

Abstract
T-bet is a master regulator for IFN-γ production and Th1 differentiation. We evaluated the roles of T-bet and IFN-γ in T-cell responses in acute graft-versus-host disease (GVHD) and found that T-bet−/− T cells induced significantly less GVHD compared to WT or IFN-γ−/− counterparts in both MHC-mismatched and MHC-matched but minor histocompatibility antigen (miHA)-mismatched models driven by CD4 T cells. T-bet−/−, but not IFN-γ−/−, CD4 T cells had a markedly reduced ability to cause tissue damage in liver and gut. This distinct outcome is reflected by the differential gene expression on donor CD4 T cells deficient for T-bet or IFN-γ. At mRNA and protein levels, we defined several T-bet-dependent molecules that may account for the impaired ability of T-bet−/− T cells to migrate into target organs and to produce Th1-related cytokines. Moreover, these molecules were independent of either endogenous IFN-γ such as CXCR3 and PD-1, or systematic IFN-γ such as NKG2D, I-Ab, and granzyme B. Although both T-bet−/− and IFN-γ−/− CD4 T cells are prone to differentiate into Th17 cells, polarized Th17 cells deficient for T-bet but not for IFN-γ had a significantly reduced ability to cause GVHD. Finally, T-bet−/− T cells had compromised graft-versus-leukemia (GVL) effect, which could be essentially reversed by neutralization of IL-17 in the recipients. We conclude that T-bet is required for Th1 differentiation and migration, as well as for optimal function of Th17 cells. Thus, targeting T-bet or regulating its downstream effectors independent of IFN-γ may be a promising strategy to control GVHD in the clinic.
Introduction
Graft-versus-host disease (GVHD) is a major limitation for the efficacy of allogeneic hematopoietic stem cell transplantation (allo-HSCT) in the treatment of hematologic malignancies because it leads to significant morbidity and mortality (1). The cytokine storm caused by conditioning and Th1-cell cytokines produced by allogeneic T cells are the driving forces for the initiation and development of GVHD (2-5). Paradoxically, the principal Th1 cytokine, IFN- γ, plays a dispensable role for GVHD development in some experimental murine BMT models (6-11), where exacerbated GVHD was observed in hosts receiving IFN-γ−/− grafts (7-9, 11) or after IFN-γ neutralization (7) following lethal irradiation. On the other hand, administration of recombinant IFN-γ showed a protective effect for CD4 T-cell mediated GVHD (10) .
T-bet, the T-box transcription factor, has a unique role in the differentiation of all three subsets (Th1, Th2, Th17) of CD4+ helper T cells by promoting Th1 differentiation, while simultaneously inhibiting Th2 and Th17 lineage commitment (12). T-bet target genes have been identified in primary human T cells, which show that T-bet is associated with genes of various functions in Th1 cells, including those with roles in transcriptional regulation, chemotaxis, and adhesion (13). T-bet is a transcriptional activator of IFN-γ (14) and orchestrates the cell-migratory program by directly controlling expression of the chemokine receptors CXCR3 and CCR5, as well as the chemokines CCL3 and CCL4 (13, 15). T-bet also has cooperative and partially redundant functions with eomesodermin (Eomes), another T-box transcription factor, to control CD8 T cell cytotoxicity and IFN-γ production (16, 17).
Previously, we observed that T cells deficient for T-bet are impaired in the induction of acute GVHD (18). However, the effect and mechanism of T-bet on T cells to induce GVHD and mediate the GVL effect has not been thoroughly studied, particularly the reason for the paradoxical outcomes of GVHD caused by T-bet−/− or IFN-γ−/− T cells. We therefore utilized T cells from T-bet−/− or IFN-γ−/− mice as donors and tested whether T-bet could be a potential target for preventing GVHD after allogeneic bone marrow transplantation (allo-BMT). We then elucidated the underlying mechanisms by which T-bet or IFN-γ differentially regulates allogeneic T-cell response after allo-BMT. We identified several molecules that depend on T-bet, but not on endogenous IFN-γ produced by donor T cells or systematic IFN-γ produced by any type of cell, which may be responsible for T-cell pathogenicity in GVHD induction. Furthermore, we define the role of T-bet in Th17 function related to GVHD and its impact on the GVL effect. Our study provides new biological insight on T-bet, as well as the rationale to target T-bet or its downstream effectors, to control GVHD after allo-BMT.
Material and Methods
Mice
C57BL/6 (B6; H-2b, CD45.2), B6.Ly5.1 (CD45.1) and BALB/c (H-2d) were purchased from National Cancer Institute (NCI, Frederick, MD). T-bet−/−, IFN-γ−/− and IFN-γR−/− mice on B6 background and founders of C.B10-H2b/LilMcdJ (BALB.B; H-2b) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). BALB.B mice were bred at H. Lee Moffitt Cancer Center (Moffitt, Tampa, FL). All animals were housed in the American Association for Laboratory Animal Care–accredited Animal Resource Center at Moffitt or Medical University of South Carolina (MUSC, Charleston, SC). Experiments were carried out under protocols approved by the Institutional Animal Care and Use Committee of University of South Florida (Tampa, FL) or MUSC.
BMT models
T-cell purification from whole spleen and lymph nodes was done by negative depletion using magnetic beads as previously described (18). MHC- and miHA-mismatched (B6→BALB/c) or MHC-matched but miHA-mismatched (B6→BALB.B) BMT models were used as previously established (19). Briefly, 8-10 week old recipient mice were conditioned with total body irradiation (TBI) based on weight at 750 to 850 cGy (single dose) for BALB/c and 900 to 950 cGy (split 3 hours apart) for BALB.B using a Shepherd Mark I Cesium Irradiator (J. L. Shepherd and Associates, San Fernando, CA) at Moffitt, or at 650 to 750 cGy (single dose) for BALB/c using an X-RAD 320 X-ray Irradiator (Precision X-ray Inc., North Branford, CT) at MUSC. Within 24 hours post-conditioning, recipients were intravenously injected with T-cell depleted (TCD) BM alone with or without T cells from different strains of donors. Recipient mice were monitored for weight loss and other clinical signs of GVHD twice/week. Clinical scores were tabulated as 5 parameters: weight loss, posture, activity, fur texture, and skin integrity. Individual mice were scored 0 to 2 for each criteria and 0 to 10 overall (20). A GVL model was established by intravenously injecting luc/neo plasmid-transduced A20 B-cell lymphoma cells (A20-luc) on BALB/c background the day of BMT (2×103 A20 cells/mouse). In addition, recipients were treated with anti-IL-17 mAb (clone: 17F3; Bio X Cell, West Lebanon, NH) or PBS control at doses and schedules specified. Tumor growth was measured with bioluminescent imaging (BLI) using Xenogen-IVIS ® 200 Pre-clinical In vivo Imaging System (Perkin-Elmer, Waltham, MA) and analyzed by Living Image Software (Perkin-Elmer) as previously described (21). Tumor and GVHD mortality were distinguished by BLI signal intensity and clinical manifestation of GVHD. Recipients at pre-moribund stage were euthanized and counted for lethality. Representative samples of GVHD target organs were excised from recipients 14 days post-BMT and subjected to pathology scoring as previously described (18, 21).
