

Abstract
Objective
The aim of this study was to characterize the genome-wide DNA methylation profile of chondrocytes from knee and hip cartilage obtained from patients with osteoarthritis (OA) and hip cartilage obtained from patients with femoral neck fracture, providing the first comparison of DNA methylation between OA and non-OA hip cartilage, and between OA hip and OA knee cartilage.
Methods
The study was performed using the Illumina Infinium HumanMethylation450 BeadChip array, which allows the annotation of ∼480,000 CpG sites. Genome-wide methylation was assessed in chondrocyte DNA extracted from 23 hip OA patients, 73 knee OA patients, and 21 healthy hip control patients with femoral neck fracture.
Results
Analysis revealed that chondrocytes from the hip cartilage of OA patients and healthy controls have unique methylation profiles, with 5,322 differentially methylated loci (DMLs) identified between the 2 groups. In addition, a comparison between hip and knee OA chondrocytes revealed 5,547 DMLs between the 2 groups, including DMLs in several genes known to be involved in the pathogenesis of OA. Hip OA samples were found to cluster into 2 groups. A total of 15,239 DMLs were identified between the 2 clusters, with an enrichment of genes involved in inflammation and immunity. Similarly, we confirmed a previous report of knee OA samples that also clustered into 2 groups.
Conclusion
We demonstrated that global DNA methylation using a high-density array can be a powerful tool in the characterization of OA at the molecular level. Identification of pathways enriched in DMLs between OA and OA-free cartilage highlight potential etiologic mechanisms that are involved in the initiation and/or progression of the disease and that could be therapeutically targeted.
Osteoarthritis (OA) is a common degenerative disease of the synovial joints, characterized by the gradual thinning and eventual loss of articular cartilage, which can be accompanied by changes in other joint tissues (1,2). Age, obesity, and genetic susceptibility are known OA risk factors (3,4).
Cartilage is made and maintained by a single cell type, the chondrocyte. The extracellular matrix (ECM) of cartilage undergoes constant remodeling, with the chondrocyte responsible for maintaining the balance between anabolic and catabolic factors (5). In OA, chondrocytes undergo a phenotypic change, characterized by clonal expansion and hypertrophy, accompanied by a shift in the balance of cartilage homeostasis toward overall matrix degradation (4).
Since the shift in the homeostasis of the cartilage ECM principally results from altered gene expression, it has been hypothesized that epigenetic alterations in chondrocytes could be a key driver of OA pathogenesis (6,7). Of the 3 epigenetic modifications, DNA methylation at CpG sites is by far the most extensively studied in common diseases. It has long been suspected that epigenetics plays a key role in the onset and progression of common diseases by providing a link between genetic and environmental risk factors (6,8).
There have been several candidate gene–based studies investigating the role of DNA methylation in OA cartilage. For example, it has previously been shown that up-regulation of the metalloproteinases matrix metalloproteinase 3 (MMP-3), MMP-9, MMP-13, MMP-14, and ADAMTS-4 in OA chondrocytes is mediated by the demethylation of the promoter regions of the genes coding for these proteins (9–11). Furthermore, demethylation of an enhancer region within the nitric oxide synthase gene leads to increased transcription through elevated binding of the transcription factor NF-κB (12). It has also been shown that the promoter of IL1B undergoes demethylation in human articular chondrocytes in response to inflammatory cytokine signaling (11,13). DNA methylation can also modulate OA genetic susceptibility loci, with altered methylation impacting the effect of the OA associated single-nucleotide polymorphism (SNP) rs143383 on GDF5 expression (14). The only genome-wide approach to methylation changes in OA cartilage reported so far was conducted by Fernández-Tajes et al (15). Those investigators used an Illumina Infinium HumanMethylation27 array, which measures the methylation level at ∼27,000 CpG sites. They measured methylation differences between knee cartilage in 23 OA patients and 18 healthy controls and discovered 91 differentially methylated loci (DMLs).
Given the clear importance of DNA methylation in disease and its emerging role in OA, we set out to characterize the cartilage DNA methylome in OA. To measure DNA methylation, we used the highest density array currently available, the Illumina Infinium HumanMethylation450 BeadChip, which encompasses ∼480,000 CpG probes throughout the genome. We conducted the first comparison between OA and healthy hip chondrocytes, as well as the first comparison between OA hip and knee chondrocytes. We identified a series of highly compelling differences relevant to both the OA disease state and to the joint site studied. We conclude that the identification of pathways enriched in DMLs may lead to more effective characterization of this common arthritis and could offer novel insights into treatment developments.
PATIENTS AND METHODS
Patients
Cartilage tissue was collected from the hip joints of patients undergoing hip replacement surgery because of primary hip OA (n = 23) or fracture of the femoral neck (n = 21). Preoperative radiographs of the joints were graded for their OA status using the Kellgren/Lawrence (K/L) scoring system (range 0–4, where ≤1 indicates no OA and ≥2 indicates OA). Patients with femoral neck fracture had no radiographic signs of hip OA (K/L score of 0 or 1), with macroscopically intact cartilage and no lesions. These cartilage samples served as controls for the hip OA cartilage. Cartilage tissue was also collected from 73 patients undergoing knee joint replacement surgery because of primary knee OA. All OA patients had a K/L score of at least 2, had visible cartilage lesions, and were screened to exclude OA due to trauma or other pathologic conditions. The cartilage was collected from the tibial plateau and from the lateral and medial femoral condyles. In all OA patients, the cartilage was collected from sites distal to the OA lesion. The cartilage represented a mix of superficial, intermediate, and deep layers of the tissue. Supplementary Table 1 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38713/abstract) provides further details regarding the 117 patients studied.
The Newcastle and North Tyneside research ethics committee granted approval for the study (REC reference number 09/H0906/72). Informed consent was obtained from each donor.
Genomic DNA isolation and bisulfite treatment
For each patient, genomic DNA was isolated from 250 mg of ground cartilage tissue using an EZNA DNA/RNA isolation kit (Omega Bio-Tek), as previously described (16). A total of 500 ng of this cartilage genomic DNA was then bisulfite converted using an EZ DNA methylation kit (Zymo Research), and eluted in 10 μl of elution buffer (50 ng/μl).
DNA methylation profiling
DNA methylation profiling was carried out on the bisulfite-converted cartilage DNA by Cambridge Genomic Services, using the Infinium HumanMethylation450 BeadChip array (Illumina). The raw data were extracted using GenomeStudio tool (Illumina). GenomeStudio provides the methylation data as β values: β = M/(M + U), where M represents the fluorescent signal of the methylation probe and U represents the methylation signal of the unmethylated probe. The β values range from 0 (no methylation) to 1 (100% methylation).
