

Abstract
L-DOPA replacement therapy has long provided the most effective treatment for Parkinson’s disease. We review how this dopamine precursor enhances dopaminergic transmission by providing a greater sphere of neurotransmitter influence, due to the confluence of increased quantal size and decreased dopamine reuptake, as well as loading dopamine as a false transmitter into surviving serotonin neuron synaptic vesicles. We further review literature on how presynaptic dysregulation of DA release following L-DOPA might trigger dyskinesias in Parkinson patients.
Since its introduction 1,2, L-DOPA has provided effective treatment for Parkinson’s disease (PD) by replacing dopamine (DA) neurotransmission following the death of substantia nigra (SN) neurons. This therapy however leads to motor side effects, L-DOPA-induced dyskinesias (LIDs), limiting the utility of the drug. Here we review literature on DA neurotransmission in healthy and DA depleted striata and on presynaptic mechanisms that may underlie the development of LIDs.
Dopamine neurotransmission
In SN neurons, DA is normally synthesized from tyrosine in a two step enzymatic reaction (Fig 1A). First, tyrosine hydroxylase (TH) attaches a hydroxyl group to tyrosine using oxygen, tetrahydrobiopterin and Fe2+ as cofactors 3 to produce L-DOPA. Next, aromatic L-amino acid decarboxylase (AADC) converts L-DOPA to DA using pyridoxal phosphate as a cofactor. Both TH and AADC are regulated by DA D2 autoreceptor-mediated second messenger systems, with enzyme activities increased by receptor antagonists and decreased by agonists 4-6. Together, these homeostatic responses decrease DA synthesis when extracellular DA is increased.
Figure 1.
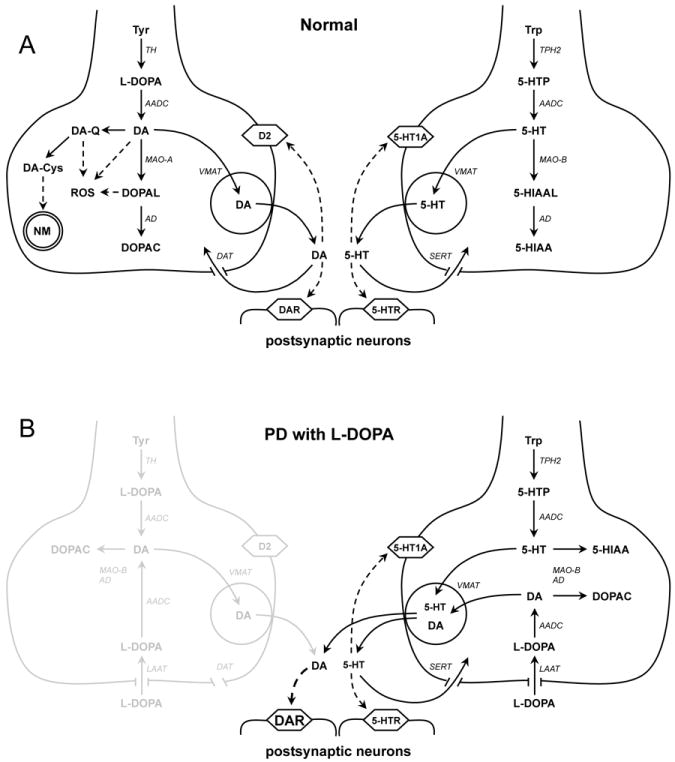
DA and 5-HT homeostasis in normal conditions (A) and Parkinson’s disease (B). (A) Schematics of DA (left) and 5-HT (right) synthesis, degradation and neurotransmission. There are substantial similarities between DA and 5-HT metabolism, including the presence of AADC, MAO and AD in the cytosol and VMAT2 on the synaptic vesicle membrane. Note that some metabolic pathways, such as neuromelanin synthesis, are more prominent in the cell bodies than in the terminals of DA neurons. (B) Exogenous L-DOPA can be taken up by the L-amino acid transporter (LAAT) and metabolized to DA in both types of neurons. In a healthy brain, very high levels of L-DOPA are required to induce dyskinesia 114. In earlier stages of PD, DA release from surviving dopaminergic terminals is augmented by exogenous L-DOPA, which provides the therapeutic benefits of the drug, while decreased DAT activity provides a larger sphere of influence. It may be that at high doses, the system is flooded with DA released from 5-HT neurons, triggering dyskinesias. A dose of L-DOPA that gives symptomatic relief without causing motor side effects provides a “therapeutic window” of DA replacement therapy. In later stages of PD, severe loss of dopaminergic terminals reduces the therapeutic window and striatal DA levels become less dependent on the activity of DA neurons and more on the availability of exogenous L-DOPA.
The cytosolic DA is loaded into synaptic vesicles to provide neurotransmission. The neuronal synaptic vesicle monoamine transporter (VMAT2) 7-9 translocates DA using energy generated by the vesicular proton pump, V-type H+-ATPase, exchanging two intravesicular protons for each cytoplasmic DA molecule. The acidic vesicular lumen prevents DA auto-oxidation and degradation by cytosolic enzymes, allowing accumulation of very high (up to molar) intravesicular transmitter concentrations for extrasynaptic release by stimulation-dependent exocytosis 10,11. VMAT2 translocates many compounds, and in addition to native transmitters - DA, norepinephrine, epinephrine, serotonin and histamine - can transport a diverse range of synthetic substrates 10,12.
An important attribute of DA and other monoamine neurotransmitter synapses is that the neurotransmitter overflows far past its release site to interact with multiple synapses, a form of volume transmission 13 labeled social neurotransmission 14. That social nature of striatal DA transmission is evident from electrochemical measurements – the probes used for microdialysis and voltammetry are thousands of times too large to be within synapses and must be measuring extrasynaptic DA.
In striatum, action of DA is terminated by diffusion and reuptake by the dopamine uptake transporter (DAT), which is fairly evenly distributed on the membrane surface of DA fibers rather than at release sites 15,16. DAT is very efficient at removing DA from the extracellular milieu, as demonstrated in striatal slices prepared from DAT-deficient mice where stimulation-evoked DA peaks exhibit a 300-fold longer duration 17; similar responses are measured following treatment with DAT antagonists, such as cocaine 18.
Midbrain DA neurons are tonically active and demonstrate autonomous pacemaking at ~4Hz frequency. In response to stimuli, which coincides with primary reinforcers such as food or environmental cues predicting reward, DA neurons respond with bursts of action potentials (phasic activity) in which the firing frequency reaches ~15Hz. Most striatal DA release sites are positioned near the neck of dendritic spines of the medium spiny neurons 19. Phasic activity, in part by saturating DAT reuptake, provides DA levels sufficient to activate pre- and post-synaptic DA receptors on a broad range of cells, including medium spiny neurons, cholinergic and GABAergic interneurons and corticostriatal synapses, 20; the majority of these receptors is found in perisynaptic zones distant from the DA release site.
