

Abstract
Age is a fundamental aspect of animal ecology, but is difficult to determine in many species. Humpback whales exemplify this as they have a lifespan comparable to humans, mature sexually as early as 4 years and have no reliable visual age indicators after their first year. Current methods for estimating humpback age cannot be applied to all individuals and populations. Assays for human age have recently been developed based on age-induced changes in DNA methylation of specific genes. We used information on age-associated DNA methylation in human and mouse genes to identify homologous gene regions in humpbacks. Humpback skin samples were obtained from individuals with a known year of birth and employed to calibrate relationships between cytosine methylation and age. Seven of 37 cytosines assayed for methylation level in humpback skin had significant age-related profiles. The three most age-informative cytosine markers were selected for a humpback epigenetic age assay. The assay has an R2 of 0.787 (P = 3.04e−16) and predicts age from skin samples with a standard deviation of 2.991 years. The epigenetic method correctly determined which of parent–offspring pairs is the parent in more than 93% of cases. To demonstrate the potential of this technique, we constructed the first modern age profile of humpback whales off eastern Australia and compared the results to population structure 5 decades earlier. This is the first epigenetic age estimation method for a wild animal species and the approach we took for developing it can be applied to many other nonmodel organisms.
Keywords: age, cetacean, DNA methylation, epigenetics, population
Introduction
Animal age is a determinant of many individual and population characteristics. Estimates of age can be used to understand traits in wild animals such as reproductive potential (Clapham 1992), developmental processes (Cook et al. 2006) and factors affecting survival and reproductive success (Chaloupka et al. 1999; Campana 2001). A common approach for age estimation in wild animals that have not previously been encountered is to count annually accrued features such as the growth rings found in fish otoliths that are widely used for producing population age estimates for informing fishery management (Campana 2001). Animals that cannot be lethally sampled provide a greater challenge as many have no outward features that change with age. In these species, individuals can be marked artificially, or records can be made of natural individual features in early life and the elapsed time determined upon re-identification. Whale age estimation was important when whales were the subject of commercial fisheries for monitoring population status (Chittleborough 1959, 1965). It continues to be important for monitoring the recovery of whale populations from the effects of past harvesting (Chaloupka et al. 1999; Baker & Clapham 2004).
Humpback whales (Megaptera novaeangliae, ‘humpbacks’) are one of the best studied cetacean species. However, they display no accurate visual markers for age after they are weaned at about 1 year (Chittleborough 1959) and lack of age information continues to limit our understanding of them. Sexual maturity is reached early in life and age at first parturition can be as young as 5 years (Chittleborough 1965; Clapham 1992; Barlow & Clapham 1997). The age of dead humpback whales can be estimated from ear plug growth layer groups (GLGs), a waxy structure within the ear (Chittleborough 1959, 1965; Lockyer 1984; Gabriele et al. 2010), and baleen plate thickness or ovary condition (Chittleborough 1959). Humpbacks have a maximum verified lifespan of 95 years (Chittleborough 1965), if single ear plug GLGs are considered to accrue annually (Gabriele et al. 2010). None of these age estimation methods can be applied to live whales.
Age estimation methods for live humpbacks are based either on repeat photographic identification or minimally invasive sampling of whale tissues. Humpbacks have natural individual markings on the underside of the tail flukes that identify them (Chaloupka et al. 1999). Repeat sightings of whales whose natural markings were photographed as calves allow age to be estimated (Gabriele et al. 2010). Humpback DNA is used for a wide range of genetic analyses including population size by mark–recapture (Palsbøll et al. 1997; Rew et al. 2011), population structure (Baker et al. 1993; Schmitt et al. 2013), population assignment of individuals (Pomilla & Rosenbaum 2005), effective population size (Roman & Palumbi 2003), kinship (Valsecchi et al. 2002) and sex determination (Morin et al. 2005). Attempts to use humpback whale DNA for age estimation have so far focused on telomere length assays (Dennis 2006; Olsen et al. 2012), but these suffer from numerous sources of measurement error (Olsen et al. 2012); factors other than age that cause telomere length changes (Dunshea et al. 2011); and a wide range of inherited telomere lengths at birth (Kappei & Londoño-Vallejo 2008). In cetaceans, there is an additional problem in that ‘telomeric’ repeat sequences occur in nontelomeric regions (Dunshea et al. 2011). The only molecular method currently available for age estimation of live humpback whales is lipid profile analysis (Herman et al. 2009).
Biological ageing is a combination of programmed processes (Berdasco & Esteller 2012; Horvath 2013) and accumulated changes caused by unrepaired environmental damage (Kujoth et al. 2005). Recent evidence suggests that epigenetic changes are both directing the process of ageing and being caused by it (Maegawa et al. 2010; Koch et al. 2011; Winnefeld & Lyko 2012; Hannum et al. 2013; Horvath 2013). The best studied class of epigenetic change in vertebrates is methyl group presence or absence at the C5 position of Cytosine residues that are adjacent to Guanidine residues (‘CpG sites’). Clusters of CpG sites are common in the 5′ regulatory region of vertebrate genes (Hannum et al. 2013). CpG methylation levels play an important role in control of gene expression, where higher methylation levels (‘hypermethylation’) generally reduce gene transcription rate. Methylation changes at specific CpGs have been linked to age in mice (Maegawa et al. 2010) and humans (Christensen et al. 2009; Grönniger et al. 2010; Bocklandt et al. 2011; Koch & Wagner 2011; Hannum et al. 2013). Epidermal methylation changes in some genes that relate to age have been shown to affect a set of CpG sites distinct from the sites that change in relation to environmental impacts such as sun exposure (Grönniger et al. 2010). This suggests that some CpG sites are linked to genetically programmed ageing and are less influenced by environmental variables. Identification of epigenetic changes in tissues that can be sampled relatively noninvasively such as blood (Koch et al. 2011), skin (Grönniger et al. 2010) and buccal cells (Bocklandt et al. 2011) provides opportunities for development of epigenetic age assays for live animals. Several recent studies have used epigenetic assays to estimate the age of humans or human tissues (Bocklandt et al. 2011; Koch & Wagner 2011; Garagnani et al. 2012; Hannum et al. 2013; Horvath 2013).
We developed an epigenetic method for estimating humpback whale age with DNA purified from skin biopsy samples, hereafter referred to as the HEAA (Humpback Epigenetic Age Assay). We identified three CpG sites in the 5′ regulatory regions of three humpback whale genes with cytosine methylation levels that have a strong age relationship. We measured methylation levels in 45 whales of known ages ranging from <1 to 30 years to characterize the statistical properties of the HEAA including its precision and accuracy. We also specifically explored the ability of the HEAA to determine which of a pair of humpback whales is older and which is younger (ordinal age estimation) because this has a specific application in close-kin population size estimation. The HEAA was then used to estimate the age profile for a sample of 63 humpback whales of unknown age of Australia. This age profile was compared to profiles from the same population 47–57 years earlier as an example of one application of this technique. Finally, we applied the methylation assays used in the HEAA to sperm whale DNA to explore cross-species application of age assays based on DNA methylation.