Data processing
Processing of the raw methylation data was performed in R (version 3.0.1) using the Watermelon package (version 2.12) (17). The data were processed and normalized as previously described (18). Batch correction of the samples was performed using the ComBat package in R (19).
Filtering of methylation data
During normalization of the β values, probes which had a detection P value greater than 0.01 were removed from the analysis. Since male and female patients were studied, the sex chromosome probes were also removed. A total of 19,697 probes were removed with detection P values greater than 0.01, and 11,713 sex chromosomes probes were removed. This filtering left a total of 454,167 probes that were used for subsequent analysis.
Differential methylation analysis
To identify DMLs, the average β value was compared between the groups of interest (for example, hip cartilage from OA versus femoral neck fracture patients). F test of equality of variances demonstrated that there was no significant difference in the variance of global methylation between the groups of samples (hip OA versus femoral neck fracture [P = 0.74]; hip OA versus knee OA [P = 0.98]), and so, P values were calculated using a t-test assuming equal variances. A locus was deemed significantly differentially methylated if there was at least a 10% difference in methylation between the 2 groups and had a Benjamini-Hochberg corrected P value of less than 0.05, as previously reported (20). Genomic annotation of DMLs was carried out using the Infinium HumanMethylation450 BeadChip annotation file (http://www.illumina.com).
Pyrosequencing
A total of 500 ng of the cartilage-extracted genomic DNA was bisulfite converted as above and eluted in 10 μl of elution buffer (50 ng/μl). Pyrosequencing assays for the region of interest were designed using PyroMark assay design software 2.0 (Qiagen), and sequencing was performed using PyroMark Q24 (Qiagen). The primers used are listed in Supplementary Table 2 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38713/abstract). DMLs selected for validation by pyrosequencing were chosen at random from the list of DMLs between cartilage samples from the hip of patients with femoral neck fracture and OA.
Pathway analysis
Gene ontology (GO) analysis was carried out using human GO term associations (http://www.geneontology.org/). Enriched GO terms with a Benjamini-Hochberg corrected P value of less than 0.05 were deemed significant. The enrichment of GO terms was calculated using the DAVID bioinformatics database functional tool (21).
Hierarchical clustering
For all unsupervised clustering, distances between the samples were measured as the Euclidean distance and were clustered using the Ward method.
Principal components analysis (PCA)
PCA was performed with the PCA function of the FactoMineR package in R, using the default parameters.
RNA isolation and reverse transcription
RNA was isolated from 250 mg of ground cartilage tissue using an RNeasy kit (Qiagen). A total of 250 ng of extracted RNA was DNase treated with 2 units of Turbo DNase (Ambion) and was reverse transcribed using a SuperScript First-Strand complementary DNA synthesis kit (Invitrogen).
Relative gene expression
Relative gene expression was measured by real-time PCR using TaqMan chemistry. Gene expression analysis was carried out using predesigned primers and probes (Integrated DNA Technologies); primers and probes were selected so that all known transcript variants of a particular gene were measured. Expression of target genes was measured relative to the housekeeping genes 18S, ribosomal RNA, GAPDH, and HPRT1. The relative expression of the target genes was calculated using the 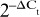 method:
method:
RESULTS
Comparison of the hip cartilage DNA methylome between OA patients and controls
Unsupervised hierarchical clustering revealed that samples from patients with hip OA and controls with femoral neck fracture can be largely distinguished by their DNA methylome (Figure 1a). Two main clusters were clearly apparent. Cluster 1 contained 11 hip OA samples and 18 femoral neck fracture samples. These 18 femoral neck fracture samples clustered separately from the OA hip samples, with only 1 hip OA sample (sample Hip.4479) among them. Within cluster 2, there were 12 hip OA samples and 3 femoral neck fracture samples, with the 3 femoral neck fracture samples (samples T141, T138, and T132) clustering separately from the OA samples.
Figure 1.

Genome-wide methylation in hip cartilage chondrocyte DNA from patients with femoral neck fracture and patients with osteoarthritis (OA). a, Unsupervised hierarchical clustering of the global β values in the 23 hip OA samples (red) and the 21 femoral neck fracture samples (blue). b, Heatmap showing the unsupervised clustering of the 5,322 differentially methylated loci (DMLs) identified between the femoral neck fracture and hip OA samples. DMLs were defined as those with at least a 10% difference in methylation between the 2 groups and with a Benjamini-Hochberg corrected P value of less than 0.05. Dendrogram at the top shows the clustering of the samples. Dendrogram at the left shows the clustering of the loci. The methylation scale is shown to the right of the heatmap (1 = 100% methylation; 0 = no methylation). c, Gene ontology pathway analysis of the 5,322 DMLs. ∗ = P ≤ 0.05; ∗∗ = P ≤ 0.01; ∗∗∗ = P ≤ 0.001, after Benjamini-Hochberg correction for multiple tests. TGFβ = transforming growth factor β; ECM = extracellular matrix.
Identification of DMLs between hip cartilage from OA patients and controls
Given that the unsupervised cluster analysis highlighted that hip OA and femoral neck fracture samples could segregate based on their methylation profile, a more detailed analysis was performed to identify individual loci that are differentially methylated between the 2 groups. A total of 5,322 DMLs were identified, of which 2,653 were hypomethylated and 2,669 hypermethylated in OA. Hierarchical clustering of the DMLs allowed us to largely distinguish the control hip samples from the OA hip samples (Figure 1b). Examples of DMLs within or near the genes of interest to the OA disease process are listed in Table1, and a complete list of DMLs are shown in Supplementary Table 3 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38713/abstract).
Table 1.