Flow cytometry and serum cytokine detection
Mononuclear cells were isolated from recipient spleen or liver as previously described (18, 21, 22) and stained for surface receptors and intracellular cytokines using standard flow cytometric protocols. Stained cells were analyzed using Diva software, LSR II (BD Biosciences, San Jose, CA), and FlowJo (TreeStar, Ashland, OR). Cytokine levels in recipient serum were quantified using a cytometric bead assay (BD Biosciences, San Jose, CA) (23).
Microarray and real-time PCR
Lethally irradiated BALB/c mice were transplanted with WT Ly5.1+ B6 TCD-BM (5 × 106/mouse) plus WT, T-bet−/− or IFN-γ−/− naïve CD4+ Ly5.1− T cells (1 × 106/mouse). Recipients were euthanized on day 7. Donor-derived T cells (CD4+ H-2Kb+ Ly5.1− DAPI−) in recipient splenocytes were isolated by cell sorting (BD FACSAria II Cell Sorter), and lysed in TRIzol (Life technologies, Grand Island, NY) to extract total RNAs. Microarray analysis was performed using GeneChip Mouse Genome 430 2.0 arrays (Affymetrix, Santa Clara, CA). Array images were analyzed and processed by robust multi-array average (RMA) procedure (24, 25). Heat map was generated by using Cluster 3.0 and Java TreeView. In order to identify potential genes significantly changed (either increased or decreased) in the T-bet−/− versus WT group, but not shared by the IFN-γ−/− group, three sets of genes were selected: 1) signal fold change of T-bet−/− vs. WT >2; 2) signal fold change of T-bet−/− vs. IFN-γ−/− >2; and 3) signal fold change of IFN-γ−/− vs. WT >2. Fold change calculation was based on the mean value of individual values from three independent experiments. The genes overlapped in set 1 and set 2 were further selected, and genes from set 3 were excluded from those selected genes. The signal value below 500 is considered as background noise. Thus, the primary pool of T-bet-dependent but endogenous IFN-γ-independent genes was established. Additional criteria were added when heat map analysis was performed. When selecting T-bet positively regulated genes, the signal fold change of T-bet−/− vs. WT, T-bet−/− vs. IFN-γ−/−, and IFN-γ−/− vs. WT is >4, >3 and <2, respectively, and the lowest signal value limit of WT group is set at 1675 (210.71). When selecting T-bet negatively regulated genes, the signal fold change of T-bet−/− vs. WT, T-bet−/− vs. IFN-γ−/−, and IFN-γ−/− vs. WT is >2, >2 and <2, respectively, and the lowest signal value limit of T-bet−/− group is set at 1024 (210). In heat map scale, signal value of 1024 (210) was termed “0.00” and represented by black color; signal value of 8192 (213) was termed “3.00” and represented by red color; signal value of 128 (27) was termed “−3.00” and represented by green color. Mouse array data can be accessed at ArrayExpress (http://www.ebi.ac.uk/arrayexpress/) under accession number E-MTAB-2198.
To quantify mRNA using real-time PCR, extracted total RNA was reverse transcribed to cDNA using a High Capacity cDNA Reverse Transcription Kit. All PCR experiments were performed using TaqMan gene expression assays (Hopx, Mm00558630_m1; Slamf1, Mm00443316_m1; Serpinb9, Mm00777163_m1; Igf2r, Mm00439576_m1; Klrd1, Mm00495182_m1; Klrc1, Mm00516111_m1; H-2Aa, Mm00439211_m1; H-2Ab1, Mm00439216_m1; Retnla, Mm00445109_m1; GAPDH, Mm99999915_g1), universal PCR Master Mix and the ABI-PRISM 7900 Sequence Detection System (Applied Biosystems, Life technologies, Grand Island, NY). Target gene expression was calculated using the comparative method for relative quantitation upon normalization to the internal GAPDH expression control.
In vitro generation of Th17 cells and Th17-mediated GVHD model
CD4+CD25− cells isolated from WT, T-bet−/− or IFN-γ−/− mice on B6 background were stimulated in the presence of syngeneic APCs with 2 μg/mL anti-CD3 mAb, 5 ng/mL TGFβ, 10 ng/mL IL-6 and 5 μg/mL anti-IFN-γ mAb. On day 3, 50U/mL mIL-2 was added. Cell phenotype (CD4+ IL-17+ %) was confirmed on day 4 by intracellular cytokine staining of IFN-γ and IL-17. On day 5, polarized T cells were collected and dead cells were removed using Ficoll separation. Equal numbers of live CD4+ IL-17+ cells derived from WT, T-bet−/− or IFN-γ−/− T cells were used to induce GVHD to compare pathogenicity.
Statistics
For comparison of recipient survival among groups in GVHD experiments, the log-rank test was used to determine statistical significance. To compare GVHD clinical scores, pathology scores, weight loss, cytokine levels, and gene expression levels, a Student t test was used.
Results
T-bet is required for CD4 T-cell mediated acute GVHD
Our previous study indicated that T-bet−/− total T cells are less pathogenic than their WT counterparts in inducing GVHD in fully MHC-mismatched BMT models (18). In contrast, IFN-γ, a major cytokine regulated by T-bet, is dispensable or even protective against GVHD development (6-11). Considering Eomes is preferentially expressed on CD8 T cells (16, 26) and CD4 T-cell differentiation is better defined (5, 27), we first focused our studies on CD4-mediated GVHD and aimed to further understand how T-bet and IFN-γ differentially affect the development of acute GVHD. As shown in figure 1, IFN-γ−/− CD4 T cells mediated comparable GVHD to WT CD4 T cells, whereas T-bet−/− CD4 T cells caused significantly milder GVHD. Recipients of T-bet−/− donor T cells showed significantly higher long-term survival rates (Figure 1A), less weight loss (Figure 1B) and lower GVHD clinical scores after 4 and 8 weeks following allo-BMT (Figure 1, C and D) compared with the recipients of either WT or IFN-γ−/− CD4 T cells. Alleviated GVHD caused by T-bet−/− CD4 T cells was also supported by significantly lower pathological scores of those recipients 14 days after allo-BMT. Recipients of T-bet−/− CD4 T cells had markedly reduced T-cell infiltration and tissue damage in GVHD target organs including the liver, gut, and skin (Figure 1, E and F). However, T-bet−/− CD4 T cells induced similar severity of pulmonary GVHD as WT CD4 T cells (Figure 1, E and F), consistent with the previous report that pulmonary GVHD is not associated with Th1-mediated response (28). Serum cytokines detected 14 days post-transplant revealed that T-bet−/− CD4 T cells produced significantly lower levels of IFN-γ compared to WT CD4 T cells (Figure 1G). Consistent with the pathological scores, T-bet−/− CD4 T cells also produced lower levels of TNF-α, but higher levels of IL-10 and IL-6 compared to either WT or IFN-γ−/− CD4 T cells (Figure 1G). Furthermore, intracellular IL-17 expression was significantly higher in T-bet−/− or IFN-γ−/− donor T cells than in WT donor T cells in recipient spleens and livers (Figure 1H). Altogether, T-bet deficient CD4 T cells produced lower levels of inflammatory cytokines and induced less damage in the recipient liver, gut and skin, indicating that T-bet, not IFN-γ, is required for CD4 T-cell mediated acute GVHD.
