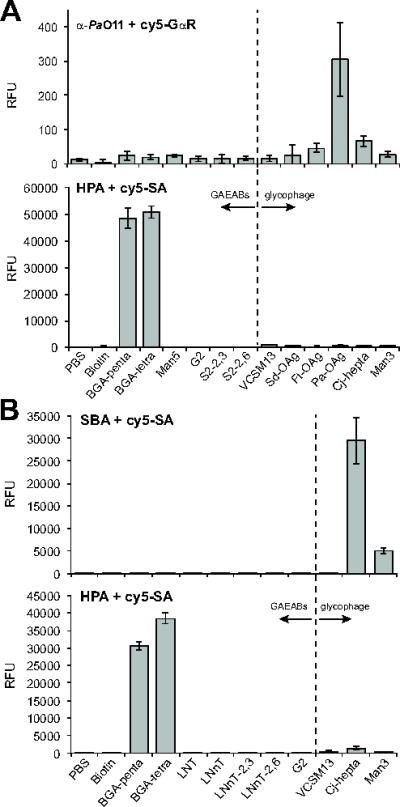
(A) Glycan binding by serum antibodies against Pa-OAg was detected with Cy5-labeled goat anti-rabbit antibodies (Cy5-GαR) while binding by lectin HPA was detected with Cy5-SA. Arrays were printed with the following: PBS, biotin, blood group A pentasaccharide (BGA-penta), BGA tetrasaccharide (BGA-tetra), Man5GlcNAc2 (Man5), biantennary non-sialylated G2 glycan (G2), biantennary disialylated glycan (α2,3 linkage; S2-2,3), biantennary disialylated glycan (α2,6 linkage; S2-2,6), helper phage control (VCSM13), and phages displaying Sd-OAg, Ft-OAg, Pa-OAg, Cj-hepta and Man3. Data reported as the average (n=6) RFU and error bars represent the standard deviation of the mean. (B) Glycan binding by the lectins SBA and HPA was detected with Cy5-SA. Arrays were printed with the following: PBS, biotin, blood-group A pentasaccharide (BGA-penta), BGA tetrasaccharide (BGA-tetra), lacto-N-tetraose (LNT), lacto-N-neotetraose (LNnT), sialyllacto-N-neotetraose (α2,3 linkage; LNnT-2,3), sialyllacto-N-neotetraose (α2,6 linkage, LNnT-2,6), biantennary non-sialylated G2 glycan (G2), helper phage control (VCSM13), and phages displaying Cj-hepta and Man3. Data reported as average (n=6) RFU and error bars represent the standard deviation of the mean.
Discussion
Glycan arrays have emerged in the last decade as a powerful tool for analyzing carbohydrate-macromolecule interactions, especially in disease related research. Microarrays are specifically preferred because they facilitate rapid screening of numerous samples and substantially reduce reagent consumption. However, unlike their DNA and protein counterparts, glycans cannot be readily amplified in vitro. Hence, acquiring a large collection of glycans in a format suitable for immobilization on the array surface remains a formidable challenge given the low amounts of glycan that can be chemically synthesized or isolated from natural sources. In this study, we demonstrate an alternative approach to glycan arrays that leverages the biosynthetic potential of engineered E. coli to covalently link tailor-made N-glycans and O-antigens to filamentous phage particles in vivo. The resulting glycan-displaying phages accumulate outside the cells and thus can be easily recovered from the culture media without the need for cell lysis or laborious purification techniques. In fact, the phage recovery method is completely independent of the glycan structure, which effectively streamlines the entire glycan library production process. In addition to facile production, the method also yields large quantities of glycans with even greater quantities of glycans possible in the future given the scalability of bacterial cell culture. Most importantly, we show that glycophages are entirely compatible with every step of the microarray fabrication process, and can be immobilized to a support surface by either noncovalent or covalent chemistries in a manner that does not disrupt their ability to be recognized by different GBPs (e.g., antibodies, lectins) after immobilization.
