

Abstract
Transforming growth factor-beta3 (TGF-β3) plays a critical role in palatal epithelial cells by inducing palatal epithelial fusion, failure of which results in cleft palate, one of the most common birth defects in humans. Recent studies have shown that Smad-dependent and Smad-independent pathways work redundantly to transduce TGF-β3 signaling in palatal epithelial cells. However, detailed mechanisms by which this signaling is mediated still remain to be elucidated. Here we show that TGF-β activated kinase-1 (Tak1) and Smad4 interact genetically in palatal epithelial fusion. While simultaneous abrogation of both Tak1 and Smad4 in palatal epithelial cells resulted in characteristic defects in the anterior and posterior secondary palate, these phenotypes were less severe than those seen in the corresponding Tgfb3 mutants. Moreover, our results demonstrate that Trim33, a novel chromatin reader and regulator of TGF-β signaling, cooperates with Smad4 during palatogenesis. Unlike the epithelium-specific Smad4 mutants, epithelium-specific Tak1:Smad4- and Trim33:Smad4-double mutants display reduced expression of Mmp13 in palatal medial edge epithelial cells, suggesting that both of these redundant mechanisms are required for appropriate TGF-β signal transduction. Moreover, we show that inactivation of Tak1 in Trim33:Smad4 double conditional knockouts leads to the palatal phenotypes which are identical to those seen in epithelium-specific Tgfb3 mutants. To conclude, our data reveal added complexity in TGF-β signaling during palatogenesis and demonstrate that functionally redundant pathways involving Smad4, Tak1 and Trim33 regulate palatal epithelial fusion.
Keywords: TGF-β3 signaling, palatogenesis, Tak1, Smad4, Trim33
Introduction
Cleft palate, one of the most common birth defects in humans, is caused by a failure in palatogenesis (Chai and Maxson, 2006). During mammalian development, the secondary palate, which separates the oral cavity from the nasal cavity, develops as bilateral outgrowths (palatal shelves) of the maxillary processes of the first pharyngeal arch (Bush and Jiang, 2012). Palatal shelves first grow vertically down along the sides of the tongue; then they rapidly elevate and fuse in the midline (Ferguson, 1987). Failure in any of these three processes can result in cleft palate. The palatal shelf growth and patterning are governed by complex interactions between the ectoderm-derived epithelium, and the underlying mesenchyme derived from the cranial neural crest. One of the later events in palatogenesis, albeit a critical one, is epithelial fusion. During this process, the medial edge epithelium (MEE) in tips of the apposing palatal shelves first forms the midline seam, which subsequently disappears. Many studies, both in humans and mice, have shown that signaling initiated by TGF-β3 is required for successful epithelial fusion (Dudas et al., 2007).
TGF-β3 binds and activates a heterotetrameric receptor complex composed of two type II and two type I receptors. Ligand-receptor interactions result in phosphorylation of TGF-β R-Smads 2 and 3 and subsequent complex formation with a common Smad (Co-Smad or Smad4). R-Smad/Co-Smad complexes then accumulate in the nucleus, where they function as transcriptional co-regulators (Shi and Massague, 2003). This so called canonical (or Smad-dependent) signaling is regulated by many proteins interacting either with receptor complexes or Smads, e.g., SARA, Axin, inhibitory Smads (I-Smads) and Trim33 (Derynck and Zhang, 2003; Lamouille et al., 2014; Massague and Xi, 2012). Previous studies have shown that Trim33 (Tif1γ, ectodermin) may regulate TGF-β superfamily signaling in different ways depending on the biological context. Dupont et al suggested that Trim33 is a negative regulator of TGF-β signaling by functioning as a monoubiquitin ligase capable of disrupting activated R-Smad/Co-Smad complexes , while He et al showed that Trim33 can bind to activated R-Smads in competition with Smad4 and mediate distinct TGF-β signaling processes (He et al., 2006). It also has been suggested that Trim33/R-Smad complexes can function as chromatin readers by making target genes accessible to R-Smad/Co-Smad complexes or by controlling the time the Smad complexes are bound to promoter sequences . In addition to the canonical (Smad-dependent) pathway, TGF-βs can also trigger non-canonical (Smad-independent) signaling processes leading to activation of various downstream mediators, such as small rho-related GTPases, TGF-β-activated kinase-1 and downstream map kinase cascades including p38 Mapk, and Ikk-α (Derynck and Zhang, 2003).
Tgfb3 is strongly and specifically expressed in the MEE (Fitzpatrick et al., 1990; Pelton et al., 1990), and mouse embryos deficient in Tgfb3, as well as epithelium-specific Tgfbr1 or Tgfbr2 mutants suffer from defective palatal epithelial fusion (Dudas et al., 2006; Kaartinen et al., 1995; Proetzel et al., 1995; Xu et al., 2006). Xu et al previously showed that, at least in palatal explant cultures in vitro, Smad-dependent (Smad4-mediated) and Smad-independent (p38Mapk-mediated) pathways act redundantly during palatal epithelial fusion (Xu et al., 2008). Here, we show that cooperation between different TGF-β downstream mediators is even more extensive. By using tissue-specific mouse mutants in conjunction with a whole-head roller culture assay, we demonstrate that both Tak1:Smad4- and Trim33:Smad4-double conditional mutants display specific palatal fusion defects, and that simultaneous deletion/inactivation of all three proteins in palatal epithelial cells results in palatal phenotypes typically seen in epithelium-specific Tgfb3 mutants.
Experimental Procedures
Mice
Tgfb3F, Tak1F IkkaF and Trim33F mice have been described earlier (Doetschman et al., 2012; Kim and Kaartinen, 2008; Liu et al., 2008; Yumoto et al., 2013). K14-Cre and Smad4F mice were obtained from S. Millar (Andl et al., 2004) and C. Deng (Yang et al., 2002), respectively. To generate mutant embryos, Cre-positive male mice heterozygous for floxed (F) gene(s) were crossed with female mice carrying corresponding homozygous floxed allele(s) (see Table I). For timed matings, the presence of a vaginal plug was designated as embryonic day 0 (E0). DNA for genotyping was prepared from tail tissues using DirectPCR lysis reagents (Viagen Biotech). Mouse lines were maintained in mixed genetic backgrounds. All experiments involving the use of animals were approved by the Institutional Animal Use and Care Committee at the University of Michigan-Ann Arbor (Protocol #00004320).
Table I.
