

Abstract
BRCA1-associated ATM activator-1 (BRAT1) was identified by our group as a DNA damage response (DDR) protein, which can bind with many DDR proteins and regulates their functions after DNA damage. However, previous study has also implicated BRAT1 as a regulator of cell growth and apoptosis. In this study, targeted gene deletion showed that BRAT1 is critical in stability and serum-induced expression of mTOR and downstream protein. Conditional deletion of BRAT1 of mouse embryonic fibroblasts suppressed serum-induced cell cycling progress. Our results suggest that BRAT1 is essential factor for PIKK signaling cascades.
Keywords: BRCA1, Human cervical carcinoma, Cell cycle, Cell signaling
INTRODUCTION
BRAT1 was originally identified as a BRCA1 interacting protein [1], however, subsequent studies showed that it also binds to ATM, DNA-PK, and SMC1, implicating the broad role in DNA repair and cell cycle regulation, as well as in DDR after DNA damage [2]. Previous our reports showed that BRAT1 is essential for the phosphorylation of ATM and DNA-PK, and these proteins-mediated DDR was significantly diminished in BRAT1 knockdown cells [3], suggesting BRAT1 is required for reaction stability of ATM and DNA-PK. Interestingly, BRAT1 knockdown induces constitutive apoptosis of mouse embryonic fibroblasts and human osteosarcoma cell line U2OS. Such results suggest that BRAT1 function is not limited to the response to DDR, and BRAT1 seems to be involved in controlling cell growth.
In this study, we hypothesize that BRAT1 is required for protein stability of mTOR and mTOR-related proteins, and cell cycle progress by growth factors. We found the protein expression of mTOR and S6-kinase, downstream substrate of mTOR, was impaired during serum starvation and delayed after serum treatment in BRAT1 deficient conditions, compared to control. Our data suggested that BRAT1 regulates the stability of PIKKs, such as ATM, DNA-PK, and mTOR.
MATERIALS AND METHODS
BRAT1 knockdown stable cell lines
Human Cervical carcinoma, HeLa cells were purchased from American Type Culture Collection (ATCC, Manassas, VA) and maintained in complete DMEM media (Invitrogen, Carlsbad, CA) containing 10% Fetal Bovine Serum (FBS) and penicillin/streptomycin (Invitrogen). Sure silencing shRNA plasmids for human C7 or f27 were purchased from SABiosciences (Valencia, CA). Control (NC) and BRAT1 knockdown hela cells were cloned as previously described [2].
Mouse Embryonic Fibroblasts culture and BRAT1 gene targeting
Primary MEFs were isolated from 13.5 day embryo of BRAT1flox/flox/129S6 strain (unpublished data). MEFs were maintained in 10% FBS DMEM. Plasmids containing Cre gene (pCAG-Cre:GFP) and GFP vector were provided from Addgene (Cambridge, MA), and used to target BRAT1 gene, transiently. MEFs were transformed by infection with retrovirus containing SV40-LT gene to be immortalized. Retrovirus containing Cre gene will be used for BRAT1 gene targeting. Retroviral vector (pBabe) was used for cloning and making control retrovirus.
Immunoprecipitation and western blot
BRAT1 GST fragments were generated by PCR from human BRAT1 full-length plasmids as described (2). #3, #5, and #6 fragments contained 302–400, 541–700, and 701–821 amino acids, respectively. 293 cells were transfected with plasmids containing GST-full length or fragments of human BRAT1 gene. After 48 h, Total lysates were prepared in ice-cold lysis buffer (50 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1 mM EDTA (pH 8.0), 20 mM NaF, 1 mM Na3VO4, 1% NP-40, 0.5 mM dithiothreitol] in the presence of protease-inhibitor mix (leupeptin, aprotinin and phenylmethylsulfonyl fluoride (PMSF) (10 μg/ml), respectively)). Twenty microgram of total lysates was loaded and separated on 6.0% SDS polyacrylamide gels. Transfer to PVDF membranes (Millipore) was carried out using a semi-dry transfer kit (Bio-Rad) in transfer buffer (25mM TrisHCl, 192 mM glycine and 10% methanol for 1h at 20 V). GSH-bead (GE Healthchare BioSciences, Pittsburgh, PA) was incubated with total lysates (1 mg/sample) to capture BRAT1 binding proteins. The beads were extensively washed with lysis buffer, boiled and loaded on SDS PAGE gel as described above. Primary antibodies used in this study were anti-BRAT1 (Abcam, Cambridge, MA), anti-mTOR, anti-S6K, anti-Raptor (Cell Signaling, Danvers, MA), anti-Akt, anti-GST, and anti-actin (Santa Cruz Biotechnology, Santa Cruz, CA). Additionally, specific anti-phosphorylation antibodies were used against phosphor-mTOR (Ser2481) and phosphor-S6K (Thr389) (Cell Signaling).