Using both microtiter plates and glass slides, significant S:N ratios were obtained for a panel of GBP-glycan pairs; however, a fair amount of unexpected or non-specific glycan binding was observed with the microtiter plate format. This background was not seen for the limited set of GBP-glycan pairs tested using the glass slide arrays, suggesting this may be the preferred format for future glycomic applications. An issue that remains unanswered is the density of glycans on the different glycophages, which is not currently known and is difficult to accurately quantify due to the relatively low level of phage glycosylation [17, 18]. For a different glycoprotein target, namely C. jejuni AcrA, the efficiency with which different recombinant O-antigens were conjugated by C. jejuni PglB was variable [45]. This is significant because GBPs typically bind to clustered glycans, and if the glycan densities vary significantly among the glycophages, conclusions about binding specificity will be compromised. Hence, future efforts must focus on techniques for quantifying glycan density and controlling/modulating the glycosylation efficiency of PglB. Another limitation of the technology at present is the fact that de novo biosynthesis of diverse glycoforms (i.e., candidate glycan library members) in engineered bacteria is in its infancy (for recent reviews on the topic, see [51, 52]). The silver lining, however, is that every newly engineered glycoform produced using E. coli in the future will potentially serve as a library member for glycophage array development. In addition, it may be possible to diversify the panel of existing glycan structures by combining in vivo glycophage production with in vitro remodeling of phage-displayed glycans using chemoenzymatic approaches as described recently [53]. In light of these possibilities, our results pave the way for future robotic printing of large panels of glycan epitopes displayed on phages for highly parallel, high-throughput profiling of GBP specificities in experimental and clinical samples.
Supplementary Material
Acknowledgements
We thank Brendan Wren, Markus Aebi, Rebecca Thomas, Renato Morona, and Chung-Jr Huang for plasmids and antiserum used in this work. We also thank Julius Lucks for help preparing figures. The National Center for Functional Glycomics is supported by NIGMS grant P41 GM103694. This work was also supported by the National Science Foundation Career Award CBET-0449080 (to M.P.D.), the New York State Office of Science, Technology and Academic Research Distinguished Faculty Award (to M.P.D.), National Institutes of Health grant # 1 R43 GM086965-01 (to A.C.F), and by a Marie Curie FP7 Integration Grant within the 7th European Union Framework Programme under REA grant agreement # 322096 (to E.C.), UNESCO-L’Oréal National Young Women in Science Award (to E.C.), and Hacettepe University (to E.C.).
Abbreviations
- AEAB
2-amino-N-(2-aminoethyl)-benzamide
- CFU
asparagine-linked (N-linked), colony-forming unit
- CT
C-terminal domain of g3p
- Cy5-SA
Cy5-streptavidin
- ELISA
enzyme-linked immunosorbent assay
- GBP
glycan-binding protein
- GAEAB
glycan-AEAB
- HPA
Helix pomatia agglutinin
- MAL
Maakia amurensis lectin
- MBP
maltose-binding protein
- Man3
Man3GlcNAc2
- NHS
N-hydroxysuccinimide
- OST
oligosaccharyltransferase
- SNA
Sambucus nigra agglutinin
- SBA
soybean agglutinin
- RFU
relative fluorescence unit
Footnotes
Conflict of Interest Statement
The authors declare no commercial or financial conflict of interest
References
- 1.Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freeze HH. Genetic defects in the human glycome. Nat Rev Genet. 2006;7:537–551. doi: 10.1038/nrg1894. [DOI] [PubMed] [Google Scholar]
- 3.Rillahan CD, Paulson JC. Glycan microarrays for decoding the glycome. Annu Rev Biochem. 2011;80:797–823. doi: 10.1146/annurev-biochem-061809-152236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oyelaran O, Gildersleeve JC. Glycan arrays: recent advances and future challenges. Curr Opin Chem Biol. 2009;13:406–413. doi: 10.1016/j.cbpa.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith DF, Song X, Cummings RD. Use of glycan microarrays to explore specificity of glycan-binding proteins. Methods Enzymol. 2010;480:417–444. doi: 10.1016/S0076-6879(10)80033-3. [DOI] [PubMed] [Google Scholar]
