

Abstract
Background
Autoantibodies to the angiotensin II type 1 receptor (AT1R) have been reported in patients with primary aldosteronism including aldosterone producing adenoma (APA) and idiopathic adrenal hyperplasia (IAH).
Methods and Results
Sera from 25 primary aldosteronism subjects (12 with IAH and 13 with APA) and 15 normotensive control subjects were assayed for AT1R autoantibodies by ELISA and an AT1R-transfected cell-based bioassay. Nine of 12 IAH subjects (75%) and 6 of 13 APA subjects (46%) were positive for AT1R autoantibodies in the bioactivity assay. The mean AT1R autoantibody activity for the IAH and APA subjects was significantly greater than controls (P<0.001 and P<0.01, respectively), and this in vitro activity was suppressed by the AT1R blocker losartan. None of the controls had significant AT1R autoantibody activity. ELISA values were less sensitive but were positive in some subjects with IAH and APA. The mean arterial pressure of these primary aldosteronism subjects correlated modestly with AT1R autoantibody activity.
Conclusion
These data confirm the presence of active AT1R autoantibodies in a high percentage of subjects with primary aldosteronism irrespective of their underlying etiology. These observations have both pathophysiological and clinical implications.
Keywords: aldosterone producing adenoma, idiopathic adrenal hyperplasia, AT1R autoantibodies, hypertension
Introduction
Primary aldosteronism has been characterized by elevated aldosterone production in the face of suppressed plasma renin activity (1). Subsequent studies have demonstrated three major subcategories, aldosterone producing adenoma (APA), idiopathic adrenal hyperplasia (IAH) and now less common forms of adrenal hyperplasia with associated germline genetic mutations (2, 3). In the last three years, numerous groups have reported somatic mutations in the potassium channel gene KCNJ5 that appears to explain in part the mechanism(s) by which these benign tumors grow and over secrete aldosterone (4-6). Recently, Rossitto et al (7) identified the presence of autoantibodies to the angiotensin II (Ang II) type 1 receptor (AT1R) in primary aldosteronism. They used a peptide-based ELISA directed toward the second extracellular loop (ECL2) of the ATIR and noted elevation of these autoantibodies predominantly in their subjects with an APA but not with IAH. They suggested the usefulness of this assay as a non-invasive discriminant for differentiating the two subtypes. Our group had independently recognized the presence of these autoantibodies in subjects with primary aldosteronism (8) and focused on examining their potential for contributing to the pathophysiology of the hyperaldosteronism and hypertension associated with this condition. We reported a high frequency of these autoantibodies in subjects with confirmed primary aldosteronism irrespective of their underlying subtype and demonstrated these autoantibodies actively would cause contraction of resistance arteries in vitro and also stimulate and/or facilitate aldosterone production in HAC15 adrenal cells in vitro (8). These mechanistic studies however were not designed to examine the prevalence of the AT1R autoantibodies in the general population of primary aldosteronism.
In the present study, we report that approximately 60% of subjects with primary aldosteronism, including both APA and IAH as characterized by adrenal venous sampling, harbor autoantibodies that activate the AT1R in transfected cells and this activity has been previously demonstrated to correlate directly with their ability to contract perfused cremaster resistant arteries and to stimulate aldosterone production in HAC15 cells in vitro. We further demonstrate these autoantibodies are uniformly suppressed by the AT1R blocker losartan.