Genes harboring differentially methylated loci between hip osteoarthritis (OA) and femoral neck fracture samples
| Gene | Mean β value | P, after Benjamini-Hochberg correction | |
|---|---|---|---|
| Femoral neck fracture patients | Hip OA patients | ||
| Hypermethylated in OA | |||
| ACVR1B | 0.42 | 0.53 | 0.00091 |
| ADAMTS2 | 0.47 | 0.58 | 0.0081 |
| ADAMTS8 | 0.49 | 0.60 | 0.00045 |
| BMPR1A | 0.60 | 0.70 | 0.038 |
| BMPR1B | 0.35 | 0.60 | 0.000031 |
| COL2A1 | 0.24 | 0.37 | 0.010 |
| COL5A3 | 0.29 | 0.42 | 0.000030 |
| COL9A1 | 0.57 | 0.69 | 0.0035 |
| COL9A3 | 0.27 | 0.38 | 0.0091 |
| COL11A2 | 0.27 | 0.47 | 0.000013 |
| COL13A1 | 0.21 | 0.31 | 0.018 |
| IL17B | 0.32 | 0.42 | 0.0060 |
| MCF2L | 0.46 | 0.56 | 0.0043 |
| MMP16 | 0.63 | 0.75 | 0.00020 |
| PTHLH | 0.15 | 0.28 | 0.000030 |
| SMAD2 | 0.32 | 0.46 | 0.0033 |
| SMAD3 | 0.69 | 0.81 | 0.000048 |
| SUPT3H | 0.55 | 0.68 | 0.0071 |
| TGFB2 | 0.32 | 0.47 | 0.032 |
| TGFBR2 | 0.61 | 0.72 | 0.00011 |
| Hypomethylated in OA | |||
| ACVR1 | 0.87 | 0.76 | 0.00096 |
| ADAMTS4 | 0.65 | 0.52 | 0.00040 |
| ADAMTS5 | 0.75 | 0.60 | 0.0000031 |
| ADAMTS10 | 0.82 | 0.65 | 0.00079 |
| ADAMTS17 | 0.59 | 0.43 | 0.00015 |
| BMP1 | 0.33 | 0.22 | 0.00000030 |
| BMP6 | 0.34 | 0.22 | 0.00060 |
| CHST11 | 0.83 | 0.70 | 0.0031 |
| COL1A2 | 0.58 | 0.42 | 0.0022 |
| COL6A3 | 0.57 | 0.44 | 0.00093 |
| COL7A1 | 0.36 | 0.24 | 0.000025 |
| COL8A1 | 0.80 | 0.66 | 0.00055 |
| COL22A1 | 0.49 | 0.38 | 0.0040 |
| ECM1 | 0.59 | 0.47 | 0.00061 |
| FILIP1 | 0.54 | 0.37 | 0.00022 |
| IL4R | 0.53 | 0.34 | 0.0015 |
| MMP13 | 0.73 | 0.60 | 0.0025 |
| PBRM1 | 0.83 | 0.68 | 0.000017 |
| TGFB1 | 0.42 | 0.27 | 0.031 |
| TGFBR3 | 0.39 | 0.28 | 0.016 |
Noteworthy DMLs included those that occur in genes coding for proteins associated with degradation of the ECM, including ADAMTS2, ADAMTS4, ADAMTS5, ADAMTS10, ADAMTS17, MMP13, and MMP16. DMLs were also located in other genes involved in the anabolic/catabolic pathways of cartilage homeostasis, including ECM1 and CHST11, as well as members of the transforming growth factor β (TGFβ) signaling pathway, including ACVR1B, SMAD2, SMAD3, TGFBR2, TGFB1, and BMP6. We also identified DMLs within genes that reside within genomic intervals that have been genetically associated with OA, including the aforementioned CHST11 and PBRM1, SUPT3H, FILIP1, and PTHLH (22). GO term analysis of genes containing DMLs indicated an enrichment of pathways known to be involved in OA (see Figure 1c for examples), predominantly pathways involved in regulating the ECM and collagen synthesis.
Validation of DMLs
Three genes with DMLs between the hip OA and femoral neck fracture samples (SIM2, TNXB, and ALX1) were selected for validation by pyrosequencing (Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38713/abstract). This revealed a strong correlation between the methylation values obtained from the array and those obtained from pyrosequencing: r2= 0.68 for SIM2, r2 = 0.75 for TNXB, and r2 = 0.84 for ALX1.
Methylation of gene promoters and/or enhancers is known to correlate with decreased gene expression, whereas methylation within nonenhancer regions of the gene body correlates with increased gene expression (23,24). We examined 2 genes (ADAMTS5 and ACVR1) that contained DMLs within the gene body (nonenhancer region) between hip OA and femoral neck fracture samples. Both showed decreased methylation in hip OA samples as compared to femoral neck fracture samples, and we observed a decrease in the expression of both genes in the hip OA samples (Supplementary Figure 2a, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38713/abstract.
We also examined 2 genes (LRP5 and CHST11) that contained DMLs within predicted enhancer regions between hip OA and femoral neck fracture samples. Both were hypomethylated in hip OA as compared to femoral neck fracture samples and, as expected, we observed an inverse relationship with the level of gene expression between the 2 groups (Supplementary Figure 2b).
Clustering of hip OA cartilage samples based on their methylome
As noted above, initial unsupervised clustering of the 454,167 probes indicated that hip OA samples segregate based on their DNA methylation profile (Figure 1a). We further investigated this observation by performing hierarchical clustering of the hip OA samples, which confirmed the clustering (Figure 2a). Two clear clusters were apparent, with cluster 1 containing 11 samples and cluster 2 containing 12. PCA of the OA hip samples confirmed this clustering (Supplementary Figure 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38713/abstract). There were 15,239 DMLs between the 2 clusters, with 8,324 hypermethylated and 6,915 hypomethylated in cluster 2. Hierarchical clustering demonstrated that the 15,239 DMLs could be used to distinguish between the 2 clusters (Figure 2b). Intriguingly, GO term analysis of genes containing DMLs revealed an enrichment of several pathways involved in the immune response and inflammation (Figure 2c), including IL2, IL3, IL4, and IL6. There were again several DMLs in genes involved in TGFβ signaling and in genes involved in cartilage degradation and homeostasis (Table2 and see Supplementary Table 4 for full list [available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38713/abstract]).
Figure 2.

Segregation of cartilage chondrocyte DNA from patients with hip osteoarthritis (OA) based on the methylation profile. a, Unsupervised hierarchical clustering of the global β values in the hip OA samples, which revealed 2 distinct clusters. b, Heatmap showing the unsupervised clustering of the 15,239 differentially methylated loci (DMLs) identified between the 2 hip OA clusters. DMLs were defined as those with at least a 10% difference in methylation between the 2 groups and with a Benjamini-Hochberg corrected P value of less than 0.05. Dendrogram at the top shows the clustering of the samples. Dendrogram at the left shows the clustering of the loci. The methylation scale is shown to the right of the heatmap (1 = 100% methylation; 0 = no methylation). c, Gene ontology pathway analysis of the 15,239 DMLs. ∗ = P ≤ 0.05; ∗∗ = P ≤ 0.01; ∗∗∗ = P ≤ 0.001, after Benjamini-Hochberg correction for multiple tests. d, Analysis of 4 selected loci found to be differentially methylated in hip OA versus femoral neck fracture (NOF) samples and between hip OA clusters 1 and 2 (OA hip 1 and 2). Each data point represents a single sample; horizontal lines and error bars show the mean ± SEM. ∗ = P ≤ 0.05; ∗∗ = P ≤ 0.01; ∗∗∗ = P ≤ 0.001, by one-way analysis of variance. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.38713/abstract.
Table 2.