Figure 1. T-bet is required for CD4 T-cell mediated acute GVHD.

Lethally irradiated BALB/c mice were transplanted with 5×106/mouse TCD-BM from WT B6 donor alone (n=7), or plus WT (n=10), T-bet−/− (n=11), or IFN-γ−/− (n=10) CD4+ T cells at 1×106/mouse. Recipient mice were monitored throughout the experimental period for survival (A), weight change (B), GVHD clinical scores at 4 weeks (C) and 8 weeks (D) post-transplant. Lethally irradiated BALB/c mice were transplanted with 5×106/mouse TCD-BM from WT B6 donor alone (n=4), or plus WT (n=7), T-bet−/− (n=7), or IFN-γ−/− (n=8) naïve CD4+ T cells at 1×106/mouse. Recipients were euthanized 14 days post-transplant and samples of liver, lung, small intestine, colon and skin were collected for H&E staining (E) and scored for microscopic GVHD severity by a pathologist blinded to the treatment groups. Photomicrographs depicting the average disease score morphology from one representative experiment out of two separate experiments. Pathological score mean ± SE (F) of GVHD target organs across two separate experiments are depicted. Sera collected at necropsy 14 days post-transplant were subjected to cytokine bead analysis (G) to quantify serum cytokine concentrations of IFN-γ, TNF-α, IL-6, and IL-10. Cytokines that were undetectable are not graphically represented. (H) Recipients were euthanized 14 days post-transplant and samples of spleen and liver were collected for intracellular staining of IL-17. FACS plots show CD4+IL-17+ cells from representative recipients in each group. Data are shown from two replicate experiments combined. Asterisk indicates statistical significance: *p<0.05, **p<0.01, ***p<0.001.
T-bet is important for GVHD induction in miHA-mismatched model
MiHA-antigen mismatches play a critical role in clinical GVHD development in human leukocyte antigen (HLA)-identical BMT conditions (29). Given the effect of T-bet−/− or IFN-γ−/− total T cells in fully MHC-mismatched models has been reported previously by us and others (8, 18, 30), we utilized a clinically relevant MHC-matched, multiple miHAs-mismatched CD4-dependent BMT model, B6→BALB.B, and found that the recipients which received splenocytes from T-bet−/− mice demonstrated prolonged survival with decreased GVHD severity compared with those that received WT or IFN-γ−/− splenocytes (Figure 2). These results suggested that T-bet plays a critical role in the development of acute GVHD regardless of BMT model.
Figure 2. T-bet is required for GVHD induction in miHA-mismatched BMT model.
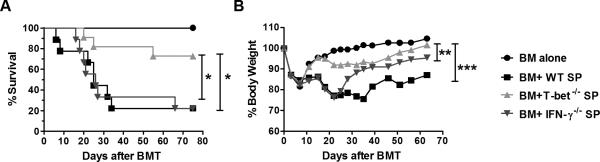
Lethally irradiated BALB.B mice were transplanted with 5×106/mouse TCD-BM from WT B6 donor alone (n=6), or plus WT (n=9), T-bet−/− (n=11), or IFN-γ−/− (n=9) splenocytes (SP) at 20×106/mouse. Recipient mice were monitored throughout the experimental period for survival (A) and weight change (B). Data are shown from two replicate experiments combined. Asterisk indicates statistical significance: *p<0.05, **p<0.01, ***p<0.001.
Differential gene profiles of T-bet−/− and IFN-γ−/− CD4 T cells after allo-BMT
To determine the mechanism of how T-bet affects T-cell pathogenicity in the induction of GVHD, we hypothesized that the molecules which are regulated by T-bet but not required for IFN-γ production are responsible for GVHD development. To identify these molecules, we isolated donor CD4 T cells from spleens of recipients at 7 days following allo-BMT (B6→BALB/c) and analyzed the gene profiles by microarray. Internal controls, Tbx21 (gene encoding T-bet) and Ifng (gene encoding IFN-γ), were essentially absent in T-bet−/− CD4 T cells and IFN-γ−/− CD4 T cells respectively (Figure 3, A and B). The microarray heat map showed that, a total of 28 genes were down-regulated and 8 genes were up-regulated only in T-bet−/− but not in IFN-γ−/− CD4 T cells compared to WT CD4 T cells (Figure 3A). Among them, we chose to present those genes potentially relevant to GVHD in a bar graph, including Cxcr3, Ccr5, Ccl3, Ccl4, Klrc1, Klrd1, Nkg7, and Pdcd1 that were positively regulated, whereas H2-Aa and H2-Ab1 were negatively regulated by T-bet independent of endogenous IFN-γ (Figure 3B). Changes in representative genes (Hopx, Slamf1, Serpinb9, Igf2r, Klrd1, Klrc1, H2-Aa, H2-Ab1 and Retnla) were confirmed by real time PCR (Figure 3C). We reason that at least some of those genes we identified are responsible for the compromised ability of T-bet−/−, not IFN-γ−/−, CD4 T cells in the induction of GVHD.
Figure 3. Differential gene profiles of T-bet−/− and IFN-γ−/− CD4 T cells after allo-BMT.

Lethally irradiated BALB/c mice were transplanted with 5×106/mouse TCD-BM from WT Ly5.1+ B6 donor plus WT, T-bet−/− or IFN-γ−/− naïve CD4+ Ly5.1− T cells at 1×106/mouse (n=4 per group per experiment). Recipients were euthanized 7 days post-transplant and their spleens were collected. By sorting different strains of donor-derived T cells (CD4+ H-2Kb+ Ly5.1− DAPI−) from recipient splenocytes, 1~2×106 cells were obtained per group, and total RNAs were extracted for microarray and real-time PCR analysis. The genes shown are significantly changed in the T-bet−/− group but not shared by the IFN-γ−/− group. Heat map (A), and mean signal value ± SE (B) of representative genes identified by microarray using either WT, T-bet−/−, or IFN-γ−/− donor CD4 T cells were shown. Selected gene expressions were confirmed by real-time PCR (C). Pooled data from three independent experiments are presented. Asterisk indicates statistical significance: **p<0.01, ***p<0.001.