Effects of L-DOPA on quantal size in DA neurons
Exogenous L-DOPA provided as a drug is accumulated by the L-amino acid transporter (LAAT) into neuronal cytosol 21. As AADC is typically not saturated, L-DOPA is efficiently converted to DA. In cultured SN neurons and the PC12 dopaminergic cell line, L-DOPA treatment rapidly increases DA levels in the cytosol by >100-fold 22,23.
The effects of L-DOPA on dopaminergic neurotransmission was extensively studied in cultured cells using amperometric recordings to provide a direct measurement of DA molecules released during single synaptic vesicle fusion 24, a parameter known as “quantal size” 14. In primary cultures of murine SNc neurons, L-DOPA produces a ~300% increase in quantal size (from 3,000 to 10,000 DA molecules) in only 30 minutes 25,26. Similar increase in quantal size occurs in large dense cored vesicles in chromaffin cells 27 and PC12 cells 28,29.
The ability of L-DOPA to increase the loading of DA into synaptic vesicles in living animals was recently confirmed using a technique known as electrochemical cytometry, in which synaptic vesicles are isolated from brain and the quantity of DA molecules is measured inside individual synaptic vesicles 30. In healthy mice, L-DOPA (50 mg/kg injected intraperitoneally 2 hours prior to sacrifice) increases vesicular DA storage by 240% (from 30,000 to 71,000 molecules). Thus, even in intact DA axons, L-DOPA can be transported into cytosol and converted to DA that is accumulated into synaptic vesicles. A surprising consequence of L-DOPA-mediated increase in vesicular DA content first noted by Andrew Ewing and colleagues is that it is accompanied by an increase in the volume of secretory vesicles 29. While this was initially controversial, as it was not clear that vesicle membrane would have sufficient elasticity, it has been confirmed by capacitance recordings in cultured cells 27 and optical measurements of vesicles isolated from animals treated with L-DOPA 31; it is possible that the increased volume is due to the fusion of additional membrane with the vesicles.
How does L-DOPA normalize basal ganglia function in PD?
These findings beg a central issue in L-DOPA therapy: as striatal DA axons degenerate in PD and only a small number of DA release sites remain that can be refilled, how can L-DOPA correct basal ganglia function? The answer stems in large part from the properties of social neurotransmission.
The motor symptoms of PD appear when ~30% of the pigmented neurons of the SNc has degenerated, and striatal terminal density is decreased by 50-70% (reviewed by 32). Similarly, in 6-OHDA lesion rat model of PD, motor function and striatal DA release is unchanged until >60% of the DAT immunoreactivity is lost and tissue DA levels are decreased by >80% 33,34. Thus, the degenerating DA system is able to maintain sufficient DA transmission to preserve motor functions until relatively late stages of the disease. This remarkable property is mostly due to two adaptive changes in presynaptic function of the spared DA terminals. First, diminished extracellular DA and reduced activation of DA autoreceptors stimulates DA synthesis 35 which may enhance DA release from the remaining terminals. Second, the degeneration of DA axons also produce a comparable loss of DAT activity, which results in a larger volume of transmission, so that extrasynaptic DA receptors are activated further from release sites 20.
These effects of L-DOPA and DAT depletion on striatal DA neurotansmission can be roughly estimated with diffusion models. Calculations by Margaret Rice and Stephanie Cragg indicate that an increase in quantal size corresponds to a commensurate increase in the sphere of influence of transmitter response, and so the increased quantal size from L-DOPA alone would roughly increase the volume that a DA release event affects by 3 or 4-fold 36. Consistently, Figure 2A shows a random walk simulation of the diffusional profile of DA following single vesicle release within the striatum 14,26. An increase in quantal size from 3,000 to 10,000 molecules exerts proportional effect on the amplitude of extrasynaptic DA peak and increases the distance at which transmitter concentration is above 10 nM - the EC50 of the activation of D2 DA receptors - from 12 to 18 μm, which corresponds to a ~3.5-fold increase of the sphere of DA influence, from 7,200 μm3 to 24,400 μm3.
Figure 2.
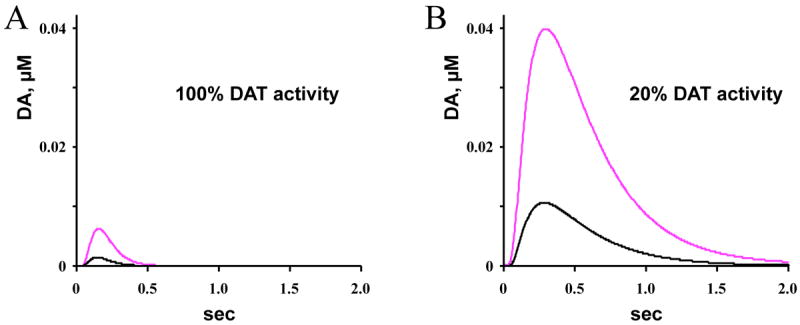
Random walk simulations of DA overflow from a quantal release event under normal (5 μM/sec) and PD-like (1 μM/sec) levels of DAT activity. The traces represent diffusional profiles of DA concentrations 20 μm from the presynaptic release site following transmitter exocytosis from a synaptic vesicle containing 3,000 (black) or 10,000 molecules (magenta), corresponding to L-DOPA’s increase of quantal size in cultured midbrain neurons 25,26. The “spheres of influence”, i.e., the volume around the release sites where DA concentrations are > 10 nM, were as follows. A: 3,000 molecules, r = 12 μm, 7,200 μm3 (100 %); 10,000 molecules, r = 18 μm, 24,400 μm3 (338 %). B: 3,000 molecules, r = 21 μm, 38,800 μm3 (536 %); 10,000 molecules, r = 32 μm, 137,300 μm3 (1896 %). Tutorials on this random walk analysis and spreadsheets with the simulation can be downloaded from http://sulzerlab.org/download.html.
In the PD patient, however, DA overflow is greatly enhanced due to the decreased DAT levels. The maximal velocity of DAT in normal striatum in mice is ~ 5 μM/sec 37 and would decrease by 80% following degeneration of striatal DA terminals in symptomatic PD. A random walk simulation indicates that such diminished DAT activity dramatically increases both the amplitude and the duration of extrasynaptic DA (Figure 2B). In pre-symptomatic PD, this may account for the ability of the remaining more sparsely distributed DA terminals to “cover” larger areas of the striatum, compensating for the loss of a large portion of SNc axons. In later stages of PD, the levels of DA overflow following L-DOPA treatment exceed those in unperturbed striatum and the overall increase in the sphere of influence due to the combination of increased quantal size and decreased DAT levels may be 20-fold (to 137,300 μm3).
The simulations above do not take into account the presence of other transport mechanisms, including SERT, OCT-3, PMAT and others 41-43 that may in part compensate for the loss of striatal DAT leading to DA accumulation in non-catecholaminergic cells. L-DOPA-mediated neurotransmission will moreover be affected differently in healthy and PD brains by the presence of D2-mediated auto-inhibition of DA terminals 44. As detailed below, in later PD it appears that the majority of L-DOPA conversion to DA, vesicle uptake, and release are likely occurring in striatal serotonin (5-HT) axons. D2-receptor mediated regulation effectively inhibits DA release from intact DA axons, but not from 5-HT terminals that lack D2 receptors. Thus, D2 autoinhibition would limit the effect of L-DOPA in a normal striatum, but would have little effect in the DA-depleted brain (Figure 1).