Materials and methods
Sample collection and processing
Skin samples were collected from humpback whales by punch biopsy darts fired from crossbows or modified rifles. Samples were preserved in 75% ethanol or frozen and later transferred to ‘RNA later ICE’ (Qiagen). DNA was purified by CTAB extraction (Stewart & Via 1993). Three populations of humpback whales were sampled at three sites: the Gulf of Maine off eastern North America (43° N, 68° W) from May to August in 2007–2011; Evans Head in eastern Australia (29° S, 153° E) from June to July 2009; and Exmouth, Western Australia (22° S, 114° E) in September 2009. The sex of the whale associated with each sample was determined with a qPCR assay (Morin et al. 2005).
Three sample sets were assembled. The ‘calibration’ samples were chosen to represent an even distribution of ages within the available range from photo-identification studies (a few weeks to 30 years) and to evenly represent both sexes. They consisted of 45 DNA samples purified from skin biopsies, 40 from the Gulf of Maine, one known-age adult from Evans Head and three calves from Exmouth. There were 21 samples from females and 24 from males. The 24 ‘mother–calf pair’ samples consisted of six pairs from Exmouth, two from Evans Head and four from the Gulf of Maine. The ‘test’ samples were purified from 63 skin biopsies collected off Evans Head, 50 of which were males and 13 female.
Age determination for ‘calibration’ population whales
The age of 40 humpbacks from the Gulf of Maine was determined by resighting of individuals first seen as dependent calves. Biopsies taken in the Gulf of Maine were linked in the field to identifying fluke photographs. Identity and year of birth was confirmed against a photographic identification catalogue curated by the Centre for Coastal Studies. The age of one humpback sampled near Evans Head, Australia was also known as it has been repeatedly resighted since it was a calf (Polanowski et al. 2011). Samples of four calves from near Exmouth, west Australia were estimated to be 4–6 weeks old based on size.
Identification of age-related epigenetic markers in humpbacks
Genes with age-related epigenetic changes in humans and mice were identified through literature searches. The genes tested and the studies demonstrating their age-related methylation are given in Table1. Candidate 5′ regulatory region sequences were taken from GenBank and used as queries for blastn (Altschul et al. 1990) searches of cetacean sequences in GenBank and BLAT searches of the dolphin (Tursiops truncatus) genome (Vollmer & Rosel 2012). Where candidate genes had a clearly orthologous 5′ regulatory regions in the T. truncatus genome, primers for amplification of humpback sequences were designed by eye based on all available homologous cetacean sequences. Humpback gene regions were amplified in 10 μL PCR reactions containing 5 μL 2× Phusion HF (NEB) master mix, 1 μm of each amplification primer, 10 ng of humpback whale DNA and milli-Q H20 with thermal cycling conditions appropriate to each primer set and predicted amplicon. The fragments were purified with Ampure magnetic beads (Agencourt) and bidirectionally sequenced by dye terminator v 3.1 chemistry on an ABI 3100 Sanger sequencer (Applied Biosystems).
Table 1.
CpG sites screened for age-related methylation in Megaptera novaengliae
| Gene | Evidence | CpG position | Age relationship |
|---|---|---|---|
| TET2 KF791963 |
Human hypermethylation (Grönniger et al. 2010) |
−12 | Hypomethylation, R2 = 0.211, P = 0.000877 |
| +16 | Hypomethylation, R2 = 0.174, P = 0.00256 | ||
| +21 | Hypomethylation, R2 = 0.189, P = 0.00159 | ||
| +31 | Hypomethylation, R2 = 0.409, P = 1.75 e−06 | ||
| +58 | None, P > 0.05 | ||
| CDKN2A KF791964 |
Human hypermethylation (Gonzalez-Zulueta et al. 1995; Krishnamurthy et al. 2006; Koch & Wagner 2011; Koch et al. 2011; Horvath 2013) Mouse hypermethylation (Maegawa et al. 2010) |
+297 | Hypermethylation, R2 = 0.409, P = 1.37 e−06 |
| +303 | None, P > 0.05 | ||
| +309 | Hypermethylation, R2 = 0.344, P = 1.39 e−05 | ||
| +327 | None, P > 0.05 | ||
| GRIA2 KF791965 |
Human hypermethylation (Chakrabarti et al. 2001; Koch et al. 2011) |
+202 | Hypermethylation, R2 = 0.469, P = 1.26 e−07 |
| TRIM58 KF791966 |
Human hypermethylation (Koch et al. 2011) |
+181 | None, P > 0.05 |
| +190 | None, P > 0.05 | ||
| +205 | None, P > 0.05 | ||
| +222 | None, P > 0.05 | ||
| +230 | None, P > 0.05 | ||
| +257 | None, P > 0.05 | ||
| +291 | None, P > 0.05 | ||
| +295 | None, P > 0.05 | ||
| +309 | None, P > 0.05 | ||
| +312 | None, P > 0.05 | ||
| +314 | None, P > 0.05 | ||
| +323 | None, P > 0.05 | ||
| +329 | None, P > 0.05 | ||
| +332 | None, P > 0.05 | ||
| +340 | None, P > 0.05 | ||
| HoxA9 KF791967 |
Human hypermethylation (Grönniger et al. 2010; Koch et al. 2011) |
+252 | None, P > 0.05 |
| +255 | None, P > 0.05 | ||
| +265 | None, P > 0.05 | ||
| DDAH2 | Human hypermethylation (Grönniger et al. 2010) |
+31 | None, P > 0.05 |
| +52 | None, P > 0.05 | ||
| +65 | None, P > 0.05 | ||
| +85 | None, P > 0.05 | ||
| TOM1L1 | Human hypermethylation (Bocklandt et al. 2011) |
+539 | None, P > 0.05 |
| +533 | None, P > 0.05 | ||
| +517 | None, P > 0.05 | ||
| Edaradd KF791968 |
Human hypermethylation (Bocklandt et al. 2011) |
−20 | None, P > 0.05 |
| −31 | None, P > 0.05 |
Regressions of CpG methylation with age for 37 CpG sites in eight Megaptera novaengliae genes. The name of the homologous gene in humans is given and the accession no. of the GenBank entry for the M. novaengiae sequence produced in this study. GenBank entries for the M. novaengiae DDAH2 and TOM1L1 sequences were not possible as these sequences were too short to be accepted by GenBank. The source and nature of the evidence for age-related CpG methylation in humans or mice is shown. The position of the 5′ Cytosine of each CpG in each humpback gene is indicated relative to the gene's start codon with negative values indicating distance in base pairs to the 5′ of the start codon and positive values 3′ of the start codon. The CpG and age regression R2 values are shown. All regression P values <0.05 were significant after Bonferroni–Holm correction.