Crosses used to generate mutant embryos
| Male | Female |
|---|---|
| Tgfb3FWT:K14Cre+ | Tgfb3FF |
| Smad4WT:K14Cre+ | Smad4FF |
| Tak1FWTSmad4FWT:K14Cre+ | Tak1FFSmad4FF |
| IkkaFWTSmad4FWT:K14Cre+ | IkkaFFSmad4FF |
| Trim33FWTSmad4FWT:K14Cre+ | Trim33FFSmad4FF |
Histology, immunohistochemistry and cell death assays
Embryos were collected into sterile DPBS and fixed at 4°C overnight in freshly prepared 4% paraformaldehyde in PBS. Samples used for wax embedding were washed, dehydrated through a graded ethanol series (20, 50, 70, 95 and 100%) and an overnight step in 50% Ethanol/50% Toluene, one hour step in 100% toluene, one hour step in 50% toluene/50% fresh Blue Ribbon Tissue Embedding/Infiltration Medium (Leica Surgipath) before being oriented and embedded in fresh Blue Ribbon Tissue Embedding/Infiltration Medium (Leica Surgipath) after three changes. 7µm sections (histology and immunohistochemistry) were cut, mounted on Superfrost plus slides (Fisher) and stained with hematoxylin and eosin according to standard protocols. Fusion% was calculated as described by (Sun et al., 1998). Briefly, the length of confluence was divided by total length of adherence and multiplied by 100. For immunohistochemistry, sections were rehydrated, and after antigen retrieval (5–20 minutes at 95–100°C in 10mM citrate buffer, pH6.0) proliferating cells were detected using Ki67 antibody (#M7249; Dako). Cells positive for phosphorylated p38Mapk were detected using p-p38 Mapk antibody (#4511; Cell Signaling). Antibody binding was visualized with Alexa Fluor 594 secondary antibody (Life Technologies). Apoptotic cells were detected using a TUNEL assay (Dead End, Promega) following manufacturer’s instructions. Fluorescent images were viewed on an inverted fluorescent Leica DMI3000B microscope and documented using an Olympus DP72 camera.
In situ hybridization
Embryos were processed and embedded in paraffin as described above. 10µm sections were cut and mounted on glass slides. RNA probes were labeled using a DIG-labeled NTP mix (Roche Applied Science) according to manufacturer’s instructions, stored at −80°C and diluted in hybridization buffer. Section ISH was performed as described (Moorman et al, 2001). After staining, sections were fixed and mounted in Immu-Mount (Thermo Scientific). Probe templates for Tgfb3 and Mmp13 were prepared as described (Blavier et al., 2001; Dudas et al., 2004).
Real-time quantitative PCR
Tissues were harvested from tips of palatal shelves from E14 and E15 mouse embryos, placed into 200µl of RLT (RNeasy mini kit, Qiagen), and RNAs were isolated by using RNeasy columns (Qiagen). cDNAs were synthesized by using Omniscript reverse transcriptase (Qiagen) according to the manufacturer’s protocols. Real time quantitative PCR experiments were done either by using Universal Probe library-based assays (Roche Applied Science) or by using TaqMan assay reagents (Applied Biosystems) (see Table II). 30µl assays were quantified using Applied Biosystems ABI7300 PCR and ViiA7 detection systems and software. Data were normalized to β-actin mRNA levels using the 2−ΔΔCt method.
Table II.
Real-time quantitative PCR
| Q-RTPCR using the Universal Probe System (Roche) | |||
|---|---|---|---|
| Gene | Forward Sequence | Reverse Sequence | Universal Probe (Roche) |
| Cytokeratin 14 (K14) | atcgaggacctgaagagcaa | tcgatctgcaggaggacatt | #83 |
| Tp63 (delta N p63) | ggaaaacaatgcccagactc | aatctgctggtccatgctgt | #45 |
| Cdkn1a (p21) | cagatccacagcgatatcca | ggcacactttgctcctgtg | #21 |
| Q-RTPCR using the TaqMan approach (Life Techologies) | |||
| Gene | TaqMan Reference (Life Technologies) | ||
| Irf6 | Mm00516797_m1 | ||
| Mmp13 | Mm00439491_m1 | ||
| Cdkn2a (p16) | Mm00494449_m1 | ||
| Cdkn1c (p57) | Mm01272135_g1 | ||
Whole-head roller culture assays
Roller culture assays were modified from (Goudy et al., 2010). Heads from E15 embryos were collected in DPBS and mandibles, tongues, and brains were removed. The resulting mid-face samples were cultured for 24 hours at 37°C in roller bottles (60ml serum bottles) (60 rotations/minute) in serum-free BGJb medium without penicillin and streptomycin. The bottles were gassed at the beginning of the culture and again every 12 hours by gently bubbling the medium for 2 minutes with O2/CO2 (95%/5%). The palatal cultures were fixed, sectioned and stained as described above. In 5Z-7-oxozeanenol (Tak1 inhibitor; 10µM; eMolecules) or SB202190 (p38 Mapk inhibitor; 10µM; Sigma-Aldrich) treated samples, the inhibitor was added directly into the culture medium.
Western-blot assays
Palatal shelf edges were removed and cultured as described above in 2ml cryovials (60 rotations/minute) at 37°C in BGJb medium for 1 hour and then stimulated with TGF-β3 (10ng/ml) (#243-B3-002; R&D Systems) for 40 minutes. Tissues were then isolated and lysed in 2x Laemmli sample buffer, ran on NuPage 4–12% Bis-Tris gradient gels (Invitrogen) and transferred by “iBlot dry blotting” (Invitrogen) onto nitrocellulose filters. Immunobloting and detection were done using standard protocols. The antibodies used were p-38 Mapk (N-20) (#SC-728; Santa Cruz Biotechnology) and p-p38 MapK (#4511; Cell Signaling).
Statistical analyses
For histological and real-time quantitative PCR analyses three or more samples were analyzed. Averages, standard error and probability (Student’s t-test, 2-tailed) were calculated. Probability (p) equal or less than 0.05 was marked as statistically significant (*).
Results
Epithelium-specific Smad4 mutants display mild defects in palatogenesis
Smad-dependent and Smad-independent pathways have been shown to play functionally redundant roles during palatal epithelial fusion (Xu et al., 2008). To confirm and expand these findings, we first compared palate phenotypes of epithelium-specific Tgfb3 mutants (Tgfb3FF:K14-Cre = Tgfb3-cKO) to those of corresponding Smad4-cKOs (Smad4FF:K14-Cre). Tgfb3-cKOs display a fusion defect in the anterior palate, failure of the anterior palate to fuse to the nasal septum, variable degree of fusion in the mid-palate, persistent midline seam and occasionally a cleft in the posterior palate (Fig. 1 A–F). These observed phenotypes are less severe than those seen in Tgfb3 germline mutants (Kaartinen et al., 1995; Proetzel et al., 1995), but are practically identical to those seen in epithelium-specific Tgfbr1 and Tgfbr2 mutants (Dudas et al., 2006; Xu et al., 2006) suggesting that the phenotypic difference between Tgfb3 germ line mutants and Tgfb3-cKO results, at least in part, from the inability of K14-Cre to recombine in the periderm as shown by (Lane et al., 2014). In contrast to phenotypes seen in Tgfb3-cKOs, palatal phenotypes displayed by Smad4-cKOs were very mild (Fig. 1G–I, M and Table III), yet consistent and previously unappreciated (Xu et al., 2008). These include posterior epithelial triangles and occasional failure of the anterior palate to fuse, and the nasal septum to fuse to the anterior palate (1/4).
Figure 1. Epithelium-specific deletion of Tgfb3, Smad4 and Tak1 together with Smad4 results in defects in palatogenesis.