Genes harboring differentially methylated loci between the 2 hip osteoarthritis clusters
| Gene | Mean β value | P, after Benjamini-Hochberg correction | |
|---|---|---|---|
| Cluster 1 | Cluster 2 | ||
| Hypermethylated in cluster 2 | |||
| ACAN | 0.68 | 0.79 | 0.0035 |
| ADAMTS2 | 0.23 | 0.37 | 0.00093 |
| ADAMTS7 | 0.41 | 0.55 | 0.0020 |
| ADAMTS9 | 0.23 | 0.37 | 0.031 |
| ADAMTS10 | 0.55 | 0.66 | 0.0012 |
| ADAMTS14 | 0.44 | 0.59 | 0.033 |
| BMP2 | 0.28 | 0.56 | 0.00058 |
| BMP4 | 0.42 | 0.54 | 0.0096 |
| BMP6 | 0.27 | 0.39 | 0.029 |
| BMP8A | 0.69 | 0.84 | 0.0069 |
| BMPR1A | 0.32 | 0.48 | 0.025 |
| COL2A1 | 0.21 | 0.34 | 0.0011 |
| GDF5 | 0.66 | 0.79 | 0.000078 |
| IL6 | 0.43 | 0.58 | 0.027 |
| IL10 | 0.15 | 0.35 | 0.017 |
| IL16 | 0.71 | 0.82 | 0.035 |
| IL17B | 0.41 | 0.58 | 0.0017 |
| MCF2L | 0.26 | 0.48 | 0.0033 |
| MMP2 | 0.39 | 0.52 | 0.00028 |
| MMP20 | 0.14 | 0.29 | 0.025 |
| MMP28 | 0.25 | 0.41 | 0.028 |
| SMAD5 | 0.62 | 0.73 | 0.018 |
| SMAD7 | 0.44 | 0.59 | 0.00019 |
| Hypomethylated in cluster 2 | |||
| ACVR1 | 0.83 | 0.71 | 0.033 |
| ADAM11 | 0.41 | 0.31 | 0.022 |
| ADAM12 | 0.89 | 0.75 | 0.021 |
| ADAM19 | 0.82 | 0.66 | 0.046 |
| ADAMTS1 | 0.52 | 0.32 | 0.017 |
| ADAMTS5 | 0.31 | 0.18 | 0.0026 |
| ADAMTS6 | 0.64 | 0.51 | 0.026 |
| ADAMTSL2 | 0.58 | 0.40 | 0.016 |
| CHST11 | 0.43 | 0.21 | 0.0052 |
| ECM1 | 0.55 | 0.39 | 0.00042 |
| IL2 | 0.51 | 0.34 | 0.031 |
| IL3 | 0.52 | 0.35 | 0.0028 |
| IL15 | 0.55 | 0.40 | 0.015 |
| IL17C | 0.45 | 0.34 | 0.032 |
| IL4R | 0.47 | 0.23 | 0.00035 |
| MMP19 | 0.39 | 0.24 | 0.0045 |
| SMAD3 | 0.60 | 0.43 | 0.020 |
| TGFB1 | 0.34 | 0.22 | 0.032 |
| SMAD3 | 0.25 | 0.15 | 0.048 |
Figure 2d presents specific examples of CpG sites that showed significant differences in β values between OA hip samples from cluster 1 and cluster 2, as well as the β values for the femoral neck fracture samples, which showed significant differences between each of the 2 OA hip clusters. The CpG sites reside at the genes BMPR1B, ADAMTS5, CHST11, and SOX5.
Clustering of knee OA cartilage samples based on their methylome
A previous study showed that knee OA samples also segregate into 2 groups (13). However, that study was performed with the relatively low-density Illumina 27K human methylation array. Unsupervised hierarchical clustering of our 73 knee OA samples also revealed 2 separate clusters, with 39 samples within cluster 1 and 34 samples within cluster 2 (Supplementary Figure 4a, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38713/abstract). PCA confirmed that the samples split into 2 clusters (Supplementary Figure 5, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38713/abstract). Comparison between the 2 knee clusters revealed 5,769 DMLs, of which 3,000 were hypermethylated in cluster 2 and 2,769 were hypomethylated in cluster 2. As for the enrichment observed for the 2 hip OA clusters described above, there was also enrichment between the 2 knee OA clusters of genes involved in immune response pathways (see Supplementary Figure 4b for examples). Table3 highlights key examples, and Supplementary Table 5 provides a full list (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38713/abstract).
Table 3.
Genes harboring differentially methylated loci between the 2 knee osteoarthritis clusters
| Gene | Mean β value | P, after Benjamini-Hochberg correction | |
|---|---|---|---|
| Cluster 1 | Cluster 2 | ||
| Hypermethylated in cluster 2 | |||
| ACVR2B | 0.52 | 0.62 | 0.0000021 |
| ADAMTSL1 | 0.47 | 0.59 | 0.0039 |
| ADAMTSL3 | 0.61 | 0.71 | 0.0000027 |
| BMP2 | 0.32 | 0.45 | 0.000026 |
| BMP5 | 0.38 | 0.51 | 0.0000055 |
| COL2A1 | 0.31 | 0.44 | 0.00000000016 |
| COL6A3 | 0.18 | 0.29 | 0.00000033 |
| FGF1 | 0.24 | 0.36 | 0.0000013 |
| IL10 | 0.14 | 0.25 | 0.000021 |
| IL16 | 0.44 | 0.58 | 0.000000016 |
| IL18 | 0.29 | 0.41 | 0.00020 |
| IL19 | 0.39 | 0.49 | 0.000046 |
| MMP28 | 0.45 | 0.56 | 0.000000064 |
| MTOR | 0.24 | 0.38 | 0.0000034 |
| SMAD2 | 0.46 | 0.58 | 0.00000011 |
| SMAD7 | 0.19 | 0.29 | 0.000020 |
| TGFB1 | 0.48 | 0.62 | 0.00000096 |
| TGFB2 | 0.39 | 0.51 | 0.00088 |
| Hypomethylated in cluster 2 | |||
| ACVR1 | 0.78 | 0.67 | 0.0000000081 |
| ADAM12 | 0.84 | 0.74 | 0.000000011 |
| ADAM32 | 0.77 | 0.65 | 0.000016 |
| ADAMTS10 | 0.70 | 0.51 | 0.00000000066 |
| ADAMTS12 | 0.54 | 0.44 | 0.00057 |
| CHST11 | 0.75 | 0.64 | 0.00000021 |
| IL2 | 0.50 | 0.39 | 0.00070 |
| IL4R | 0.42 | 0.27 | 0.000000000078 |
| IL6R | 0.63 | 0.49 | 0.000084 |
| MMP19 | 0.37 | 0.26 | 0.000000010 |
| RUNX1 | 0.65 | 0.46 | 0.000000037 |
| RUNX2 | 0.69 | 0.55 | 0.000011 |
| RUNX3 | 0.71 | 0.59 | 0.0000034 |
| SMAD3 | 0.63 | 0.48 | 0.0000000020 |
| TGFBR2 | 0.67 | 0.48 | 0.0000000025 |
| TGFBR3 | 0.56 | 0.41 | 0.0000000023 |
| TIMP2 | 0.87 | 0.76 | 0.000011 |
Comparison of the DNA methylome between hip OA and knee OA cartilage
Having demonstrated that hip OA and knee OA each segregates into clusters, we next investigated whether there was a commonality of DMLs accounting for the stratification observed in the hip clusters and in the knee clusters. A total of 3,496 shared DMLs (23% of hip OA cluster DMLs, 61% of knee OA cluster DMLs) were identified (data not shown). GO term analysis again revealed an enrichment of genes involved in the immune response (data not shown). However, particularly in the case of the hip OA samples, a large number of the DMLs did not overlap between the 2 joints, which suggests that alternative genes/pathways may be involved in the observed stratification. A full list of the shared DMLs between the hip OA and knee OA clusters is shown in Supplementary Table 6 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38713/abstract).