- 6.de Paz JL, Seeberger PH. Recent advances and future challenges in glycan microarray technology. Methods Mol Biol. 2012;808:1–12. doi: 10.1007/978-1-61779-373-8_1. [DOI] [PubMed] [Google Scholar]
- 7.Song X, Heimburg-Molinaro J, Cummings RD, Smith DF. Chemistry of natural glycan microarrays. Curr Opin Chem Biol. 2014;18:70–77. doi: 10.1016/j.cbpa.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park S, Gildersleeve JC, Blixt O, Shin I. Carbohydrate microarrays. Chem Soc Rev. 2013;42:4310–4326. doi: 10.1039/c2cs35401b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang D, Liu S, Trummer BJ, Deng C, Wang A. Carbohydrate microarrays for the recognition of cross-reactive molecular markers of microbes and host cells. Nat Biotechnol. 2002;20:275–281. doi: 10.1038/nbt0302-275. [DOI] [PubMed] [Google Scholar]
- 10.Arthur CM, Cummings RD, Stowell SR. Using glycan microarrays to understand immunity. Curr Opin Chem Biol. 2014;18:55–61. doi: 10.1016/j.cbpa.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stowell SR, Arthur CM, McBride R, Berger O, et al. Microbial glycan microarrays define key features of host-microbial interactions. Nat Chem Biol. 2014;10:470–476. doi: 10.1038/nchembio.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adams EW, Ratner DM, Bokesch HR, McMahon JB, et al. Oligosaccharide and glycoprotein microarrays as tools in HIV glycobiology; glycan-dependent gp120/protein interactions. Chem Biol. 2004;11:875–881. doi: 10.1016/j.chembiol.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 13.Lawrie CH, Marafioti T, Hatton CS, Dirnhofer S, et al. Cancer-associated carbohydrate identification in Hodgkin's lymphoma by carbohydrate array profiling. Int J Cancer. 2006;118:3161–3166. doi: 10.1002/ijc.21762. [DOI] [PubMed] [Google Scholar]
- 14.Oyelaran O, Gildersleeve JC. Application of carbohydrate array technology to antigen discovery and vaccine development. Expert Rev Vaccines. 2007;6:957–969. doi: 10.1586/14760584.6.6.957. [DOI] [PubMed] [Google Scholar]
- 15.Wang D, Carroll GT, Turro NJ, Koberstein JT, et al. Photogenerated glycan arrays identify immunogenic sugar moieties of Bacillus anthracis exosporium. Proteomics. 2007;7:180–184. doi: 10.1002/pmic.200600478. [DOI] [PubMed] [Google Scholar]
- 16.Song X, Xia B, Stowell SR, Lasanajak Y, et al. Novel fluorescent glycan microarray strategy reveals ligands for galectins. Chem Biol. 2009;16:36–47. doi: 10.1016/j.chembiol.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Celik E, Fisher AC, Guarino C, Mansell TJ, DeLisa MP. A filamentous phage display system for N-linked glycoproteins. Protein Sci. 2010;19:2006–2013. doi: 10.1002/pro.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Durr C, Nothaft H, Lizak C, Glockshuber R, Aebi M. The Escherichia coli glycophage display system. Glycobiology. 2010;20:1366–1372. doi: 10.1093/glycob/cwq102. [DOI] [PubMed] [Google Scholar]
- 19.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 20.Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR. Making antibodies by phage display technology. Annu Rev Immunol. 1994;12:433–455. doi: 10.1146/annurev.iy.12.040194.002245. [DOI] [PubMed] [Google Scholar]
- 21.Rader C, Barbas CF., 3rd Phage display of combinatorial antibody libraries. Curr Opin Biotechnol. 1997;8:503–508. doi: 10.1016/s0958-1669(97)80075-4. [DOI] [PubMed] [Google Scholar]
- 22.Smith GP. Surface presentation of protein epitopes using bacteriophage expression systems. Curr Opin Biotechnol. 1991;2:668–673. doi: 10.1016/0958-1669(91)90032-z. [DOI] [PubMed] [Google Scholar]
- 23.Viti F, Nilsson F, Demartis S, Huber A, Neri D. Design and use of phage display libraries for the selection of antibodies and enzymes. Methods Enzymol. 2000;326:480–505. doi: 10.1016/s0076-6879(00)26071-0. [DOI] [PubMed] [Google Scholar]
- 24.Watters AL, Baker D. Searching for folded proteins in vitro and in silico. Eur J Biochem. 2004;271:1615–1622. doi: 10.1111/j.1432-1033.2004.04072.x. [DOI] [PubMed] [Google Scholar]
- 25.Forrer P, Jung S, Pluckthun A. Beyond binding: using phage display to select for structure, folding and enzymatic activity in proteins. Curr Opin Struct Biol. 1999;9:514–520. doi: 10.1016/S0959-440X(99)80073-6. [DOI] [PubMed] [Google Scholar]
