

Abstract
Objectives
GPR40 (FFAR1), a clinically proven anti-diabetes target, is a Gq-coupled receptor for long chain fatty acids (LCFA) stimulating insulin secretion directly and mediating a major part of the dietary triglyceride-induced secretion of the incretins GLP-1 and GIP. In phase-II studies the GPR40 agonist TAK-875 decreased blood glucose but surprisingly without stimulating incretins.
Methods and results
Here we find that GPR40 can signal through not only Gq and IP3 but also Gs and cAMP when stimulated with certain agonists such as AM-1638 and AM-5262 in contrast to the endogenous LCFA ligands and agonists such as TAK-875 and AM-837, which only signal through Gq. In competition binding against [3H]AM-1638 and [3H]L358 the Gq + Gs and the Gq-only agonists either competed for or showed positive cooperativity by increasing the binding of the two different radio-ligands, in opposite ways. Nevertheless, both the Gq-only and the Gq + Gs agonists all docked surprisingly well into the binding site for TAK-875 in the X-ray structure of GPR40. In murine intestinal primary cell-cultures the endogenous LCFAs and the Gq-only agonists stimulated GLP-1 secretion with rather poor efficacy as compared with the high efficacy Gq + Gs GPR40 agonists and a prototype GPR119 agonist. Similarly, in fasting both male and female mice the Gq + Gs agonists showed significantly higher efficacy than the Gq-only agonists in respect of increasing plasma GLP-1 and plasma GIP in a GPR40-dependent manner.
Conclusions
It is concluded that stimulation of GPR40 by endogenous LCFAs or by Gq-only synthetic agonists result in a rather limited incretin response, whereas Gq + Gs GPR40 agonists stimulate incretin secretion robustly.
Keywords: G protein-coupled receptor, Glucagon like peptide 1 (GLP-1), Biased signaling, Ago-allosteric agonist, Long chain fatty acids (LCFAs)
1. Introduction
GLP-1 mimetics, such as exenatide and liraglutide have over the last years become widely used in the treatment of type 2 diabetes. The success of GLP-1 mimetics is based on the fact that they function not only as incretins, i.e. compounds which stimulate insulin secretion in a glucose dependent manner, but that they also suppress glucagon secretion, delay gastric emptying, decrease appetite and promote β-cell survival [1,2].
However GLP-1 itself is just one out of a handful of gut hormones with similar beneficial metabolic properties, which all are released in response to food ingestion from closely related enteroendocrine cells scattered along the small and large intestine [3,4]. The co-released gut hormones act in symphony and in several cases even synergistically [5]. Thus, it could be tempting to try to develop compounds, which stimulate the secretion of the powerful mixture of endogenous gut hormones [6,7]; compounds, which may not even enter the body as such, but act locally in the gut [6].
Metabolites of dietary triglycerides, i.e. free fatty acids (FFA) and 2-monoacyl glycerol (2-MAG) are among the most efficacious gut hormone secretagogues [8,9] conceivably acting through the G protein-coupled receptors GPR40 [10–14] and GPR119 [9,15], respectively. Since these receptors are highly expressed on β-cells of the endocrine pancreas, they rapidly became high priority drug discovery targets for new anti-diabetes agents. A number of GPR119 agonists reached phase II [9,16], but, although the compounds stimulated GLP-1 and insulin secretion in preclinical models they apparently did not deliver the required clinical efficacy perhaps due to their ability to also stimulate the secretion of the counter regulatory hormone glucagon [9].
GPR40 was originally deorphanized as a receptor for medium to long chain fatty acids being almost exclusively expressed in the β-cells and potentially responsible for the glucose-dependent stimulation of insulin secretion by long chain fatty acids (LCFA) [10–12,17]. However, the notion that GPR40 potentially was involved in lipotoxicity made it unclear whether you wanted agonists or antagonists and whether GPR40 would at all be a good drug target [17,18]. However, transgenic overexpression of GPR40 and administration of selective agonists later demonstrated that GPR40 activation augments insulin secretion and improves glucose tolerance and that GPR40 may even protect islets from the toxic effect of fatty acids [19–21]. Importantly, GPR40 was shown to be highly expressed and enriched also on enteroendocrine cells and to mediate GLP-1 and GIP secretion [22,23].
In respect of clinical development Takeda has pioneered the field with the GPR40 agonist TAK-875 or fasiglifam (Figure 2A), which in phase-II studies decreased HbA1c as efficiently as sulfonylurea without signs of hypoglycemia [24]. This was obtained with surprisingly little effect on systemic insulin and no effect on plasma GLP-1 [25,26]. Recently the TAK-875 program was, however stopped in phase-III due to liver toxicity,5 which probably is not GPR40-mediated as the receptor is not expressed in the liver. A new series of GPR40 agonists were by the Amgen group shown to be positive ago-allosteric modulators [27–29], i.e. compounds which both act as agonists and as allosteric modulators increasing the potency and/or the efficacy of other agonists, for example the endogenous agonists [30]. Interestingly, this new class of ago-allosteric modulators improved glucose tolerance not only through direct stimulation of insulin secretion but also through stimulation of GLP-1 [28,29,31].
Figure 2.
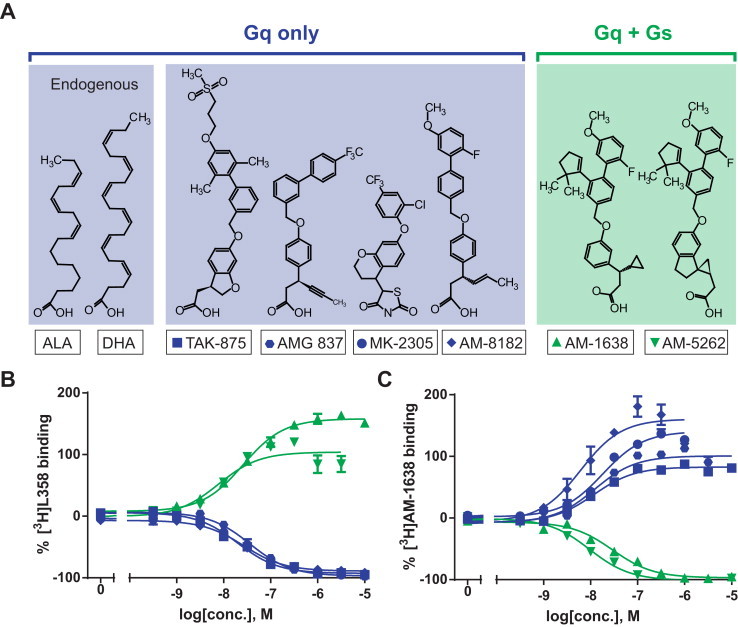
Chemical structures and binding properties of GPR40 agonists. (A) Chemical structures of endogenous agonists α-linolenic acid (ALA) and docosahexaenoic acid (DHA) as well as synthetic agonists TAK-875, AMG 837, MK-2305, AM-8182, AM-1638 and AM-5262. Agonists are grouped according to G-protein activation and competition binding is shown here with the Gq only agonist [3H]L358 (B) or the Gq+Gs agonist [3H]AM-1638 (C) to human GPR40. Data represent mean ± SEM of two independent experiments performed in duplicates/triplicates.
In the present study we compare a series of GPR40 agonists in respect of their signal transduction and binding properties and their ability to stimulate GLP-1 and GIP release ex vivo and increase plasma concentrations of these incretins in vivo. Surprisingly, we find that GPR40 can couple efficiently not only through Gq but also through Gs to increase cAMP concentrations, which however is a property restricted to a sub-set of GPR40 agonists and is associated with the ability to stimulate incretin secretion in a robust manner as compared to the agonists, which like the endogenous lipid ligands only are able to signal through Gq. Unexpectedly both Gq-only and Gq + Gs agonists docked well into the binding site of the Gq-only agonist, TAK-875 in the recently published high resolution X-ray structure of GPR40 [32].