Frontal sections (E18.0) on the anterior level (A, D, G, J), mid anterior (B, E, H, K) and posterior (C, F, I, L) levels (see N for levels of sections along the anterior-posterior axis). A-C, Control; D-F, Tgfb3-cKO, G-I, Smad4-cKO, J-L, Tak1:Smad4-dcKO. Asterisks in A, B, C, G illustrate the palatal mesenchymal confluence, black arrows in D and J point to the sites of failed fusion in the anterior palate, black arrows in E and K point to a gap between the nasal septum and the secondary palate, the black arrow in F points to an epithelial bridge in the posterior palate and black arrowheads in H, I, K and L point to persistent epithelial seams. Scale bar, 200 µm. Bar graph (M) illustrates the degree of fusion (mesenchymal confluence); Error bars, SEM; *, p<0.05; Ant, anterior level; Post, posterior level; NS, fusion between the nasal septum and secondary palate. N, mouse mouth roof at E18; superior view; red lines illustrate the levels of frontal sections shown in A-L (n=3).
Table III.
Summary of palatal defects in Tgfb3, Smad4, Tak1 cKOs and Tak1:Smad4-dcKOs (E18.0).
| Genotype | 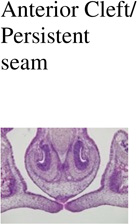 |
 |
 |
 |
|---|---|---|---|---|
| Control (Cre -); n=3 | 0/3 | 0/3 | 0/3 | 0/3 |
| Tgfb3FFK14-Cre; n=3 | 3/3 | 3/3 | 2/3 | 1/3 |
| Smad4FFK14-Cre+; n=3 | 1/4 | 1/4 | 4/4 | 0/4 |
| Tak1FFK14-Cre+; n=3 | 0/3 | 0/3 | 0/3 | 0/3 |
| Tak1FWTSmad4FFK14-Cre+; n=3 | 1/3 | 2/3 | 2/3 | 0/3 |
| Tak1FWTSmad4FWTK14-Cre+;n=3 | 0/3 | 0/3 | 0/3 | 0/3 |
| Tak1FFSmad4FFK14-Cre+; n=3 | 4/4 | 4/4 | 4/4 | 1/4 |
Simultaneous deletion of TGF-β activated kinase-1 and Smad4 results in anterior and posterior palate defects
To further examine cooperation between Smad-independent and Smad-dependent pathways during palatal epithelial fusion in vivo, we generated an allelic series of double mutants lacking one or both alleles of Tak1 (which, at least in some cell and/or tissue types, is an upstream activator of p38 Mapk (Yamashita et al., 2008)) and Smad4 in epithelial cells by crossing transgenic K14-Cre male mice (Andl et al., 2004) that also were heterozygous for both Tak1F and Smad4F alleles, with double homozygote Tak1FFSmad4FF female mice (Table III). Epithelium-specific Tak1 mutants (Supplementary Fig. 1) and double heterozygotes showed no palatal phenotypes, while corresponding Tak1:Smad4 double mutants (herein called Tak1:Smad4-dcKO) displayed specific and consistent defects in palatogenesis (Fig. 1J–L, M, and Table III), e.g., persistent epithelial seam, anterior fusion defect and a failure of the nasal septum to fuse to the anterior secondary palate. Cre-positive embryos heterozygous for the floxed Tak1 allele and homozygous for the floxed Smad4 allele showed similar but milder defects as the dcKOs with variable penetrance (Table III).
Additional modifiers of TGF-β signaling during palatal epithelial fusion
Our results suggested that Tak1 plays a redundant regulatory role in TGF-β signaling, perhaps by functioning upstream of p38Mapk in palatal epithelial cells. Yet, the Tak1:Smad4-dcKO palatal phenotypes (Fig. 1J–L) were noticeably less severe than those seen in corresponding Tgfb3-cKOs (Fig. 1D–F) or Tgfbr1-cKOs (Dudas et al., 2006)). Therefore we wondered whether, in addition to the Tak1-mediated pathway, other signaling mediators could be involved in TGF-β3-induced palatal epithelial fusion. As candidates, we chose Ikk-α (Chuk) and Trim33 (Tif1γ; ectodermin), because both proteins have been shown to be able to modulate TGF-β signaling both by Smad-dependent and Smad-independent mechanisms contingent on the biological context (He et al., 2006; Liu et al., 2008). Our results showed that epithelium-specific single mutants (i.e., Ikka-cKOs and Trim33-cKOs) did not show palatal phenotypes (Supplementary Fig. 1, and data not shown), and that Ikka:Smad4-dcKO mutant palates did not significantly differ from those of Smad4-cKOs (Fig. 2A–F,M) suggesting that Ikk-α does not contribute to TGF-β-triggered MEE removal. However, Trim33:Smad4-dcKO mutants consistently showed a persistent midline epithelial seam both in the anterior and in the posterior palate, and between the nasal septum and the anterior palate (Fig. 2G–L,M and Table IV). This phenotype differs from that seen in Smad4-cKOs or in Tak1:Smad4-dcKOs (compare Fig.1 and 2) suggesting that Trim33 and Smad4 cooperate in palatal epithelial fusion that are at least in part distinct from that of a combined role of Tak1 and Smad4.
Figure 2. Simultaneous deletion of Trim33 and Smad4 in epithelial cells leads to persistence of the midline seam.

Frontal sections (E18.0) on the anterior level (A, D, G, J), mid anterior (B, E, H, K) and posterior (C, F, I, L) levels (see Fig 1N). A–C and G–I, Control; D–F, Ikka:Smad4-dcKO, J-L, Trim33:Smad4-dcKO. Asterisks in A, B, C, D, E, G, H, I illustrate the palatal mesenchymal confluence, black arrowheads in E point to epithelial islands between the nasal septum and the posterior palate, arrowhead in F points to the persistent oral epithelial angle and black arrows in J, K, and L point to persistent epithelial seams. Scale bar, 200 µm. Bar graph (M) illustrates the degree of fusion (mesenchymal confluence); Error bars, SEM; *, p<0.05; Ant, anterior level; Post, posterior level; NS, fusion between the nasal septum and secondary palate (n=3).
Table IV.
Summary of palatal defects in Ikka- and Trim33-cKOs and Ikka:Smad4- and Trim33:Smad4-dcKOs (E18.0).