Measurement of cytosine methylation levels
Cytosine methylation levels were measured with Qiagen PyroMark assays. The pyrosequencing assays were designed using pyromark Assay Design Software (Version 2.0.1, Qiagen). Humpback DNA was converted using the Epitect Bisulphite Conversion Kit (Qiagen). The assay regions were PCR amplified using a biotin-labelled, HPLC-purified primer and standard sequencing grade primer (Table S1, Supporting information). Amplification reactions consisted of 12.5 μL pyromark mastermix, 2.5 μL Coral Load, 1 μL each of 5 μm forward and 5 μm reverse primers, 2 μL of bisulphite converted template DNA and 6 μL of water. Thermocycling conditions were 15 min at 95 °C followed by 45 cycles of 30 s at 95 °C, 30 s at 56 °C and 30 s at 72 °C and a final extension step of 10 min at 72 °C. Pyrosequencing was performed on a pyromark 24 Pyrosequencing System (Qiagen). The pyromark Q24 software gave percentage methylation values for each CpG site.
Selection of sites for the HEAA
Cytosine methylation percentages for 37 CpG sites were compared to the ages of the 45 whales in the ‘calibration’ sample set. Linear regression was used to show how much of the variation in CpG methylation was explained by age differences. To correct P values for multiple age-methylation comparisons, a Bonferroni–Holm correction procedure was applied (Holland & Copenhaver 1987). The CpG sites that had significant relationships with age were considered for incorporation into the HEAA. Twenty different combinations of either two or three CpG sites that were found in separate gene regions were combined into multiple linear regression models. The combination of sites that produced a multiple regression with maximum predictive power was selected by scores for the Akaike Information Criterion (AIC) (Table S2, Supporting information).
Measurement of HEAA accuracy and precision
The accuracy of the HEAA was assessed with multiple linear regression. The overall precision of the HEAA was assessed with a Leave One Out Cross Validation (LOOCV) (Picard & Cook 1984). For each of the 45 samples in the ‘calibration’ set, age was estimated with the model using the other 44 samples to calculate the multiple linear regression. The difference between the known and estimated age value was recorded for each sample. The distribution of these residuals was assessed for normality and an assessment of leverage of individual points made.
An estimate of the proportion of assay error that could be attributed to the PyroMark assay was made by repeated measurement of CpG methylation levels in the same DNA samples. Four samples with known ages of 0.4, 5.5, 7.5 and 22.3 years were assayed six times each. The mean standard deviation for all 24 measurements was calculated from differences between known and estimated age.
Estimate of whale ages in a test population
The age distribution estimated by the HEAA for the 63 Evan's Head whales (21% female, 79% male) was compared to age distributions determined by ear plug GLG counts and ovarian measurement for east coast Australian humpbacks in 1952–1962 (Chittleborough 1965). These age estimates were doubled to conform to more recent evidence that one ear plug GLG accrues annually (Gabriele et al. 2010). The numbers of animals with age estimates for 1952–1962 were, respectively 598, 696, 718, 720, 723, 720, 800, 810, 732, 173 and 88. The sex ratio was 32% female and 68% male. For comparison to the 2009 HEAA age estimates, ages were grouped into 4-year categories starting at age 2.
Estimates of ordinal age differentiation performance
The performance of the HEAA in correctly determining the age order of whales was estimated for a range of age differences. The exact ages for the older and younger age were converted into estimates that the HEAA might produce by selecting a value at random from a normal distribution centred on the exact age with a standard deviation of 2.991 as estimated with LOOCV. This was done 10 000 times for all age differences from 0 to 100 (Fig. S1A, Supporting information).
To estimate how often age order in parents and offspring in a real population would be correct, population age distributions were simulated as negative exponential distributions with the λ parameter estimated from the mean age of the 2009 HEAA results for a growing population (λ = 1/10.01 = 0.0999) and from mean age recorded in 1952 (Chittleborough 1965) for the same population (λ = 1/21.8 = 0.0459) (Fig. S1B, Supporting information). Ten thousand parental ages were selected at random from the portion of these distributions greater than the minimum parturition age for humpbacks of 5 years. An offspring age was also selected at random with a maximum age limit of the parental age minus 5 years. This gave the distribution of different age intervals present in the population (Fig. S1C, Supporting information). These were multiplied by the proportion that would be correctly aged and the results integrated across all ages to give an overall proportion that would be correctly age ordered.
The HEAA's ordinal age estimation performance was tested empirically on 12 samples from mothers and calves. The pairs were initially identified visually as mothers with dependent young and their relationship confirmed by microsatellite genotyping following previously published methods (Schmitt et al. 2013). The four mother–calf pairs collected from the Gulf of Maine (real ages 17.4 + 0.4, 10.3 + 2.5, 25.3 + 3.6, 25.8 + 2.2) had their age estimated with the HEAA calibrated with the other 44 (total n – 1) samples in the calibration sample set as for the LOOCV analysis, where all other pairs used the full HEAA calibration.
Test of HEAA assays on sperm whale DNA
Samples of sperm whale (Physeter catodon) skin and teeth were collected from whales that died in mass strandings at Perkins Island off northern Tasmania, Australia (42°2 S, 145°14 E) in December 2004 and January 2009. Ages were estimated at 0 years for a foetus, 1 year for a calf and 15, 20, 28 and 38 years for four individuals by standard tooth growth ring counting methods (Evans & Hindell 2004). The HEAA was applied to these samples as for humpbacks.
Results
Development of the HEAA
Humpback whale 5′ regulatory regions were isolated for eight genes that had evidence of CpG methylation changes in other mammals. Thirty-seven CpGs in these genes were assayed for correspondence between CpG methylation levels and age (Table1). CpG sites considered for the HEAA were selected from the seven CpG sites that had a significant regression relationship with age. Multiple linear regression models were made for all 20 combinations of CpG sites that were not from the same gene region. The models were ranked by Akaike Information Criterion (AIC) score to identify which combination of two or three sites had the best ability to predict age, as shown in Table S3 (Supporting information). The model with the best AIC score had the sites TET2_CpG+31, CDKN2A_CpG+297 and GRIA2_CpG+202. These three sites were also those with the strongest regression relationship with age (Table1). The regressions of age and CpG methylation levels in the three sites selected for the assay are shown in Fig.1. There was no significant difference in the regressions for female and male CpG levels and age (analysis of covariance P > 0.05 in all cases). This was also true for the other four sites that had significant relationships between age and CpG methylation.
Figure 1.
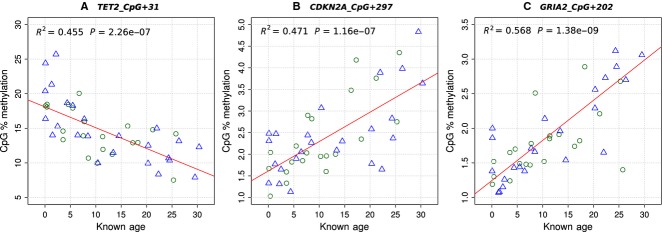
Regressions of CpG methylation and age at sites selected for the HEAA. CpG methylation was measured at each site by a PyroMark assay in N = 45 whales. Females are shown by a green circle and males by blue triangles. CpG sites shown were as follows: (A) TET2_CpG+31, (B) CDKN2A_CpG+297 and (C) GRIA2_CpG+202.