2. Materials and methods
2.1. Compounds
DHA, ALA and GW9508 were purchased from Sigma–Aldrich (Steinheim, Germany). Other GPR40 ligands were synthesized as published TAK-875 [33], AM-8182, AM-1638 and [3H]AM-1638 [27,34,35], AM-5262 [28], MK-2305 and [3H]L358 [36].
2.2. Molecular biology
Receptor cDNAs were cloned into the eukaryotic expression vector pCMV-Tag(2B) (Stratagene, CA, USA) (IP-turnover and cAMP accumulation) and pcDNA3.1+ (Life Technologies, CA, USA) (binding assay). All constructs were verified by DNA sequence analysis.
2.3. Cell culture and transfections
COS-7 cells were grown in Dulbecco's modified Eagle's medium 1885 (DMEM) supplemented with 10% fetal calf serum, 2 mM glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. COS7 cells were transiently transfected (20 μg/75 cm2) using the calcium phosphate precipitation method with chloroquine addition. CHO-k1 cells were stably transfected with human GPR40 using FuGENE6 transfection reagent (Roche Diagnostics) according to manufacturer's protocol. Stably expressing cells were cultured in Minimum Essential Medium Eagle (Sigma–Aldrich) supplemented with 10% FBS, 2 mM glutamine, 0.01 mg/ml gentamicin and 0.05 mg/ml zeocin and 1 mg/ml G418 for selection of GPR40 containing cells.
2.4. Binding assay
5 μg/well membrane together with 200 μg/well of PVT WGA SPA beads (GE #RPNG-0001) were suspended in a white 96 well plate with a final concentration of 50 μl/well in assay buffer (50 mM Tris, 10 mM MgCl2 & 2 mM EDTA). The plate was shaken aggressively for 30 min before adding 20 μl/well of compound in 0.1% BSA assay buffer and 30 μl/well of radio-ligand in 0.5% assay buffer yielding a final concentration of 15,000 cpm/well. The plate is briefly mixed and incubated for 3–18 h at room temperature, before it is read on a MicroBeta plate counter (Perkin Elmer, MS, USA).
2.5. IP turnover assay
One day after transfection COS-7 cells were plated into poly-d-lysine-coated 96-well plates (20,000 cells/well) and incubated with 0.5 μCi/ml myo[3H]inositol (Perkin Elmer) in 100 μl growth medium. The following day cells were washed twice with HBSS (Gibco, Life Technologies) and incubated for 30 min 37 °C in 100 μl buffer supplemented with 10 mM LiCl prior to ligand addition followed by 90 min incubation 37 °C. Cells were lysed with 50 μl 10 mM formic acid followed by incubation on ice for 30–60 min 20 μl of the extract were transferred to a white 96-well plate and 80 μl of 1:8 diluted YSi poly-d-lysine coated beads (Perkin Elmer) were added. After vigorously shaking the plate was centrifuged for 5 min at 1500 rpm, and γ-radiation was counted on a Packard Top Count NXT counter after an 8 h delay. Determinations were made in duplicates.
2.6. cAMP accumulation assay
Transient transfected COS-7 cells were plated into poly-d-lysine-coated white 96-well plates (20,000 cells/well). The following day, the cells were washed twice in HBS and incubated in HBS containing isobutyl-1-methylxanthine for 30 min at 37 °C. Varying concentrations of ligands were added and incubated in 30 min at 37 °C, followed by cAMP accumulation assay using the DiscoveRx HitHunter™ cAMP XS-kit (Birmingham, UK) according to manufacturer's protocol. Determinations were made in duplicates. Chemiluminescense was read on an EnVison 2104 Multilabel Reader (Perkin Elmer).
2.7. Molecular modeling
The structure of the human GPR40 receptor (PDB code: 4PHU, solved to 2.3 Å resolution in complex with TAK-875) [32] was imported into the internal coordinate mechanics software package (ICM 3.7-3b, Molsoft L.L.C., San Diego, CA, USA). Crystallization water and co-crystal molecules were deleted and the structure was converted into an ICM object to optimize the position of hydrogen atoms. The compounds DHA, AMG-837, TAK-875, MK-2305, AM-8182, AM-5262, AM-1638 and TUG-761 were docked to the TAK-875 binding site (pocket #1) and two additional pockets (pocket #3 and Pocket #4 – see Figure 3) identified by the icmPocketFinder method (default threshold 4.6) using the step-by-step docking menu employed in ICM [37]. The docking procedure included: (i) Setup receptor; for each pocket the grid box was adjusted to cover all atoms within 15 Å from the center of the identified pockets. (ii) Setup batch ligand; (iii) run docking batch; thoroughness 10.0; assigning 3D ligand conformations, MMFF atom types and formal charges to ionizeable groups. The ligand docking poses predicted for the three different pockets were ranked based on ICMs empiric ligand docking scoring function weighted according to the following parameters: (i) internal energy of the ligand, (ii) entropy loss of the ligand between bound and unbound states, (iii) ligand-receptor hydrogen bond interactions, (iv) polar and non-polar solvation energy differences between bound and unbound states, (v) electrostatic energy, (vi) hydrophobic energy, and (vii) hydrogen bond donor or acceptor desolvation – and therefore unitless. Generally a score below −32 is regarded as a good docking score based on assessments of ligand docking scores of high resolution receptor and protein ligand complexes. Furthermore, the ligand conformational strain of all the best-scored docking poses was computed to confirm that the conformations of the bound ligands were accepted in low energy minimums. The final docking complexes were not refined further e.g. by including side chain flexibility to certain side chains.
Figure 3.
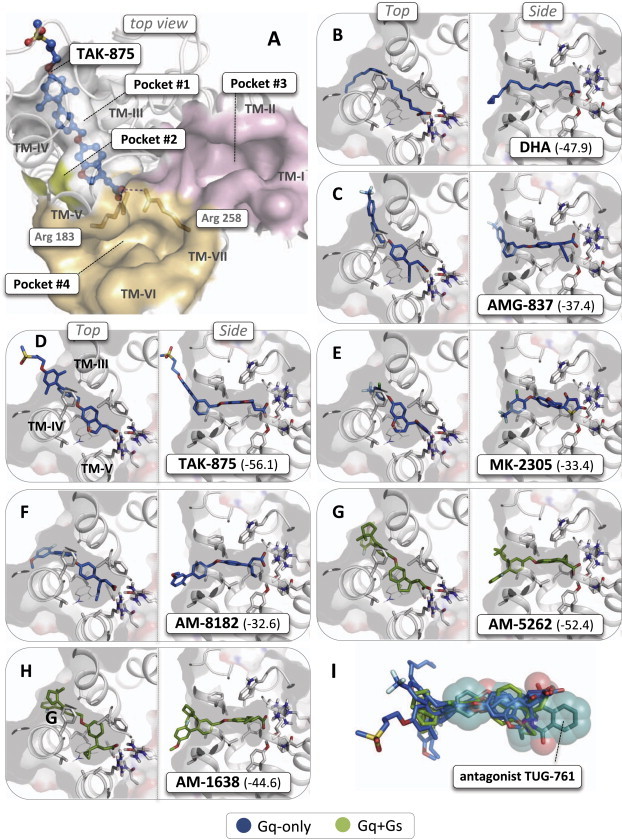
Molecular docking of GPR40 agonists in crystal structure of the human GPR40. (A) Overview of possible ligand binding pockets in the crystal structure of the human GPR40 receptor in complex with TAK-875 (PDB code: 4PHU, solved to 2.3 Å resolution) as viewed from the extracellular side [32]. The binding site for TAK-875 is covered by residues mainly from extracellular loop 3 (transparent) and the location of TM-I to TM-VII are indicated as well as the two Arg residues forming the anchor point for the carboxylic acid moiety of TAK-875. (B–H) Best scored docking poses of the Gq-only agonist shown in blue stick models, DHA, AMG-837, TAK-875, MK-2305, AM-8182 and the Gq-Gs agonists shown in gree stick models, AM-1638 and AM5262 to the TAK-875 binding pocket (docking scores are specified in brackets – see Section 2). (I) Superimposition of the antagonist TUG-761 (transparent spheres and dark green stick model) onto the agonists in their docked binding conformations.