Methods
Study Subjects
Sera from a group of subjects with a history of hypertension, some with occasional hypokalemia and a plasma aldosterone/plasma renin activity (PA/PRA) ratio of >30 were examined in Italy. Their diagnosis of primary aldosteronism was established by an abnormal captopril suppression test [2 hr post captopril plasma aldosterone ≥ 12 ng/dl and/or PA/PRA ratio ≥12] (9). All underwent a serial 2 mm section CT of the adrenals to identify existing nodules. Each of the patients underwent adrenal vein sampling to confirm lateralization (ipsilateral/contralateral adrenal vein plasma aldosterone/cortisol >3/1). Sera from 10 subjects with a diagnosis of APA and 10 subjects with a diagnosis of IAH were selected and sent to our laboratory for analysis (Table 1). Sera from 10 normotensive subjects were sent as control sera. The identity of each sample was blinded to our laboratory until after the assay values were reported to the originating physicians (M.V.C. and F.M.) who then confirmed the pathophysiological subgroup associated with each numerical identifier. Five other primary aldosteronism subjects (2 with IAH and 3 with APA) that were similarly defined as to subgroup analysis in our clinics as well as 5 additional normotensive control subjects were added. This study was approved by the University of Padua and by the OUHSC Institutional Review Board. All subjects provided informed consent.
Table 1. Clinical characteristics of the 25 studied subjects with primary aldosteronism.
| Diagnosis | Age/Sex | MAP | K/Na | PA/PRA (p-CEI) | CT (cm) | Adrenal Vein A/C (I/C) |
|---|---|---|---|---|---|---|
| APA | 50/M | 111 | 3.5/144 | 20.7/0.1 | L 1.0 | R 2.9, L 9.1 (0.3) |
| APA | 39/F | 116 | 3.3/137 | 36.4/0.1 | R 2.0 | R 1.9, L 8.2 (0.2) |
| APA | 38/M | 127 | 3.0/142 | 22.8/0.21 | L 2.5 | R 1.22, L 8 (0.15) |
| APA | 64/F | 112 | 3.8/142 | 48/0.33 | R 1.0 | R 0.26, L 10.67 (0.02) |
| APA | 43/F | 133 | 2.4/145 | 22.6/0.1 | L 1.5 | R 11.2, L 1.53 (7.32) |
| APA | 51/M | 106 | 4.1/136 | 20/0.1 | L 1.0 | R 1.48, L 0.5 (2.96) |
| APA | 69/M | 126 | 2.7/142 | 34.4/0.51 | R 1.0 | R 7.1, L 0.3 (23.6) |
| APA | 72/M | 106 | 3.2/144 | 41.2/0.59 | L 1.5 | R 4.25, L 0.26 (16.3) |
| APA | 45/F | 117 | 3.3/138 | 52.3/1.8 | R 2.0 | R 0.02, L 0.08 (0.25) |
| APA | 61/M | 101 | 4.0/139 | 34.5/1.1 | R 1.0 | R 17.6, L 5 (3.5) |
| APA | 56/F | 92 | 2.7/142 | 21/0.2 p-sal | L 2.0 | L 23, R 2 (11.5) |
| APA | 32/M | 134 | 2.9/142 | 27/1.6 | L 2.5 | Not done |
| APA | 53/M | 118 | 3.2/139 | 13.5/1.1 | L 2.2 | L 3.9, R 0.6 (6.5) |
| IAH | 47/F | 125 | 4.0/138 | 35.5/0.2 | Bil hyper | R 4, L 72 (0.05) |
| IAH | 66/F | 120 | 3.3/140 | 20/0.1 | Bil hyper | R 5.9, L 18.6 (0.31) |
| IAH | 75/M | 117 | 3.5/142 | 35/0.6 | L 1.0 | R 3.3, L 2.6 (1.26) |
| IAH | 57/M | 117 | 3.9/143 | 28.8/0.24 | L 0.9, R 1.0 | R 0.07, L 0.4 (0.17) |
| IAH | 64/M | 110 | 3.5/144 | 36.6/0.29 | Bil hyper | R 145.5, L 2.8 (52) |
| IAH | 46/M | 133 | 2.4/145 | 49/0.21 | L 1.4, R hyper | R 1.7, L 41.6 (0.04) |
| IAH | 50/F | 103 | 3.4/139 | 21.6/0.6 | R 1.2, L hyper | R 0.3, L 0.9 (0.3) |
| IAH | 52/F | 103 | 3.3/146 | 48/0.1 | R 1.5, L 2 | R 2.6, L 0.3 (8.6) |
| IAH | 47/M | 117 | 2.9/138 | 39.4/0.8 | Bil hyper | R 0.36, L 1.72 (0.21) |
| IAH | 70/M | 100 | 3.7/144 | 30.7/1.1 | L 1.0, R 1.5 | R 0.8, L 0.9 (0.89) |
| IAH | 79/M | 107 | 2.9/132 | 11.8/0.15 | L 0.8 (Nod hyper) | L 3.1, R 1.3 (2.4) |