The main reason for selecting only one CpG site from each gene was that sites within the same region that have been methylated or demethylated as part of the same process would not provide independent biological age estimates. Concerted methylation changes are often found among CpG sites in the same human gene regions (Grönniger et al. 2010; Koch & Wagner 2011) and we found this in all sites within the same regions of humpback TET2 and CDKN2A (Fig. S2 and Table S2, Supporting information). Sites from the same region would also be subject to similar experimental error when assayed in the same PCR amplifications and PyroMark assays. This strategy follows those taken for developing human epigenetic age assays (Bocklandt et al. 2011; Koch & Wagner 2011).
Characteristics of the HEAA
The accuracy of the HEAA can be assessed from the multiple linear regression for its three CpG markers and known age shown in Fig.2A. The regression R2 of 0.787 indicates that although most of the response can be attributed to age, there are other factors affecting CpG levels at the HEAA sites. The significant y intercept of 2.395 means that young whales will have their age slightly overestimated. The gradient of the HEAA regression shows that the age of older whales will be slightly underestimated.
Figure 2.
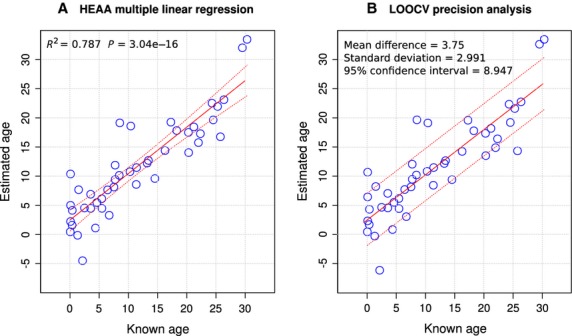
Accuracy and precision of the HEAA. (A) Multiple linear regressions for predicted ages of N = 45 whales from measurement of CpG methylation at three CpG sites. 95% confidence limits of the placement of the regression line are shown. (B) Results of ‘Leave One Out Cross Validation’ (LOOCV) analysis. The estimated ages of every whale in the ‘calibration’ population when the predictive model is based on data for the other N = 44 whales are plotted. 95% confidence limits for age prediction are shown.
The precision of the HEAA as assessed by Leave One Out Cross Validation (LOOCV) is shown in Fig.2B. The overall precision of the HEAA was estimated as the standard deviation of the mean difference between known and estimated ages, which was 2.991 years. The distribution of the differences in known and estimated age was approximately normally distributed (Fig. S3A, Supporting information). A Shapiro–Wilk normality test demonstrates that the difference from normality is not significant (W = 0.984, P = 0.785) and this can be seen in a quantile–quantile plot (Fig. S3B, Supporting information). The differences in known and estimated age show little heteroscedasticity, meaning that variance in the differences is similar throughout the range of ages assayed (Fig. S3C, Supporting information) and there is an even dispersion of these values around the mean (Fig. S3D, Supporting information). The leverage effect of outlying points on the multiple regression was only minor, with all values for Cook's Distance < 0.5 (Fig. S3E, Supporting information). The residuals of the 45 estimates had a mean of 3.575 years and the 95% confidence interval for age estimates was 8.947 years.
The precision of the PyroMark system alone was estimated from the standard deviation of six repeated measurements of the same four DNA samples to be 2.205 years. This indicates that a reasonably high proportion of the precision error is attributable to error in measurement of methylation levels. This is likely to be because the percentage methylation differences measured in the CDKN2A_CpG+297 and GRIA2_CpG+202 assays are small (Fig.1B and C).
HEAA estimation of age in east coast Australian humpbacks
The ages of 63 humpbacks sampled near Evans Head on their northbound migration along the east coast of Australia were measured with the HEAA. The results of these estimates are summarized in Fig.3A. The estimates had a mean age of 10.01 and a range of 0–52. The decreasing number of animals in each age class approximated a negative exponential decrease (two sample Kolmogorov–Smirnov D = 0.141, P = 0.245). An equivalent negative exponential distribution of ages with a λ rate parameter calculated by treating the observed mean as the expected value of the distribution (λ = 1/10.01 = 0.0999) is shown in Fig.3B. This indicates that the HEAA produces age distributions that are close to the distribution expected for noncalf animals in a population where interannual fecundity is reasonably constant and there is little difference in mortality rate among adult age classes (Beverton, Holt, 1956). The proportion of the test population that was within the age range of the calibration samples (0–30 years) was calculated from this distribution to be 95.6%.
Figure 3.
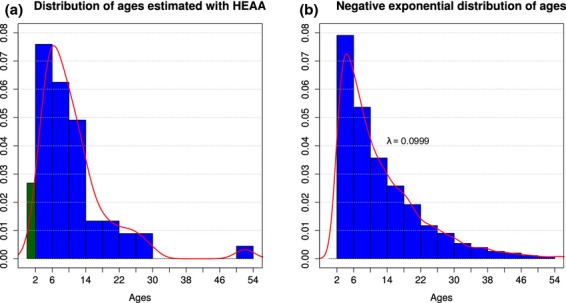
Age estimates generated by the HEAA for east Australian humpback whales. (A) Population age distribution estimated with the HEAA for N = 63 noncalf whales samples near Evans Head. Ages are grouped into categories of 4 years. The mean observed age of 10.01 years was used for estimation of the negative exponential distribution of age shown in (B). Whales with an estimated age of <2 years are indicated in green and were not included in this comparison.
Ages estimated for east coast Australian humpbacks sampled in 2009 were compared to ages for humpbacks recorded during the operation of the Byron Bay whaling station from 1952 to 1962 and estimated by measurement of ear plug GLGs and ovaries (Chittleborough 1965) as shown in Fig.4. The age structure from 1952 to 1960 represents an almost unexploited age structure for migratory whales found in this region (Chittleborough 1965), which is the same region where the Evans Head samples were collected. The humpback fishery in this region began to collapse in 1961 and 1962 was the final year of operations.
Figure 4.