2.8. Primary colonic crypt cultures
Colonic crypts were prepared from male C57BL/6 WT, Gpr40 KO or Gpr40/120 double KO littermate mice (obtained from Taconics, NY). The crypts were isolated by collagenase digestion as described by Reimann et al. [23] and seeded into 48-well plates coated with Matrigel (BD Biosciences). The following day, cells were incubated for 2 h with ligands (triplicates) at 37 °C in standard solution [23] containing 0.1% fatty acid-free BSA (Sigma–Aldrich) and 10 mM glucose. Synthetic ligands were dissolved in DMSO and ALA or DHA were complexed with free fatty acid BSA in a 6:1 molar ratio. Total GLP-1 was determined using Total GLP-1 (ver. 2) assay kit (Meso Scale Discovery, Gaithersburg, USA) (model number K150JVC-1) according to manufacturer's protocol.
2.9. In vivo
Adult (11–18 weeks) C57BL/6 Gpr40 KO mice and WT littermate mice were grouphoused with up to 8 mice in each cage at 24 °C on a 12:12 h light–dark cycle. After an overnight fast a retro orbital blood sample was taken (app. 200 μl) and mice were orally gavaged with either vehicle solution containing 0.5% (w/v) carboxymethylcellulose sodium salt, medium viscosity, (Sigma–Aldrich) or 30 mg/kg of TAK-875, MK-2305, AM-1638 or AM-5262 dissolved in vehicle solution. PK experiments had shown the following Cmax concentrations: TAK-875 – 2 μM; MK-2305 – 22 μM; AM-1638 – 11 μM; and AM5262 – 19 μM. Each mouse was given 10 ml/kg of the test solution. 30 min after the oral gavage, the mice were decapitated and blood was collected in EDTA coated microvettes (Sarstedt AG & Co, Nümbrecht, Germany) containing 0.5 KIU aprotinin/μl blood (Sigma Chemical, St. Louis, MO) and 1 μM DPP4 inhibitor (a kind gift from Novo Nordisk, Denmark). All blood samples were kept on ice at all times. The blood samples were centrifuged at 4 °C for 10 min at 9.6 g in a centrifuge (Heraeus Fresco 21 Microcentrifuge, Thermo Scientific, Denmark), plasma was collected and stored at −80 °C. Total GLP-1 was determined using Total GLP-1 (ver. 2) assay kit (Meso Scale Discovery, Gaithersburg, USA; model number K150JVC-1) according to manufacturer's protocol. GIP was measured using a Rat/Mouse Total GIP ELISA (Millipore, St. Charles, MO).
2.10. Calculations and statistical analyses
Data were visualized and tested for significance using the Prism 6.0 software (GraphPad Software, San Diego). Emax values, IC50 and EC50 values were determined by non-linear regression (variable slope). Differences in tGLP-1 release from primary colon cultures compared to vehicle were determined by non-parametric one-way ANOVA (Kruskal–Wallis) with Dunn's posthoc test of multiple comparisons. Statistical significance between WT and Gpr40 KO colonic crypt cultures was determined by a non-parametric unpaired two-tailed t-test (Mann–Whitney). In vivo, incretin release between groups of mice receiving compound compared to vehicle was determined by a parametric unpaired t-test with Welch's correction. Statistical significance is denoted by *(p < 0.05), **(p < 0.01), ***(p < 0.001).
3. Results
3.1. In vitro signal transduction properties of GPR40 ligands
GPR40 is normally considered to be a Gq-coupled receptor. However, based on the knowledge that receptors often can signal through more than one pathway and the fact that Gs signaling very efficiently stimulates GLP-1 secretion, for example through activation by GPR119, we decided to study the effect of a series of six different synthetic GPR40 agonists and two endogenous lipid ligands in respect of stimulation of both IP accumulation and cAMP production in COS-7 cells transiently transfected with the human GPR40 receptor.
In the Gq mediated IP-turnover experiments the classical GPR40 agonist GW9508 [13] was used as a reference compound. The two endogenous ligands ALA and DHA stimulated IP-accumulation efficiently, but with somewhat lower Emax values than GW9508, i.e. 53 ± 4% and 56 ± 6% of the Emax of GW9508, and with EC50 values of 17 μM and 12 μM, respectively (Figure 1A).
Figure 1.

In vitro signaling properties of GPR40 agonists in transiently transfected COS7 cells. Effects on IP-turnover (black curves, left panels using GW9508 as positive control – stippled line) and cAMP accumulation (red curves, right panels) were determined for: (A) Endogenous lipid agonists α-linolenic acid (ALA) and docosahexaenoic acid (DHA); (B) synthetic agonists TAK-875, AMG 837, MK-2305, AM-8182, AM-1638 and AM-5262. Black curves in the cAMP accumulation panels for AM-1638 and AM-5262 represent empty vector control. Data are normalized to empty vector (0%) and Emax for GW9508 (100%) in IP-turnover. Concentrations of cAMP were interpolated based on cAMP standard curves for each experiment. Data represents mean ± SEM and represents a minimum of three experiments.
The six synthetic GPR40 agonists stimulated IP-accumulation with rather similar potencies (Table 1) and with maximal efficacies ranging from 68 ± 5 (MK-2305) to 179 ± 14%, (AM-5262) of the Emax for GW9508 (Figure 1B).
Table 1.
Summary of GPR40 ligand pharmacology and ex vivo response.
| Ligand | Binding (in vitro) |
IP3 (in vitro) |
cAMP (in vitro) |
tGLP-1 (ex vivo) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| [3H]L358 LogIC50 | [3H]AM-1638 LogIC50 | ||||||||||
| LogEC50 | Emax | LogEC50 | Emax | WT | GPR40 (−/−) | ||||||
| Endogenous | ALA | n.d. | n.d. | −4.8 ± 0.19 | 53 ± 4 | >−3 | 1.39 ± 0.4 | 1.4 ± 0.06a | 1.0 ± 0.04a | ||
| DHA | n.d. | n.d. | −4.9 ± 0.24 | 56 ± 6 | >−3 | 1.50 ± 0.5 | n.d | n.d | |||
| Orthosteric | TAK-875 | −7.7 ± 0.05 | (pos) | −7.9 ± 0.07 | (neg) | −7.9 ± 0.14 | 98 ± 4 | >−5 | 0.13 ± 0.6 | 1.5 ± 0.08 | 1.1 ± 0.12 |
| MK-2305 | −7.5 ± 0.05 | (pos) | −7.7 ± 0.07 | (neg) | −7.9 ± 0.24 | 68 ± 5 | >−5 | 0.70 ± 0.4 | 2.8 ± 0.42 | 1.1 ± 0.06 | |
| AM-8182 | −7.6 ± 0.03 | (pos) | −8.2 ± 0.17 | (neg) | −7.3 ± 0.11 | 171 ± 9 | >−5 | 1.48 ± 0.6 | 1.4 ± 0.19 | 1.4 ± 0.14 | |
| Ago-allosteric | AM-1638 | −7.6 ± 0.05 | (neg) | −7.5 ± 0.07 | (pos) | −7.6 ± 0.20 | 170 ± 14 | −6.8 ± 0.13 | 13.01 ± 1.1 | 3.0 ± 0.54 | 1.1 ± 0.07 |
| AM-5262 | −8.1 ± 0.18 | (neg) | −8.0 ± 0.05 | (pos) | −7.7 ± 0.19 | 179 ± 14 | −7.0 ± 0.13 | 12.63 ± 0.1 | 4.6 ± 0.77 | 1.2 ± 0.13 | |
Data are representative of two or more experiments, and expressed as mean ± SEM. n.d; not determined – binding was not carried out with the endogenous ligands, and ex vivo tGLP-1 release was not carried out with DHA. (pos) and (neg) indicates positive or negative competitive binding with radio labeled ligand. Ex vivo tGLP-1 release (fold change) as determined with 1 μM ligand, aexcept for ALA which was 30 μM.