Overall, these changes in the volume of neurotransmission may help explain why L-DOPA has minimal effect on striatal extracellular DA levels in normal animals but increases it dramatically in animals with severe DA lesion 33,38,39. Similarly, L-DOPA has little or no effect in healthy people and is not abused as are other drugs that enhance extracellular DA in intact striatum, but is sometimes consumed compulsively by PD patients 40.
Neuronal stress due to increased cytosolic DA
There has long been a controversy on whether L-DOPA treatment may enhance neurodegeneration. Metabolic turnover of DA is tightly regulated by its synthesis, degradation and compartmentalization and high cytosolic DA levels following L-DOPA treatment have been shown to induce the formation of neuromelanin 47, a pigment derived from the oxidized metabolites of DA and localized to autophagic vacuoles 48. L-DOPA also promotes alpha-synuclein-dependent neurodegeneration 22 and antigen presentation by SN neurons in culture 49. We recently found that AADC activity is dramatically enhanced by Ca2+ 22, creating much higher cytosolic DA levels and oxidative stress in L-DOPA-treated neurons that rely on Ca2+ - driven pacemaking, i.e., the SNc and LC neurons 50,51, which are compromised in PD. Additionally, non-catecholaminergic cells, including serotonergic and glutamatergic neurons, exposed to L-DOPA accumulate cytosolic DA and undergo degeneration as a result 22,52. These studies, however, have been performed on cultured DA neurons with high L-DOPA exposures intended to greatly increase cytosolic DA, and there is no clear evidence at this time that similar neurotoxicity occurs with doses of L-DOPA used in patients.
More broadly, the selective vulnerability of neuromelanin-containing neurons and the ability of cytosolic DA to produce oxidative stress and protein damage have long suggested that a dysregulation of DA homeostasis may play a role in PD 37,53-58. Following cleavage by MAO, DA generates the highly reactive compound DOPAL 59 the presence of which has been demonstrated both in vitro and in vivo 60,61. The cytosolic catechols, DA, L-DOPA and DOPAC, can auto-oxidize and further react with cysteine residues of proteins and glutathione. 5-S-cysteine conjugated catechol derivatives are often used as markers of excess cytosolic DA and oxidative stress in vivo 62,63. Increased loading of DA from cytosol to vesicles following overexpression of VMAT2 can provide neuroprotection from L-DOPA 22 and other interventions that increase cytosolic DA, such as exposure to MPTP or methamphetamine 58,64,65. A recent report suggests that PD patients have low levels of VMAT2 on striatal DA neuron synaptic vesicles 57, although as these patients were almost certainly treated with L-DOPA, a decrease in VMAT expression could be a compensatory response rather than a cause of PD.
L-DOPA-induced dyskinesia (LIDs)
By the 5th year of L-DOPA treatment, more than 50% of patients develop debilitating motor side-effects, collectively defined here as L-DOPA induced dyskinesia (LID) 66,67. Several forms of LID have been characterized. “Peak-dose” LID is associated with high plasma L-DOPA concentrations and is expressed as involuntary spasmodic twitching or jerking in muscle (choreic movements). “Diphasic on/off” dyskinesias coincide with rising and decreasing plasma concentrations of L-DOPA and may include both chorea and dystonia (uncontrollable repetitive or twisting movements). “Off” dyskinesia presents during unmedicated states with low L-DOPA and is characterized by an often painful dystonic posture. There is a gradual increase in incidence and severity of LIDs during prolonged therapy, and by the 10th year of L-DOPA medication, LID is experienced by ~ 80% of the patients 68. The risk factors for LID include the age of PD onset, duration of L-DOPA treatment and dose, indicating that both the progressive loss of DA neurons and L-DOPA exposure play roles in the establishment of LID 69-71. Consistently, several studies report decreased LIDs with continuous rather than intermittent exposure to L-DOPA (reviewed in 72), and a recent clinical study reported that continuous delivery of L-DOPA using an intestinal gel increased on-time without dyskinesia more than immediate-release oral L-DOPA 73.
Several animal models of “peak-dose” LID have been developed based on lesion of SNc DA neurons by MPTP or 6-OHDA followed by repeated administration of L-DOPA or synthetic DA receptor agonists 74. These studies suggest a wide spectrum of metabolic and neuroadaptive changes at multiple synaptic connections of the basal ganglia 75,76. Hypotheses on the cause of LID can be divided into two non-exclusive categories 69. Postsynaptic hypotheses ascribe LID to the changes in DA receptor sensitivity in denervated striatum, which may occur in virtually all striatal neurons. These alterations entail a dysregulation of signaling cascades activated by DA receptors, and consequent effects on gene transcription, protein translation, and post-translational modifications in striatal neurons that are beyond the scope of this review [see 77 for a recent review]. A new study by Paul Greengard and colleagues demonstrates changes in several hundred genes in the striatum following DA depletion and L-DOPA reinstatement creating an extraordinarily high number of potential molecular candidates 78.
Presynaptic hypotheses of LID are directly relevant to the effects of L-DOPA on DA neurotransmission, as they attempt to explain LID in terms of dysregulated presynaptic control of vesicular loading, uncontrolled release and decreased reuptake of DA 69,79. A presynaptic cause of LIDs is supported by imaging studies in PD that demonstrate a positive correlation between the ability of L-DOPA to increase synaptic DA levels and the severity of “peak-dose” LID 80-82. Similarly, L-DOPA-induced increase in striatal extracellular DA in animals with severe DA lesion is higher than non-lesioned animals 33,38,39, and still higher in lesioned animals with LID 83,84. Because L-DOPA mediated DA efflux is particularly prominent in advanced PD patients and animals with severe DA lesions, it has been suggested that LID is linked to an inability of the striatum to maintain presynaptic control of DA released following L-DOPA administration.
Strong evidence supporting a presynaptic locus for LID comes from a rat model where severe striatal DA deficit was produced by silencing TH using short-hairpin RNA (shRNA), resulting in a reduction of DA levels comparable to a 6-OHDA lesion 85. Repeated L-DOPA did not produce LID in these animals, apparently because the DA terminals and synaptic vesicles were preserved and DA produced by L-DOPA could be released by normal SN neuronal activity. We note, however, that patients carrying genetic TH deficiency still develop severe LIDs in response to L-DOPA 86.
DA release from striatal 5-HT terminals
In advanced PD, most of the L-DOPA conversion to DA, vesicular uptake, and release appear to occur within striatal 5-HT axons, which are low in number compared to DA innervation, but survive longer in the disease 87, and their density may be substantially increased due to sprouting in advanced stages of PD 88. The initial evidence that L-DOPA can be decarboxylated to DA in 5-HT neurons was obtained with the Falck-Hillarp histochemistry technique, which provides visualization of catecholaminergic neurotransmitters 89; administration of L-DOPA to rats increased the fluorescent signal in both DA and 5-HT neurons. Consistently, in lesioned mice, L-DOPA’s increase in extracellular DA levels is substantially reduced following destruction of 5-HT neurons with the neurotoxin 5,7 dihyroxytryptamine 90-92, confirming 5-HT neurons as a source of L-DOPA-mediated DA overflow. DA efflux in 6-OHDA lesioned animals treated with L-DOPA is also abolished by the sodium channel blocker tetrodotoxin, which inhibits neuronal activity, or the VMAT inhibitor reserpine 38,93 which indicates that DA released by 5-HT terminals requires neuronal activity and accumulation of DA into monoamine synaptic vesicles. Conversely, in DA lesioned striatum, inhibition of DAT or activation of D2 receptors has no effect on L-DOPA-mediated DA release 38,94, consistent with the loss of presynaptic regulatory control of DA neurotransmission.