We next compared DNA methylation between hip OA and knee OA samples. A total of 5,547 DMLs were identified, with 2,598 hypermethylated in hip OA and 2,949 hypomethylated in hip OA. A large number of the genes containing DMLs encode proteins that are involved in development; selected pathways relevant to OA are shown in Supplementary Figure 4c (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38713/abstract). In addition, genes coding for proteins involved in OA pathogenesis were again represented by the DMLs, including ADAM12, ADAMTS5, CHST11, GDF5, and MCF2L (Supplementary Table 7, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38713/abstract). Several key DMLs are listed in Table4.
Table 4.
Genes harboring differentially methylated loci between hip and knee osteoarthritis (OA) samples
| Gene | Mean β value | P, after Benjamini-Hochberg correction | |
|---|---|---|---|
| Knee OA patients | Hip OA patients | ||
| Hypermethylated in hip OA | |||
| ADAM12 | 0.70 | 0.83 | 0.00000000015 |
| ADAM7 | 0.17 | 0.28 | 0.000033 |
| ADAMTS2 | 0.77 | 0.88 | 0.00000016 |
| ADAMTS5 | 0.51 | 0.66 | 0.000080 |
| ADAMTS17 | 0.27 | 0.42 | 0.0000049 |
| COL6A3 | 0.43 | 0.63 | 0.00000000083 |
| IL17RD | 0.48 | 0.65 | 0.000000034 |
| IL18 | 0.35 | 0.46 | 0.0047 |
| IL1RN | 0.47 | 0.59 | 0.0000016 |
| RUNX3 | 0.59 | 0.74 | 0.000000000025 |
| TGFB2 | 0.21 | 0.32 | 0.00073 |
| Hypomethylated in hip OA | |||
| ADAMTS9 | 0.42 | 0.31 | 0.00036 |
| BMP6 | 0.31 | 0.19 | 0.0011 |
| BMP7 | 0.46 | 0.33 | 0.0000022 |
| CHST11 | 0.65 | 0.53 | 0.0000026 |
| COL4A2 | 0.60 | 0.44 | 0.00000020 |
| COL18A1 | 0.68 | 0.58 | 0.00000000018 |
| FGF1 | 0.66 | 0.56 | 0.0084 |
| FGFR3 | 0.66 | 0.55 | 0.00014 |
| GDF5 | 0.85 | 0.73 | 0.000049 |
| IL10 | 0.69 | 0.56 | 0.000018 |
| MCF2L | 0.51 | 0.40 | 0.0013 |
| PCOLCE | 0.46 | 0.33 | 0.00015 |
| SMAD7 | 0.78 | 0.62 | 0.0000018 |
| TGFBR2 | 0.59 | 0.49 | 0.0065 |
Analysis of genome features enriched within the DMLs
It has recently become apparent that the location of a CpG has important implications in the effect of DNA methylation on gene expression (24). We therefore investigated whether there is enrichment for certain genome features within the DMLs. We studied all of the DMLs that we have identified and present those data here; the same results were obtained when we looked at the hip DMLs and the knee DMLs separately (data not shown). Accounting for the distribution of the CpG probes on the 450 BeadChip array, Supplementary Figure 6a demonstrates that there was a decrease in DMLs within CpG islands, but an increase within CpG shores and shelves. As shown in Supplementary Figure 6b, the increased frequency of DMLs within enhancers was confirmed.
Recent studies have challenged the dogma that CpG island–associated promoters are more likely to be differentially regulated. Instead, it has now become apparent that relatively CpG-poor regions in distal regulatory elements are much more likely to show methylation alterations in response to environmental effects and that these methylation changes are more strongly associated with differential gene expression (25,26). Likewise, our results show an increase in DMLs within less dense CpG regions.
DISCUSSION
In the present report, we describe the characterization of the cartilage chondrocyte DNA methylome in a cohort of 96 OA patients and 21 non-OA controls. For the first time, we have characterized the DNA methylome of OA and non-OA hip chondrocytes and identified several DMLs between the 2 groups that may play a role in the disease. Furthermore, we revealed that hip OA samples can be separated into 2 groups based on their methylation profile. This has previously been observed in knee OA and has clear implications for future diagnostic and therapeutic approaches to the disease. Similarly, knee OA and hip OA samples have distinct methylation profiles, emphasizing the importance of separating a study of OA by skeletal site. Likewise, a previous microarray study has shown that knee and hip OA samples show discordance at the gene expression level (27).
A previous investigation of the cartilage DNA methylome in knee OA samples used the Illumina Infinium HumanMethylation27 array (15). By using the Illumina Infinium HumanMethylation450 array, our study provides a much more comprehensive analysis. Not only does this array contain significantly more probes, it also contains probes within a range of genomic features, including enhancers, promoters, and the gene body, that are not covered by the promoter-centric Infinium HumanMethylation27 array. Including probes within different genome features is important, as it is now well established that the effect of differential methylation is dependent on the location of the CpG dinucleotide (23,24,28–30).
Our comparison of the DNA methylome of hip cartilage samples from OA patients and controls revealed a total of 5,322 DMLs. Several reside in genes that have previously been shown via candidate gene studies to be differentially methylated in OA (9,10,31). However, we identified a large number of DMLs in hip OA that have not previously been described, including within genes coding for proteins active in the catabolic and anabolic pathways of cartilage homeostasis. This observation suggests that DNA methylation is a key regulatory step in the shift in the balance of cartilage maintenance in the direction of overall degradation.