In respect of Gs signaling, the two endogenous lipid ligands ALA and DHA were as expected not able to stimulate cAMP accumulation meaningfully through GPR40 (Figure 1A). Like the endogenous lipid ligands, four of the synthetic GPR40 agonists, TAK-875, AM-837, MK-2305, and AM-8182, showed no or very limited stimulation of cAMP production (Figure 1B).
However, surprisingly GPR40 could in fact signal through Gs, but only when stimulated with a subset of synthetic agonists, here represented by AM-1836 and AM-5262. Thus, in cells transfected with GPR40 as opposed to cells transfected with empty vector AM-1638 and AM-5256 stimulated cAMP accumulation robustly (Figure 1). The EC50 values for cAMP stimulation by AM-1638 and AM-5162 were 160 and 100 nM, respectively, which is 7 and 6 fold lower than the potencies for IP-accumulation (Table 1). In both the IP3 and cAMP assay, AM-1638 and AM-5262 displayed bell-shaped dose–response curves with a signal decline between 1 and 10 μm (Figure 1B). This phenomenon was also observed for several of the other GPR40 agonists in the IP3 assays (Figure 1B).
It is concluded that GPR40 can signal not only through Gq as observed with the endogenous lipid ligands but also in a ligand-biased fashion through Gs. In the following we differentiate between ‘Gq-only’ agonists, which here are TAK-875, AM-837, MK-2305 and AM-8182 and ‘Gq + Gs’ agonists, i.e. AM-1638 and AM-5262.
3.2. Binding properties of GPR40 agonists
Competition binding experiments were performed in membranes from stably transfected CHO-K1 cells using two different radio ligands: [3H]L358, which is a close analog of the Gq-only agonist MK2305, and the Gq + Gs agonist [3H]AM-1638.
As shown in Figure 2B, the Gq-only agonists TAK-875, AM-837, AM-8182 and MK-2305 all displaced [3H]L358 efficiently and with surprisingly similar affinities ranging from 20 to 36 nM. In contrast, the two Gq + Gs agonists AM-1638 and AM-5262 did not compete for [3H]L358 binding but instead displayed positive co-operativity by increasing [3H]L358 binding with apparent affinities of 26 and 8 nM, respectively (Figure 2B). However, when the Gq + Gs agonist [3H]AM-1638 was used as radio-ligand both AM-1638 and AM-5262 competed efficiently for binding with affinities of 29 and 10 nM, respectively (Figure 2C). In contrast, the Gq-only agonists TAK-875, AM-837, AM-8182 and MK-2305 did not displace but instead all increased the binding of the Gq + Gs agonist [3H]AM-1638 with apparent affinities ranging from 6 to 17 nM (Figure 2C).
These binding data indicate that the Gq + Gs agonists bind to GPR40 in a very different manner than the Gq-only agonists and that the use of the two distinct radio ligands, [3H]L358 and [3H]AM-1638, differentiate between these binding modes in a reciprocal manner.6
3.3. Molecular modeling of the binding mode of Gq-only versus Gq + Gs agonists in GPR40
The high resolution X-ray structure of GPR40 in complex with the Gq-only agonist TAK-875 was recently published in Ref. [32], which provides a unique basis for computational chemistry analysis of potential binding modes of other GPR40 ligands. As shown in Figure 3, TAK-875 binds in a pocket or channel extending out into the lipid bilayer between TM-III and TM-IV from the anchor point for the carboxylic acid group centered around the guanidinium groups of Arg183 (ArgV:05) and Arg258 (ArgVII:02). As viewed from the extracellular side, this binding pocket is totally covered by residues from TM-III, -IV and -V and extracellular loop 2. A small, empty ‘side-pocket’ is found between TM-IV and TM-V (originally called ‘pocket #2’) close to the carboxylic acid moiety of TAK-875. Two other relatively large, empty pockets are found on the opposite side of the carboxylic acid anchor point, i.e. an elongated pocket extending between TM-III and TM-VII over towards TM-I and TM-II (pocket#3) and a smaller pocket located between TM-V, -VI and -VII (pocket #4).
Using the ICM docking and scoring software all the GPR40 agonists employed in the signaling and binding assays described above docked well into the binding site for TAK-875 (pocket #1) using the unmodified GPR40 X-ray structure from which TAK-875 had been removed. In view of the clearly cooperative binding properties it was very surprising that both Gq-only and Gq + Gs agonists all docked into the same binding site (Figure 1B–H). As expected, TAK-875 itself was the top scorer (−56.1), whereas the docking score for the other agonists ranged from −52.4 (AM-5262) to −32.6 (AM-8182) which all are regarded as good scores. Surprisingly, the two Gq + Gs agonists, AM-1638 (−44.6) and AM5262 (−52.4) docked better into the TAK-875 pocket than the three Gq-only agonists, AM-8182 (−32.6), MK-2305 (−33.4) and AMG-837 (−37.4) (Figure 3B–H). Importantly, all the GPR40 agonists docked into the TAK-875 site with rather low intra-molecular strain of the ligand i.e. accepted in low energy conformations. Interestingly, the different chemical moieties extending from the propionic acid group in several of the agonists nicely fitted into the small side-pocket (pocket #2) not used by TAK-875, for example the propyne, propene and cyclopropyl moiety of AMG-837, AM-8182 and AM-1638, respectively (Figure 3C,F and H). In contrast to the agonists, the potent dihydro-isoquinoline based GPR40 antagonist TUG-761 could not be docked into the TAK-875 binding pocket in any meaningful way, do to major structural differences compared to the agonists as illustrated in Figure 3I, where TUG-761 is overlaid on the structures of the collection of agonists as they docked into the TAK-875 site. Attempts to dock the agonists or the antagonist into the very polar pocket #3 or the shorter pocket #4 were also unsuccessful.
It is concluded that all the agonists in contrast to the antagonist TUG-761 dock very well into the TAK-875 binding site in the high resolution X-ray structure of GPR40 and that the Gq + Gs agonists surprisingly dock at least as good if not better than the Gq-only agonists in this site.