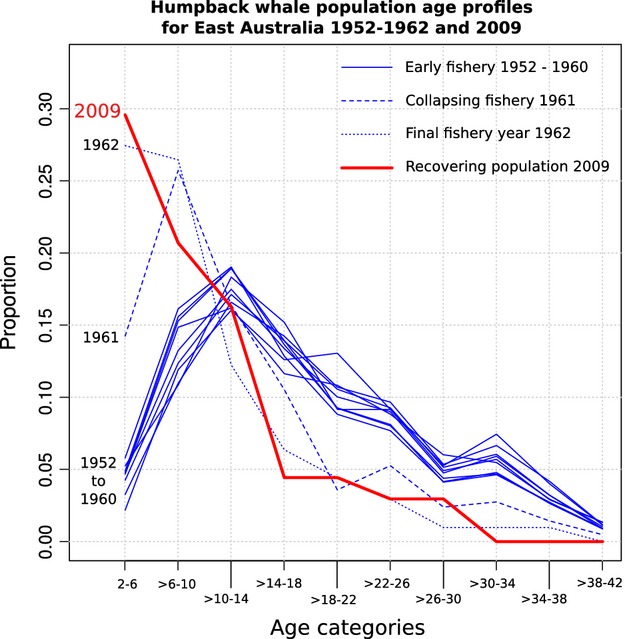
Population age profiles for humpback whales from east coast Australia. Ten profiles for each year from 1952 to 1962 were produced from ear plug growth layer measurement. The HEAA was used to estimate the profile for 2009.
Parent–offspring ordinal age prediction
The ability of the HEAA to correctly order the age of a parent and its progeny was tested by simulation. A negative exponential population age distribution was simulated for the 2009 east coast Australian humpback whales based on HEAA data (Fig.3B) and a distribution also simulated from the 1952 data to provide an example distribution from an unexploited population. Parent–offspring age intervals with a minimum of 5 years were estimated from these distributions and the age estimate errors that would be produced by the HEAA simulated from its standard deviation. In 10 000 estimates of parent–offspring age order, the proportion in which the age order was correctly determined was 93.7% and 99.1% of cases for the 2009 and 1952 age profiles respectively (Fig. S3, Supporting information).
An empirical test of HEAA ordinal age estimation was made with eight mother–calf pairs from the ‘test’ sample set. The ‘calibration’ sample set also contained four mother–offspring pairs, which had age order estimated with the LOOCV approach. In all 12 cases, the correct mother–offspring age order was found.
Cross-species testing of the HEAA
We successfully assayed the three CpG sites in HEAA in six samples of Sperm Whale (Physeter catodon) DNA with ages ranging from 0 to 38 years. Of the three HEAA sites, the P. catodon orthologue of TET2_CpG+31 was the only one with a significant age-related DNA methylation regression (R2 = 0.927, P = 0.0013). Increased age was associated with hypomethylation, as found in humpbacks.
Discussion
Molecular methods for estimating animal age are improving as understanding of the processes of biological ageing deepens. Telomere length assays are the most extensively studied molecular ageing method for animals, but with a few exceptions, these have not proven useful for population studies (Dunshea et al. 2011). In many animals, average telomere length varies extensively at birth, so although changes in individuals can be tracked over time, they are not useful for cross-sectional studies of population age (Kappei & Londoño-Vallejo 2008; Karlsson et al. 2008). Other recent molecular age estimation methods that have performed well are ‘T Cell Receptor Excision Circle’ (TREC) qPCR for humans (Zubakov et al. 2010); changes in levels of specific mRNAs in mosquitos (Cook et al. 2006); assays for specific CpG site methylation levels in humans (Bocklandt et al. 2011; Koch & Wagner 2011; Garagnani et al. 2012); and accumulation of specific lipids in whale blubber (Herman et al. 2009). All of these methods estimate a ‘biological’ age as the features they measure do not change with an annual trigger, but change with constant biological processes such as immunological insults in TREC qPCR (Zubakov et al. 2010); mRNA expression relating to developmental stage (Cook et al. 2006); telomere length changes resulting from number of mitotic cycles (Karlsson et al. 2008) or accumulation of dietary lipids in adipose (Herman et al. 2009). There is almost always population-wide variability in correlation between ‘biological’ age estimates and ‘chronological’ age because individuals within a population have different genotypes and experience different life histories. Even an extremely thorough age estimation assay such as genome-wide measurement of methylation at 70 387 age-related CpG sites (Hannum et al. 2013) results in an assay that has a standard deviation in age prediction of approximately 5% of the lifespan of the animal (humans) which is similar to the HEAA's standard deviation = 3.0/lifespan of ∼95 ≈ 3.1%. This indicates that the underlying relationship between chronological age and proxy markers for age is the limiting factor for achieving precision. Adding extra CpG sites to the HEAA would therefore not necessarily improve its predictive ability. In fact, two of the human epigenetic age assays used combinations of three CpG sites with the best relationships with age selected from a large number of possible age-related sites (Bocklandt et al. 2011; Koch & Wagner 2011). The differences in percentage methylation found in CDKN2A_CpG+297 and GRIA2_CpG+202 were quite small and repeated measurements of the same samples demonstrated that some of the variation in age-related methylation change can be attributed to the measurement error of the PyroMark system. Technologies for measuring DNA methylation are constantly improving and this suggests that measurement of these sites with deeper sequencing or other more accurate methodologies could further improve the precision of the HEAA.
Humpback whales are an excellent example of a species where age estimation is difficult. The prevalent method for age estimation in this species during the commercial whaling era was counting ear plug GLGs (Chittleborough 1959). However, there is measurement error in this method (Chittleborough 1959) and historical lack of consensus on how many growth layers accrue annually (Lockyer 1984). It was initially thought that two GLGs were produced each year (Chittleborough 1959) but more recent evidence suggests only one group (Gabriele et al. 2010), which is also consistent with the accrual rate in other baleen whales (Lockyer 1984; Gabriele et al. 2010) and odd-numbered ear plug GLG counts (Chittleborough 1965; Gabriele et al. 2010). The most reliable method currently used to determine humpback age is visual re-identification of individuals that were first seen as calves. This approach clearly requires substantial effort and cannot be applied to populations without extensive records of past sightings. The only available method for estimating the age of live, previously unencountered humpback whales is analysis of blubber lipid profiles (Herman et al. 2009). This method has a similar estimated precision (standard deviation = 3.1–5.3 in different populations) to our HEAA (standard deviation = 3.0). A limitation of this approach is that it requires calibration for each population as many of the accumulated lipids are derived from dietary items that are not uniformly consumed in different places. The HEAA does not require population-specific calibration and may therefore be particularly useful for comparing age data among populations.
Methods for measuring the age of long lived animals such as humpback whales are difficult to calibrate for the older portion of their range because there are few data available for validation. In this study, the oldest sample in the calibration set was from a 30-year old, which approaches the span of photo-identification research but is only about a third of the potential lifespan of the species. This relationship should be further evaluated for older whales as validating data become available, but all of the age-methylation relationships identified in other mammals so far are approximately linear (e.g. Maegawa et al. 2010; Bocklandt et al. 2011; Koch & Wagner 2011) and the relationships shown in Fig.1 are also, which suggests that age estimates outside the calibration range will be correct relative to estimates within the calibration range. Even with currently available samples for calibration, the age range over which the HEAA was calibrated was estimated to cover more than 95% of humpbacks that will be encountered in the wild because older whales are expected to be rare (see Fig.3).