T-bet regulates T-cell pathogenicity through IFN-γ-independent manners
Our microarray data provides potential explanations for why T-bet−/− and IFN-γ−/− T cells induce distinct outcomes of GVHD at the gene level. However, protein expression is typically better correlated with biological function. To confirm our microarray study and further investigate the underlying mechanism, we utilized a MHC-mismatched BMT model (B6→BALB/c), and examined the expression of several molecules, which represent multiple aspects of allogeneic T-cell function: activation (I-Ab and NKG2D), migration (CXCR3), cytotoxicity (CD94 and GZMB), and exhaustion (PD-1), of donor T cells by flow cytometry 7 days after allo-BMT. We observed that T-bet−/− donor T cells significantly impaired proliferation in secondary lymphoid organs (spleen) and migration into target organs (liver), when compared to their WT counterparts (Figure 4A). This was consistent with their significantly decreased IFN-γ production, but increased IL-17 and IL-10 secretion (Figure 4B). T-bet−/− donor T cells also showed significantly less infiltration into the liver compared with IFN-γ−/− T cells (Figure 4A). The expansion defect of T-bet−/− T cells in the spleen is mostly displayed in the CD8 but not CD4 subpopulation (Figure 4A), which indicated that absence of T-bet affects multiple aspects of CD4 T cell function independent of cell expansion. Moreover, in contrast to WT or IFN-γ−/− T cells, T-bet−/− T cells significantly decreased the expression of CXCR3, NKG2D, PD-1, and CD94, but increased the expression of I-Ab and GZMB in CD4 or CD8 constituent subpopulations (Supplemental Table 1), or both (Figure 4C).
Figure 4. T-bet regulates T-cell migration and cytokine production through IFN-γ-independent manners.

Lethally irradiated BALB/c mice were transplanted with 5×106/mouse TCD-BM from WT Ly5.1+ B6 donor plus WT, T-bet−/−, IFN-γ−/− or IFN-γR−/− naïve CD4+ Ly5.1− T cells at 1×106/mouse (n=3~4 per group). Recipients were euthanized 7 days post-transplant and their spleens and livers were collected and absolute numbers of donor total T cell, CD4+ or CD8+ T cell were shown (A). Production of IFN-γ, IL-17 and IL-10, and expression of CXCR3, NKG2D, I-Ab, PD-1, CD94 and granzyme B (GZMB) by WT, T-bet−/−, IFN-γ−/− or IFN-γR−/− donor T cell in recipient spleen were shown in bar graph (B), and histograms (C), respectively. Data from one representative experiment were presented. Asterisk indicates statistical significance: *p<0.05, **p<0.01, ***p<0.001.
Given IFN-γ can be produced by other cell types besides donor T cells, we aimed to distinguish the dependence of endogenous or systematic IFN-γ by including IFN-γR−/− T cells as additional controls, which are able to produce IFN-γ (Figure 4B) but unable to respond to IFN-γ. We found that CXCR3 and PD-1 were expressed in similarly low levels on the T cells deficient for T-bet or IFN-γR, but not IFN-γ. This suggests that their protein expression depends on T-bet, not endogenous IFN-γ, although it can be regulated by IFN-γ produced by other types of cells in the recipient. On the other hand, the expression of NKG2D, CD94, I-Ab and GZMB depends on T-bet, but not systematic IFN-γ, because their protein profiles are distinct on the T cells deficient for T-bet versus either IFN-γ or IFN-γR. Therefore, we have identified several T-bet-dependent, but endogenous or systematic IFN-γ-independent molecules, that likely contributed to the impaired target organ migration and Th1-cytokine production by T-bet−/− T cells.
T-bet controls the optimal function of Th17 cells in GVHD induction
It is known that Th1-differentiation antagonizes Th17-differentiation (31, 32). Our data shows that either T-bet−/− or IFN-γ−/− T cells are prone to differentiate into Th17 cells both in vitro (Supplemental figure 1) and in vivo (Figure 1H), which is consistent with previous reports (18, 33). Given the Th17 subset, per se, is capable of causing GVHD (34, 35), we hypothesized that optimal activity of Th17 cells may require T-bet but not IFN-γ, which may attribute to a distinct pathogenicity of T-bet versus IFN-γ in GVHD induction. Therefore, we examined the role of T-bet or IFN-γ in the pathogenicity of Th17 cells to cause GVHD using optimized Th17-polarizing culture conditions. We generated > 70% CD4+ IL-17+ cells from naïve CD4 T cells of WT, T-bet−/− or IFN-γ−/− B6 mice (Figure 5A) and then transferred equal numbers of these T cells into lethally irradiated BALB/c recipients. We found that T-bet−/−, not IFN-γ−/−, Th17 cells had a significantly reduced ability to induce GVHD. This is reflected by ameliorated morbidity and mortality of the recipients (Figure 5, B and C). Because bone marrow stromal niche is considered a sensitive target of allogeneic T cells, and the defective donor bone marrow-derived B lymphopoiesis is correlated with GVHD severity (36), we examined the percentage of donor-derived B cells at the end of observation periods (around day 85 after allo-BMT), which was found to be significantly higher in the recipients of T-bet−/− Th17 cells than those of WT or IFN-γ−/− Th17 cells (Figure 5E), despite a similar number of total splenocytes among these groups (Figure 5D). These data strongly suggest that T-bet also affects the pathogenicity of Th17 cells in GVHD induction. Moreover, Th17 pathogenic and nonpathogenic signature molecules are previously defined in autoimmune models, such as experimental autoimmune encephalomyelitis (EAE) (37-39). Based on the similar requirement of Th1 and/or Th17 cells and on T-bet expression in the induction of EAE and GVHD (18, 40), we used the genes identified in EAE model as a reference, to indirectly reflect the effects of T-bet on regulating Th17 pathogenicity in allo-BMT settings. Our microarray results indicate that T-bet positively regulated Th17 pathogenic signature genes on CD4 T cells after allo-BMT, such as Lrmp, Ccl5, ICOS, Stat4, Lgals3, Malt1 and GM-CSF, while negatively regulating Th17 nonpathogenic signature genes such as IL-10 (Supplemental figure 2). The observation is in-line with our hypothesis that T-bet contributes to the optimal function of Th17 cells after allo-BMT.
Figure 5. T-bet controls the optimal function of Th17 cells in GVHD induction.

Lethally irradiated BALB/c mice were transplanted with 5×106/mouse TCD-BM from WT B6 donor alone (n=8), or plus in vitro polarized WT (n=12), T-bet−/− (n=17) or IFN-γ−/− (n=12) Th17 cells (A) at 0.5~1×106 /mouse after normalization of IL-17 producing CD4+ cells. Recipient mice were monitored throughout the experimental period for survival (B) and weight change (C). Upon completion of the experiment on day 80-90, absolute number (D) and percentage of donor-derived B-cell (B220+) and T-cell (CD4+ or CD8+) reconstitution (E) of the spleen cells from remaining recipients were analyzed. Pooled data from three independent experiments are presented. Asterisk indicates statistical significance: *p<0.05, **p<0.01, ***p<0.001.