| Genotype | Anterior Cleft/ Persistent seam |
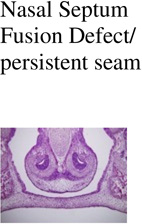
Nasal Septum Fusion Defect/ persistent seam |
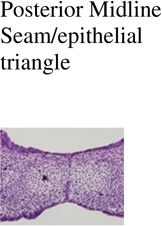
Posterior Midline Seam/epithelial triangle |
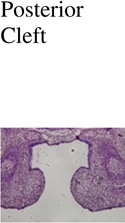
Posterior Cleft |
|---|---|---|---|---|
| Control (Cre -); n=3 | 0/3 | 0/3 | 0/3 | 0/3 |
| IkkaFFK14-Cre+; n=3 | 0/3 | 0/3 | 0/3 | 0/3 |
| Trim33FFK14-Cre+; n=3 | 0/3 | 0/3 | 0/3 | 0/3 |
|
IkkaFFSmad4FFK14- Cre+; n=3 |
0/3 | 1/3 | 2/3 | 0/3 |
|
Trim33FFSmad4FFK14- Cre+; n=3 |
3/3 | 2/3 | 2/3 | 1/3 |
Expression of a TGF-β signaling target Mmp13 is affected in Tak1:Smad4- and Trim33:Smad4 - double conditional mutants
It has been previously shown that Mmp13 expression is rapidly induced by TGF-β3-signaling in the midline seam during palatogenesis (Blavier et al., 2001; Xu et al., 2006). To analyze whether Tak1 and Smad4- or Trim33 and Smad4-mediated pathways are involved in regulation of Mmp13 expression, we first harvested palatal shelves from control, Tgfb3-cKO, Smad4-cKO, Tak1:Smad4-dcKO and Trim33:Smad4-dcKO embryos at E15.0 and used qRT-PCR to quantify their Mmp13 mRNA levels. As shown in Fig. 3A, Mmp13 expression was significantly reduced in Tgfb3, Tak1:Smad4-dcKO and Trim33:Smad4-dcKO samples, while the reduction in Smad4-cKO samples was rather modest and not statistically significant (Fig. 3A). We also used section ISH to analyze Mmp13 and Tgfb3 expression in the MES and nasal septal epithelium (Fig. 3B–I). Cre-negative control and Smad4-cKO samples displayed strong Tgfb3 and Mmp13 expression both in the MES and in the nasal septal epithelium. In Tak1:Smad4-dcKOs there was a notable reduction in Mmp13 expression, while in Trim33:Smad4-dcKOs Mmp13 expression was barely detectable. In both samples, Tgfb3 mRNA levels did not differ from those seen in controls. These results suggest that a known TGF-β signaling target Mmp13 is regulated both by Tak1:Smad4- and Trim33:Smad4-mediated pathways in palatal medial edge epithelial cells.
Figure 3. Gene expression differences in prefusion and fusing palatal shelves between controls and TGF-β pathway mutants.

A, QRT-PCR analysis of Mmp13 expression in palatal tissues of control (blue), Tgfb3cKO (red), Smad4-cKO (green), Tak1:Smad4-dcKO (purple) and Trim33:Smad4-dcKO (turquoise) at E15 (Error bars, SEM; *, p<0.05, n>3 or more). B-I, In situ hybridization for Tgfb3 (B, C, D, E) and for Mmp13 (F, G, H, I). B, F, Control; C, G, Smad4-cKO; D, H, Tak1:Smad4-dcKO; E, I, Trim33:Smad4-dcKO. Frontal sections of E15 embryos; anterior level. Scale bar in I, 100µm (for B-I). J, QRT-PCR analysis of Irf6, dNP63 and p21 expression in palatal tissues from control, Tgfb3-cKO, Smad4-cKO, Tak1:Smad4-dcKO and Trim33:Smad4-dcKO embryos harvested at E14 and E15 (Error bars, SEM; *, p<0.05, n>3). K, QRT-PCR analysis of p16, and p57 expression in palatal tissues from control, Tgfb3-cKO, Smad4-cKO, Tak1:Smad4-dcKO and Trim33:Smad4-dcKO embryos harvested at E14 and E15 (Error bars, SEM; *, p<0.05; n>3).
Altered gene expression in TGF-β pathway mutants
Iwata et al recently reported that signaling via TgfβRII stimulates Irf6 expression, which in turn suppresses dNp63 allowing appropriate p21 (Cdkn1a, p21Cip1) expression (Iwata et al., 2013). In prefusion palatal shelves at E14, we could not detect any differences in Irf6 or dNP63 expression levels between the control, Tgfb3-cKO, Smad4-cKO, Tak1:Smad4-dcKO or Tak1:Smad4-dcKO samples (Fig.3J). In the fusing palatal shelves at E15, Irf6 was reduced only in Tak1:Smad4-dcKO samples, while dNP63 levels were increased in Tgfb3- and Smad4-cKO samples. In contrast to the lack of differences in Irf6 and dNP63 expression at E14, p21 expression levels were reduced in all samples lacking Smad4 (i.e., Smad4-cKO, Tak1:Smad4-dcKO and Trim33:Smad4-dcKO), but not in Tgfb3-cKO samples (Fig. 3J). In addition to p21, TGF-β signaling has been shown to regulate expression of other cyclin-dependent kinase inhibitors in the MEE (Iordanskaia and Nawshad, 2011). Therefore, we compared expression levels of p15 (Cdkn2b, p15Ink4b), p16 (Cdkn2a, p16Ink4a) and p57 (Cdkn1c, p57Kip2) as outlined above (Fig. 3K and data not shown). At E14, p16 expression was significantly reduced in Tgfb3-cKOs and in Tak1:Smad4-dcKOs, but not in Trim33:Smad4-dcKOs, whereas Tgfb3-cKO samples showed a dramatic increase in p57 expression at E15 (expression levels of p15 were very low and did not show detectable differences between controls and different mutants; data not shown). Taken together, differences in expression level of candidate genes between Tak1:Smad4-dcKOs and Tgfb3-cKOs suggest that Tak1 and TGF-β3 can affect, at least, in part distinct pathways that act together to promote palatal epithelial fusion. Similarly, differences in gene expression between Trim33:Smad4 and Tak1:Smad4 double mutants imply that these molecules mediate distinct arms of TGF-β signaling.
Epithelial cell proliferation and apoptosis are variably affected in Tak1:Smad4- and Trim33:Smad4- dcKOs
Previous studies showed that TGF-β signaling in the palatal epithelium induces cell cycle arrest that is a prerequisite for the disappearance of the midline seam by apoptosis and other possible mechanisms (Cui et al., 2003; Iwata et al., 2013; Nawshad, 2008). To examine whether cell proliferation in the MES was affected in Tak1:Smad4- or Trim33:Smad4-dcKOs, we used Ki67 immunostaining on anterior frontal sections obtained from control (Cre-negative) and dcKO embryos at E14.5 E15.0 (Fig. 4A–C). Both Tak1:Smad4- and Trim33:Smad4-dcKOs showed an increased number of positively staining epithelial cells in the tip of the nasal septum and in epithelial tips of prefusion palatal shelves when compared to the controls (Fig. 4A–C, J). In contrast, the number of TUNEL-positive apoptotic cells was significantly reduced in the control and mutant samples in the anterior palate on the level of the nasal septum (Fig. 4D, F, H). MEE cells of Tak1:Smad4-dcKOs showed reduced phosphorylation (activation) of apoptosis-associated TGF-β-induced signaling molecule p38 Mapk, while Trim33:Smad4-dcKOs did not demonstrate comparable changes (Fig. 4 E, G, I).
Figure 4. Proliferation and cell death are affected in Tak1:Smad4 and Trim33:Smad4-dcKOs.

Immunostaining for Ki67 (red) in control (A, A ), Tak1:Smad4- (B, B ) and Trim33:Smad4-dcKO (C, C ) at E14.5-E15. Frontal sections, anterior level. Counterstaining with DAPI (blue nuclei). In A, B and C palatal shelves are forming a contact with each other, and in A , B C the midline seam is forming. White arrowheads in A-C point to Ki67-positive cells in the MEE and nasal septum epithelium. TUNEL staining to detect apoptotic/necrotic cells (white arrows in D, F, H; green nuclei) in control (D), Tak1:Smad4:dcKOs (F) and in Trim33:Smad4-dcKO(H) samples (E15.0). Frontal sections, anterior level; counter staining with DAPI (blue). Immunostaining for phosphorylated p38 Mapk (p-p38, red staining) in control (E), Tak1:Smad4:dcKOs (G) and in Trim33:Smad4-dcKO (I) samples (E15.0). White arrows in E, G, and I point to positively-staining cells. Frontal sections, anterior level; counter staining with DAPI (blue). Scale bar in A, 50µm (for A-I). J, Bar graph shows quantification of proliferation (Ki67, positively staining cells both in the nasal septum and MEE were counted) apoptosis (TUNEL) and phopho-p38 Mapk assays (Error bars, SEM; *, p<0.05; n>3).