The CpG sites that we use in the HEAA are in the first exons of humpback whale genes homologous to genes with known functions in other mammals. TET2 (ten eleven translocation 2) is a member of a multigene family that encodes DNA-binding proteins. It is involved in regulation of cytosine methylation of other genes and is a proto-oncogene (Branco et al. 2012). In humans, TET2 becomes hypermethylated with age (Grönniger et al. 2010), so it was interesting to observe significant, concerted age-related hypomethylation in humpback whales in four of the five CpG sites that we assayed and hypomethylation in the single sperm whale TET2 site assayed. CDKN2A (cyclin dependent kinase inhibitor 2A) is part of a gene complex with various names (CDKN2A/CDKN2B, p16INK4a/p16INK4b, ARF) for which there is also widespread evidence of age-related methylation changes (Gonzalez-Zulueta et al. 1995; Maegawa et al. 2010; Koch et al. 2011; Horvath 2013). CDKN2A mRNA expression levels have been proposed as a biomarker for human age, which is likely to be at least partly regulated by CpG methylation (Krishnamurthy et al. 2006). Glutamate receptors such as GRIA2 (glutamate receptor Ia2/AMPA2) are the predominant receptors in the mammalian brain and have a role in neuronal death associated with ageing (Chakrabarti et al. 2001). GRIA2 shows strong age-associated hypermethylation in humans (Koch & Wagner 2011).
As an example of the value of this technique, we estimated age structure for a suite of samples of unknown age obtained off the east coast of Australia and compared those results to historical whaling data. The population of humpbacks that migrate past the east coast of Australia was largely unexploited before 1952, so the age profiles for that time give an estimate of an unperturbed age structure (Chittleborough 1965). The fishery initially produced consistent annual yields and from 1952 to 1960 the average hunting time required to catch each whale was less than 2 h. In 1961 this increased to 4.5 h and in 1962 it had extended to 15 h as the fishery collapsed, ceasing to operate after 1962 (Chittleborough 1965) The population age structures for 1961 and 1962 reflect this depletion of adult whales. The 2009 population age structure estimated with the HEAA from this region is more similar to the age structures recorded in the final 2 years of the fishery than to those of the population encountered at the commencement of whaling. Our results suggest that 47 years after the cessation of whaling, the east coast Australian humpback population has not yet returned to a stable state. However, the modern sample size was small, the samples were not obtained for this purpose and the sampling did not necessarily replicate that of historical whaling operations. Biopsy samples were obtained during a portion of the northbound migration and humpback whales exhibit seasonal variation in migratory timing that varies with age, sex and reproductive status (Dawbin 1966). There may also be differences in selectivity between biopsy sampling and historical whaling operations. Despite these caveats, the strong bias towards young animals being represented and older ones being rare in our results is interesting and warrants further study. This analysis is included as an example of the sort of population comparisons that the HEAA allows, which will of course be more informative with larger sample sizes that are collected with the intent of assessing age structure. This population is growing rapidly (Chaloupka et al. 1999; Noad et al. 2011), so the 2009 age structure has probably resulted from high fecundity and survival, in contrast to the very similar age profiles for 1961 and 1962 that were caused by the size-dependent mortality imposed by the whale fishery. This illustrates a limitation of single ‘snapshot’ population age profiles in estimating underlying demographic parameters such as mortality and fecundity, as similar age profiles can result from changes in different parameters (De La Mare 1985).
The HEAA is more useful for improving demographic analysis methods based on mark–recapture or close-kin recognition. An assumption of many of the commonly used mark–recapture models is that all animals have an equal chance of recapture (Seber 1982). This assumption will be violated in many populations where there is age-specific mortality. The HEAA enables estimation of mortality rates in different age classes. Ordinal age determination is especially useful for improving estimates of population size from close-kin recognition analyses (Skaug et al. 2010). Parent–offspring pairs can be identified by genotyping individuals with multiple markers such as microsatellites or SNPs, but it is often not possible to know which individual is the parent and which the offspring. The power of close-kin population size estimation is diminished when the order of age is not known (Skaug et al. 2010). HEAA determination of age order from the same DNA sample that is used for genotyping will be particularly useful in this application.
Cytosine methylation is reasonably chemically stable and has been successfully assayed in ancient DNA purified from ∼ 38 000-year-old Neanderthal bones (Briggs et al. 2010) and ∼ 60 000-year-old bison bones (Llamas et al. 2012). This suggests potential for epigenetic age estimation from degraded DNA such as faecal samples, which would enable noninvasive sampling for age estimation material from whales or other vertebrates. Another clear application is age estimation from deceased animals. Age-specific population mortality rates could be derived from animals that die of natural causes (Sinclair 1977) as occurs in mass strandings of whales. It is often not possible to generate baleen whale age estimates from stranded whales by ear plug GLG measurement as this tissue typically degrades quickly (Gabriele et al. 2010).
The approach we took to develop the HEAA is widely applicable. Targeted searches for whale orthologues of genes with known age-related methylation patterns in other mammals were quite successful with 19% (≈ 7/37) of CpG sites screened showing significant age-related methylation. Epigenetic regulation mechanisms are not as highly conserved among mammal species as their associated gene and regulatory region DNA sequences are (Horvath 2013), so we had to screen more sites than were included in the final assay. The conservation of PCR priming sites in the sperm whale application of the HEAA, but only partial conservation of age relationship in markers follows this pattern. However, this approach should identify age-related CpG methylation in most vertebrates where ‘calibration’ samples with known ages are available and it appears more efficient than a genome-wide search in nonmodel animals. Genome-wide methylation has been shown to decrease with age in a bird (Gryzinska et al. 2013), a reptile (Parrott et al. 2014) and a plant (Yuan et al. 2014), so it is likely that specific CpG methylation in age-related sites can be identified for them as well. Epigenetic age estimation has great potential for expanding the scope of molecular studies of nonmodel organisms.
Acknowledgments
This work was funded by Australian Antarctic Science grant 4014. We thank Bruce Deagle, Mark Bravington, Bill De La Mare, Nick Gales, Peter Jarman, Phillip Zeigler and two anonymous referees for comments that improved the manuscript. David Mattila, Scott Landry, Christine McMillan, Amy Kennedy and Jennifer Tackaberry assisted with sample collection in the Gulf of Maine. Dave Paton, Sarah Laverick and Simon Childerhouse helped collect samples from near Evans Head. Curt Jenner provided samples from Exmouth. Sperm whale samples and age estimates were provided by Rachael Alderman and Kris Carlyon and this work was supported by the Princess Melikoff Trust Marine Mammal Conservation Program of the Tasmanian DPIPWE.
A.P. performed experiments to develop the HEAA. J.R. collected samples for the calibration population. D.C. performed the PyroMark assays. A.P. and S.J. collected samples for the test population and jointly developed the concept for the HEAA. S.J. performed the data analyses. All authors contributed to writing the paper.