Cell cycle analysis
MEFs were starved in DMEM media without FBS for 24h, and then incubated in complete DMEM containing 10% FBS for indicated times. Cell cycle was assessed by DNA staining with propidium Iodide (PI) as previously described [4].
RESULTS AND DISCUSSION
The activation and expression of mTOR were reduced in BRAT1 deficient cells
Because BRAT1 can bind to PIKKs, such ATM and DNA-PK, we hypothesized that BRAT1 can also bind to mTOR, and this binding is critical step for growth and cell cycle progress. In order to investigate the role of BRAT1 in mTOR signaling pathway, control and BRAT1 knockdown HeLa cells were cultured in serum-free media for starvation. Cells were restored in complete DMEM containing serum, and the expression and phosphorylation of mTOR were examined (Figure 1A). The protein expression of mTOR and p70 S6 kinase was not significant changed by BRAT1 deletion, but the protein level of mTOR and S6K was markedly reduced after serum-starvation in these cells compared to control HeLa cells. Furthermore, serum-induced restoration of protein level was suppressed in knockdown cells, suggesting that the stability and serum-induced expression of mTOR was diminished under BRAT1 deficient condition. To confirm this data, we prepared BRAT-deficient MEFs from BRAT1flox/flox/129S6 strain (Figure 1B and 1C). In MEFs, the reduction of mTOR level was already evident in BRAT1 deficient MEFs (lane 1 vs. lane 3 in Figure 1C). Enforced expression of BRAT1 increased the level of mTOR protein in both control and BRAT1 deficient MEFs, indicating that BRAT1 upregulates the level of mTOR protein in MEFs.
Figure 1. BRAT1 is a possible member of mTOR pathways and critical for cell growth.
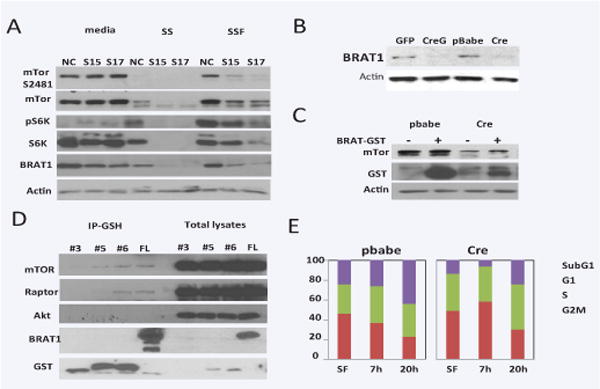
A. Control (NC) and BRAT1 knockdown (S15 and S17) Hela cells were cultured in 10% FBS/DMEM (media) or serum-free DMEM for 24 h (SS), followed by 10% complete DMEM for 2 h (SSF). Total lysates were isolated and subjected to immunoblotting with indicated antibodies. Actin was used as internal control to validate protein loading. B. Mouse embryonic fibroblasts (MEFs) were isolated from E15 of BRAT1flox/flox female mouse as described in Materials and Methods. Cells were transfected with GFP only (V) or Cre-GFP plasmid (CreG). C. Immortalized MEFs infected with pBabe retrovirus (pbabe) or Cre gene retrovirus (Cre) were transfected with BRAT-GST plasmid (+) or GST vector plasmid (−). After 48 hour, Total lysates were subjected to immunoblot. D. 293 cells were transfected with plasmids expressing GST-fusion fragments (#3, 5, 6) or full-length (FL) of BRAT1. Total lysates (1 mg/sample) were subjected to immunoprecipitation with GSH-sepharose beads. After immunoprecipitation, samples were blotted with the indicated antibodies. E. Control (pbabe) and BRAT1 knockout (Cre) MEFs were culture in plain DMEM for 24 hr, and then re-cultured in 10% complete DMEM for 7 or 20 h. Cell cycle was quantified by single-parameter flow cytometry after PI staining. Graphs represent percentages of cells in sub-G1, G1, S, and G2/M phases. Data shown here are representative of three independent experiments performed.