Inactivation of Tak1 in Trim33:Smad4-dcKOs phenocopies the palate defects seen in Tgfb3-cKOs
As outlined above, the palatal phenotypes of both Tak1:Smad4- and Trim33:Smad4-dcKOs, while consistent and clear, were still less severe than those seen in Tgfb3-cKOs. Therefore, we wondered whether combined loss of Tak1, Smad4 and Trim33 would result in palatal phenotypes similar to those seen in Tgfb3-cKOs. To this end, we used a potent Tak1 inhibitor, 5Z-7-oxozeaenol, in conjunction with Trim33:Smad4-dcKOs ex vivo by using a whole-head roller culture assay. Control cultures treated with 5Z-7-oxozeaenol, which efficiently blocked p38 Mapk phosphorylation (Fig. 5I), showed consistent and reliable fusion identical to that seen in untreated cultures (Fig. 5A–D). Furthermore, Smad4-cKO cultures did not demonstrate detectable phenotypes, and with addition of Tak1 inhibitor resembled the Tak1:Smad4-dcKO phenotype (Supplementary Fig. 2). Under similar culture conditions, Trim33:Smad4-dcKOs without the Tak1 inhibitor showed a persistent midline seam both in the anterior and posterior palate, comparable to that seen in Trim33:Smad4-dcKOs in vivo (Fig. 5E,F). However, treatment of double-cKO cultures with the Tak1 inhibitor resulted in palatal phenotypes practically identical to those seen in Tgfb3-cKOs, i.e., an anterior hole and a posterior epithelial bridge (Fig. 5G,H). These data suggest that Tak1, Smad4 and Trim33 are redundantly transducing TGF-β3 signaling to remove the midline epithelial seam, and to facilitate palatal anterior and posterior fusion during palatogenesis (Fig. 6).
Figure 5. Conditional removal of Trim33 and Smad4 along with Tak1 inhibition recapitulates the palatal phenotype seen in Tgfb3-cKO embryos.

A whole-head roller culture assay of Cre-negative controls (A-D) and Trim33:Smad4-dcKOs (E-H) in the absence (A,B, E, F) and presence of Tak1 inhibitor (C, D, G, H). A, C, E, G, frontal sections, anterior level; B, D, F, H, frontal sections, posterior level. Asterisks in A, B, C, D illustrate mesenchymal confluence, black arrow in D points to oral epithelial triangle, black arrows in E point to intact MEE cells in palatal shelves and nasal septum, black arrow in F points to the persistent epithelial seam, black arrows in G point to the anterior cleft and black arrow in H points to the posterior epithelial bridge. Scale bar, 100µm. I, Tak1 inhibitor 5Z-7-oxozeaenol (5Z) prevents p38 Mapk phosphorylation in palatal shelves harvested from whole-head roller cultures. J, bar graph shows degree of palatal fusion in control and Trim33:Smad4-cKO samples cultured in the presence or absence of 5Z in a whole-head roller culture assay. Error bars, SEM; *, p<0.05; n=3). Blue columns, anterior palate; red columns, posterior palate. 0% fusion represents a full cleft or the presence of a fully intact epithelial seam.
Figure 6. Schematic drawing illustrating cooperation between Tak1, Smad4 and Trim33 during palatal epithelial fusion.

MEE, medial edge epithelium; MES, midline seam.
Discussion
Palatogenesis is a complex developmental process involving appropriate growth, elevation and fusion of palatal shelves (Ferguson, 1988). While the role of TGF-β3 in the palatal epithelium (MEE) is well-established (Kaartinen et al., 1995; Proetzel et al., 1995), recent studies have shown that mechanisms by which the downstream signaling is transduced are surprisingly complicated, involving redundantly acting canonical and non-canonical arms of TGF-β signaling (Iwata et al., 2013; Xu et al., 2008). Here we show that in MEE cells, redundancy of TGF-β signal transduction is even more extensive, since recapitulation of the Tgfb3-cKO palatal phenotype required inactivation of three separate genes/gene products (Tak1, Smad4, Trim33) mediating different arms of TGF-β signaling.
Developing palatal shelves are composed of the mesenchyme (largely derived from the cranial neural crest with a minor contribution coming from the cranial paraxial mesoderm) and from the ectoderm-derived surface epithelium, which is covered by a thin layer of peridermal cells (Lane and Kaartinen, 2014). Recent studies have shown that peridermal cells play a key role in palatal shelf adherence, and that this layer has to be removed for normal palatal adhesion and fusion to take place (Wu et al., 2013a). Tgfb3 is expressed in both cell types (Lane et al., 2014) and interestingly, the cleft palate phenotype of Tgfb3 null mutants can be partially rescued by transducing Tgfb3 exclusively in peridermal cells (Wu et al., 2013a). Likewise, Tgfb3-cKO mice, which lack Tgfb3 in epithelial cells including the MEE but not in peridermal cells, display significantly milder palatal phenotypes than those seen in systemic Tgfb3 mutants ((Lane et al., 2014) and the present study), suggesting that TGF-β3 signaling is important for peridermal cell removal and is thus required for adhesion and fusion between the palatal shelves in the mid-palate. In fact, Tgfb3-cKO palatal phenotypes are practically identical to those seen in epithelium-specific Tgfbr1 or Tgfbr2 mutants (Dudas et al., 2006; Xu et al., 2006). Moreover, this distinct palatal phenotype can be seen in mice expressing Tgfb1 in the Tgfb3 locus (Yang and Kaartinen, 2007) suggesting that Tgfb1 can functionally replace Tgfb3 in peridermal cells but not in MEE cells. Nevertheless, the consistent defects of Tgfb3-cKO embryos in the anterior and posterior palate (anterior hole, nasal septum fusion defect and posterior epithelial bridge/submucous cleft) show that TGF-β3-mediated signaling is also crucial in the MEE, and that the K14-Cre driver line is a useful tool to study gene functions in this cell type during palatogenesis.
A previous study on epithelium-specific Tgfbr2:K14-Cre mice suggested that TGF-β via TgfbRII triggers a Smad4-Irf6 signaling cascade, which downregulates dNP63 resulting in p21 induction, cell cycle arrest and removal of MEE cells (Iwata et al., 2013). Using the same K14-Cre transgenic line (Andl et al., 2004) we demonstrate that Tgfb3-cKO embryos, which display clear and consistent palatal defects similar to those seen in Tgfbr2:K14-Cre mutants, do not show differences in p21 expression in prefusion or fusing palatal shelves. In addition, Smad4-cKOs, which display a very mild phenotype in the posterior palate, consistently show reduced levels of p21 expression. Based on these results we conclude that changes in p21 expression are not causally related to the observed palatal phenotypes. Despite the obvious reduction in p21 expression in Smad4-cKOs as well as in Tak1:Smad4- and Trim33:Smad4-dcKOs, we could not see significant differences in Irf6 or dNP63 expression in prefusion palatal shelves at E14. In fusing palatal shelves at E15, Tak1:Smad4-dcKOs palatal shelves showed reduction in Irf6 expression while palatal tissues harvested from Tgfb3-cKOs at E15 showed an increase in dNP63. Whether these discrepancies between our current findings and those reported by others (Iwata et al., 2013) are caused by experimental variables, e.g., timing and/or technique of tissue harvest and differences in genetic backgrounds, or whether they are caused by other currently unknown causes, remains to be shown.