Data Accessibility
All R scripts, DNA sequence data and CpG methylation data were deposited in the DRYAD database archive for this study (doi: 10.5061/dryad.h4b48). Megaptera novaengliae 5′ regulatory region sequences determined for six genes were deposited in GenBank and have accessions KF791963 – KF791968 (Table1).
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Fig. S1 Simulations for estimating performance of the HEAA in estimating age order.
Fig. S2 Co-plot of CpG methylation levels with age among seven sites considered for the HEAA.
Fig. S3 Assessment of normality of distribution of differences in known ages and ages predicted by our epigenetic age assay.
Table S1 Primers for the HEAA PyroMark assays.
Table S2 Akaike Information Criterion assessments of models with 25 different combinations of sites with CpG methylation levels relating to age in humpback whales.
Table S3 Correlation of age-related methylation in 18 pairwise combinations of CpG sites.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Beverton RJH, Holt SJ. A review of methods for estimating mortality rates in exploited fish populations, with special reference to sources of bias in catch sampling. Rapp. P.-V. Réun. Ciem. 1956;140:67–83. [Google Scholar]
- Baker CS, Clapham PJ. Modelling the past and future of whales and whaling. Trends in Ecology & Evolution. 2004;19:365–371. doi: 10.1016/j.tree.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Baker CS, Perry A, Bannister JL, et al. Abundant mitochondrial DNA variation and world-wide population structure in humpback whales. Proceedings of the National Academy of Sciences of the USA. 1993;90:8239–8243. doi: 10.1073/pnas.90.17.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow J, Clapham PJ. A new birth-interval approach to estimating demographic parameters of humpback whales. Ecology. 1997;78:535–546. [Google Scholar]
- Berdasco M, Esteller M. Hot topics in epigenetic mechanisms of aging: 2011. Aging Cell. 2012;11:181–186. doi: 10.1111/j.1474-9726.2012.00806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocklandt S, Lin W, Sehl ME, et al. Epigenetic predictor of age. PLoS ONE. 2011;6:e14821. doi: 10.1371/journal.pone.0014821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco MR, Ficz G, Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nature Reviews Genetics. 2012;13:7–13. doi: 10.1038/nrg3080. [DOI] [PubMed] [Google Scholar]
- Briggs AW, Stenzel U, Meyer M, Krause J, Kircher M, Pääbo S. Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Research. 2010;38:e87–e87. doi: 10.1093/nar/gkp1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campana SE. Accuracy, precision and quality control in age determination, including a review of the use and abuse of age validation methods. Journal of Fish Biology. 2001;59:197–242. [Google Scholar]
- Chakrabarti L, Bandyopadhyay BC, Poddar MK. Is age-induced decline in immune response associated with hypothalamic glutamate receptor density and dietary protein? Nutritional Neuroscience. 2001;4:375–387. doi: 10.1080/1028415x.2001.11747374. [DOI] [PubMed] [Google Scholar]
- Chaloupka M, Osmond M, Kaufman G. Estimating seasonal abundance trends and survival probabilities of humpback whales in Hervey Bay (east coast Australia) Marine Ecology Progress Series. 1999;184:291–301. [Google Scholar]
- Chittleborough RG. Determination of age in the humpback whale, Megaptera nodosa (Bonnaterre) Marine and Freshwater Research. 1959;10:125–143. [Google Scholar]
- Chittleborough R. Dynamics of two populations of the humpback whale, Megaptera novaeangliae (Borowski) Marine and Freshwater Research. 1965;16:33–128. [Google Scholar]
- Christensen BC, Houseman EA, Marsit CJ, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG Island context. PLoS Genetics. 2009;5:e1000602. doi: 10.1371/journal.pgen.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham PJ. Age at attainment of sexual maturity in humpback whales, Megaptera novaeangliae. Canadian Journal of Zoology. 1992;70:1470–1472. [Google Scholar]
- Cook PE, Hugo LE, Iturbe-Ormaetxe I, et al. The use of transcriptional profiles to predict adult mosquito age under field conditions. Proceedings of the National Academy of Sciences of the USA. 2006;103:18060–18065. doi: 10.1073/pnas.0604875103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawbin WH. The seasonal migratory cycle of humpback whales. In: Norris KS, editor. Whales, Dolphins and Porpoises. Berkeley, California: University of California Press; 1966. pp. 145–169. [Google Scholar]
- De La Mare WK. On the estimation of mortality rates from whale age data, with particular reference to minke whales (Balaenoptera acutorostrata) in the Southern Hemisphere. Report of the International Whaling Commission. 1985;35:239–250. [Google Scholar]
- Dennis C. Conservation at a distance: a gentle way to age. Nature. 2006;442:507–508. doi: 10.1038/442507a. [DOI] [PubMed] [Google Scholar]
- Dunshea G, Duffield D, Gales N, Hindell M, Wells RS, Jarman SN. Telomeres as age markers in vertebrate molecular ecology. Molecular Ecology Resources. 2011;11:225–235. doi: 10.1111/j.1755-0998.2010.02976.x. [DOI] [PubMed] [Google Scholar]
- Evans K, Hindell MA. The age structure and growth of female sperm whales (Physeter macrocephalus) in southern Australian waters. Journal of Zoology. 2004;263:237–250. [Google Scholar]
- Gabriele CM, Lockyer C, Straley JM, Jurasz CM, Kato H. Sighting history of a naturally marked humpback whale (Megaptera novaeangliae) suggests ear plug growth layer groups are deposited annually. Marine Mammal Science. 2010;26:443–450. [Google Scholar]
- Garagnani P, Bacalini MG, Pirazzini C, et al. Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell. 2012;11:1132–1134. doi: 10.1111/acel.12005. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Zulueta M, Bender CM, Yang AS, et al. Methylation of the 5′ CpG island of the p16/CDKN2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer Research. 1995;55:4531–4535. [PubMed] [Google Scholar]
- Grönniger E, Weber B, Heil O, et al. Aging and chronic sun exposure cause distinct epigenetic changes in human skin. PLoS Genetics. 2010;6:e1000971. doi: 10.1371/journal.pgen.1000971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryzinska M, Blaszczak E, Strachecka A, Jezewska-Witkowska G. Analysis of age-related global DNA methylation in chicken. Biochemical Genetics. 2013;51:554–563. doi: 10.1007/s10528-013-9586-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannum G, Guinney J, Zhao L, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Molecular Cell. 2013;49:359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman D, Ylitalo G, Robbins J, et al. Age determination of humpback whales Megaptera novaeangliae through blubber fatty acid compositions of biopsy samples. Marine Ecology Progress Series. 2009;392:277–293. [Google Scholar]
- Holland BS, Copenhaver MD. An improved sequentially rejective Bonferroni test procedure. Biometrics. 1987;43:417. [Google Scholar]