Serotonergic neurons use AADC for the biosynthesis of 5-HT 90,91,95,96 [see however 97]. Both 5-HT and DA neurons use VMAT2 for the accumulation of neurotransmitters into synaptic vesicles (Figure 1), enabling vesicle loading and stimulation-dependent release of DA from 5-HT axons 98,99. Furthermore, high doses of L-DOPA reduce brain 5-HT levels in mice, confirming that DA displaces endogenous 5-HT from synaptic vesicles 89,100. Thus, L-DOPA therapy may cause changes in 5-HT transmission 101, not only in striatum but also in SN pars reticulata, prefrontal cortex and the hippocampus 102, as well as in histamine neurons 103.
A role for DA release from 5-HT neurons in causing “peak-dose” LID has been suggested because neurotransmission would be dependent on the availability of exogenous L-DOPA rather than DA neuronal activity 96,104. In support of this, lesion of the neurons with 5,7 dihyroxytryptamine virtually abolished LID in 6-OHDA lesioned rats, while still providing effective L-DOPA treatment 105,106. Agonists of inhibitory 5-HT1A and 5-HT1B auto-receptors on 5-HT terminals and cell bodies moreover decreased L-DOPA mediated increase in DA efflux, and inhibited LID in rodents 84,99,105,107,108. It is possible that such agonists may provide future therapy.
DA release from systemically administered L-DOPA does not occur exclusively in the striatum, and the brain regions that receive serotonergic innervation, including prefrontal cortex, hippocampus and SNr, display increased extrasynaptic DA following L-DOPA treatment in both healthy and DA lesioned brains 45. It is possible that extrastriatal DA release or the action of L-DOPA itself 46 contribute to the development and manifestation of LIDs.
DA derived from L-DOPA may also be released from other cells, as behavioral effects and DA efflux following very high doses of L-DOPA (100 mg/kg) in 6-OHDA lesioned animals were not affected by 5-HT lesions 92,97. A possible source for L-DOPA induced DA release may include histamine neurons as well as striatal interneurons and glia cells that may have AADC expression 97,109-111, although this is controversial 112,113. Thus, multiple neuronal and non-neuronal cells may be candidates for DA release following L-DOPA, particularly under conditions where cytosolic DA levels are so high that means of release occur that are independent of VMAT expression.
Concluding remarks
L-DOPA in early stages of PD provides an elegant therapy, as the “social” nature of DA transmission and decreased DAT-mediated uptake allow fewer DA release sites to engage distant receptors and provide replacement of normal DA signaling. In the later stages of PD, however, DA denervation with subsequent L-DOPA treatment leads to unregulated DA release that produces larger and a longer-lasting diffusional cloud of the transmitter.
Understanding more precisely how L-DOPA reinstatement leads to over activity of basal ganglia circuitry is of central importance in developing PD therapies that avoid LID. If the loss of controlled DA release in more advanced PD genuinely causes LID, elucidating the steps involved will be important even if replacement of DA by transplantation becomes a standard treatment, as these cells will also not participate in normal circuitry to drive their activity. It seems most likely, however, that a relatively complex combination of presynaptic and postsynaptic alterations are involved, in which case there may be multiple possible therapeutic avenues that could interrupt the circuits that lead to LID.
Acknowledgments
Supported by an Udall Center of Excellence award, the JPB and Parkinson’s Disease Foundations, and #R01NS075222 from the National Institute of Neurological Disorders And Stroke (EM). AB received fellowships from Swedish Research Council and Sweden-America Foundation.
Abbreviations
- AADC
aromatic L-amino acid decarboxylase
- AD
aldehyde dehydrogenase
- DA
dopamine
- DAcyt
cytosolic DA
- DAT
DA uptake transporter
- DAR
DA receptors
- D2
DA autoreceptor 2
- DA-Cys
5-S-cysteinyl-DA
- DA-Q
DA-o-quinone
- DOPAC
3,4-dihydroxyphenylacetate
- DOPAL
3,4-dihydroxyphenylacetaldehyde
- 5-HIAA
5-hydroxyindole acetic acid
- 5-HIAAL
5-hydroxyindole acetaldehyde
- 5-HT
5-hydroxytryptophan (serotonin)
- 5-HT1A
serotonin autoreceptor 1A
- 5-HTR
serotonin receptors
- LAAT
L-amino acid transporter
- LC
locus coeruleus
- L-DOPA
L-3,4-dihydroxyphenylalanine (levodopa)
- MAO
monoamine oxidase
- MPP+
1-methyl-4-phenylpyridinium
- MSN
medium spiny neurons
- NM
neuromelanin
- PD
Parkinson’s disease
- SNc
substantia nigra pars compacta
- ROS
reactive oxygen species
- SERT
serotonin uptake transporter
- TH
tyrosine hydroxylase
- TPH2
tryptophan hydroxylase 2
- Trp
tryptophan
- VTA
ventral tegmental area
- VMAT
vesicular monoamine transporter
References
- 1.Birkmayer W, Hornykiewicz O. Der L-3,4-dioxyphenylalanin (DOPA) Effekt bei der Parkinson-akinesia. Wien Klin Wochenschr. 1961;73:787–788. [PubMed] [Google Scholar]
- 2.Cotzias GC, Van Woert MH, Schiffer LM. Aromatic amino acids and modification of parkinsonism. The New England journal of medicine. 1967;276:374–379. doi: 10.1056/nejm196702162760703. [DOI] [PubMed] [Google Scholar]
- 3.Ramsey AJ, Fitzpatrick PF. Effects of phosphorylation on binding of catecholamines to tyrosine hydroxylase: specificity and thermodynamics. Biochemistry. 2000;39:773–778. doi: 10.1021/bi991901r. [DOI] [PubMed] [Google Scholar]
- 4.Hadjiconstantinou M, Neff NH. Enhancing aromatic L-amino acid decarboxylase activity: implications for L-DOPA treatment in Parkinson’s disease. CNS Neurosci Ther. 2008;14:340–351. doi: 10.1111/j.1755-5949.2008.00058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolf ME, Roth RH. Autoreceptor regulation of dopamine synthesis. Annals of the New York Academy of Sciences. 1990;604:323–343. doi: 10.1111/j.1749-6632.1990.tb32003.x. [DOI] [PubMed] [Google Scholar]
- 6.Lindgren N, et al. Dopamine D(2) receptors regulate tyrosine hydroxylase activity and phosphorylation at Ser40 in rat striatum. The European journal of neuroscience. 2001;13:773–780. doi: 10.1046/j.0953-816x.2000.01443.x. [DOI] [PubMed] [Google Scholar]
- 7.Peter D, et al. Differential expression of two vesicular monoamine transporters. J Neurosci. 1995;15:6179–6188. doi: 10.1523/JNEUROSCI.15-09-06179.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weihe E, Schafer MK, Erickson JD, Eiden LE. Localization of vesicular monoamine transporter isoforms (VMAT1 and VMAT2) to endocrine cells and neurons in rat. Journal of Molecular Neuroscience. 1994:149–164. doi: 10.1007/BF02736730. [DOI] [PubMed] [Google Scholar]