| IAH | 61/M | 96 | 3.4/144 | 40/0.15 | L 1.5 | R3.3, L 1.3 (2.5) |
MAP, mean arterial pressure; PA/PRA, plasma aldosterone/plasma renin activity ratio; p-CEI, post converting enzyme inhibitor; p-Sal, post saline infusion; CT, CAT scan of adrenal; cm, size of adrenal adenoma; L, left; R, right; hyper, hyperplasia; bil, bilateral; nod, nodular; A/C, aldosterone/cortisol ratio; I/C, ratio of the ipsilateral and contralateral adrenal vein A/C ratios
ELISA
ELISA assays to screen for AT1R autoantibodies were performed as previously described (8). Briefly, microtiter plates were coated with a multiple antigenic peptide (Pi Proteomics) containing the amino acids AFHYESQ, a sequence from the AT1R ECL2 identified as the functional epitope of AT1R-activating autoantibodies (10). Sera were diluted 1:100, and goat anti-human IgG conjugated with alkaline phosphatase and its substrate para-nitrophenyl-phosphate 104 were used to detect antibody binding. The optical density (OD) values were read at 405 nm at 60 min.
Cell-Based AT1R Assay
Ang II-like autoantibody activity was measured in AT1R-transfected Chinese hamster ovary (CHO) cells using β-arrestin activation as the parameter (DiscoveRx, Fremont, CA) as previously described (8). Negative (buffer) and positive Ang II controls were included in each assay. Serum samples (1:100) were run in the absence and presence of the orthosteric AT1R blocker losartan (10 mM). The β-arrestin recruitment levels by the sera were expressed as Ang II “equivalent” values. These were obtained by running a concurrent Ang II standard curve and comparing serum sample relative luminescence units to those on the standard curve. The intra-assay and inter-assay coefficient of variation were 6.1% (n=9) and 9.8% (n=9), respectively.
Statistics
Data are expressed as mean±SEM. Student's t test and χ2 test were used for comparison of age and gender between patient and normotensive control groups. Autoantibody positivity by ELISA was defined as OD values above the mean + 2SD from the control group. The positivity of bioactive autoantibodies was defined as bioactivity values above the mean + 2SD from the control group. Spearman rank correlation was performed to examine the relationship between ELISA and bioactivity values and also the relationship between the mean arterial pressure of each patient and their serum autoantibody bioactivity. Comparison between patient and normotensive control groups was performed using the nonparametric Mann-Whitney test. A P value of <0.05 was considered statistically significant.
Results
Patient Characteristics
Twenty five subjects with primary aldosteronism and 15 normotensive control subjects were included in the study. The clinical data of the primary aldosteronism subjects are shown in Table 1. The primary aldosteronism patients were older than the normotensive controls (55±12 years vs. 36±13 years, P<0.001). There was no significant difference in gender between the primary aldosteronism and control groups (female/male: 9/16 vs. 6/9, P=1.0, Fisher's exact test).
Autoantibody Screening by ELISA
Sera were examined by ELISA for autoantibodies directed toward the ECL2 of AT1R. Five of 12 (42%) of the IAH subjects, 3 of 13 (23%) of the APA subjects and 1 of 15 (7%) of the normotensive controls were positive for AT1R autoantibodies (Figure 1). Overall 32% (8 of 25) of the primary aldosteronism patients showed antibody positivity.