In epithelial cells, TGF-βs are well-known growth inhibitory cytokines (Siegel et al., 2003). It has been suggested that inhibition of MEE proliferation by TGF-β signaling triggers a cascade of events that results in the removal of MEE via apoptosis (and to some extent by transdifferentiation/migration) (Cui et al., 2003; Iwata et al., 2013; Nawshad, 2008). Despite some differences in gene expression patterns between Tak1:Smad4-dcKOs and Trim33:Smad4-dcKOs, we show that both of these epithelium-specific double mutants show increased levels of proliferation, particularly in the epithelial tips of the nasal septum, while a smaller number of TUNEL-positive nuclei could be seen both in the epithelial tips of palatal shelves and nasal septum. Increased cell proliferation is consistent with our finding that p21 expression levels are reduced in both dcKOs. However, it is likely that expression of other negative cell cycle regulators, e.g., p16, is also regulated by TGF-β, as p21 expression levels are not reduced in Tgfb3 mutants, although they display a well-documented failure to suppress cell proliferation in the prefusion MEE (Cui et al., 2003; Dudas et al., 2004; Martinez-Alvarez et al., 2000).
A role of Tak1 in regulation of TGF-β3-induced p38Mapk activation is currently controversial . Several studies have suggested that Traf6-Tak1 signaling module mediates TGF-β-induced p38Mapk activation (Kim et al., 2009; Sorrentino et al., 2008; Yamashita et al., 2008), while other studies have proposed that instead of Tak1, other Map3Ks, e.g., Map3K4 and Map3K10, play the key role in transducing TGF-β signaling that results in p38 Mapk phosphorylation (Sapkota, 2013). We previously showed that neural crest-derived ectomesenchymal cells lacking Tak1 fail to demonstrate TGF-β-induced p38 Mapk activation (Yumoto et al., 2013). Our present results imply that also in MEE cells Tak1 is at least partially responsible for mediating p38 Mapk activation, since we could see reduction in p38Mapk phosphorylation in Tak1:Smad4-dcKOs when compared to controls or Trim33:Smad4-dcKOs. Moreover, similar to palatal enxplant cultures with inactivated p38 Mapk , inactivation of Tak1 alone, which totally abolished the p38Mapk phosphorylation (Fig. 5I), did not result in defects in palatogenesis (Fig. 5), while simultaneous inactivation of Tak1 and deletion of Smad4 in explant cultures resulted in palatal phenotypes similar to that seen in Tak1:Smad4-dcKOs in vivo (Supplementary Fig. 1). In the roller culture assay, Tak:Wnt1-Cre mutant palatal shelves were able to go through normal palatogenesis (Yumoto et al., 2013) suggesting that lack of Tak1 in the palatal mesenchyme does not alter the ability of the palatal shelves to go through normal fusion. In our study, the Tak1 inhibitor was added at the time of palatal shelf adhesion and does not cause visible phenotypes within the mesenchyme that would alter the way analysis is done in the presence of a persistent epithelial seam. In addition, upon simultaneous inactivation of Tak1 and deletion of Smad4 in the roller culture system, we see a palatal phenotype similar to that seen in Tak1:Smad4-dcKOs in vivo. The Tak1 inhibitor 5Z-7-oxozeaenol also inhibits several other protein kinases when tested with a kinase panel (Wu et al., 2013b), and therefore we cannot fully exclude a possibility that (in addition to Tak1), other protein kinases would be partially responsible for p38 Mapk inhibition and for the observed palate phenotypes in the roller culture system. However, notable similarities between Tak1 mutant palate phenotypes in vivo and corresponding phenotypes in inhibitor-treated samples in vitro (as shown above), together with the finding that the inhibitor displays remarkable specificity in inhibition of Tak1 pathway in cell lines (Wu et al., 2013b), alleviate these concerns.
Mutations in the IRF6 gene cause van der Woude syndrome, an autosomal dominant syndrome characterized by cleft lip and/or palate (Kondo et al., 2002). Mouse embryos lacking Irf6 display early differentiation defects of the ectoderm, which are practically identical to those seen in Ikka knockout mice (Ingraham et al., 2006; Liu et al., 2008; Richardson et al., 2006). Therefore, it is surprising that deletion of Ikka in epithelial cells including the MEE does not result in noticeable defects in palatogenesis. Instead, we discovered that in MEE cells, concomitant deletion of a novel chromatin reader Trim33 and Smad4 led to defective palatal fusion. Several recent reports have demonstrated that epigenetic mechanisms involving appropriate histone modifications are crucial for successful craniofacial development and palatogenesis. In humans, haploinsufficiency of histone-3 demethylases, e.g., PHF8 and KATB6, has been shown to result in cleft palate (Clayton-Smith et al., 2011; Laumonnier et al., 2005), while neural crest specific deletion of Hdac3 resulted in severe craniofacial defects including cleft palate in mouse embryos (Singh et al., 2013). Trim33 is a multifunctional protein (Dupont et al., 2005), which has been shown to both facilitate and antagonize TGF-β signaling. In differentiating stem cells, Trim33, when complexed with Smad2, is able to bind to specific poised chromatin marks found in the promoter regions of master regulators, rendering them accessible to R-Smad/Co-Smad complexes (Xi et al., 2011). In other cellular systems, Trim33 has been shown to mono-ubiquitinate Smad4 disrupting Smad4-chromatin complexes and limiting their residence time at TGF-β-responsive enhancers (Agricola et al., 2011; Dupont et al., 2009). Here we show that in MEE cells Trim33 does not function as a negative regulator of canonical TGF-β signaling, but rather that Trim33 and Smad4 are functionally redundant during palatal fusion (Fig. 6). A similar redundant role of Smad4 and Trim33 in control of neural stem cell proliferation was recently shown to take place in the developing cortex (Falk et al., 2014).
In conclusion, our data indicate that complex and largely redundant mechanisms involving both non-canonical and canonical arms of TGF-β signaling control disappearance of MEE cells during palatal fusion. These involve Tak1, Smad4 and a novel Smad2 binding partner and a putative chromatin reader Trim33 (Fig. 6). While the roles of Tak1 and Trim33 appear fully redundant with Smad4, our results also reveal a subtle but consistent, and previously unappreciated, non-redundant role for Smad4 in MEE cells during palatogenesis.
Supplementary Material
Highlights.