T-bet−/− donor T cells largely preserve the GVL effect by neutralizing IL-17 in allo-BMT recipients
The ultimate goal of allo-HSCT is to prevent GVHD while preserving the GVL effect. Cytotoxic CD8 T cells are known to play a predominate role in mediating GVL effects (41, 42). GVL effects of IFN-γ−/− T cells have been shown to correlate inversely with their GVHD-inducing activity in a CD8-dependent model (11). We hypothesized that T-bet−/− T cells may maintain their GVL activity due to the presence of Eomes, which preserves CD8 T-cell cytotoxic activity (16, 17). We tested the expression of Eomes and IFN-γ on CD4 and CD8 T cells in the presence or absence of T-bet and found that Eomes was mainly expressed on CD8 T cells compared to CD4 T cells. T-bet−/− CD8 T cells highly expressed Eomes and maintained IFN-γ production (Supplemental figure 3). To determine whether T-bet is required for T-cell mediated GVL effect, we used a B6 → BALB/c BMT model and infused a low dose of A20-luc B-cell lymphoma on the day of allo-BMT, which mimics clinical setting where a small number of malignant cells survive in patients after pre-conditioning regimen. We found that WT T cells at 0.25~0.5 × 106/mouse could effectively reject tumor, but the majority of recipients (79%) quickly died of GVHD (Figure 6A, B and C). However, the recipients of 0.25~0.5 × 106 T-bet−/− T cells showed delayed tumor growth compared to those of BM alone, but eventually the majority of these recipients (86%) died of tumor relapse (Figure 6A, B and C). These data suggest that although T-bet deficient T cells still express Eomes and had partially preserved ability to produce IFN-γ, their GVL activity was largely compromised.
Figure 6. T-bet−/− donor T cells largely preserve GVL effect upon neutralizing IL-17 in allo-BMT recipients.

BALB/c mice were lethally irradiated and transplanted with 5×106/mouse TCDBM from WT B6 donor alone, or plus WT or T-bet−/− purified T cells at 0.25~0.5×106/mouse. Additionally, recipients were intravenously injected with 2×103/mouse A20 luciferasetransduced lymphoma cells at the time of BMT. Recipients were i.p. injected with α-IL-17 mAb (500μg/mouse for first dose, and 200μg/mouse for the following doses) or PBS control for 4 weeks (at day 0, 3, 7, 10, 14, 18, 21, 25 and 28), and were monitored throughout the experimental period for survival (A), weight change (B), and tumor growth by luciferin i.p. injection and whole body BLI (n=4~7 per group per experiment). Recipient BLI image (C) represents one of the 2 replicated experiments and average radiance intensity of BLI (D) pooled from all experiments was shown. Asterisk indicates statistical significance: *p<0.05.
T cells deficient for T-bet produce elevated levels of IL-17 (Figure 1H and 4B). Substantial evidence supports that IL-17 enhances tumor resistance to anti-angiogenesis therapy (43), promoting tumor progression through an IL-6/Stat3 signaling pathway (44, 45). Our previous study indicates that additional RORγt (transcription factor of Th17 cells) deficiency reverses the enhanced ability of T-bet-deficient T cells to produce IL-17. Furthermore, T-bet/RORγt-deficient T cells have largely preserved GVL activity (18). Therefore, we further hypothesized that elevated IL-17 contributed to the compromised GVL effect of T-bet−/− T cells. To test this, we used the same MHC-mismatched B6→BALB/c model with IL-17 neutralizing mAb (clone: 17F3), and found that treatment with anti-IL-17 mAb significantly reduced tumor relapse in the recipients of T-bet−/− T cells. This is reflected by improved survival (Figure 6A) and reduction of tumor signal intensity by BLI (Figure 6 C and D). In contrast, the same treatment did not affect the survival of the recipients transplanted with WT T cells. Our data suggests that the compromised GVL activity of T-bet−/− T cells was, at least in part, due to elevated IL-17 produced by these T cells.
Discussion
Here we show that T-bet deficient donor T cells, in contrast to IFN-γ deficient donor T cells, are impaired in their ability to induce acute GVHD in allogeneic recipients in fully MHC-mismatched (Figure 1 and 5) or MHC-matched but miHA-mismatched (Figure 2) murine BMT models. We identified distinct genetic profiles of T cells deficient for T-bet or IFN-γ to account for this difference (Figure 3) and further elucidated several T-bet downstream molecules, independent of either endogenous or systematic IFN-γ, contributed to GVHD pathogenicity (Figure 4). Furthermore, we found that T-bet deficient donor T cells have preserved GVL activity when the resulting increased IL-17 is neutralized (Figure 6).
Acute GVHD has been considered a Th1-type disease dominated by cytotoxic T cell-mediated pathology and increased production of Th1-type cytokines, including IFN-γ (2-4). Our data indicates that T-bet is required for CD4 T-cell mediated GVHD by controlling the differentiation and migration of Th1 cells (Figure 1) and the optimal function of Th17 cells (Figure 5). T-bet−/− CD4 T cells produced significantly lower levels of the pathogenic cytokine TNF-α, but higher levels of the anti-inflammatory cytokine IL-10 in recipient sera after adoptive transfer, which lead to reduced tissue damage in the liver and gut, compared to either WT or IFN-γ−/− CD4 T cells (Figure 1). Conversely, the Th17-related cytokine IL-6 was elevated in recipient sera if the donor CD4 T cells were T-bet−/− (Figure 1G). In addition, increased IL-17 production by T-bet−/− donor CD4 T cells was present in recipient spleens and livers (Figure 1H). Our previous study also indicates that T-bet−/− CD4 T cells produced higher levels of Th2-related cytokines, IL-4 and IL-5, in recipient spleens, livers, and lungs (18). Consistent with previous reports, donor CD4 T cells can reciprocally differentiate into Th1, Th2, and Th17 cells, and each Th subset contributes to specific GVHD target organ tissue damage. Liver and gut are the primary target organs for Th1 cells, and skin is the primary target organ for Th17 cells, whereas pulmonary GVHD is mainly mediated by Th17 and Th2 cells (33). We observed that T-bet−/− CD4 T cells induced severe pulmonary GVHD similar to WT counterparts (Figure 1, E and F). We attribute the severe lung pathology mediated by T-bet−/− CD4 T cells to the augmented Th2 and Th17 cells and to the minimal IFN-γ production which leads to decrased PD-L1 expression on lung parenchyma (33). Moreover, due to the direct cytotoxicity of IFN-γ to the gastrointestinal tract (6), absence of T-bet or IFN-γ in donor CD4 T cells induced similar severity of colon GVHD but was less than that induced by WT CD4 T cells (Figure 1, E and F).