Figure 1. ELISA detection of AT1R autoantibodies in primary aldosteronism patients and normotensive control subjects.
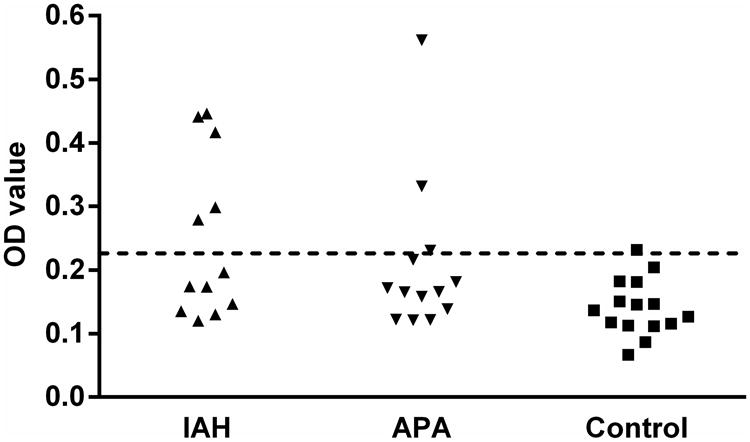
IAH, idiopathic adrenal hyperplasia (n=12), closed upward triangles; APA, aldosterone producing adenoma (n=13), closed downward triangles; control, normotensive control (n=15), closed squares. The dashed line is the threshold derived from the mean optical density (OD) values + 2SD of the normotensive controls.
Autoantibody Activation of AT1R
Sera from each patient and control subject were assayed for the ability to activate AT1R in transfected CHO cells in vitro. Nine of 12 IAH subjects (75%) and 6 of 13 APA subjects (46%) were positive for AT1R autoantibodies in this bioactivity assay (Figure 2). AT1R autoantibody activity was observed in ELISA-negative patient samples. Overall 60% (15 of 25) of the primary aldosteronism patients showed positivity for active AT1R autoantibodies. None of the controls had significant AT1R autoantibody activity.
Figure 2. The effect of sera from each primary aldosteronism patient and normotensive control subject on AT1R activation in transfected Chinese hamster ovary (CHO) cells.
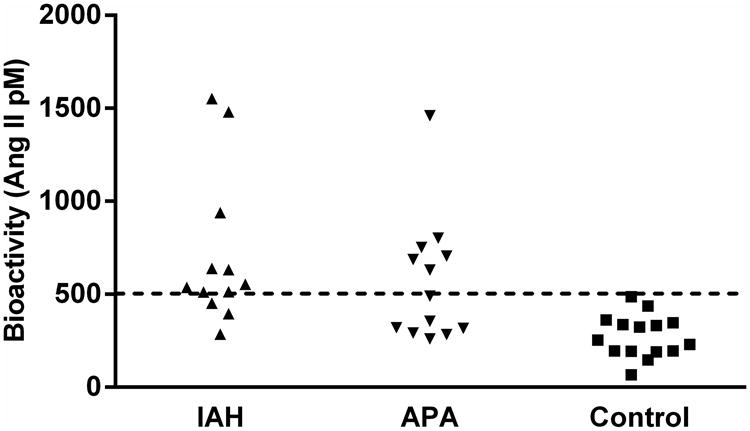
IAH (n=12), closed upward triangles; APA (n=13), closed downward triangles; control (n=15), closed squares. Data are expressed in Ang II equivalent dosages (mean±SEM). The dashed line is the threshold derived from the mean values + 2SD of the normotensive controls.
The mean AT1R autoantibody activity for the IAH and APA groups was significantly greater than the controls (708±118 and 566±93 vs. control: 273±29 in pM Ang II equivalent activity; P<0.001 and P<0.01, respectively) (Figure 3). No significant difference in antibody activity was observed between the IAH and APA groups (P=0.35). The AT1R blocker losartan (10 mM) markedly suppressed patient sera-induced AT1R activation to control levels.