3.4. GPR40 agonist-mediated GLP-1 secretion ex vivo
In mouse colonic crypt cultures direct treatment with the endogenous LCFAs ALA or DHA led to 20- to 30-fold large increases in GLP-1 release above basal, which however were also observed in cultures from Gpr40-deficient mice (data not shown). However, treatment of the primary cell cultures with BSA-conjugated ALA resulted in small but GPR40-dependent GLP-1 release of 1.4 ± 0.06 fold above basal at 30 μM (Figure 4A). Slightly larger increases in GLP-1 secretion of 2.0 ± 0.2 and 2.6 ± 0.3 fold above basal were observed with concentrations of 0.1 and 1 mM of BSA-conjugated ALA (Figure 4A). However, at these concentrations a large fraction of the responses was also observed in cells from Gpr40-deficient mice and even in cells from Gpr40/Gpr120 double-deficient mice (Figure 4A), indicating that even when using BSA as a carrier ALA cause unspecific damage in this cell-based assay. Similar results were obtained with BSA-conjugated DHA (data not shown). Importantly, the small GPR40-dependent GLP-1 response to ALA was only a fraction of the 4.7 ± 0.3 fold increase in GLP-1 observed with the positive control, the GPR119 agonist AR231453.
Figure 4.

GPR40 agonist-dependent GLP-1 release ex vivo – in murine colonic crypt cultures. (A) Total GLP-1 (tGLP-1) release from primary cell cultures from WT (solid), GPR40(−/−) mice (grid) or GPR40/GPR120 double deficient mice (stripes) treated with 1 μM GPR119 agonist AR231453 or varying concentrations of α-linolenic acid (ALA) as indicated in complex with BSA. One out of two experiments performed in triplicates is shown. (B) As panel A but treated with 1 μM GPR40 synthetic agonists TAK-875, MK-2305, AM-8182, AM-1638 or AM-5262. Numbers in brackets indicate number of repeated experiments in triplicates. Data are normalized to the basal secretion of tGLP-1 from vehicle treated cells and are shown as mean ± SEM. Statistical significance of WT compared to GPR40(−/−) (crotchets) or vehicle compared to GPR40(−/−) (above) is denoted by *(p < 0.05),**(p < 0.01), ***(p < 0.001) as determined by non-parametric unpaired two-tailed t-test (Mann–Whitney).
In contrast, the two synthetic Gq-only GPR40 agonists, TAK-875 and MK-2305, both stimulated GLP-1 secretion from the colonic crypt cultures in a GPR40-dependent way, albeit still with efficacies less than the GPR119 agonist, AR231453, i.e. with responses of 1.5 ± 0.08 and 2.8 ± 0.4 fold above basal, respectively (Figure 4B). However, surprisingly the third Gq-only agonist, AM-8182, which in respect of stimulation of IP turnover in vitro acted as a highly efficacious GPR40 agonist only gave a small GLP-1 response of 1.4 ± 0.2 fold above vehicle treated cells and a similar response, was observed in cells from Gpr40-deficient animals (Figure 4).
In contrast to the Gq-only the two Gq + Gs GPR40 agonists AM-1638 and AM-5262 both stimulated GLP-1 secretion from the mouse colonic crypt cultures with efficacies similar to and even higher than the GPR119 positive control agonist, i.e. 3.0 ± 0.5 fold and 4.6 ± 0.8 fold, respectively, which in the case of AM-5262 is the highest receptor medicated efficacy we have yet observed in this preparation. For both of the Gq + Gs agonists the GLP-1 stimulation was GPR40-dependent (Figure 4B).
3.5. Effect of GPR40 agonists on GLP-1 and GIP release in vivo
The two Gq-only GPR40 agonists, which were efficacious in the ex vivo colonic crypt cultures, TAK-875 and MK-2305 and the two Gq + Gs agonists, AM-1638 and AM-5262 were tested in vivo in lean mice after an overnight fast dosed orally with 30 mg/kg based on initial dose-range finding experiments. The four GPR40 agonists were all tested head-to-head and against vehicle with 6–8 mice in each group in two individual experiments one of which is shown in Figure 5A. In total, three to five experiments were done with each compound in various combinations and a summation of all the experiments is shown in Figure 5B.
Figure 5.

GLP-1 and GIP plasma responses to GPR40 agonists in vivo in mice. tGLP-1 and GIP plasma concentrations were measured 30 min after an oral administration of 30 mg/kg of the orthosteric agonists TAK-875, MK-2305 (in blue) and the ago-allosteric agonists, AM-1638 or AM-5262 (in green). (A) A representative head-to-head experiment in male B6 mice; (B) Summation of multiple experiment (X–Y) using the four ligands in various combinations, column indicate the median of incretin response in all experiments and symbols indicate response in each experiment (black squares – experiment in ‘A’); (C) Effect of the GPR40 agonists in female WT versus GPR40(−/−) littermate mice (this experiment is shown in open circle in ‘B’). Statistical significance compared to vehicle (A) or GPR40(−/−) (C) is denoted by *(p < 0.05), **(p < 0.01), ***(p < 0.001) as determined by unpaired t-test assuming Gaussian distribution with Welch's correction.
Concerning the Gq-only agonists, TAK-875 increased plasma GLP-1 significantly in three experiments but only with a relatively small median increase from 5.1 to 10.8 pmol/L (Figure 5B left panel). MK-2305 increased plasma GLP-1 levels significantly in all five experiments with a median increase from 5 to 12 pmol/L (Figure 5B). The Gq + Gs GPR40 agonists both increased plasma GLP-1 significantly in all experiments and to a larger degree, i.e. both AM-1638 and AM-5262 increased plasma GLP-1 from 5 to 29 pmol/L (Figure 5B). In the head-to-head experiments the Gq + Gs agonists increased plasma GLP-1 significantly more than the Gq-only agonists (Figure 5A).
Concerning effects on GIP, TAK-875 only increased plasma GIP significantly in one of the three experiments, whereas MK-2305 increased plasma GIP in four out of the five experiments giving a median increase from 53 to 108 pmol/L (Figure 5B right panel). Also in the case of GIP, the two Gq + Gs agonists were more efficacious and increased plasma GIP to 383 (AM-1638) and 345 pmol/L (AM-5262) (Figure 5B). In the head-to-head experiments the increases in plasma GIP were significantly higher for the Gq + Gs agonists than for the Gq-only agonists (Figure 5A). The in vivo stimulatory effects of AM-5262 on both GLP-1 and GIP were GPR40-dependent, (Figure 5C). The in vivo stimulatory effects were similar in male and female mice (Figure 5B,C).
4. Discussion
In the present study we find that GPR40, which generally is considered to be a Gq coupled receptor surprisingly can signal efficiently through Gs to increase intracellular cAMP; however not in response to the endogenous lipid ligands but in a ligand-dependent biased way, i.e. in response to some but not other synthetic agonists. Importantly, compounds which are capable of activating both the Gq and the Gs signaling pathway are particular efficient incretin secretagogues, i.e. these compound stimulate GLP-1 and GIP secretion robustly both ex vivo and in vivo.
4.1. Gq and Gs co-action on hormone secretion
Traditionally, both Gq and Gs signaling have been strongly implemented in peptide hormone secretion. However, the relative importance and precise mechanisms vary from system to system. In general, the main routes from Gq to hormone secretion goes through phospholipase C (PLC), IP3 and increase in intracellular Ca2+ released from ER stores which acts directly on the molecular exocytosis machinery for the hormone-containing secretory vesicles (Figure 6). On the other hand, the pathway from Gs classically goes through adenylyl cyclase, increase in cAMP and either PKA or Epac (exchange protein directly activated by cAMP) [38,39].
Figure 6.

Schematic overview of signal transduction pathways connecting receptors sensing triglyceride metabolites with incretin secretion. In the physiological setting triglyceride ingestion will lead to concomitant LCFAs stimulation of the Gq-coupled GPR40 and potentially but probably not GPR120[14]; 2-monoacyl glyceride stimulation of Gs through GPR119 and bile acid stimulation of Gs through TGR5 resulting in strong incretin secretion. In the pharmacological setting orthosteric GPR40 agonists (blue) will like LCFAs only stimulate Gq pathways resulting in a rather limited incretin response on their own. In contrast, ago-allosteric agonists (blue and green) will stimulate both Gq and Gs pathways leading to a robust incretin response.