- Horvath S. DNA methylation age of human tissues and cell types. Genome Biology. 2013;14:R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappei D, Londoño-Vallejo JA. Telomere length inheritance and aging. Mechanisms of Ageing and Development. 2008;129:17–26. doi: 10.1016/j.mad.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Karlsson AO, Svensson A, Marklund A, Holmlund G. Estimating human age in forensic samples by analysis of telomere repeats. Forensic Science International: Genetics Supplement Series. 2008;1:569–571. [Google Scholar]
- Koch CM, Wagner W. Epigenetic-aging-signature to determine age in different tissues. Aging. 2011;3:1018. doi: 10.18632/aging.100395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch CM, Suschek CV, Lin Q, et al. Specific age-associated DNA methylation changes in human dermal fibroblasts. PLoS ONE. 2011;6:e16679. doi: 10.1371/journal.pone.0016679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy J, Ramsey MR, Ligon KL, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Llamas B, Holland ML, Chen K, Cropley JE, Cooper A, Suter CM. High-resolution analysis of cytosine methylation in ancient DNA. PLoS ONE. 2012;7:e30226. doi: 10.1371/journal.pone.0030226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockyer C. Age determination by means of the earplug in baleen whales. Report of the International Whaling Commission. 1984;34:692–696. [Google Scholar]
- Maegawa S, Hinkal G, Kim HS, et al. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Research. 2010;20:332–340. doi: 10.1101/gr.096826.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin PA, Nestler A, Rubio-Cisneros NT, Robertson KM, Mesnick SL. Interfamilial characterization of a region of the ZFX and ZFY genes facilitates sex determination in cetaceans and other mammals. Molecular Ecology. 2005;14:3275–3286. doi: 10.1111/j.1365-294X.2005.02651.x. [DOI] [PubMed] [Google Scholar]
- Noad MJ, Dunlop RA, Paton D, Cato DH. Absolute and relative abundance estimates of Australian east coast humpback whales (Megaptera novaeangliae. Journal of Cetacean Research and Management. 2011:243–252. Special Issue 3. [Google Scholar]
- Olsen MT, Bérubé M, Robbins J, Palsbøll PJ. Empirical evaluation of humpback whale telomere length estimates; quality control and factors causing variability in the singleplex and multiplex qPCR methods. BMC Genetics. 2012;13:77. doi: 10.1186/1471-2156-13-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsbøll PJ, Allen J, Bérubé M, et al. Genetic tagging of humpback whales. Nature. 1997;388:767–769. doi: 10.1038/42005. [DOI] [PubMed] [Google Scholar]
- Parrott BB, Bowden JA, Kohno S, et al. Influence of tissue, age, and environmental quality on DNA methylation in Alligator mississippiensis. Reproduction. 2014;147:503–513. doi: 10.1530/REP-13-0498. (Cambridge, England) [DOI] [PubMed] [Google Scholar]
- Picard RR, Cook RD. Cross-validation of regression models. Journal of the American Statistical Association. 1984;79:575–583. [Google Scholar]
- Polanowski AM, Robinson-Laverick SM, Paton D, Jarman SN. Variation in the tyrosinase gene associated with a white humpback whale (Megaptera novaeangliae. Journal of Heredity. 2011;103:130–133. doi: 10.1093/jhered/esr108. [DOI] [PubMed] [Google Scholar]
- Pomilla C, Rosenbaum HC. Against the current: an inter-oceanic whale migration event. Biology Letters. 2005;1:476–479. doi: 10.1098/rsbl.2005.0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rew MB, Robbins J, Mattila D, Palsbøll PJ, Bérubé M. How many genetic markers to tag an individual? An empirical assessment of false matching rates among close relatives. Ecological Applications. 2011;21:877–887. doi: 10.1890/10-0348.1. [DOI] [PubMed] [Google Scholar]
- Roman J, Palumbi SR. Whales before whaling in the North Atlantic. Science. 2003;301:508–510. doi: 10.1126/science.1084524. [DOI] [PubMed] [Google Scholar]
- Schmitt NT, Double MC, Jarman SN, et al. Low levels of genetic differentiation characterize Australian humpback whale (Megaptera novaeangliae) populations. Marine Mammal Science. 2013;30:221–241. [Google Scholar]
- Seber GAF. The Estimation of Animal Abundance and Related Parameters. London: Charles Griffin & Co; 1982. [Google Scholar]
- Sinclair A. The African Buffalo. Chicago, Illinois: University of Chicago Press; 1977. [Google Scholar]
- Skaug HJ, Bérubé M, Palsbøll PJ. Detecting dyads of related individuals in large collections of DNA-profiles by controlling the false discovery rate. Molecular Ecology Resources. 2010;10:693–700. doi: 10.1111/j.1755-0998.2010.02833.x. [DOI] [PubMed] [Google Scholar]
- Stewart CN, Jr, Via LE. A rapid CTAB DNA isolation technique useful for RAPD fingerprinting and other PCR applications. BioTechniques. 1993;14:748–750. [PubMed] [Google Scholar]
- Valsecchi E, Hale P, Corkeron P, Amos W. Social structure in migrating humpback whales (Megaptera novaeangliae. Molecular Ecology. 2002;11:507–518. doi: 10.1046/j.0962-1083.2001.01459.x. [DOI] [PubMed] [Google Scholar]
- Vollmer NL, Rosel PE. Developing genomic resources for the common bottlenose dolphin (Tursiops truncatus): isolation and characterization of 153 single nucleotide polymorphisms and 53 genotyping assays. Molecular Ecology Resources. 2012;12:1124–1132. doi: 10.1111/1755-0998.12008. [DOI] [PubMed] [Google Scholar]
- Winnefeld M, Lyko F. The aging epigenome: DNA methylation from the cradle to the grave. Genome Biology. 2012;13:165. doi: 10.1186/gb-2012-13-7-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J-L, Sun H-M, Guo G-P, Yue J-J, Gu X-P. Correlation between DNA methylation and chronological age of Moso bamboo (Phyllostachys heterocycla var. pubescens. Botanical Studies. 2014;55:4. doi: 10.1186/1999-3110-55-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubakov D, Liu F, van Zelm MC, et al. Estimating human age from T-cell DNA rearrangements. Current Biology. 2010;20:R970–R971. doi: 10.1016/j.cub.2010.10.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Simulations for estimating performance of the HEAA in estimating age order.
Fig. S2 Co-plot of CpG methylation levels with age among seven sites considered for the HEAA.
Fig. S3 Assessment of normality of distribution of differences in known ages and ages predicted by our epigenetic age assay.
Table S1 Primers for the HEAA PyroMark assays.
Table S2 Akaike Information Criterion assessments of models with 25 different combinations of sites with CpG methylation levels relating to age in humpback whales.
Table S3 Correlation of age-related methylation in 18 pairwise combinations of CpG sites.
Data Availability Statement
All R scripts, DNA sequence data and CpG methylation data were deposited in the DRYAD database archive for this study (doi: 10.5061/dryad.h4b48). Megaptera novaengliae 5′ regulatory region sequences determined for six genes were deposited in GenBank and have accessions KF791963 – KF791968 (Table1).