Multiple preclinical studies have investigated a paradoxical protective role of IFN-γ in GVHD development under lethal conditioning (6-11). In our current settings, IFN-γ−/− T cell induced comparable, not more severe GVHD, as their WT counterparts, which is likely due to the type and dose of donor T cells given was different from other studies (6-11). Taken together, the consensus is that IFN-γ produced by donor T cells is not required for the development of acute GVHD after lethal TBI and allo-BMT. However, the current work showed that T-bet, a transcriptional activator of IFN-γ, was required for GVHD induction. To further define the underlying mechanisms, we identified the differential gene profiles of donor T cells deficient for T-bet or IFN-γ after allo-BMT. The potential key mediators include but are not limited to the following genes: Cxcr3, Ccr5, Ccl3, Ccl4, Klrc1, Klrd1, Nkg7, Pdcd1, H2-Aa and H2-Ab1 (Figure 3). These targets are either positively or negatively regulated by T-bet and represent different aspects of allogeneic T-cell activity: activation (H2-Aa and H2-Ab1), migration (Cxcr3, Ccr5, Ccl3, and Ccl4), cytotoxic function (Klrc1, Klrd1, and Nkg7) or exhaustion (Pdcd1).
It is interesting that expression of MHC-II genes H2-Aa and H2-Ab1 were increased on T-bet−/− CD4 T cells. The protein level of I-Ab (gene: H2-Ab1) was also significantly increased in T-bet−/− T cells, compared to either WT, IFN-γ−/−, or IFN-γR−/− T cells (Figure 4C and Supplemental table 1), which we classified as a T-bet-dependent but systematic IFN-γ-independent molecule. Given that transfer of MHC molecules from APCs to T cells does exist, and the MHC-II-expressing T cells can engage in T: T interactions leading to increased apoptosis and hyporesponsiveness (46, 47), it is possible that overexpression of MHC-II molecules (such as I-Ab) on T-bet−/− donor T cells may result in increased cell death due to fratricide of allogeneic T cells. Alternatively, those MHC-II-expressing T-bet−/− T cells may compete with professional dendritic cells to present antigen, but not effectively induce T-cell proliferation due to lack of costimulatory signals; the regulatory mechanism utilized by T-bet−NKp46−RORγt+ innate lymphoid cells as demonstrated in a recent report (48).
Chemokine receptors and chemokines play important roles in T-cell migration to GVHD target organs (49, 50). CXCR3 is a direct target of T-bet (13), and targeting CXCR3 using its mAb can inhibit CD8-mediated GVHD in murine allo-BMT models (51). Targeting CCR5 using a small molecule inhibitor (Maraviroc) has recently been reported to be beneficial in patients with visceral GVHD in early clinical trials (52). Additionally, gene expression of CCL3 and CCL4, ligands of CCR5 (53), were also found to be positively regulated by T-bet. Our data further indicates that CXCR3 is likely one of the key effectors contributing to the severe GVHD caused by IFN-γ−/− but not T-bet−/− T cells (Figure 1); in that the presence of systematic IFN-γ preserves the high expression of CXCR3 in IFN-γ−/− T cells via IFN-γR signaling, which was significantly reduced in T-bet−/− T cells at gene level (Data not shown). This was also consistent with previous reports that the addition of exogenous IFN-γ to cultures of IFN-γ−/− T effector cells rescued CXCR3 expression (30). Therefore, in contrast to WT or IFN-γ−/− T cells, T-bet−/− T cells expressed significantly lower levels of Th1-related chemokine receptor genes (Cxcr3 and Ccr5) and chemokine genes (Ccl3 and Ccl4), as well as the CXCR3 protein. This impedes the migration and infiltration of allogeneic T cells to GVHD target organs (Figure 1 and 4).
Administration of an agonistic mAb of NKG2A inhibited donor T cell expansion and ameliorated acute GVHD in mice (54). However, we show that T-bet−/− donor T cells express extremely low levels of CD94/NKG2A genes (Klrd1and Klrc1, respectively) (Figure 3), and CD94 protein (Figure 4), in concert with ameliorated GVHD, suggesting that CD94/NKG2A could be biomarkers to positively predict GVHD severity. Lower expression levels of inhibitory molecules CD94, NKG2A, and T cell exhaustion marker PD-1 (Figure 3 and 4), on the other hand, may suggest that T-bet−/− T cells can still preserve their cytotoxic function to overcome tumor growth, which was additionally supported by enhanced production of GZMB by T cells deficient for T-bet but not IFN-γ (Figure 4C). Nkg7, a promoter of the T and NK cell surface cytotoxic molecule (55), has been considered as a Th1 cell-specific gene of which expression was previously shown to be regulated by T-bet (13, 56), and is positively correlated to cytotoxic T-cell destruction of epidermal cells in human GVHD (57).
In murine EAE models, deficiency in IFN-γ leads to exacerbated disease (58, 59). However, T-bet-deficient mice are protected from developing EAE (40, 60). We have observed similar results in GVHD models, which suggest that silencing Tbx21 has therapeutic potential. Because transcription factors are difficult to be pharmacologically targeted, an alternative approach is targeting T-bet downstream molecules that are independent of endogenous or systematic IFN-γ (Figure 4), such as NKG2D, which is expressed on a variety of immune cells and plays a costimulatory role in activating CD8 T cells (61). Indeed, we classified NKG2D as a T-bet-dependent but systematic IFN-γ-independent molecule (Figure 4), consistent with the unpublished data from us and others (62), which indicates that blockade of NKG2D alleviated GVHD induced by CD8 or total T cells.
Th17 cells are capable of inducing GVHD in mice (34, 35). Both T-bet−/− and IFN-γ−/− CD4 T cells are prone to Th17-differentiation with high levels of IL-17 production (Supplemental figure 1). In contrast to in vitro polarized WT or IFN-γ−/− Th17 T cells, T-bet−/− Th17 cells caused very mild GVHD (Figure 5, B-E). Consistently, our microarray data reveals that T-bet controls the optimal function of Th17 cells in GVHD possibly through regulating the expression of multiple genes representing Th17 pathogenic or nonpathogenic signatures (Supplemental figure 2). Our study showed T-bet−/− and IFN-γ−/− CD4 T cells have similar expression patterns of IFN-γ and IL-17 but opposing GVHD severities, which indicated that the culprit for GVHD development may not be IFN-γ or IL-17, but likely downstream effectors of T-bet we identified.
Although IL-17 seems to have less effect on GVHD development, it influences the tumor microenvironment, especially when shielding the GVL response of T-bet deficient donor T cells (Figure 6). T-bet−/− T cells produce more IL-17 than WT T cells (Figure 1H and Supplemental figure 1), and IL-17 promotes tumor progression in a variety of tumors (43-45). Consistently, neutralizing IL-17 benefits overall survival of recipients transplanted with T-bet−/−, not WT T cells. However, the GVL preservation of T-bet−/− T cells is reduced after stopping anti-IL-17 treatment. The tumor gradually relapsed, which further supports the detrimental role of IL-17 against preserved GVL effects of T-bet−/− T cells. We conclude that T-bet deficient T cells induce less GVHD and may still preserve GVL effects if the elevated levels of IL-17 are reversed. This is also supported by our previous findings that double deficiency of T-bet and RORγt prevents GVHD while sparing the GVL effect (18). Targeting T-bet itself is still far from translational application due to a lack of a specific inhibitor. Recently, the small molecule inhibitors for other transcription factors, such as c-Rel and RORγt, have been developed and showed promising results in alleviating GVHD (63) or cutaneous inflammatory disorders (64), respectively. The c-Rel inhibitor also permits the maintenance of the GVL effect. Once T-bet inhibitors are developed, the mechanism we defined in the current study together with our previous report (18) will become applicable, e.g. a combinational treatment of T-bet inhibitor and anti-IL-17 mAb may be beneficial in controlling GVHD while maintaining the GVL effect.