Figure 3. The mean effect of sera from primary aldosteronism patients and normotensive control subjects in the absence and presence of AT1R blockade on AT1R activation in transfected CHO cells.
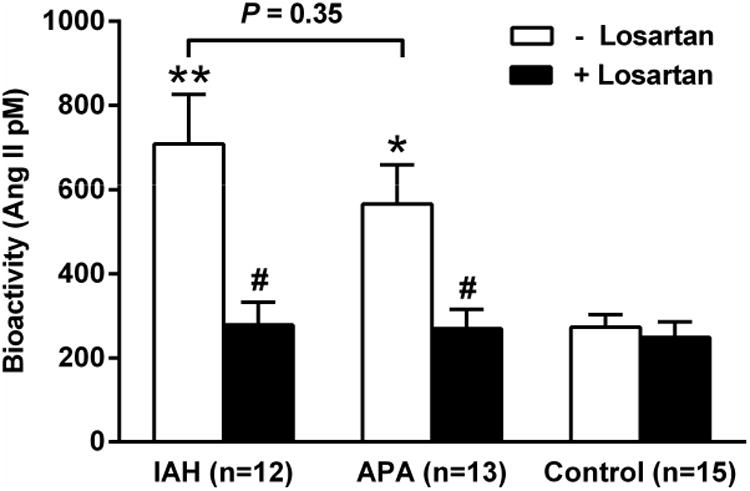
Both IAH and APA had a highly significant increase of AT1R activity compared to the normotensive controls (*P<0.01, **P<0.001). There was no significant difference in AT1R activity between the two primary aldosteronism groups. The AT1R blocker losartan (10 μM) effectively blocked the autoantibody activity in each group of the primary aldosteronism patients (#P<0.01). Although the activity in IAH and APA dropped significantly with losartan, there was no significant change in the low level of activity in the control subjects.
We have plotted the correlation of the individual primary aldosteronism subjects with their bioactivity measured by the receptor activation assay compared to their ELISA OD values in Figure 4. Although individuals with elevated ELISA values more frequently had significant bioactivity, others with low ELISA values also showed increased antibody activity and the Spearman correlation coefficient failed to reach significance. There was a significant correlation of the mean arterial pressure for each primary aldosteronism patient with their serum autoantibody bioactivity (r=0.43, P=0.03) as shown in Figure 5.
Figure 4. Correlation analysis between ELISA and cell-based bioassay for AT1R autoantibodies.
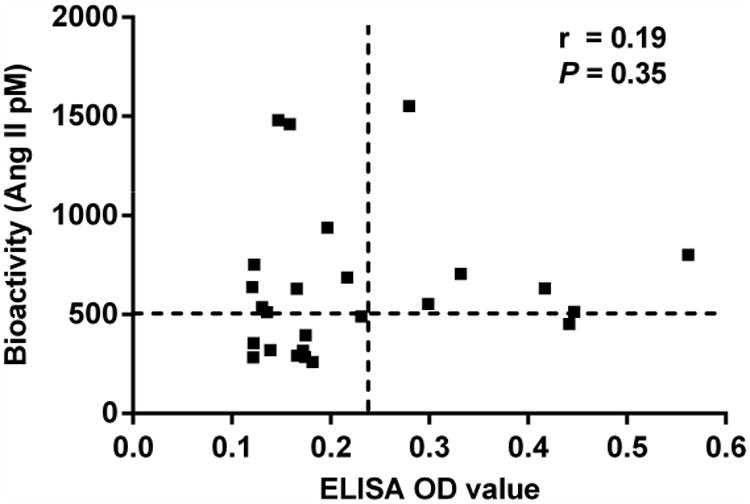
The vertical and horizontal dashed lines represent the cut-off values (mean + 2SD of controls) for ELISA and bioassay positivity, respectively. No significant correlation was detected between ELISA OD values and bioactivity values.