- 9.Erickson JD, Schafer MK, Bonner TI, Eiden LE, Weihe E. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:5166–5171. doi: 10.1073/pnas.93.10.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wimalasena K. Vesicular monoamine transporters: structure-function, pharmacology, and medicinal chemistry. Med Res Rev. 2011;31:483–519. doi: 10.1002/med.20187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guillot TS, Miller GW. Protective actions of the vesicular monoamine transporter 2 (VMAT2) in monoaminergic neurons. Mol Neurobiol. 2009;39:149–170. doi: 10.1007/s12035-009-8059-y. [DOI] [PubMed] [Google Scholar]
- 12.Sames D, Dunn M, Karpowicz RJ, Jr, Sulzer D. Visualizing neurotransmitter secretion at individual synapses. ACS chemical neuroscience. 2013;4:648–651. doi: 10.1021/cn4000956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zoli M, Jansson A, Sykova E, Agnati LF, Fuxe K. Volume transmission in the CNS and its relevance for neuropsychopharmacology. Trends Pharmacol Sci. 1999;20:142–150. doi: 10.1016/s0165-6147(99)01343-7. [DOI] [PubMed] [Google Scholar]
- 14.Sulzer D, Pothos EN. Regulation of quantal size by presynaptic mechanisms. Rev Neurosci. 2000;11:159–212. doi: 10.1515/revneuro.2000.11.2-3.159. [DOI] [PubMed] [Google Scholar]
- 15.Nirenberg MJ, Chan J, Liu Y, Edwards RH, Pickel VM. Ultrastructural localization of the vesicular monoamine transporter-2 in midbrain dopaminergic neurons: potential sites for somatodendritic storage and release of dopamine. J Neurosci. 1996;16:4135–4145. doi: 10.1523/JNEUROSCI.16-13-04135.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pickel VM, Nirenberg MJ, Milner TA. Ultrastructural view of central catecholaminergic transmission: immunocytochemical localization of synthesizing enzymes, transporters and receptors. J Neurocytol. 1996;25:843–856. doi: 10.1007/BF02284846. [DOI] [PubMed] [Google Scholar]
- 17.Jones SR, et al. Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc Natl Acad Sci U S A. 1998;95:4029–4034. doi: 10.1073/pnas.95.7.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sulzer D. How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron. 2011;69:628–649. doi: 10.1016/j.neuron.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freund TF, Powell JF, Smith AD. Tyrosine hydroxylase-immunoreactive boutons in synaptic contact with identified striatonigral neurons, with particular reference to dendritic spines. Neuroscience. 1984;13:1189–1215. doi: 10.1016/0306-4522(84)90294-x. [DOI] [PubMed] [Google Scholar]
- 20.Venton BJ, et al. Real-time decoding of dopamine concentration changes in the caudate-putamen during tonic and phasic firing. Journal of neurochemistry. 2003;87:1284–1295. doi: 10.1046/j.1471-4159.2003.02109.x. [DOI] [PubMed] [Google Scholar]
- 21.Vieira-Coelho MA, Soares-Da-Silva P. Uptake and intracellular fate of L-DOPA in a human intestinal epithelial cell line: Caco-2. Am J Physiol. 1998;275:C104–112. doi: 10.1152/ajpcell.1998.275.1.C104. [DOI] [PubMed] [Google Scholar]
- 22.Mosharov EV, et al. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62:218–229. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mosharov EV, et al. Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J Neurosci. 2006;26:9304–9311. doi: 10.1523/JNEUROSCI.0519-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wightman RM, et al. Temporally resolved catecholamine spikes correspond to single vesicle release from individual chromaffin cells. Proc Natl Acad Sci U S A. 1991;88:10754–10758. doi: 10.1073/pnas.88.23.10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pothos EN, Davila V, Sulzer D. Presynaptic recording of quanta from midbrain dopamine neurons and modulation of the quantal size. J Neurosci. 1998;18:4106–4118. doi: 10.1523/JNEUROSCI.18-11-04106.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Staal RG, Mosharov EV, Sulzer D. Dopamine neurons release transmitter via a flickering fusion pore. Nat Neurosci. 2004;7:341–346. doi: 10.1038/nn1205. [DOI] [PubMed] [Google Scholar]
- 27.Gong LW, Hafez I, Alvarez de Toledo G, Lindau M. Secretory vesicles membrane area is regulated in tandem with quantal size in chromaffin cells. J Neurosci. 2003;23:7917–7921. doi: 10.1523/JNEUROSCI.23-21-07917.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pothos E, Desmond M, Sulzer D. L-3,4-dihydroxyphenylalanine increases the quantal size of exocytotic dopamine release in vitro. Journal of neurochemistry. 1996;66:629–636. doi: 10.1046/j.1471-4159.1996.66020629.x. [DOI] [PubMed] [Google Scholar]
- 29.Colliver TL, Hess EJ, Pothos EN, Sulzer D, Ewing AG. Quantitative and statistical analysis of the shape of amperometric spikes recorded from two populations of cells. J Neurochem. 2000;74:1086–1097. doi: 10.1046/j.1471-4159.2000.741086.x. [DOI] [PubMed] [Google Scholar]
- 30.Omiatek DM, et al. The real catecholamine content of secretory vesicles in the CNS revealed by electrochemical cytometry. Scientific reports. 2013;3:1447. doi: 10.1038/srep01447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Omiatek DM, Dong Y, Heien ML, Ewing AG. Only a Fraction of Quantal Content is Released During Exocytosis as Revealed by Electrochemical Cytometry of Secretory Vesicles. ACS chemical neuroscience. 2010;1:234–245. doi: 10.1021/cn900040e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burke RE, O’Malley K. Axon degeneration in Parkinson’s disease. Experimental neurology. 2013;246:72–83. doi: 10.1016/j.expneurol.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abercrombie ED, Bonatz AE, Zigmond MJ. Effects of L-dopa on extracellular dopamine in striatum of normal and 6-hydroxydopamine-treated rats. Brain research. 1990;525:36–44. doi: 10.1016/0006-8993(90)91318-b. [DOI] [PubMed] [Google Scholar]
- 34.Lee J, et al. Sprouting of dopamine terminals and altered dopamine release and uptake in Parkinsonian dyskinaesia. Brain : a journal of neurology. 2008;131:1574–1587. doi: 10.1093/brain/awn085. [DOI] [PubMed] [Google Scholar]
- 35.Zigmond MJ. Do compensatory processes underlie the preclinical phase of neurodegenerative disease? Insights from an animal model of parkinsonism. Neurobiology of disease. 1997;4:247–253. doi: 10.1006/nbdi.1997.0157. [DOI] [PubMed] [Google Scholar]