Potentiating interaction between the cAMP and Ca2+ signaling pathways in hormone secretion is probably best described in the β-cell where, for example the GLP-1 receptor-mediated increase in cAMP potentiates glucose-induced insulin secretion elicited by intracellular Ca2+, which in this case is initiated through the ATP-sensitive potassium channel and opening of voltage-dependent calcium channels in the cell membrane [40]. Thus, a combined increase in intracellular cAMP and Ca2+, which can be achieved in several different manners, is a highly efficient way of stimulating hormone secretion. In the case of incretin secretion this can be achieved physiologically through concomitant activation of different Gq- and Gs-coupled receptors and pharmacologically through activation of, for example GPR40 by Gq + Gs agonists as described in the present study.
4.2. GPR40 acts jointly with Gs-coupled receptors during physiological sensing of dietary triglycerides
When it originally was proposed that GPR40 mediates LCFA stimulation of incretin secretion, this was not based on LCFA challenges but only inferred from studies with triglycerides in wild type versus knock out animals [22,29,41]. Here we find that LCFAs like the Gq-only synthetic agonists stimulate GLP-1 secretion with low efficacy ex vivo and only signals through GPR40 via Gq and not Gs in vitro (Figure 2A). Interestingly, in order to get appropriate stimulation of GIP secretion by, for example linoleic acid, Parker and coworkers had to pre-treat the primary cell cultures with forskolin and IBMX, which increase cAMP concentrations [42]. Importantly, the selective orthosteric GPR40 agonist TAK-875 is also rather poor in stimulating incretin secretion and did not increase plasma GLP-1 at therapeutic doses in clinical trials [25]. Thus, it appears that stimulation of GPR40 by LCFAs or by orthosteric agonists is not an efficient stimulus for incretin release – on its own. However, under physiological circumstances dietary triglycerides are sensed by the enteroendocrine cells not only in the form of LCFAs but also by 2-MAG acting on the Gs coupled receptor GPR119 (Figure 6) [9]. Moreover, another important part of the physiological absorption and digestion of dietary triglycerides is their formation of micelles with bile acids, which are liberated in the distal small intestine to be sensed by the Gs-coupled TGR5 receptor (Figure 6). Thus, although the LCFAs by themselves only result in a rather inefficient Gq signaling through GPR40, dietary triglycerides are in fact highly efficient stimulators of incretins conceivably as a result of a joint action of LCFAs though the Gq-coupled GPR40 plus 2-MAG and bile acids acting through the Gs-coupled GPR119 and TGR5, respectively (Figure 6).
4.3. Pharmacological activation of both Gq and Gs by certain synthetic GPR40 agonists
As demonstrated in the present study, efficient pharmacological stimulation of both Gq and Gs can be obtained through GPR40 alone with agonists such as AM-1638 and AM-5262 and we propose that this dual signaling property is the molecular pharmacological property associated with efficient stimulation of incretin release both ex vivo and in vivo. However, although these compounds are clearly able to stimulate cAMP in vitro in transfected ‘test tube cells’, we do not yet know whether such compounds actually exploit the Gs signaling pathway in vivo in the enteroendocrine GLP-1 and GIP cells or for that matter in the pancreatic β-cells. We just know that GPR40 agonists, who are able to increase cAMP also, are efficacious in stimulating incretin secretion.
AM-1638 and AM-5262 have previously been referred to as ‘full agonists’, which is based on the fact that in certain in vitro Gq measuring systems such as Ca2+, they display efficacies which are similar to ALA and DHA as opposed to, for example AMG-837 and TAK-875, which accordingly have been called ‘partial agonists’, i.e. in particular in relation to stimulation of intracellular Ca2+ [26,27,31]. In the present study we also find that AM-1638 and AM-5262 have higher maximal efficacies in IP3 signaling than, for example TAK-875 and AMG-837. However, in our hands all of these ligands display efficacies above the efficacy of the endogenous ligands ALA and DHA (Figure 1). Importantly, there is not a direct correlation between high efficacy in Gq signaling and high signaling through Gs and there is not a direct correlation between high efficacy in Gq signaling and robust incretin secreting effect, as exemplified by AM-8182, which is as efficient as AM-1638 and AM-5262 in respect of Gq signaling but very poorly if at all in signaling through Gs and does not release GLP-1. However, there are compounds such as MK-2305, which does not signal through Gs, but in certain experiments does increase plasma GLP-1 and GIP fairly well, albeit in total not as well as AM1638 and AM5262 (Figure 5).
4.4. Complex allosteric properties of GPR40 ligands in binding assays
The clear positive cooperative effects of the Gq-only and the Gq-Gs agonists on each other as determined in our radio ligand binding assays confirm the complex allosteric properties of GPR40 ligands [26,27,31]. Swaminath and coworkers have pioneered the field of developing and characterizing ago-allosteric modulators for GPR40 where they recently showed that certain second generation ago-allosteric agonists can improve glucose tolerance not only through direct stimulation of insulin secretion but also through stimulation of incretins [27,29,31].7 They suggested that there may be even two allosteric sites or binding modes for the structurally closely related agonists besides the orthosteric binding site for the endogenous lipid ligands [27]. As observed in other systems the properties of the GPR40 ligands are probe-dependent and for example give very different allosteric pictures dependent upon which radio-ligand is used. Thus, in the present study we find that both AM-8182 and AM-837 compete with very similar high potency and full efficacy against [3H]-L358 indicating that all three ligands share a common binding mode (Figure 2). In contrast, Lin and coworkers found that AM-8182 displaced [3H]-AM837 with several orders of magnitude lower affinity and based on these binding data and on functional data, they suggest that AM-8182 binds to another site than AM-837 [27]. From a functional point-of-view positive cooperativity has been demonstrated between different types of synthetic agonists and between the synthetic agonists and the endogenous lipid ligands, such as DHA or ALA, even in cases where the ligands show negative cooperativity in binding experiments [27]. For example, although TAK-875 only stimulates insulin secretion with a rather low efficacy on its own, it has robust glucose-dependent stimulatory effects on insulin secretion in combination with the endogenous LCFA γ-linoleic acid [26].
4.5. Apparent simple, common binding modes for agonists in the GPR40 X-ray structure
In view of the fact that the different types of agonists show strong signs of allosteric binding properties, it was highly surprising that all agonists docked nicely with minimal internal strain and with high energy scores into a common binding site in the X-ray structure of GPR40 (Figure 3). Importantly, although several other potential binding pockets could be identified in the ‘main ligand-binding pocket’ of the receptor structure where ligands bind in other 7TM GPCRs [46–48] (Figure 3), it was not possible to dock any of the known agonists into these alternative pockets in any meaningful way.
It should be noted that in order to obtain the crystal structure a number of termo-stabilizing mutants were introduced in GPR40 one of which was an Ala-substitution of Phe88 (PheIII:10) in TM-III located just below the binding pocket for TAK-875. Most importantly, although the five termo-stabilizing mutants did not affect ligand binding, the receptor could not be activated by the agonists [32]. In other words, we are docking ligands into an X-ray structure of GPR40 which has been ‘frozen’ in a conformation where the allosteric mechanism between the agonist binding site and the G proteins, in this case Gq or Gs is no longer present and where a large Phe residue, which perhaps could interchange between different rotameric forms, has been exchanged with an Ala residue – close to the binding pocket. Thus, although the present GPR40 structure is a great starting point, it is only the first picture of a very complex allosteric receptor system, where agonists and allosteric modulators could stabilize active conformations at different or at more or less overlapping orthosteric and allosteric sites as previously proposed for ago-allosteric agonists [30,49]. GPR40 appears to be a particularly interesting system to study allosteric mechanisms in and with a wealth of pharmacological tools. With all the many X-ray structures available today, we are starting to get a picture of the structural basis for allosteric binding modes and for biased signaling in ‘multi-state’-models allowing multiple ligand-specific conformations [50,51].