Figure 5. Correlation analysis between patient mean arterial pressure (MAP) and AT1R antibody activity measured in AT1R-trasnfected CHO cells.
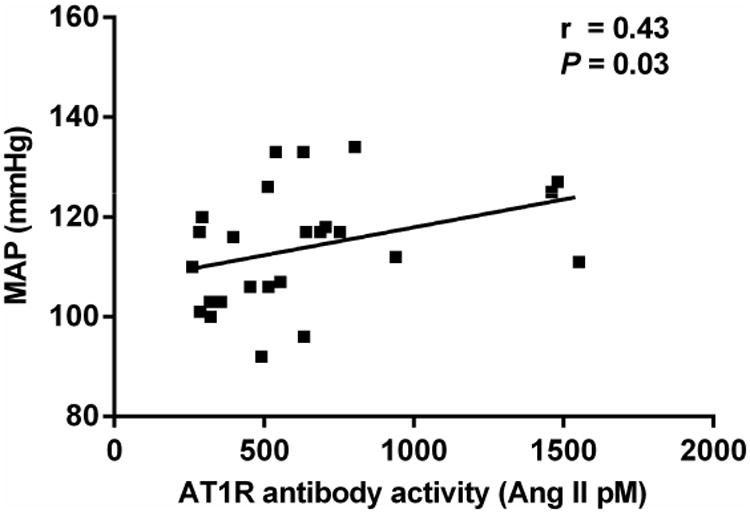
MAP was computed from blood pressures taken at the time of bilateral adrenal vein sampling. There was a significant correlation between the patient MAP and their concurrent serum AT1R antibody activity.
Discussion
The etiology and pathophysiology of primary aldosteronism and its subtypes has long been puzzling and a challenge. Recently, a number of studies have explored the presence of several related mutations in the KCNJ5 gene controlling K access to the adrenal glomerulosa cells (4-6). These studies suggest up to 50% of the adenomas are related to the presence of one or more mutations leading to hypertrophy and aldosterone overproduction by these cells. These studies however fail to fully establish the genesis of the hypertension associated with their presence and do not address the genesis of aldosterone hypersecretion in the more prevalent IAH subtype. The presence of autoantibodies directed toward the AT1R in patients with primary aldosteronism has been established independently by two groups and raise several issues relevant to the diagnosis and pathophysiology of primary aldosteronism (7, 8). Rossitto et al used an ELISA-based assay to demonstrate the presence of autoantibodies predominantly in subjects harboring an APA and a markedly lower occurrence in IAH. They were unable to determine the potential role of these autoantibodies in the pathophysiology of the hypertension since the ELISA technique inherently fails to demonstrate any potential activity of the autoantibodies on the vasculature or adrenal aldosterone secretion (8). We have used three different bioactivity assays and demonstrated both serum and IgG purified from the serum from a small group of subjects including those with either APA and/or IAH contained AT1R autoantibodies capable of: 1) contracting small resistance arteries in vitro; 2) directly stimulated aldosterone production in HAC15 adrenal cells in vitro and 3) facilitated the effects of low dosages of Ang II on these same cells when co-incubated. These in vitro effects were all blocked by the AT1R blockers including losartan and candesartan (8). In vivo mechanistic studies have been performed in AT1R ECL2 target epitope-immunized rabbits (11). These rabbits became markedly hypertensive six weeks after immunization with elevated autoantibody titers. This elevated blood pressure returned to nearly normal within 90 minutes after intravenous infusion of a decoy peptide directed toward the AT1R ECL2 target epitope of the autoantibodies. While it is premature to claim direct evidence these active autoantibodies cause hypertension in man, it is of interest that the present study demonstrates a modest but significant correlation of the mean arterial pressure and AT1R antibody activity in the sera of the primary aldosteronism patients. It is already accepted that AT1R-activating autoantibodies activate the receptor through allosteric changes and mimic the contractile and endocrine functions of Ang II (8). These studies provide a mechanistic basis for the association of the activating autoantibodies and the elevated blood pressures observed herein.