Tak1 and Smad4 interact genetically during palatal epithelial fusion
Trim33 and Smad4 cooperate during palatal epithelial fusion
Inactivation of Tak1 in Trim33:Smad4 double mutants results in Tgfb3-like phenotype
Acknowledgements
We thank Manas K. Ray and Greg Scott for technical assistance, Junichi Iwata for sharing unpublished data, and Ben Allen, Deneen Wellik, Penny Thomas and Rogerio Castilho for helpful discussions. This work was supported by the NIH RO1 grants RO1DE020843 (to YM), RO1GM068812 (JN) and RO1DE013085 (to VK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agricola E, Randall RA, Gaarenstroom T, Dupont S, Hill CS. Recruitment of TIF1gamma to chromatin via its PHD finger-bromodomain activates its ubiquitin ligase and transcriptional repressor activities. Molecular cell. 2011;43:85–96. doi: 10.1016/j.molcel.2011.05.020. [DOI] [PubMed] [Google Scholar]
- Andl T, Ahn K, Kairo A, Chu EY, Wine-Lee L, Reddy ST, Croft NJ, Cebra-Thomas JA, Metzger D, Chambon P, Lyons KM, Mishina Y, Seykora JT, Crenshaw EB, Millar SE., 3rd Epithelial Bmpr1a regulates differentiation and proliferation in postnatal hair follicles and is essential for tooth development. Development. 2004;131:2257–2268. doi: 10.1242/dev.01125. [DOI] [PubMed] [Google Scholar]
- Blavier L, Lazaryev A, Groffen J, Heisterkamp N, DeClerck YA, Kaartinen V. TGF-beta3-induced palatogenesis requires matrix metalloproteinases. Mol.Biol.Cell. 2001;12:1457–1466. doi: 10.1091/mbc.12.5.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush JO, Jiang R. Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development. 2012;139:231–243. doi: 10.1242/dev.067082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Maxson RE., Jr Recent advances in craniofacial morphogenesis. Dev.Dyn. 2006;235:2353–2375. doi: 10.1002/dvdy.20833. [DOI] [PubMed] [Google Scholar]
- Clayton-Smith J, O'Sullivan J, Daly S, Bhaskar S, Day R, Anderson B, Voss AK, Thomas T, Biesecker LG, Smith P, Fryer A, Chandler KE, Kerr B, Tassabehji M, Lynch SA, Krajewska-Walasek M, McKee S, Smith J, Sweeney E, Mansour S, Mohammed S, Donnai D, Black G. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. American journal of human genetics. 2011;89:675–681. doi: 10.1016/j.ajhg.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui XM, Chai Y, Chen J, Yamamoto T, Ito Y, Bringas P, Shuler CF. TGF31 beta3-dependent SMAD2 phosphorylation and inhibition of MEE proliferation during palatal fusion. Dev.Dyn. 2003;227:387–394. doi: 10.1002/dvdy.10326. [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Doetschman T, Georgieva T, Li H, Reed TD, Grisham C, Friel J, Estabrook MA, Gard C, Sanford LP, Azhar M. Generation of mice with a conditional allele for the transforming growth factor beta3 gene. Genesis. 2012;50:59–66. doi: 10.1002/dvg.20789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas M, Kim J, Li WY, Nagy A, Larsson J, Karlsson S, Chai Y, Kaartinen V. Epithelial and ectomesenchymal role of the type I TGF-beta receptor ALK5 during facial morphogenesis and palatal fusion. Dev.Biol. 2006;296:298–314. doi: 10.1016/j.ydbio.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas M, Li WY, Kim J, Yang A, Kaartinen V. Palatal fusion - Where do the midline cells go? A review on cleft palate, a major human birth defect. Acta Histochem. 2007;109:1–14. doi: 10.1016/j.acthis.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Dudas M, Nagy A, Laping NJ, Moustakas A, Kaartinen V. Tgf-beta3-induced palatal fusion is mediated by Alk-5/Smad pathway. Dev.Biol. 2004;266:96–108. doi: 10.1016/j.ydbio.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Dupont S, Mamidi A, Cordenonsi M, Montagner M, Zacchigna L, Adorno M, Martello G, Stinchfield MJ, Soligo S, Morsut L, Inui M, Moro S, Modena N, Argenton F, Newfeld SJ, Piccolo S. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell. 2009;136:123–135. doi: 10.1016/j.cell.2008.10.051. [DOI] [PubMed] [Google Scholar]
- Dupont S, Zacchigna L, Cordenonsi M, Soligo S, Adorno M, Rugge M, Piccolo S. Germ-layer specification and control of cell growth by Ectodermin, a Smad4 ubiquitin ligase. Cell. 2005;121:87–99. doi: 10.1016/j.cell.2005.01.033. [DOI] [PubMed] [Google Scholar]
- Falk S, Joosten E, Kaartinen V, Sommer L. Smad4 and trim33/tif1gamma redundantly regulate neural stem cells in the developing cortex. Cerebral cortex. 2014;24:2951–2963. doi: 10.1093/cercor/bht149. [DOI] [PubMed] [Google Scholar]
- Ferguson MW. Palate development: mechanisms and malformations. Ir. J.Med.Sci. 1987;156:309–315. doi: 10.1007/BF02951261. [DOI] [PubMed] [Google Scholar]
- Ferguson MW. Palate development. Development. 1988;103(Suppl):41–60. doi: 10.1242/dev.103.Supplement.41. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick DR, Denhez F, Kondaiah P, Akhurst RJ. Differential expression of TGF beta isoforms in murine palatogenesis. Development. 1990;109:585–595. doi: 10.1242/dev.109.3.585. [DOI] [PubMed] [Google Scholar]
- Goudy S, Law A, Sanchez G, Baldwin HS, Brown C. Tbx1 is necessary for palatal elongation and elevation. Mech Dev. 2010;127:292–300. doi: 10.1016/j.mod.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Dorn DC, Erdjument-Bromage H, Tempst P, Moore MA, Massague J. Hematopoiesis controlled by distinct TIF1gamma and Smad4 branches of the TGFbeta pathway. Cell. 2006;125:929–941. doi: 10.1016/j.cell.2006.03.045. [DOI] [PubMed] [Google Scholar]
- Ingraham CR, Kinoshita A, Kondo S, Yang B, Sajan S, Trout KJ, Malik MI, Dunnwald M, Goudy SL, Lovett M, Murray JC, Schutte BC. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6) Nat Genet. 2006;38:1335–1340. doi: 10.1083/ng1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iordanskaia T, Nawshad A. Mechanisms of transforming growth factor beta induced cell cycle arrest in palate development. Journal of cellular physiology. 2011;226:1415–1424. doi: 10.1002/jcp.22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata J, Suzuki A, Pelikan RC, Ho TV, Sanchez-Lara PA, Urata M, Dixon MJ, Chai Y. Smad4-Irf6 genetic interaction and TGFbeta-mediated IRF6 signaling cascade are crucial for palatal fusion in mice. Development. 2013;140:1220–1230. doi: 10.1242/dev.089615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat.Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- Kim J, Kaartinen V. Generation of mice with a conditional allele for Trim33. Genesis. 