- 36.Rice ME, Cragg SJ. Dopamine spillover after quantal release: rethinking dopamine transmission in the nigrostriatal pathway. Brain Res Rev. 2008;58:303–313. doi: 10.1016/j.brainresrev.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmitz Y, Lee CJ, Schmauss C, Gonon F, Sulzer D. Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J Neurosci. 2001;21:5916–5924. doi: 10.1523/JNEUROSCI.21-16-05916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller DW, Abercrombie ED. Role of high-affinity dopamine uptake and impulse activity in the appearance of extracellular dopamine in striatum after administration of exogenous L-DOPA: studies in intact and 6-hydroxydopamine-treated rats. Journal of neurochemistry. 1999;72:1516–1522. doi: 10.1046/j.1471-4159.1999.721516.x. [DOI] [PubMed] [Google Scholar]
- 39.Sarre S, De Klippel N, Herregodts P, Ebinger G, Michotte Y. Biotransformation of locally applied L-dopa in the corpus striatum of the hemi-parkinsonian rat studied with microdialysis. Naunyn-Schmiedeberg’s archives of pharmacology. 1994;350:15–21. doi: 10.1007/BF00180005. [DOI] [PubMed] [Google Scholar]
- 40.Weintraub D, Nirenberg MJ. Impulse control and related disorders in Parkinson’s disease. Neurodegenerative diseases. 2013;11:63–71. doi: 10.1159/000341996. [DOI] [PubMed] [Google Scholar]
- 41.Duan H, Wang J. Selective transport of monoamine neurotransmitters by human plasma membrane monoamine transporter and organic cation transporter 3. The Journal of pharmacology and experimental therapeutics. 2010;335:743–753. doi: 10.1124/jpet.110.170142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sader-Mazbar O, Loboda Y, Rabey MJ, Finberg JP. Increased L-DOPA-derived dopamine following selective MAO-A or -B inhibition in rat striatum depleted of dopaminergic and serotonergic innervation. British journal of pharmacology. 2013;170:999–1013. doi: 10.1111/bph.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Larsen MB, et al. Dopamine transport by the serotonin transporter: a mechanistically distinct mode of substrate translocation. J Neurosci. 2011;31:6605–6615. doi: 10.1523/JNEUROSCI.0576-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmitz Y, Schmauss C, Sulzer D. Altered dopamine release and uptake kinetics in mice lacking D2 receptors. J Neurosci. 2002;22:8002–8009. doi: 10.1523/JNEUROSCI.22-18-08002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Navailles S, Lagiere M, Contini A, De Deurwaerdere P. Multisite intracerebral microdialysis to study the mechanism of L-DOPA induced dopamine and serotonin release in the parkinsonian brain. ACS chemical neuroscience. 2013;4:680–692. doi: 10.1021/cn400046e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Porras G, et al. L-dopa-induced dyskinesia: beyond an excessive dopamine tone in the striatum. Scientific reports. 2014;4:3730. doi: 10.1038/srep03730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sulzer D, et al. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc Natl Acad Sci U S A. 2000;97:11869–11874. doi: 10.1073/pnas.97.22.11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zucca FA, et al. Neuromelanin of the human substantia nigra: an update. Neurotoxicity research. 2014;25:13–23. doi: 10.1007/s12640-013-9435-y. [DOI] [PubMed] [Google Scholar]
- 49.Cebrian C, et al. MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nature communications. 2014;5:3633. doi: 10.1038/ncomms4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Surmeier DJ, Guzman JN, Sanchez-Padilla J, Goldberg JA. What causes the death of dopaminergic neurons in Parkinson’s disease? Prog Brain Res. 2010;183:59–77. doi: 10.1016/S0079-6123(10)83004-3. [DOI] [PubMed] [Google Scholar]
- 51.Hetzenauer A, Sinnegger-Brauns MJ, Striessnig J, Singewald N. Brain activation pattern induced by stimulation of L-type Ca2+-channels: contribution of Ca(V)1.3 and Ca(V)1.2 isoforms. Neuroscience. 2006;139:1005–1015. doi: 10.1016/j.neuroscience.2006.01.059. [DOI] [PubMed] [Google Scholar]
- 52.Stansley BJ, Yamamoto BK. L-dopa-induced dopamine synthesis and oxidative stress in serotonergic cells. Neuropharmacology. 2013;67:243–251. doi: 10.1016/j.neuropharm.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edwards RH. Neural degeneration and the transport of neurotransmitters. Ann Neurol. 1993;34:638–645. doi: 10.1002/ana.410340504. [DOI] [PubMed] [Google Scholar]
- 54.Gainetdinov RR, et al. Increased MPTP neurotoxicity in vesicular monoamine transporter 2 heterozygote knockout mice. Journal of neurochemistry. 1998;70:1973–1978. doi: 10.1046/j.1471-4159.1998.70051973.x. [DOI] [PubMed] [Google Scholar]
- 55.Lotharius J, Brundin P. Impaired dopamine storage resulting from alpha-synuclein mutations may contribute to the pathogenesis of Parkinson’s disease. Hum Mol Genet. 2002;11:2395–2407. doi: 10.1093/hmg/11.20.2395. [DOI] [PubMed] [Google Scholar]
- 56.Uhl GR. Hypothesis: the role of dopaminergic transporters in selective vulnerability of cells in Parkinson’s disease. Ann Neurol. 1998;43:555–560. doi: 10.1002/ana.410430503. [DOI] [PubMed] [Google Scholar]
- 57.Pifl C, et al. Is Parkinson’s disease a vesicular dopamine storage disorder? Evidence from a study in isolated synaptic vesicles of human and nonhuman primate striatum. J Neurosci. 2014;34:8210–8218. doi: 10.1523/JNEUROSCI.5456-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lohr KM, et al. Increased vesicular monoamine transporter enhances dopamine release and opposes Parkinson disease-related neurodegeneration in vivo. Proc Natl Acad Sci U S A. 2014;111:9977–9982. doi: 10.1073/pnas.1402134111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blaschko H. Amine oxidase and amine metabolism. Pharmacol Rev. 1952;4:415–458. [PubMed] [Google Scholar]
- 60.Burke WJ, et al. Neurotoxicity of MAO metabolites of catecholamine neurotransmitters: role in neurodegenerative diseases. Neurotoxicology. 2004;25:101–115. doi: 10.1016/S0161-813X(03)00090-1. [DOI] [PubMed] [Google Scholar]
- 61.Goldstein DS, Kopin IJ, Sharabi Y. Catecholamine autotoxicity. Implications for pharmacology and therapeutics of Parkinson disease and related disorders. Pharmacology & therapeutics. 2014 doi: 10.1016/j.pharmthera.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Caudle WM, et al. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci. 2007;27:8138–8148. doi: 10.1523/JNEUROSCI.0319-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hastings TG, Berman SB. In: Role of Catechol Quinone Species in Cellular Toxicity. Creveling CR, editor. F.P. Graham Publishing, Inc.; 1999. pp. 69–89. [Google Scholar]