One highly relevant pathway that emerged from our analysis is the TGFβ pathway (32). Downstream signaling of TGFβ is altered during the development of OA, with a shift from the anabolic SMAD2/3 pathway to the catabolic SMAD1/5/8 pathway (33,34). Interestingly, we observed that both SMAD2 and SMAD3 were differentially methylated in OA patients compared to controls, indicating that epigenetics is a driving force in this shift. Furthermore, genes encoding receptors of members of the TGFβ superfamily were also differentially methylated. A role of DNA methylation in switching TGFβ signaling is particularly appealing, as the switch is associated with aging, which in turn, is associated with aberrant DNA methylation (35,36).
We also observed DMLs that fall within OA genetic-association signals discovered by the Arthritis Research UK Osteoarthritis Genetics (arcOGEN) Consortium and by other studies (22). It will be intriguing to assess whether the genotype at the associated SNPs correlates with differences in methylation at CpG sites encompassed by the association signals. Several of the genes that contain DMLs within our study have also been shown to be differentially expressed between hip OA and femoral neck fracture cartilage in a previous study, including ACVR1, ADAMTS1, ADAMTS5, ADAMTS6, BMP6, COL2A1, and FGFR3 (27).
Within our analysis, we observed that 2 clusters formed within the OA hip samples based on their genome-wide methylation profiles. Subsequent analysis revealed 15,239 DMLs between the 2 clusters. There was a striking enrichment of genes containing DMLs in pathways involved in the immune response and inflammation. This suggests that inflammation, at least for a subgroup of OA patients, may play a more integral role in the disease process than has previously been envisioned. This is consistent with proteomic and transcriptomic data (37). The femoral neck fracture samples also formed 2 clusters; however, the majority of these (18 of 21 [86%]) grouped together in cluster 1. Nevertheless, our data may also point to methylation differences within the cartilage of the femoral neck fracture phenotype that may also be informative.
It has previously been shown that knee OA samples also cluster based on their DNA methylation profile (13). This result was derived from an analysis of 23 knee OA patients. Our analysis of 73 knee OA patients revealed the same effect, with 2 distinct clusters apparent. There were a total of 5,769 DMLs between the 2 groups. This is greater than the 1,357 DMLs discovered previously, which is presumably a reflection of the larger sample size we used combined with the greater density of the 450 array. Nevertheless, the observations were similar to those in the previous report, with an enrichment of genes coding for proteins involved in the inflammatory response. However, we identified additional genes that discriminate between the 2 groups, including several coding for interleukins, as well as genes coding for members of the TGFβ pathway. This clearly emphasizes the greater sensitivity that is derived from investigating a larger number of CpG sites in more patients.
A comparison between the DMLs identified in the hip cluster and knee cluster analyses revealed an overlap of inflammatory genes. This indicates that there is a shared molecular mechanism that can segregate patients, whether they present with knee OA or with hip OA. OA is strongly associated with aging, while OA chondrocytes can acquire a senescent state (38). In the senescence-associated secretory phenotype (SASP), senescent cells undergo a change in protein expression and secretory activity (39). Intriguingly, several of the genes in our study harboring DMLs between the hip OA clusters and the knee OA clusters code for SASP proteins, which raises the possibility that in a subset of samples, the clustering may be partly a result of an SASP phenotype.
A comparison between the knee and hip OA samples revealed 5,547 DMLs. A large number of these were within developmental genes, most notably homeobox genes, and in OA pathogenesis and/or cartilage homeostasis genes. This suggests that there may be unique pathways that distinguish OA of the knee with OA of the hip. This is consistent with genetic association and gene expression studies, which indicate that OA-associated signals are often joint-specific (22,27).
One of the greatest challenges in epigenetic studies is in establishing causality. We used cartilage samples from patients with end-stage OA, and we selected intact cartilage from sites distal to the OA lesion. An alternative would have been to study cartilage from the site of the lesion and to compare this with distal cartilage from the same patient. This may shed light on key events involved in the latter stages of cartilage degeneration. If such data become available, it would be intriguing to compare and contrast DMLs between these studies and ours as a means of potentially untangling the key causal and responsive DNA methylation events.
It would also have been interesting to compare the methylome of knee OA samples with knee control samples and to further compare any methylation differences observed with those identified between hip OA and femoral neck fracture samples. However, this was not possible due to the lack of a suitable control for the knee. Another limitation of our study is the older age of the femoral neck fracture patients compared to the hip OA patients (average of 12 years), since it is known that epigenetic changes occur during aging. However, such epigenetic differences are observed over many years between young individuals and the elderly (e.g., between newborns and centenarians) (40). We suspect that the relatively small difference in the age of the femoral neck fracture and OA hip samples would have minimal impact on the cartilage DNA methylome, but we cannot discount its potential effect.
In conclusion, we demonstrate that OA hip cartilage chondrocyte DNA can be distinguished from healthy hip cartilage chondrocyte DNA by their methylomes. We show that several genes involved in OA-specific pathways are differentially methylated and may therefore offer potential therapeutic targets in the treatment of this common disease. In addition, we provide evidence that DNA methylation profiling can be a powerful diagnostic tool, allowing the identification of a subcluster of knee samples and a subcluster of hip samples characterized by differences in the DNA methylomes of genes coding for inflammatory factors. Analysis of the DNA methylome is clearly therefore an extremely useful tool in our efforts to understand the molecular basis of this complex arthritis.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Rushton had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Rushton, Reynard, Barter, Young, Loughlin.
Acquisition of data. Rushton, Refaie, Rankin.
Analysis and interpretation of data. Rushton.
Supplementary Table 1-2
Supplementary Table 3
Supplementary Table 4
Supplementary Table 5
Supplementary Table 6
Supplementary Table 7
Supplementary Table 8
Supplementary Figure 1. Validation of DMLs by Pyrosequencing. Six NOF and six OA hip samples from amongst the patients analyzed with the 450 BeadChip array were selected for validation by pyrosequencing. Data is plotted as the level of methylation as determined by pyrosequencing (y-axis) against the β value obtained from the array multiplied by 100 to obtain a percentage value (x-axis). The CpG site analyzed for each gene was as follows: SIM2 (CG10682155), TNXB (CG16385684), ALX1 (CG16413687). In the array analysis each of these sites showed significant differential methylation between NOF and OA hip.
Supplementary Figure 2. The effect of differential methylation on gene expression. Gene expression was measured in seven NOF and six OA hip samples from amongst the patients analysed with the 450 BeadChip array, and for four genes (ACVR1, ADAMTS5, CHST11 and LRP5) that showed differential methylation between OA hip and NOF samples in the array analysis. A) Gene expression of genes that are differentially methylated within the gene body. B) Gene expression of genes that are differentially methylated within promoter/enhancer regions. Data is presented as 2−ΔCt (Ct Target gene – Ct average of control genes (18s, GAPDH and HPRT1)) and is shown as the mean and the standard error of the mean.