BRAT1 binds to mTORC1 complex
Next, we tested whether BRAT1 bind into mTOR and mTOR-related proteins. Previous immunoprecipitation data revealed both ATM and DNA-PK strongly bound to C-terminal fragment of BRAT1 (#5 and #6) than any other fragments [2]. Using these fragment and full-length BRAT1 (FL), we tested whether mTOR and other mTOR-related protein (Raptor and Akt) bind to BRAT1 (Figure 1D). BRAT1 fragments and full length protein were expressed in 293 cells and immunoprecipitated by GSH-bead. As shown in figure, mTOR and Raptor bound to BRAT1, but Akt was not present in the BRAT1 complex. This result suggests that BRAT1 can bind to proteins of TOCR1, rather than upstream or TOCR2 complex.
BRAT1 is required for progression of cell cycle by growth factor
Our western blot analysis shown in Figure 1 and 2 indicated that PI3/Akt/mTOR signaling pathways by growth factor was impaired in BRAT1 deficient cells for reduced level of mTOR protein. As well known, growth factor-induced activation of PI3K/Akt/mTOR cascades is critical for cellular growth control, such as cell cycle progress, inhibiting autophagy, and increasing cell mass [5]. To test whether BRAT1 deficiency down-regulates growth factor induced cell cycle progress, both control and BRAT1 deficient MEFs were synchronized by serum-starvation, and serum was added as general growth activator into media to induce cell proliferation. As shown in Figure E, population of G2/M-phase markedly increased at 20 h after serum addition in control MEFs, but not in BRAT1 deficient MEFs, indicating BRAT1 is required for sensing growth signal and induces cell cycle progress.
Our data suggested a potential role of BRAT1 in protein stability and regulation of mTOR signaling. Also, we can consider BRAT1 functions in cell growth through PI3K/Akt/mTOR cascades. It has been reported that mTOR controls mitochondrial oxidative function through an YY1-PGC-1 transcriptional complex [6]. ATM plays roles in cell growth control, lymphoid differentiation, and neural stem cell differentiation [7–9]. Therefore, further extended study about BRAT1 roles in PIKK-mediated signaling and relevance in cell growth and mitochondrial functions will give us not only basic understanding for collaboration between BRAT1 and PIKKs, but also one of solutions to develop therapeutic target for cancer treatment.
Acknowledgments
We thank all the members of the Ouchi laboratory for discussion of the results. This work is supported by NIH R01CA90631, Susan G Komen Breast Cancer Grant and Matsutani America Cancer Research Fund.
References
- 1.Aglipay JA, Martin SA, Tawara H, Lee SW, Ouchi T. ATM activation by ionizing radiation requires BRCA1-associated BAAT1. J Biol Chem. 2006;281:9710–8. doi: 10.1074/jbc.M510332200. [DOI] [PubMed] [Google Scholar]
- 2.So EY, Ouchi T. Functional interaction of BRCA1/ATM-associated BAAT1 with the DNA-PK catalytic subunit. Exp Ther Med. 2011;2:443–447. doi: 10.3892/etm.2011.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ouchi M, Ouchi T. Regulation of ATM/DNA-PKcs Phosphorylation by BRCA1-Associated BAAT1. Genes Cancer. 2010;1:1211–4. doi: 10.1177/1947601911404222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.So EY, Ausman M, Saeki T, Ouchi T. Phosphorylation of SMC1 by ATR is required for desferrioxamine (DFO)-induced apoptosis. Cell Death Dis. 2011;2:e128. doi: 10.1038/cddis.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–40. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- 7.Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 1996;10:2411–22. doi: 10.1101/gad.10.19.2411. [DOI] [PubMed] [Google Scholar]
- 8.Starczynski J, Simmons W, Flavell JR, Byrd PJ, Stewart GS, Kullar HS, et al. Variations in ATM protein expression during normal lymphoid differentiation and among B-cell-derived neoplasias. Am J Pathol. 2003;163:423–32. doi: 10.1016/S0002-9440(10)63672-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carlessi L, De Filippis L, Lecis D, Vescovi A, Delia D. DNA-damage response, survival and differentiation in vitro of a human neural stem cell line in relation to ATM expression. Cell Death Differ. 2009;16:795–806. doi: 10.1038/cdd.2009.10. [DOI] [PubMed] [Google Scholar]