These studies however fail to address the prevalence of these active autoantibodies. The ELISA assay using linear peptides associated with the targeted AT1R extracellular loop are relatively insensitive in our laboratory compared to the studies using the intact receptor and assessing its bioactivity (8). As expected, the correlation of antibody bioactivity with the ELISA data overall was not significant. In addition, studies of prevalence between the two major primary aldosteronism subgroups now require the use of bilateral adrenal venous sampling to better differentiate the two groups since nodularity and coexistent hyperplasia are common (12, 13). Even with this expensive and invasive testing, a final diagnostic division is not possible without surgical confirmation and subsequent biological testing. Residual hypertension following surgery in up to 50% of APA subjects further complicates this differentiation. Moreover, improvements in medical therapy have lessened the need for adrenal venous catheterization in our hospital as well as many others.
To better answer the question of prevalence, we have collaborated with a group in Padua wherein relatively complete evaluation of primary aldosteronism subjects is routinely performed. They agreed to provide an initial cohort of 20 subjects with primary aldosteronism including 10 with an apparent APA and 10 whose diagnosis of IAH was established by CT and a non-lateralizing adrenal venous sampling procedure. Ten normotensive control subjects were also included. Our laboratory was completely blinded to the groups and diagnoses of the 30 subjects until the assays were performed and reported to these investigators. Five additional primary aldosteronism subjects (2 with IAH and 3 with APA) and 5 additional normotensive control subjects from our clinics were added to the study. These data suggest that up to 60% of the primary aldosteronism subjects demonstrated active autoantibodies that were measured using the AT1R-transfected CHO cells in vitro. These values are lower than observed in the smaller study previously reported by our laboratory but these subjects were not all characterized by their subtype (8). There was a significant difference in age between the overall primary aldosteronism and control groups. However, it is unlikely this affected interpretation of the data since a Spearman correlation test of age and antibody bioactivity did not show a significant correlation (r=0.17, P=0.42).
The present study demonstrates that a significant number of primary aldosteronism subjects harbor AT1R-activating autoantibodies and the activity of sera containing these activating autoantibodies is markedly suppressible by AT1R blockade treatment in vitro. There are several consequences to this discovery. It will be important to broaden the number of subjects to provide a more accurate estimate of their presence in primary aldosteronism. It will also be important to determine the effect of AT1R blockade therapy on the hallmark overproduction of aldosterone in these subjects. This impact may be quite different from that by converting enzyme inhibitors or direct renin inhibitors. Since AT1R autoantibodies appear to facilitate the effects of low concentrations of Ang II in primary aldosteronism (8), this may explain a partial impact of converting enzyme inhibitors on aldosterone values reported by Rossitto et al (7). We have not determined the mutational status of the adrenal adenoma tissues in the current group of APA subjects so cannot comment on any possible relationships of AT1R autoantibodies with these mutational changes. Confirmation of the impact of these activating autoantibodies on the current and residual elevated post-operative blood pressures observed in the large number of patients whose APA has been resected will also be of interest. The role of AT1R-activating autoantibodies in patients with hypertension of unknown etiology has not been systematically studied. Isolated studies have suggested a small number of subjects harbor these and other activating autoantibodies. Based on the known effects of the AT1R autoantibodies in vivo and in vitro (8, 11), we have hypothesized that patients with low-renin essential hypertension may harbor such autoantibodies at a higher percentage. We are currently carrying out such a study at our institution.
AT1R autoantibodies are frequently present in primary aldosteronism
These autoantibodies activated AT1R in vitro and were blocked by an AT1R blocker
The mean arterial blood pressure correlated with AT1R autoantibody activity
AT1R autoantibodies have sufficient activity to produce and facilitate hypertension
Acknowledgments
This work was supported by funding from a VA Merit Review grant (D.C.K. and X.Y.), NIH HL056267 (M.W.C. and D.C.K), an American Heart Association Postdoctoral Fellowship (H.L.) and individual grant support from Will and Helen Webster.