- 64.Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci. 2002;22:8951–8960. doi: 10.1523/JNEUROSCI.22-20-08951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fon EA, et al. Vesicular transport regulates monoamine storage and release but is not essential for amphetamine action. Neuron. 1997;19:1271–1283. doi: 10.1016/s0896-6273(00)80418-3. [DOI] [PubMed] [Google Scholar]
- 66.Nutt JG. Clinical pharmacology of levodopa-induced dyskinesia. Ann Neurol. 2000;47:S160–164. discussion S164-166. [PubMed] [Google Scholar]
- 67.Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Movement disorders : official journal of the Movement Disorder Society. 2001;16:448–458. doi: 10.1002/mds.1090. [DOI] [PubMed] [Google Scholar]
- 68.Fahn S. The spectrum of levodopa-induced dyskinesias. Ann Neurol. 2000;47:S2–9. discussion S9-11. [PubMed] [Google Scholar]
- 69.Cenci MA, Lundblad M. Post- versus presynaptic plasticity in L-DOPA-induced dyskinesia. Journal of neurochemistry. 2006;99:381–392. doi: 10.1111/j.1471-4159.2006.04124.x. [DOI] [PubMed] [Google Scholar]
- 70.Bargiotas P, Konitsiotis S. Levodopa-induced dyskinesias in Parkinson’s disease: emerging treatments. Neuropsychiatric disease and treatment. 2013;9:1605–1617. doi: 10.2147/NDT.S36693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Olanow CW, et al. Levodopa in the treatment of Parkinson’s disease: current controversies. Movement disorders : official journal of the Movement Disorder Society. 2004;19:997–1005. doi: 10.1002/mds.20243. [DOI] [PubMed] [Google Scholar]
- 72.Olanow CW, Obeso JA, Stocchi F. Continuous dopamine-receptor treatment of Parkinson’s disease: scientific rationale and clinical implications. The Lancet Neurology. 2006;5:677–687. doi: 10.1016/S1474-4422(06)70521-X. [DOI] [PubMed] [Google Scholar]
- 73.Olanow CW, et al. Continuous intrajejunal infusion of levodopa-carbidopa intestinal gel for patients with advanced Parkinson’s disease: a randomised, controlled, double-blind, double-dummy study. The Lancet Neurology. 2014;13:141–149. doi: 10.1016/S1474-4422(13)70293-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Iderberg H, Francardo V, Pioli EY. Animal models of L-DOPA-induced dyskinesia: an update on the current options. Neuroscience. 2012;211:13–27. doi: 10.1016/j.neuroscience.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 75.Huot P, Johnston TH, Koprich JB, Fox SH, Brotchie JM. The pharmacology of L-DOPA-induced dyskinesia in Parkinson’s disease. Pharmacol Rev. 2013;65:171–222. doi: 10.1124/pr.111.005678. [DOI] [PubMed] [Google Scholar]
- 76.Zhuang X, Mazzoni P, Kang UJ. The role of neuroplasticity in dopaminergic therapy for Parkinson disease. Nature reviews Neurology. 2013;9:248–256. doi: 10.1038/nrneurol.2013.57. [DOI] [PubMed] [Google Scholar]
- 77.Cenci MA, Konradi C. Maladaptive striatal plasticity in L-DOPA-induced dyskinesia. Prog Brain Res. 2010;183:209–233. doi: 10.1016/S0079-6123(10)83011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Heiman M, et al. Molecular adaptations of striatal spiny projection neurons during levodopa-induced dyskinesia. Proc Natl Acad Sci U S A. 2014;111:4578–4583. doi: 10.1073/pnas.1401819111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Carta M, Bezard E. Contribution of pre-synaptic mechanisms to L-DOPA-induced dyskinesia. Neuroscience. 2011;198:245–251. doi: 10.1016/j.neuroscience.2011.07.070. [DOI] [PubMed] [Google Scholar]
- 80.Tedroff J, et al. Levodopa-induced changes in synaptic dopamine in patients with Parkinson’s disease as measured by [11C]raclopride displacement and PET. Neurology. 1996;46:1430–1436. doi: 10.1212/wnl.46.5.1430. [DOI] [PubMed] [Google Scholar]
- 81.Pavese N, et al. Clinical correlates of levodopa-induced dopamine release in Parkinson disease: a PET study. Neurology. 2006;67:1612–1617. doi: 10.1212/01.wnl.0000242888.30755.5d. [DOI] [PubMed] [Google Scholar]
- 82.de la Fuente-Fernandez R, et al. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain : a journal of neurology. 2004;127:2747–2754. doi: 10.1093/brain/awh290. [DOI] [PubMed] [Google Scholar]
- 83.Meissner W, et al. Increased slow oscillatory activity in substantia nigra pars reticulata triggers abnormal involuntary movements in the 6-OHDA-lesioned rat in the presence of excessive extracellular striatal dopamine. Neurobiology of disease. 2006;22:586–598. doi: 10.1016/j.nbd.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 84.Lindgren HS, Andersson DR, Lagerkvist S, Nissbrandt H, Cenci MA. L-DOPA-induced dopamine efflux in the striatum and the substantia nigra in a rat model of Parkinson’s disease: temporal and quantitative relationship to the expression of dyskinesia. Journal of neurochemistry. 2010;112:1465–1476. doi: 10.1111/j.1471-4159.2009.06556.x. [DOI] [PubMed] [Google Scholar]
- 85.Ulusoy A, Sahin G, Kirik D. Presynaptic dopaminergic compartment determines the susceptibility to L-DOPA-induced dyskinesia in rats. Proc Natl Acad Sci U S A. 2010;107:13159–13164. doi: 10.1073/pnas.1003432107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pons R, et al. Levodopa-induced dyskinesias in tyrosine hydroxylase deficiency. Movement disorders : official journal of the Movement Disorder Society. 2013;28:1058–1063. doi: 10.1002/mds.25382. [DOI] [PubMed] [Google Scholar]
- 87.Sulzer D, Surmeier DJ. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society. 2013;28:41–50. doi: 10.1002/mds.25095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rylander D, et al. Maladaptive plasticity of serotonin axon terminals in levodopa-induced dyskinesia. Ann Neurol. 2010;68:619–628. doi: 10.1002/ana.22097. [DOI] [PubMed] [Google Scholar]
- 89.Butcher L, Engel J, Fuxe K. L-dopa induced changes in central monoamine neurons after peripheral decarboxylase inhibition. The Journal of pharmacy and pharmacology. 1970;22:313–316. doi: 10.1111/j.2042-7158.1970.tb08529.x. [DOI] [PubMed] [Google Scholar]
- 90.Tanaka H, et al. Role of serotonergic neurons in L-DOPA-derived extracellular dopamine in the striatum of 6-OHDA-lesioned rats. Neuroreport. 1999;10:631–634. doi: 10.1097/00001756-199902250-00034. [DOI] [PubMed] [Google Scholar]