5. Conclusion
Under physiological conditions GPR40 is probably a strictly Gq-coupled receptor being activated by LCFAs which alone only provides a rather poor incretin response but conceivably act in symphony with the Gs-coupled receptors GPR119 sensing the other major triglyceride metabolite 2-MAG and TGR5 being stimulated by bile acids to collectively elicit a strong incretin response. A similar robust incretin response can be obtained pharmacologically through GPR40 alone with Gq + Gs agonists, which in contrast to LCFAs and synthetic Gq-only agonists can bias GPR40 to signal through both Gq and Gs at the same time. This biased signaling and strong incretin response by ago-allosteric agonists could make GPR40 an even more interesting target for the future treatment of diabetes..
Acknowledgments
We are grateful for the expert technical assistance from Susanne Hummelgaard and for statistical support from Frank Eriksson. The Novo Nordisk Foundation Center for Basic Metabolic Research (http://www.metabol.ku.dk) is supported by an unconditional grant from the Novo Nordisk Foundation to University of Copenhagen. The project was also supported by the UNIK project for Food, Fitness & Pharma (http://www.foodfitnesspharma.ku.dk) from the Danish Ministry of Science, Technology and Innovation. M.H. and M.A.V. received PhD scholarships from the Faculty of Health and Medical Sciences, University of Copenhagen.
Footnotes
Takeda press release, December 27, 2013; http://www.takeda.com/news/2013/20131227_6117.html.
In the present report we do not use the terms orthosteric and allosteric binding modes and do not try to differentiate between the two GPR40 allosteric binding modes proposed by Lin and coworkers because we use [3H]L358 which gives a more simple competition binding picture where the different ligands basically fall into two rather well defined groups in contrast to the more complex picture obtained with [3H]AM-837 as used by the Amgen group [27].
According to classical pharmacological concepts and definitions, an agonist could not be an allosteric modulator [43,44]. However, many synthetic agonists are in fact able to increase the potency and/or the efficacy of the endogenous agonist, i.e. they are ago-allosteric modulators [30], which also now is acknowledged by IUPHAR [45].
Conflict of interest
None declared.
References
- 1.Holst J.J. The physiology of glucagon-like peptide 1. Physiological Reviews. 2007;87:1409–1439. doi: 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- 2.Irwin N., Flatt P.R. Enteroendocrine hormone mimetics for the treatment of obesity and diabetes. Current Opinion in Pharmacology. 2013;13:989–995. doi: 10.1016/j.coph.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 3.Engelstoft M.S., Egerod K.L., Lund M.L., Schwartz T.W. Enteroendocrine cell types revisited. Current Opinion in Pharmacology. 2013;13:912–921. doi: 10.1016/j.coph.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 4.Bohorquez D.V., Liddle R.A. Axon-like basal processes in enteroendocrine cells: characteristics and potential targets. Clinical and Translational Science. 2011;4:387–391. doi: 10.1111/j.1752-8062.2011.00299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmidt J.B., Gregersen N.T., Pedersen S.D., Arentoft J.L., Ritz C., Schwartz T.W. Effects of PYY3-36 and GLP-1 on energy intake, energy expenditure and appetite in overweight men. American Journal of Physiology. Endocrinology and Metabolism. 2014;306:E1248–E1256. doi: 10.1152/ajpendo.00569.2013. [DOI] [PubMed] [Google Scholar]
- 6.Engelstoft M.S., Egerod K.L., Holst B., Schwartz T.W. A gut feeling for obesity: 7TM sensors on enteroendocrine cells. Cell Metabolism. 2008;8:447–449. doi: 10.1016/j.cmet.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 7.Reimann F., Tolhurst G., Gribble F.M. G-protein-coupled receptors in intestinal chemosensation. Cell Metabolism. 2012;15:421–431. doi: 10.1016/j.cmet.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 8.Elliott R.M., Morgan L.M., Tredger J.A., Deacon S., Wright J., Marks V. Glucagon-like peptide-1 (7–36)amide and glucose-dependent insulinotropic polypeptide secretion in response to nutrient ingestion in man: acute post-prandial and 24-h secretion patterns. Journal of Endocrinology. 1993;138:159–166. doi: 10.1677/joe.0.1380159. [DOI] [PubMed] [Google Scholar]
- 9.Hansen H.S., Rosenkilde M.M., Holst J.J., Schwartz T.W. GPR119 as a fat sensor. Trends in Pharmacological Sciences. 2012;33:374–381. doi: 10.1016/j.tips.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 10.Itoh Y., Kawamata Y., Harada M., Kobayashi M., Fujii R., Fukusumi S. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature. 2003;422:173–176. doi: 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- 11.Briscoe C.P., Tadayyon M., Andrews J.L., Benson W.G., Chambers J.K., Eilert M.M. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. Journal of Biological chemistry. 2003;278:11303–11311. doi: 10.1074/jbc.M211495200. [DOI] [PubMed] [Google Scholar]
- 12.Kotarsky K., Nilsson N.E., Flodgren E., Owman C., Olde B. A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochemical and Biophysical Research Communications. 2003;301:406–410. doi: 10.1016/s0006-291x(02)03064-4. [DOI] [PubMed] [Google Scholar]
- 13.Briscoe C.P., Peat A.J., McKeown S.C., Corbett D.F., Goetz A.S., Littleton T.R. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. British Journal of Pharmacology. 2006;148:619–628. doi: 10.1038/sj.bjp.0706770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paulsen S.J., Larsen L.K., Hansen G., Chelur S., Larsen P.J., Vrang N. Expression of the fatty acid receptor GPR120 in the gut of diet-induced-obese rats and its role in GLP-1 secretion. PLoS One. 2014;9:e88227. doi: 10.1371/journal.pone.0088227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Overton H.A., Babbs A.J., Doel S.M., Fyfe M.C., Gardner L.S., Griffin G. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metabolism. 2006;3:167–175. doi: 10.1016/j.cmet.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 16.Jones R.M., Leonard J.N., Buzard D.J., Lehmann J. GPR119 agonists for the treatment of type 2 diabetes. Expert Opinion on Therapeutic Patents. 2009;19:1339–1359. doi: 10.1517/13543770903153878. [DOI] [PubMed] [Google Scholar]
- 17.Mancini A.D., Poitout V. The fatty acid receptor FFA1/GPR40 a decade later: how much do we know? Trends in Endocrinology & Metabolism. 2013;24:398–407. doi: 10.1016/j.tem.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 18.Steneberg P., Rubins N., Bartoov-Shifman R., Walker M.D., Edlund H. The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metabolism. 2005;1:245–258. doi: 10.1016/j.cmet.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 19.Latour M.G., Alquier T., Oseid E., Tremblay C., Jetton T.L., Luo J. GPR40 is necessary but not sufficient for fatty acid stimulation of insulin secretion in vivo. Diabetes. 2007;56:1087–1094. doi: 10.2337/db06-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wagner R., Kaiser G., Gerst F., Christiansen E., Due-Hansen M.E., Grundmann M. Reevaluation of fatty acid receptor 1 as a drug target for the stimulation of insulin secretion in humans. Diabetes. 2013;62:2106–2111. doi: 10.2337/db12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christiansen E., Hansen S.V., Urban C., Hudson B.D., Wargent E.T., Grundmann M. Discovery of TUG-770: a highly potent free fatty acid receptor 1 (FFA1/GPR40) agonist for treatment of type 2 diabetes. ACS Medicinal Chemistry Letters. 2013;4:441–445. doi: 10.1021/ml4000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edfalk S., Steneberg P., Edlund H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes. 2008;57:2280–2287. doi: 10.2337/db08-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reimann F., Habib A.M., Tolhurst G., Parker H.E., Rogers G.J., Gribble F.M. Glucose sensing in L cells: a primary cell study. Cell Metabolism. 2008;8:532–539. doi: 10.1016/j.cmet.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burant C.F., Viswanathan P., Marcinak J., Cao C., Vakilynejad M., Xie B. TAK-875 versus placebo or glimepiride in type 2 diabetes mellitus: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2012;379:1403–1411. doi: 10.1016/S0140-6736(11)61879-5. [DOI] [PubMed] [Google Scholar]