Collectively, using genetic knockout mice, we prove that T-bet is critical for the development of acute GVHD through controlling the differentiation and migration of Th1 cells as well as the pathogenicity of Th17 cells. Given that T-bet is a transcription factor which is unavailable to be pharmacologically targeted currently, we identify potential molecular targets downstream of T-bet, and define the underlying mechanisms accounting for the distinct GVHD outcomes caused by T-bet-versus IFN-γ-deficient donor T cells. This provides the rationale to target those T-bet-dependent, but endogenous or systematic IFN-γ-independent molecules, for the control of acute GVHD in clinical settings.
Supplementary Material
Acknowledgements
We would like to recognize the technical assistance provided by Jodi Kroeger and Kate Shapland of the Moffitt Flow Cytometry Core Facility, Chaomei Zhang and Xiaotao Qu of the Moffitt Microarray Core and Bioinformatics Core, as well as Jennifer Morse of the Moffitt Vivarium Facilities. We also appreciate the technical support provided by the Department Lab Animal Research (DLAR), Flow Cytometry Core and Imaging Core at MUSC. Thanks for Dr. Tomomi Toubai to make scientific comments and revise the manuscript and Hiromi Tobai to provide the technical support.
This work was supported in part by National Institutes of Health Grants R01s CA143812 and CA169116 to X.-Z.Y.
Abbreviations
- GVHD
graft-versus-host disease
- GVL
graft-versus-leukemia
- allo-BMT
allogeneic bone marrow transplantation
- miHA
minor histocompatibility antigen
- TBI
total body irradiation
- BLI
bioluminescent imaging
- GZMB
granzyme B
References
- 1.Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. 2009;373:1550–1561. doi: 10.1016/S0140-6736(09)60237-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antin JH, Ferrara JL. Cytokine dysregulation and acute graft-versus-host disease. Blood. 1992;80:2964–2968. [PubMed] [Google Scholar]
- 3.Ferrara JL, Krenger W. Graft-versus-host disease: the influence of type 1 and type 2 T cell cytokines. Transfus Med Rev. 1998;12:1–17. doi: 10.1016/s0887-7963(98)80085-0. [DOI] [PubMed] [Google Scholar]
- 4.Lu Y, Sakamaki S, Kuroda H, Kusakabe T, Konuma Y, Akiyama T, Fujimi A, Takemoto N, Nishiie K, Matsunaga T, Hirayama Y, Kato J, Kon S, Kogawa K, Niitsu Y. Prevention of lethal acute graft-versus-host disease in mice by oral administration of T helper 1 inhibitor, TAK-603. Blood. 2001;97:1123–1130. doi: 10.1182/blood.v97.4.1123. [DOI] [PubMed] [Google Scholar]
- 5.Fu J, Heinrichs J, Yu XZ. Helper T-Cell Differentiation in Graft-Versus-Host Disease After Allogeneic Hematopoietic Stem Cell Transplantation. Archivum immunologiae et therapiae experimentalis. 2014 doi: 10.1007/s00005-014-0284-z. [DOI] [PubMed] [Google Scholar]
- 6.Robb RJ, Hill GR. The interferon-dependent orchestration of innate and adaptive immunity after transplantation. Blood. 2012;119:5351–5358. doi: 10.1182/blood-2012-02-368076. [DOI] [PubMed] [Google Scholar]
- 7.Murphy WJ, Welniak LA, Taub DD, Wiltrout RH, Taylor PA, Vallera DA, Kopf M, Young H, Longo DL, Blazar BR. Differential effects of the absence of interferon-gamma and IL-4 in acute graft-versus-host disease after allogeneic bone marrow transplantation in mice. The Journal of clinical investigation. 1998;102:1742–1748. doi: 10.1172/JCI3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang YG, Dey BR, Sergio JJ, Pearson DA, Sykes M. Donor-derived interferon gamma is required for inhibition of acute graft-versus-host disease by interleukin 12. The Journal of clinical investigation. 1998;102:2126–2135. doi: 10.1172/JCI4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Welniak LA, Blazar BR, Anver MR, Wiltrout RH, Murphy WJ. Opposing roles of interferon-gamma on CD4+ T cell-mediated graft-versus-host disease: effects of conditioning. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2000;6:604–612. doi: 10.1016/s1083-8791(00)70025-5. [DOI] [PubMed] [Google Scholar]
- 10.Brok HP, Heidt PJ, van der Meide PH, Zurcher C, Vossen JM. Interferon-gamma prevents graft-versus-host disease after allogeneic bone marrow transplantation in mice. Journal of immunology (Baltimore, Md. : 1950) 1993;151:6451–6459. [PubMed] [Google Scholar]
- 11.Yang YG, Qi J, Wang MG, Sykes M. Donor-derived interferon gamma separates graft-versus-leukemia effects and graft-versus-host disease induced by donor CD8 T cells. Blood. 2002;99:4207–4215. doi: 10.1182/blood.v99.11.4207. [DOI] [PubMed] [Google Scholar]
- 12.Lazarevic V, Glimcher LH. T-bet in disease. Nat Immunol. 2011;12:597–606. doi: 10.1038/ni.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jenner RG, Townsend MJ, Jackson I, Sun K, Bouwman RD, Young RA, Glimcher LH, Lord GM. The transcription factors T-bet and GATA-3 control alternative pathways of T-cell differentiation through a shared set of target genes. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:17876–17881. doi: 10.1073/pnas.0909357106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 15.Lord GM, Rao RM, Choe H, Sullivan BM, Lichtman AH, Luscinskas FW, Glimcher LH. T-bet is required for optimal proinflammatory CD4+ T-cell trafficking. Blood. 2005;106:3432–3439. doi: 10.1182/blood-2005-04-1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, Banica M, DiCioccio CB, Gross DA, Mao CA, Shen H, Cereb N, Yang SY, Lindsten T, Rossant J, Hunter CA, Reiner SL. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science. 2003;302:1041–1043. doi: 10.1126/science.1090148. [DOI] [PubMed] [Google Scholar]
- 17.Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, Mullen AC, Gasink CR, Kaech SM, Miller JD, Gapin L, Ryan K, Russ AP, Lindsten T, Orange JS, Goldrath AW, Ahmed R, Reiner SL. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nature immunology. 2005;6:1236–1244. doi: 10.1038/ni1268. [DOI] [PubMed] [Google Scholar]