- 91.Navailles S, Bioulac B, Gross C, De Deurwaerdere P. Serotonergic neurons mediate ectopic release of dopamine induced by L-DOPA in a rat model of Parkinson’s disease. Neurobiology of disease. 2010;38:136–143. doi: 10.1016/j.nbd.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 92.Lopez A, Munoz A, Guerra MJ, Labandeira-Garcia JL. Mechanisms of the effects of exogenous levodopa on the dopamine-denervated striatum. Neuroscience. 2001;103:639–651. doi: 10.1016/s0306-4522(00)00588-1. [DOI] [PubMed] [Google Scholar]
- 93.Kannari K, et al. Reserpine pretreatment prevents increases in extracellular striatal dopamine following L-DOPA administration in rats with nigrostriatal denervation. Journal of neurochemistry. 2000;74:263–269. doi: 10.1046/j.1471-4159.2000.0740263.x. [DOI] [PubMed] [Google Scholar]
- 94.Maeda T, Kannari K, Suda T, Matsunaga M. Loss of regulation by presynaptic dopamine D2 receptors of exogenous L-DOPA-derived dopamine release in the dopaminergic denervated striatum. Brain research. 1999;817:185–191. doi: 10.1016/s0006-8993(98)01248-7. [DOI] [PubMed] [Google Scholar]
- 95.Arai R, Karasawa N, Geffard M, Nagatsu T, Nagatsu I. Immunohistochemical evidence that central serotonin neurons produce dopamine from exogenous L-DOPA in the rat, with reference to the involvement of aromatic L-amino acid decarboxylase. Brain research. 1994;667:295–299. doi: 10.1016/0006-8993(94)91511-3. [DOI] [PubMed] [Google Scholar]
- 96.Yamada H, et al. Immunohistochemical detection of L-DOPA-derived dopamine within serotonergic fibers in the striatum and the substantia nigra pars reticulata in Parkinsonian model rats. Neuroscience research. 2007;59:1–7. doi: 10.1016/j.neures.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 97.Melamed E, Hefti F, Liebman J, Schlosberg AJ, Wurtman RJ. Serotonergic neurones are not involved in action of L-dopa in Parkinson’s disease. Nature. 1980;283:772–774. doi: 10.1038/283772a0. [DOI] [PubMed] [Google Scholar]
- 98.Ng KY, Chase TN, Colburn RW, Kopin IJ. Dopamine: stimulation-induced release from central neurons. Science. 1971;172:487–489. doi: 10.1126/science.172.3982.487. [DOI] [PubMed] [Google Scholar]
- 99.Nevalainen N, Af Bjerken S, Gerhardt GA, Stromberg I. Serotonergic nerve fibers in L-DOPA-derived dopamine release and dyskinesia. Neuroscience. 2014;260:73–86. doi: 10.1016/j.neuroscience.2013.12.029. [DOI] [PubMed] [Google Scholar]
- 100.Everett GM, Borcherding JW. L-dopa: effect on concentrations of dopamine, norepinephrine, and serotonin in brains of mice. Science. 1970;168:849–850. doi: 10.1126/science.168.3933.849. [DOI] [PubMed] [Google Scholar]
- 101.Miguelez C, Morera-Herreras T, Torrecilla M, Ruiz-Ortega JA, Ugedo L. Interaction between the 5-HT system and the basal ganglia: functional implication and therapeutic perspective in Parkinson’s disease. Frontiers in neural circuits. 2014;8:21. doi: 10.3389/fncir.2014.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Navailles S, Bioulac B, Gross C, De Deurwaerdere P. Chronic L-DOPA therapy alters central serotonergic function and L-DOPA-induced dopamine release in a region-dependent manner in a rat model of Parkinson’s disease. Neurobiology of disease. 2011;41:585–590. doi: 10.1016/j.nbd.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 103.Yanovsky Y, et al. L-Dopa activates histaminergic neurons. The Journal of physiology. 2011;589:1349–1366. doi: 10.1113/jphysiol.2010.203257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Carta M, Tronci E. Serotonin System Implication in l-DOPA-Induced Dyskinesia: From Animal Models to Clinical Investigations. Frontiers in neurology. 2014;5:78. doi: 10.3389/fneur.2014.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Carta M, Carlsson T, Kirik D, Bjorklund A. Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain : a journal of neurology. 2007;130:1819–1833. doi: 10.1093/brain/awm082. [DOI] [PubMed] [Google Scholar]
- 106.Eskow KL, et al. The role of the dorsal raphe nucleus in the development, expression, and treatment of L-dopa-induced dyskinesia in hemiparkinsonian rats. Synapse. 2009;63:610–620. doi: 10.1002/syn.20630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tomiyama M, et al. A serotonin 5-HT1A receptor agonist prevents behavioral sensitization to L-DOPA in a rodent model of Parkinson’s disease. Neuroscience research. 2005;52:185–194. doi: 10.1016/j.neures.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 108.Munoz A, et al. Serotonin neuron-dependent and -independent reduction of dyskinesia by 5-HT1A and 5-HT1B receptor agonists in the rat Parkinson model. Experimental neurology. 2009;219:298–307. doi: 10.1016/j.expneurol.2009.05.033. [DOI] [PubMed] [Google Scholar]
- 109.Tashiro Y, et al. Striatal neurons with aromatic L-amino acid decarboxylase-like immunoreactivity in the rat. Neuroscience letters. 1989;100:29–34. doi: 10.1016/0304-3940(89)90655-1. [DOI] [PubMed] [Google Scholar]
- 110.Mura A, Jackson D, Manley MS, Young SJ, Groves PM. Aromatic L-amino acid decarboxylase immunoreactive cells in the rat striatum: a possible site for the conversion of exogenous L-DOPA to dopamine. Brain research. 1995;704:51–60. doi: 10.1016/0006-8993(95)01104-8. [DOI] [PubMed] [Google Scholar]
- 111.Lopez-Real A, Rodriguez-Pallares J, Guerra MJ, Labandeira-Garcia JL. Localization and functional significance of striatal neurons immunoreactive to aromatic L-amino acid decarboxylase or tyrosine hydroxylase in rat Parkinsonian models. Brain research. 2003;969:135–146. doi: 10.1016/s0006-8993(03)02291-1. [DOI] [PubMed] [Google Scholar]
- 112.Nakamura K, Ahmed M, Barr E, Leiden JM, Kang UJ. The localization and functional contribution of striatal aromatic L-amino acid decarboxylase to L-3,4-dihydroxyphenylalanine decarboxylation in rodent parkinsonian models. Cell transplantation. 2000;9:567–576. doi: 10.1177/096368970000900502. [DOI] [PubMed] [Google Scholar]
- 113.Kang UJ, Park DH, Wessel T, Baker H, Joh TH. Dopa-decarboxylation in the striata of rats with unilateral substantia nigra lesions. Neuroscience letters. 1992;147:53–57. doi: 10.1016/0304-3940(92)90773-z. [DOI] [PubMed] [Google Scholar]
- 114.Togasaki DM, et al. Levodopa induces dyskinesias in normal squirrel monkeys. Ann Neurol. 2001;50:254–257. doi: 10.1002/ana.1099. [DOI] [PubMed] [Google Scholar]