- 26.Bi Q, Cen X, Wang W, Zhao X, et al. A protein microarray prepared with phage-displayed antibody clones. Biosens Bioelectron. 2007;22:3278–3282. doi: 10.1016/j.bios.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 27.Cekaite L, Haug O, Myklebost O, Aldrin M, et al. Analysis of the humoral immune response to immunoselected phage-displayed peptides by a microarray-based method. Proteomics. 2004;4:2572–2582. doi: 10.1002/pmic.200300768. [DOI] [PubMed] [Google Scholar]
- 28.Arnaud MC, Gazarian T, Palacios Rodriguez Y, Gazarian K, Sakanyan V. Array assessment of phage-displayed peptide mimics of Human Immunodeficiency Virus type 1 gp41 immunodominant epitope: binding to antibodies of infected individuals. Proteomics. 2004;4:1959–1964. doi: 10.1002/pmic.200300725. [DOI] [PubMed] [Google Scholar]
- 29.Zhong L, Hidalgo GE, Stromberg AJ, Khattar NH, et al. Using protein microarray as a diagnostic assay for non-small cell lung cancer. Am J Respir Crit Care Med. 2005;172:1308–1314. doi: 10.1164/rccm.200505-830OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baba T, Ara T, Hasegawa M, Takai Y, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:0008. doi: 10.1038/msb4100050. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hertveldt K, Belien T, Volckaert G. General M13 phage display: M13 phage display in identification and characterization of protein-protein interactions. Methods Mol Biol. 2009;502:321–339. doi: 10.1007/978-1-60327-565-1_19. [DOI] [PubMed] [Google Scholar]
- 32.Fisher AC, Haitjema CH, Guarino C, Celik E, et al. Production of secretory and extracellular N-linked glycoproteins in Escherichia coli. Appl Environ Microbiol. 2011;77:871–881. doi: 10.1128/AEM.01901-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andris-Widhopf J, Rader C, Steinberger P, Fuller R, Barbas CF., 3rd Methods for the generation of chicken monoclonal antibody fragments by phage display. J Immunol Methods. 2000;242:159–181. doi: 10.1016/s0022-1759(00)00221-0. [DOI] [PubMed] [Google Scholar]
- 34.Kowarik M, Young NM, Numao S, Schulz BL, et al. Definition of the bacterial N-glycosylation site consensus sequence. EMBO J. 2006;25:1957–1966. doi: 10.1038/sj.emboj.7601087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ollis AA, Fisher AC, Merritt JH, Zhang S, DeLisa MP. Engineered oligosaccharyltransferases with greatly relaxed acceptor site specificity. Nat Chem Biol. 2014 doi: 10.1038/nchembio.1609. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwarz F, Lizak C, Fan YY, Fleurkens S, et al. Relaxed acceptor site specificity of bacterial oligosaccharyltransferase in vivo. Glycobiology. 2011;21:45–54. doi: 10.1093/glycob/cwq130. [DOI] [PubMed] [Google Scholar]
- 37.Valderrama-Rincon JD, Fisher AC, Merritt JH, Fan YY, et al. An engineered eukaryotic protein glycosylation pathway in Escherichia coli. Nat Chem Biol. 2012;8:434–436. doi: 10.1038/nchembio.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cuccui J, Thomas RM, Moule MG, D'Elia RV, et al. Exploitation of bacterial N-linked glycosylation to develop a novel recombinant glycoconjugate vaccine against Francisella tularensis. Open Biol. 2013;3:130002. doi: 10.1098/rsob.130002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldberg JB, Hatano K, Meluleni GS, Pier GB. Cloning and surface expression of Pseudomonas aeruginosa O antigen in Escherichia coli. Proc Natl Acad Sci U S A. 1992;89:10716–10720. doi: 10.1073/pnas.89.22.10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sturm S, Timmis KN. Cloning of the rfb gene region of Shigella dysenteriae 1 and construction of an rfb-rfp gene cassette for the development of lipopolysaccharide-based live anti-dysentery vaccines. Microb Pathog. 1986;1:289–297. doi: 10.1016/0882-4010(86)90054-9. [DOI] [PubMed] [Google Scholar]
- 41.Sidhu SS, Lowman HB, Cunningham BC, Wells JA. Phage display for selection of novel binding peptides. Methods Enzymol. 2000;328:333–363. doi: 10.1016/s0076-6879(00)28406-1. [DOI] [PubMed] [Google Scholar]