- 25.Leifke E., Naik H., Wu J., Viswanathan P., Demanno D., Kipnes M. A multiple-ascending-dose study to evaluate safety, pharmacokinetics, and pharmacodynamics of a novel GPR40 agonist, TAK-875, in subjects with type 2 diabetes. Clinical Pharmacology & Therapeutics. 2012;92:29–39. doi: 10.1038/clpt.2012.43. [DOI] [PubMed] [Google Scholar]
- 26.Yabuki C., Komatsu H., Tsujihata Y., Maeda R., Ito R., Matsuda-Nagasumi K. A novel antidiabetic drug, fasiglifam/TAK-875, acts as an ago-allosteric modulator of FFAR1. PLoS One. 2013;8:e76280. doi: 10.1371/journal.pone.0076280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin D.C., Guo Q., Luo J., Zhang J., Nguyen K., Chen M. Identification and pharmacological characterization of multiple allosteric binding sites on the free fatty acid 1 receptor. Molecular Pharmacology. 2012;82:843–859. doi: 10.1124/mol.112.079640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y., Liu J., Dransfield P.J., Zhu L., Wang Z., Du X. Discovery and optimization of potent GPR40 full agonists containing tricyclic spirocycles. ACS Medicinal Chemistry Letters. 2013;4:551–555. doi: 10.1021/ml300427u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiong Y., Swaminath G., Cao Q., Yang L., Guo Q., Salomonis H. Activation of FFA1 mediates GLP-1 secretion in mice. Evidence for allosterism at FFA1. Molecular and Cellular Endocrinology. 2013;369:119–129. doi: 10.1016/j.mce.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 30.Schwartz T.W., Holst B. Allosteric enhancers, allosteric agonists and ago-allosteric modulators: where do they bind and how do they act? Trends in Pharmacological Sciences. 2007;28:366–373. doi: 10.1016/j.tips.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 31.Luo J., Swaminath G., Brown S.P., Zhang J., Guo Q., Chen M. A potent class of GPR40 full agonists engages the enteroinsular axis to promote glucose control in rodents. PLoS One. 2012;7:e46300. doi: 10.1371/journal.pone.0046300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srivastava A., Yano J., Hirozane Y., Kefala G., Gruswitz F., Snell G. High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature. 2014;513:124–127. doi: 10.1038/nature13494. [DOI] [PubMed] [Google Scholar]
- 33.Negoro N., Sasaki S., Mikami S., Ito M., Suzuki M., Tsujihata Y. Discovery of TAK-875: a potent, selective, and orally bioavailable GPR40 agonist. ACS Medicinal Chemistry Letters. 2010;1:290–294. doi: 10.1021/ml1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown S.P., Dransfield P.J., Vimolratana M., Jiao X., Zhu L., Pattaropong V. Discovery of AM-1638: a potent and orally bioavailable GPR40/FFA1 full agonist. ACS Medicinal Chemistry Letters. 2012;3:726–730. doi: 10.1021/ml300133f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown S.P., Cao Q., Dransfield P.J., Du X., Fu Z., Houze J. Amgen Inc; 2011. Substituted biphenyl GPR40 modulators. US8030354 B2. [Google Scholar]
- 36.Ge M., Yang L., Zhou C., Lin S., Tang H., Cline E.D. Merck and Co. Inc; 2006. Antidiabetic bicyclic compounds. WO2006083781 A1. [Google Scholar]
- 37.Neves M.A., Totrov M., Abagyan R. Docking and scoring with ICM: the benchmarking results and strategies for improvement. Journal of Computer-Aided Molecular Design. 2012;26:675–686. doi: 10.1007/s10822-012-9547-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kadamur G., Ross E.M. Mammalian phospholipase C. Annual Review of Physiology. 2013;75:127–154. doi: 10.1146/annurev-physiol-030212-183750. [DOI] [PubMed] [Google Scholar]
- 39.Seino S., Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiological Reviews. 2005;85:1303–1342. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- 40.Gloerich M., Bos J.L. Epac: defining a new mechanism for cAMP action. Annual Review of Pharmacology and Toxicology. 2010;50:355–375. doi: 10.1146/annurev.pharmtox.010909.105714. [DOI] [PubMed] [Google Scholar]
- 41.Liou A.P., Lu X., Sei Y., Zhao X., Pechhold S., Carrero R.J. The G-protein-coupled receptor GPR40 directly mediates long-chain fatty acid-induced secretion of cholecystokinin. Gastroenterology. 2011;140:903–912. doi: 10.1053/j.gastro.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parker H.E., Habib A.M., Rogers G.J., Gribble F.M., Reimann F. Nutrient-dependent secretion of glucose-dependent insulinotropic polypeptide from primary murine K cells. Diabetologia. 2009;52:289–298. doi: 10.1007/s00125-008-1202-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neubig R.R., Spedding M., Kenakin T., Christopoulos A. International Union of Pharmacology Committee on receptor N, drug C. International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on terms and symbols in quantitative pharmacology. Pharmacological Reviews. 2003;55:597–606. doi: 10.1124/pr.55.4.4. [DOI] [PubMed] [Google Scholar]
- 44.Christopoulos A., Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacological Reviews. 2002;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- 45.Christopoulos A., Changeux J.P., Catterall W.A., Fabbro D., Burris T.P., Cidlowski J.A. International Union of Basic and Clinical Pharmacology. XC. Multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacological Reviews. 2014;66:918–947. doi: 10.1124/pr.114.008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nygaard R., Frimurer T.M., Holst B., Rosenkilde M.M., Schwartz T.W. Ligand binding and micro-switches in 7TM receptor structures. Trends in Pharmacological Sciences. 2009;30:249–259. doi: 10.1016/j.tips.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 47.Kobilka B., Schertler G.F. New G-protein-coupled receptor crystal structures: insights and limitations. Trends in Pharmacological Sciences. 2008;29:79–83. doi: 10.1016/j.tips.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 48.Hanson M.A., Stevens R.C. Discovery of new GPCR biology: one receptor structure at a time. Structure. 2009;17:8–14. doi: 10.1016/j.str.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwartz T.W., Rosenkilde M.M. Is there a 'lock' for all agonist 'keys' in 7TM receptors? Trends in Pharmacological Sciences. 1996;17:213–216. doi: 10.1016/0165-6147(96)10017-1. [DOI] [PubMed] [Google Scholar]
- 50.Wisler J.W., Xiao K., Thomsen A.R.B., Lefkowitz R.J. Recent developments in biased agonism. Current Opinion in Cell Biology. 2014;27:18–24. doi: 10.1016/j.ceb.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reiter E., Ahn S., Shukla A.K., Lefkowitz R.J. Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annual Review of Pharmacology and Toxicology. 2012;52:179–197. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]