2008;46:329–333. doi: 10.1002/dvg.20401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SI, Kwak JH, Na HJ, Kim JK, Ding Y, Choi ME. Transforming growth factor-beta (TGF-beta1) activates TAK1 via TAB1-mediated autophosphorylation, independent of TGF-beta receptor kinase activity in mesangial cells. J Biol Chem. 2009;284:22285–22296. doi: 10.1074/jbc.M109.007146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Schutte BC, Richardson RJ, Bjork BC, Knight AS, Watanabe Y, Howard E, de Lima RL, Daack-Hirsch S, Sander A, McDonald-McGinn DM, Zackai EH, Lammer EJ, Aylsworth AS, Ardinger HH, Lidral AC, Pober BR, Moreno L, Arcos-Burgos M, Valencia C, Houdayer C, Bahuau M, Moretti-Ferreira D, Richieri-Costa A, Dixon MJ, Murray JC. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet. 2002;32:285–289. doi: 10.1038/ng985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nature reviews. Molecular cell biology. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane J, Kaartinen V. Signaling networks in palate development. Wiley interdisciplinary reviews. Systems biology and medicine. 2014;6:271–278. doi: 10.1002/wsbm.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane J, Yumoto K, Pisano J, Azhar M, Thomas PS, Kaartinen V. Control elements targeting Tgfb3 expression to the palatal epithelium are located intergenically and in introns of the upstream Ift43 gene. Frontiers in physiology. 2014;5:258. doi: 10.3389/fphys.2014.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumonnier F, Holbert S, Ronce N, Faravelli F, Lenzner S, Schwartz CE, Lespinasse J, Van Esch H, Lacombe D, Goizet C, Phan-Dinh Tuy F, van Bokhoven H, Fryns JP, Chelly J, Ropers HH, Moraine C, Hamel BC, Briault S. Mutations in PHF8 are associated with X linked mental retardation and cleft lip/cleft palate. Journal of medical genetics. 2005;42:780–786. doi: 10.1136/jmg.2004.029439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Xia X, Zhu F, Park E, Carbajal S, Kiguchi K, DiGiovanni J, Fischer SM, Hu Y. IKKalpha is required to maintain skin homeostasis and prevent skin cancer. Cancer cell. 2008;14:212–225. doi: 10.1016/j.ccr.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Alvarez C, Tudela C, Perez-Miguelsanz J, O'Kane S, Puerta J, Ferguson MW. Medial edge epithelial cell fate during palatal fusion. Dev.Biol. 2000;220:343–357. doi: 10.1006/dbio.2000.9644. [DOI] [PubMed] [Google Scholar]
- Massague J, Xi Q. TGF-beta control of stem cell differentiation genes. FEBS letters. 2012;586:1953–1958. doi: 10.1016/j.febslet.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawshad A. Palatal seam disintegration: to die or not to die? that is no longer the question. Dev Dyn. 2008;237:2643–2656. doi: 10.1002/dvdy.21599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelton RW, Dickinson ME, Moses HL, Hogan BL. In situ hybridization analysis of TGF beta 3 RNA expression during mouse development: comparative studies with TGF beta 1 and beta 2. Development. 1990;110:609–620. doi: 10.1242/dev.110.2.609. [DOI] [PubMed] [Google Scholar]
- Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MW, Doetschman T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat.Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RJ, Dixon J, Malhotra S, Hardman MJ, Knowles L, Boot-Handford RP, Shore P, Whitmarsh A, Dixon MJ. Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nat Genet. 2006;38:1329–1334. doi: 10.1038/ng1894. [DOI] [PubMed] [Google Scholar]
- Sapkota GP. The TGFbeta-induced phosphorylation and activation of p38 mitogen34 activated protein kinase is mediated by MAP3K4 and MAP3K10 but not TAK1. Open biology. 2013;3:130067. doi: 10.1098/rsob.130067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Siegel PM, Shu W, Massague J. Mad upregulation and Id2 repression accompany transforming growth factor (TGF)-beta-mediated epithelial cell growth suppression. J Biol Chem. 2003;278:35444–35450. doi: 10.1074/jbc.M301413200. [DOI] [PubMed] [Google Scholar]
- Singh N, Gupta M, Trivedi CM, Singh MK, Li L, Epstein JA. Murine craniofacial development requires Hdac3-mediated repression of Msx gene expression. Dev Biol. 2013;377:333–344. doi: 10.1016/j.ydbio.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N, Zhang S, Heldin CH, Landstrom M. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nature cell biology. 2008;10:1199–1207. doi: 10.1038/ncb1780. [DOI] [PubMed] [Google Scholar]
- Sun D, Vanderburg CR, Odierna GS, Hay ED. TGFbeta3 promotes transformation of chicken palate medial edge epithelium to mesenchyme in vitro. Development. 1998;125:95–105. doi: 10.1242/dev.125.1.95. [DOI] [PubMed] [Google Scholar]
- Wu C, Endo M, Yang BH, Radecki MA, Davis PF, Zoltick PW, Spivak RM, Flake AW, Kirschner RE, Nah HD. Intra-amniotic transient transduction of the periderm with a viral vector encoding TGFbeta3 prevents cleft palate in Tgfbeta3(−/−) mouse embryos. Molecular therapy : the journal of the American Society of Gene Therapy. 2013a;21:8–17. doi: 10.1038/mt.2012.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Powell F, Larsen NA, Lai Z, Byth KF, Read J, Gu RF, Roth M, Toader D, Saeh JC, Chen H. Mechanism and in vitro pharmacology of TAK1 inhibition by (5Z)-7-Oxozeaenol. ACS chemical biology. 2013b;8:643–650. doi: 10.1021/cb3005897. [DOI] [PubMed] [Google Scholar]
- Xi Q, Wang Z, Zaromytidou AI, Zhang XH, Chow-Tsang LF, Liu JX, Kim H, Barlas A, Manova-Todorova K, Kaartinen V, Studer L, Mark W, Patel DJ, Massague J. A poised chromatin platform for TGF-beta access to master regulators. Cell. 2011;147:1511–1524. doi: 10.1016/j.cell.2011.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Han J, Ito Y, Bringas P, Jr, Deng C, Chai Y. Ectodermal Smad4 and p38 MAPK are functionally redundant in mediating TGF-beta/BMP signaling during tooth and palate development. Dev Cell. 2008;15:322–329. doi: 10.1016/j.devcel.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Han J, Ito Y, Bringas P, Jr, Urata MM, Chai Y. Cell autonomous requirement for Tgfbr2 in the disappearance of medial edge epithelium during palatal fusion. Dev.Biol. 2006;297:238–248. doi: 10.1016/j.ydbio.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Molecular cell. 2008;31:918–924. doi: 10.1016/j.molcel.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LT, Kaartinen V. Tgfb1 expressed in the Tgfb3 locus partially rescues the cleft palate phenotype of Tgfb3 null mutants. Dev.Biol. 2007;312:384–395. doi: 10.1016/j.ydbio.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Li C, Herrera PL, Deng CX. Generation of Smad4/Dpc4 conditional knockout mice. Genesis. 2002;32:80–81. doi: 10.1002/gene.10029. [DOI] [PubMed] [Google Scholar]
- Yumoto K, Thomas PS, Lane J, Matsuzaki K, Inagaki M, Ninomiya-Tsuji J, Scott GJ, Ray MK, Ishii M, Maxson R, Mishina Y, Kaartinen V. TGF-beta-activated kinase 1 (Tak1) mediates agonist-induced Smad activation and linker region phosphorylation in embryonic craniofacial neural crest-derived cells. J Biol Chem. 2013;288:13467–13480. doi: 10.1074/jbc.M112.431775. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.