- 42.DeLisa MP, Tullman D, Georgiou G. Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. Proc Natl Acad Sci U S A. 2003;100:6115–6120. doi: 10.1073/pnas.0937838100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Remoortere A, Bank CM, Nyame AK, Cummings RD, et al. Schistosoma mansoni-infected mice produce antibodies that cross-react with plant, insect, and mammalian glycoproteins and recognize the truncated biantennaryN-glycan Man3GlcNAc2-R. Glycobiology. 2003;13:217–225. doi: 10.1093/glycob/cwg025. [DOI] [PubMed] [Google Scholar]
- 44.Wacker M, Linton D, Hitchen PG, Nita-Lazar M, et al. N-linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science. 2002;298:1790–1793. doi: 10.1126/science.298.5599.1790. [DOI] [PubMed] [Google Scholar]
- 45.Feldman MF, Wacker M, Hernandez M, Hitchen PG, et al. Engineering N-linked protein glycosylation with diverse O antigen lipopolysaccharide structures in Escherichia coli. Proc Natl Acad Sci U S A. 2005;102:3016–3021. doi: 10.1073/pnas.0500044102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Larocca D, Jensen-Pergakes K, Burg MA, Baird A. Receptor-targeted gene delivery using multivalent phagemid particles. Mol Ther. 2001;3:476–484. doi: 10.1006/mthe.2001.0284. [DOI] [PubMed] [Google Scholar]
- 47.Lowman HB, Bass SH, Simpson N, Wells JA. Selecting high-affinity binding proteins by monovalent phage display. Biochemistry. 1991;30:10832–10838. doi: 10.1021/bi00109a004. [DOI] [PubMed] [Google Scholar]
- 48.Barbas CF, 3rd, Kang AS, Lerner RA, Benkovic SJ. Assembly of combinatorial antibody libraries on phage surfaces: the gene III site. Proc Natl Acad Sci U S A. 1991;88:7978–7982. doi: 10.1073/pnas.88.18.7978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ihssen J, Kowarik M, Dilettoso S, Tanner C, et al. Production of glycoprotein vaccines in Escherichia coli. Microb Cell Fact. 2010;9:61. doi: 10.1186/1475-2859-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blixt O, Head S, Mondala T, Scanlan C, et al. Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proc Natl Acad Sci U S A. 2004;101:17033–17038. doi: 10.1073/pnas.0407902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Merritt JH, Ollis AA, Fisher AC, DeLisa MP. Glycans-by-design: engineering bacteria for the biosynthesis of complex glycans and glycoconjugates. Biotechnol Bioeng. 2013;110:1550–1564. doi: 10.1002/bit.24885. [DOI] [PubMed] [Google Scholar]
- 52.Baker JL, Celik E, DeLisa MP. Expanding the glycoengineering toolbox: the rise of bacterial N-linked protein glycosylation. Trends Biotechnol. 2013;31:313–323. doi: 10.1016/j.tibtech.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 53.Schwarz F, Huang W, Li C, Schulz BL, et al. A combined method for producing homogeneous glycoproteins with eukaryotic N-glycosylation. Nat Chem Biol. 2010;6:264–266. doi: 10.1038/nchembio.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Young NM, Brisson JR, Kelly J, Watson DC, et al. Structure of the N-linked glycan present on multiple glycoproteins in the Gram-negative bacterium, Campylobacter jejuni. J Biol Chem. 2002;277:42530–42539. doi: 10.1074/jbc.M206114200. [DOI] [PubMed] [Google Scholar]
- 55.Jansson PE, Lindberg B, Widmalm G, Leontein K. Structural studies of the Escherichia coli O78 O-antigen polysaccharide. Carbohydr Res. 1987;165:87–92. doi: 10.1016/0008-6215(87)80080-0. [DOI] [PubMed] [Google Scholar]
- 56.Feng L, Perepelov AV, Zhao G, Shevelev SD, et al. Structural and genetic evidence that the Escherichia coli O148 O antigen is the precursor of the Shigella dysenteriae type 1 O antigen and identification of a glucosyltransferase gene. Microbiology. 2007;153:139–147. doi: 10.1099/mic.0.2006/001107-0. [DOI] [PubMed] [Google Scholar]
- 57.Vinogradov EV, Shashkov AS, Knirel YA, Kochetkov NK, et al. Structure of the O-antigen of Francisella tularensis strain 15. Carbohydr Res. 1991;214:289–297. doi: 10.1016/0008-6215(91)80036-m. [DOI] [PubMed] [Google Scholar]
- 58.Knirel Yu A, Vinogradov EV, Kocharova NA, Paramonov NA, et al. The structure of O-specific polysaccharides and serological classification of Pseudomonas aeruginosa (a review). Acta Microbiol Hung. 1988;35:3–24. [PubMed] [Google Scholar]
- 59.Dmitriev BA, Knirel YA, Kochetkov NK. Somatic antigens of shigella. Structural investigation on the O-specific polysaccharide chain of Shigella dysenteriae type 1 lipopolysaccharide. Eur J Biochem. 1976;66:559–566. doi: 10.1111/j.1432-1033.1976.tb10582.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.