Supplementary Figure 3. Principal component analysis of the DNA methylome of OA hip samples. PCA of global β values in OA hip samples reveals two distinct groups corresponding to those identified through the unsupervised hierarchical clustering. The red squares correspond to the 11 samples from cluster 1, whilst the black circles correspond to the 12 samples present in cluster 2.
Supplementary Figure 4. OA knee cartilage chondrocyte DNA samples segregate based on their methylation profile. A) Unsupervised hierarchical clustering of the global β values in OA knee patient samples, which revealed two distinct groups. B) Gene ontology pathway analysis of the 5769 DMLs. P values are Benjamini-Hochberg corrected to take account of the multiple tests performed***P≤0.001, **P≤0.01, *P≤0.05. C) Gene ontology pathway analysis of the 5547 DMLs identified between OA knee and OA hip. P values are Benjamini-Hochberg corrected to take account of the multiple tests performed ***P≤0.001, **P≤0.01, *P≤0.05.
Supplementary Figure 5. Principal component analysis of the DNA methylome of OA knee samples. PCA of global β values in OA knee samples reveals two distinct groups corresponding to those identified through the unsupervised hierarchical clustering. The red squares correspond to the 39 samples from cluster 1, whilst the black circles correspond to the 34 samples present in cluster 2.
Supplementary Figure 6. Enrichment of genome features within identified DMLs. A) Frequency distribution of the DMLs between CpG islands, shelves and shores (black columns) compared to the actual frequency distribution of the 458 577 CpGs probes from the 450 BeadChip array (450K, red columns). B) Frequency of the DMLs that are present within enhancer regions (black column) compared to the actual frequency of the 458 577 CpGs probes within these regions (450K, red column).
REFERENCES
- Kuettner KE, Cole AA. Cartilage degeneration in different human joints. Osteoarthritis Cartilage. 2005;13:93–103. doi: 10.1016/j.joca.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Dieppe P, Lohmander L. Pathogenesis and management of pain in osteoarthritis. Lancet. 2005;365:965–73. doi: 10.1016/S0140-6736(05)71086-2. [DOI] [PubMed] [Google Scholar]
- Felson DT, Zhang Y. An update on the epidemiology of knee and hip osteoarthritis with a view to prevention. Arthritis Rheum. 1998;41:1343–55. doi: 10.1002/1529-0131(199808)41:8<1343::AID-ART3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Blanco FJ, Rego I, Ruiz-Romero C. The role of mitochondria in osteoarthritis. Nat Rev Rheumatol. 2011;7:161–9. doi: 10.1038/nrrheum.2010.213. [DOI] [PubMed] [Google Scholar]
- Lohmander LS, Lark MW, Dahlberg L, Walakovits LA, Roos H. Cartilage matrix metabolism in osteoarthritis: markers in synovial fluid, serum, and urine. Clin Biochem. 1992;25:167–74. doi: 10.1016/0009-9120(92)90250-v. [DOI] [PubMed] [Google Scholar]
- Reynard LN, Loughlin J. Genetics and epigenetics of osteoarthritis. Maturitas. 2012;71:200–4. doi: 10.1016/j.maturitas.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Barter MJ, Bui C, Young DA. Epigenetic mechanisms in cartilage and osteoarthritis: DNA methylation, histone modifications and microRNAs. Osteoarthritis Cartilage. 2012;20:339–49. doi: 10.1016/j.joca.2011.12.012. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17:330–9. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- Roach HI, Yamada N, Cheung KS, Tilley S, Clarke NM, Oreffo RO, et al. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 2005;52:3110–24. doi: 10.1002/art.21300. [DOI] [PubMed] [Google Scholar]
- Cheung KS, Hashimoto K, Yamada N, Roach HI. Expression of ADAMTS-4 by chondrocytes in the surface zone of human osteoarthritic cartilage is regulated by epigenetic DNA de-methylation. Rheumatol Int. 2009;29:525–34. doi: 10.1007/s00296-008-0744-z. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Otero M, Imagawa K, de Andres MC, Coico JM, Roach HI, et al. Regulated transcription of human matrix metalloproteinase 13 (MMP13) and interleukin-1β (IL1B) genes in chondrocytes depends on methylation of specific proximal promoter CpG sites. J Biol Chem. 2013;288:10061–72. doi: 10.1074/jbc.M112.421156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Andres MC, Imagawa K, Hashimoto K, Gonzalez A, Roach HI, Goldring MB, et al. Loss of methylation in CpG sites in the NF-κB enhancer elements of inducible nitric oxide synthase is responsible for gene induction in human articular chondrocytes. Arthritis Rheum. 2013;65:732–42. doi: 10.1002/art.37806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Oreffo RO, Gibson MB, Goldring MB, Roach HI. DNA demethylation at specific CpG sites in the IL1B promoter in response to inflammatory cytokines in human articular chondrocytes. Arthritis Rheum. 2009;60:3303–13. doi: 10.1002/art.24882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynard LN, Bui C, Canty-Laird EG, Young DA, Loughlin J. Expression of the osteoarthritis-associated gene GDF5 is modulated epigenetically by DNA methylation. Hum Mol Genet. 2011;20:3450–60. doi: 10.1093/hmg/ddr253. [DOI] [PubMed] [Google Scholar]
- Fernandez-Tajes J, Soto-Hermida A, Vazquez-Mosquera ME, Cortes-Pereira E, Mosquera A, Fernandez-Moreno M, et al. Genome-wide DNA methylation analysis of articular chondrocytes reveals a cluster of osteoarthritic patients. Ann Rheum Dis. 2014;73:668–77. doi: 10.1136/annrheumdis-2012-202783. [DOI] [PubMed] [Google Scholar]
- Wilkins JM, Southam L, Price AJ, Mustafa Z, Carr A, Loughlin J. Extreme context specificity in differential allelic expression. Hum Mol Genet. 2007;16:537–46. doi: 10.1093/hmg/ddl488. [DOI] [PubMed] [Google Scholar]
- Pidsley R, Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14:293. doi: 10.1186/1471-2164-14-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touleimat N, Tost J. Complete pipeline for Infinium Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics. 2012;4:325–41. doi: 10.2217/epi.12.21. [DOI] [PubMed] [Google Scholar]
- Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–27. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Whitaker JW, Shoemaker R, Boyle DL, Hillman J, Anderson D, Wang W, et al. An imprinted rheumatoid arthritis methylome signature reflects pathogenic phenotype. Genome Med. 2013;5:40. doi: 10.1186/gm444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Reynard LN, Loughlin J. Insights from human genetic studies into the pathways involved in osteoarthritis. Nat Rev Rheumatol. 2013;9:573–83. doi: 10.1038/nrrheum.2013.121. [DOI] [PubMed] [Google Scholar]
- Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK. On the presence and role of human gene-body DNA methylation. Oncotarget. 2012;3:462–74. doi: 10.18632/oncotarget.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41:1350–3. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Barter MJ, Swan DC, Rankin KS, Rowan AD, Santibanez-Koref M, et al. Identification of the pathogenic pathways in osteoarthritic hip cartilage: commonality and discord between hip and knee OA. Osteoarthritis Cartilage. 2012;20:1029–38. doi: 10.1016/j.joca.2012.05.006. [DOI] [PubMed] [Google Scholar]
- Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378–85. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Cortez CC, Yang X, Nichols PW, Jones PA, Liang G. DNA methylation directly silences genes with non-CpG island promoters and establishes a nucleosome occupied promoter. Hum Mol Genet. 2011;20:4299–310. doi: 10.1093/hmg/ddr356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal-Yam EN, Egger G, Iniguez L, Holster H, Einarsson S, Zhang X, et al. Frequent switching of polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc Natl Acad Sci U S A. 2008;105:12979–84. doi: 10.1073/pnas.0806437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui C, Barter MJ, Scott JL, Xu Y, Galler M, Reynard LN, et al. cAMP response element-binding (CREB) recruitment following a specific CpG demethylation leads to the elevated expression of the matrix metalloproteinase 13 in human articular chondrocytes and osteoarthritis. FASEB J. 2012;26:3000–11. doi: 10.1096/fj.12-206367. [DOI] [PubMed] [Google Scholar]
- Bush JR, Beier F. TGF-β and osteoarthritis: the good and the bad. Nat Med. 2013;19:667–9. doi: 10.1038/nm.3228. [DOI] [PubMed] [Google Scholar]
- Van der Kraan PM, Blaney Davidson EN, Blom A, van den Berg WB. TGF-β signaling in chondrocyte terminal differentiation and osteoarthritis: modulation and integration of signaling pathways through receptor-Smads. Osteoarthritis Cartilage. 2009;17:1539–45. doi: 10.1016/j.joca.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Zhen G, Wen C, Jia X, Li Y, Crane JL, Mears SC, et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat Med. 2013;19:704–12. doi: 10.1038/nm.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Kraan PM, Blaney Davidson EN, van den Berg WB. A role for age-related changes in TGFβ signaling in aberrant chondrocyte differentiation and osteoarthritis. Arthritis Res Ther. 2010;12:201. doi: 10.1186/ar2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Kraan PM, Goumans MJ, Blaney Davidson E, ten Dijke P. Age-dependent alteration of TGF-β signaling in osteoarthritis. Cell Tissue Res. 2012;347:257–65. doi: 10.1007/s00441-011-1194-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Rozelle AL, Lepus CM, Scanzello CR, Song JJ, Larsen DM, et al. Identification of a central role for complement in osteoarthritis. Nat Med. 2011;17:1674–9. doi: 10.1038/nm.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JS, Waters JG, Darrah C, Pennington C, Edwards DR, Donell ST, et al. The role of chondrocyte senescence in osteoarthritis. Aging Cell. 2002;1:57–65. doi: 10.1046/j.1474-9728.2002.00008.x. [DOI] [PubMed] [Google Scholar]
- Young AR, Narita M. SASP reflects senescence. EMBO Rep. 2009;10:228–30. doi: 10.1038/embor.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, et al. Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci U S A. 2012;109:10522–7. doi: 10.1073/pnas.1120658109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1-2
Supplementary Table 3
Supplementary Table 4
Supplementary Table 5
Supplementary Table 6
Supplementary Table 7
Supplementary Table 8
Supplementary Figure 1. Validation of DMLs by Pyrosequencing. Six NOF and six OA hip samples from amongst the patients analyzed with the 450 BeadChip array were selected for validation by pyrosequencing. Data is plotted as the level of methylation as determined by pyrosequencing (y-axis) against the β value obtained from the array multiplied by 100 to obtain a percentage value (x-axis). The CpG site analyzed for each gene was as follows: SIM2 (CG10682155), TNXB (CG16385684), ALX1 (CG16413687). In the array analysis each of these sites showed significant differential methylation between NOF and OA hip.
Supplementary Figure 2. The effect of differential methylation on gene expression. Gene expression was measured in seven NOF and six OA hip samples from amongst the patients analysed with the 450 BeadChip array, and for four genes (ACVR1, ADAMTS5, CHST11 and LRP5) that showed differential methylation between OA hip and NOF samples in the array analysis. A) Gene expression of genes that are differentially methylated within the gene body. B) Gene expression of genes that are differentially methylated within promoter/enhancer regions. Data is presented as 2−ΔCt (Ct Target gene – Ct average of control genes (18s, GAPDH and HPRT1)) and is shown as the mean and the standard error of the mean.
Supplementary Figure 3. Principal component analysis of the DNA methylome of OA hip samples. PCA of global β values in OA hip samples reveals two distinct groups corresponding to those identified through the unsupervised hierarchical clustering. The red squares correspond to the 11 samples from cluster 1, whilst the black circles correspond to the 12 samples present in cluster 2.
Supplementary Figure 4. OA knee cartilage chondrocyte DNA samples segregate based on their methylation profile. A) Unsupervised hierarchical clustering of the global β values in OA knee patient samples, which revealed two distinct groups. B) Gene ontology pathway analysis of the 5769 DMLs. P values are Benjamini-Hochberg corrected to take account of the multiple tests performed***P≤0.001, **P≤0.01, *P≤0.05. C) Gene ontology pathway analysis of the 5547 DMLs identified between OA knee and OA hip. P values are Benjamini-Hochberg corrected to take account of the multiple tests performed ***P≤0.001, **P≤0.01, *P≤0.05.
Supplementary Figure 5. Principal component analysis of the DNA methylome of OA knee samples. PCA of global β values in OA knee samples reveals two distinct groups corresponding to those identified through the unsupervised hierarchical clustering. The red squares correspond to the 39 samples from cluster 1, whilst the black circles correspond to the 34 samples present in cluster 2.
Supplementary Figure 6. Enrichment of genome features within identified DMLs. A) Frequency distribution of the DMLs between CpG islands, shelves and shores (black columns) compared to the actual frequency distribution of the 458 577 CpGs probes from the 450 BeadChip array (450K, red columns). B) Frequency of the DMLs that are present within enhancer regions (black column) compared to the actual frequency of the 458 577 CpGs probes within these regions (450K, red column).