Footnotes
Presented in part at the 40th Meeting of the International Aldosterone Conference, June 2014, Chicago, IL
Conflict of interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Conn JW, Louis LH. Primary aldosteronism, a new clinical entity. Ann Intern Med. 1956;44:1–15. doi: 10.7326/0003-4819-44-1-1. [DOI] [PubMed] [Google Scholar]
- 2.Funder JW. The genetic basis of primary aldosteronism. Curr Hypertens Rep. 2012;14:120–4. doi: 10.1007/s11906-012-0255-x. [DOI] [PubMed] [Google Scholar]
- 3.Al-Salameh A, Cohen R, Desailloud R. Overview of the genetic determinants of primary aldosteronism. Appl Clin Genet. 2014;7:67–79. doi: 10.2147/TACG.S45620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi M, Scholl UI, Yue P, Bjorklund P, Zhao B, Nelson-Williams C, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science. 2011;331:768–72. doi: 10.1126/science.1198785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azizan EA, Murthy M, Stowasser M, Gordon R, Kowalski B, Xu S, et al. Somatic mutations affecting the selectivity filter of KCNJ5 are frequent in 2 large unselected collections of adrenal aldosteronomas. Hypertension. 2012;59:587–91. doi: 10.1161/HYPERTENSIONAHA.111.186239. [DOI] [PubMed] [Google Scholar]
- 6.Oki K, Plonczynski MW, Luis Lam M, Gomez-Sanchez EP, Gomez-Sanchez CE. Potassium channel mutant KCNJ5 T158A expression in HAC-15 cells increases aldosterone synthesis. Endocrinology. 2012;153:1774–82. doi: 10.1210/en.2011-1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rossitto G, Regolisti G, Rossi E, Negro A, Nicoli D, Casali B, et al. Elevation of angiotensin-II type-1-receptor autoantibodies titer in primary aldosteronism as a result of aldosterone-producing adenoma. Hypertension. 2013;61:526–33. doi: 10.1161/HYPERTENSIONAHA.112.202945. [DOI] [PubMed] [Google Scholar]
- 8.Kem DC, Li H, Velarde-Miranda C, Liles C, Vanderlinde-Wood M, Galloway A, et al. Autoimmune mechanisms activating the angiotensin AT1 receptor in ‘primary’ aldosteronism. J Clin Endocrinol Metab. 2014;99:1790–7. doi: 10.1210/jc.2013-3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lyons DF, Kem DC, Brown RD, Hanson CS, Carollo ML. Single dose captopril as a diagnostic test for primary aldosteronism. J Clin Endocrinol Metab. 1983;57:892–6. doi: 10.1210/jcem-57-5-892. [DOI] [PubMed] [Google Scholar]
- 10.Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest. 1999;103:945–52. doi: 10.1172/JCI4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kem DC, Li H, Yu X, Benbrook A, Liles DC, Velarde-Miranda C, et al. Blockade of AT1R-activating autoantibodies using a new retro-inverso decoy peptide. Endocr Rev. 2014;35(03_MeetingAbstracts):PP14–4. [Google Scholar]
- 12.Rossi GP. Prevalence and diagnosis of primary aldosteronism. Curr Hypertens Rep. 2010;12:342–8. doi: 10.1007/s11906-010-0134-2. [DOI] [PubMed] [Google Scholar]
- 13.Rossi GP, Auchus RJ, Brown M, Lenders JW, Naruse M, Plouin PF, et al. An expert consensus statement on use of adrenal vein sampling for the subtyping of primary aldosteronism. Hypertension. 2014;63:151–60. doi: 10.1161/HYPERTENSIONAHA.113.02097. [DOI] [PubMed] [Google Scholar]