- 18.Yu Y, Wang D, Liu C, Kaosaard K, Semple K, Anasetti C, Yu XZ. Prevention of GVHD while sparing GVL effect by targeting Th1 and Th17 transcription factor T-bet and RORgammat in mice. Blood. 2011;118:5011–5020. doi: 10.1182/blood-2011-03-340315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu Y, Wang D, Kaosaard K, Liu C, Fu J, Haarberg K, Anasetti C, Beg AA, Yu XZ. c-Rel is an essential transcription factor for the development of acute graft-versus-host disease in mice. European journal of immunology. 2013;43:2327–2337. doi: 10.1002/eji.201243282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J, Jr., Crawford JM, Ferrara JL. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88:3230–3239. [PubMed] [Google Scholar]
- 21.Liang Y, Liu C, Djeu JY, Zhong B, Peters T, Scharffetter-Kochanek K, Anasetti C, Yu XZ. Beta2 integrins separate graft-versus-host disease and graft-versus-leukemia effects. Blood. 2008;111:954–962. doi: 10.1182/blood-2007-05-089573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valenzuela JO, Iclozan C, Hossain MS, Prlic M, Hopewell E, Bronk CC, Wang J, Celis E, Engelman RW, Blazar BR, Bevan MJ, Waller EK, Yu XZ, Beg AA. PKCtheta is required for alloreactivity and GVHD but not for immune responses toward leukemia and infection in mice. The Journal of clinical investigation. 2009;119:3774–3786. doi: 10.1172/JCI39692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu XZ, Albert MH, Anasetti C. Alloantigen affinity and CD4 help determine severity of graft-versus-host disease mediated by CD8 donor T cells. Journal of immunology (Baltimore, Md. : 1950) 2006;176:3383–3390. doi: 10.4049/jimmunol.176.6.3383. [DOI] [PubMed] [Google Scholar]
- 24.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 26.Banerjee A, Gordon SM, Intlekofer AM, Paley MA, Mooney EC, Lindsten T, Wherry EJ, Reiner SL. Cutting edge: The transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. Journal of immunology (Baltimore, Md. : 1950) 2010;185:4988–4992. doi: 10.4049/jimmunol.1002042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gowdy KM, Nugent JL, Martinu T, Potts E, Snyder LD, Foster WM, Palmer SM. Protective role of T-bet and Th1 cytokines in pulmonary graft-versus-host disease and peribronchiolar fibrosis. Am J Respir Cell Mol Biol. 2012;46:249–256. doi: 10.1165/rcmb.2011-0131OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 29.Goulmy E. Minor histocompatibility antigens: from transplantation problems to therapy of cancer. Hum Immunol. 2006;67:433–438. doi: 10.1016/j.humimm.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 30.Choi J, Ziga ED, Ritchey J, Collins L, Prior JL, Cooper ML, Piwnica-Worms D, DiPersio JF. IFNgammaR signaling mediates alloreactive T-cell trafficking and GVHD. Blood. 2012;120:4093–4103. doi: 10.1182/blood-2012-01-403196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo S, Cobb D, Smeltz RB. T-bet inhibits the in vivo differentiation of parasite-specific CD4+ Th17 cells in a T cell-intrinsic manner. Journal of immunology (Baltimore, Md. : 1950) 2009;182:6179–6186. doi: 10.4049/jimmunol.0803821. [DOI] [PubMed] [Google Scholar]
- 32.Lazarevic V, Chen X, Shim JH, Hwang ES, Jang E, Bolm AN, Oukka M, Kuchroo VK, Glimcher LH. T-bet represses T(H)17 differentiation by preventing Runx1-mediated activation of the gene encoding RORgammat. Nature immunology. 2011;12:96–104. doi: 10.1038/ni.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yi T, Chen Y, Wang L, Du G, Huang D, Zhao D, Johnston H, Young J, Todorov I, Umetsu DT, Chen L, Iwakura Y, Kandeel F, Forman S, Zeng D. Reciprocal differentiation and tissue-specific pathogenesis of Th1, Th2, and Th17 cells in graft-versus-host disease. Blood. 2009;114:3101–3112. doi: 10.1182/blood-2009-05-219402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iclozan C, Yu Y, Liu C, Liang Y, Yi T, Anasetti C, Yu XZ. T helper17 cells are sufficient but not necessary to induce acute graft-versus-host disease. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2010;16:170–178. doi: 10.1016/j.bbmt.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carlson MJ, West ML, Coghill JM, Panoskaltsis-Mortari A, Blazar BR, Serody JS. In vitro-differentiated TH17 cells mediate lethal acute graft-versus-host disease with severe cutaneous and pulmonary pathologic manifestations. Blood. 2009;113:1365–1374. doi: 10.1182/blood-2008-06-162420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shono Y, Ueha S, Wang Y, Abe J, Kurachi M, Matsuno Y, Sugiyama T, Nagasawa T, Imamura M, Matsushima K. Bone marrow graft-versus-host disease: early destruction of hematopoietic niche after MHC-mismatched hematopoietic stem cell transplantation. Blood. 2010;115:5401–5411. doi: 10.1182/blood-2009-11-253559. [DOI] [PubMed] [Google Scholar]
- 37.Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A, Kuchroo VK. Induction and molecular signature of pathogenic TH17 cells. Nature immunology. 2012;13:991–999. doi: 10.1038/ni.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brustle A, Brenner D, Knobbe CB, Lang PA, Virtanen C, Hershenfield BM, Reardon C, Lacher SM, Ruland J, Ohashi PS, Mak TW. The NF-kappaB regulator MALT1 determines the encephalitogenic potential of Th17 cells. The Journal of clinical investigation. 2012;122:4698–4709. doi: 10.1172/JCI63528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGeachy MJ. GM-CSF: the secret weapon in the T(H)17 arsenal. Nature immunology. 2011;12:521–522. doi: 10.1038/ni.2044. [DOI] [PubMed] [Google Scholar]
- 40.Yang Y, Weiner J, Liu Y, Smith AJ, Huss DJ, Winger R, Peng H, Cravens PD, Racke MK, Lovett-Racke AE. T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. The Journal of experimental medicine. 2009;206:1549–1564. doi: 10.1084/jem.20082584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bleakley M, Riddell SR. Molecules and mechanisms of the graft-versus-leukaemia effect. Nat Rev Cancer. 2004;4:371–380. doi: 10.1038/nrc1365. [DOI] [PubMed] [Google Scholar]
- 42.Orsini E, Bellucci R, Alyea EP, Schlossman R, Canning C, McLaughlin S, Ghia P, Anderson KC, Ritz J. Expansion of tumor-specific CD8+ T cell clones in patients with relapsed myeloma after donor lymphocyte infusion. Cancer Res. 2003;63:2561–2568. [PubMed] [Google Scholar]
- 43.Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, Vernes JM, Jiang Z, Meng YG, Peale FV, Ouyang W, Ferrara N. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat Med. 2013;19:1114–1123. doi: 10.1038/nm.3291. [DOI] [PubMed] [Google Scholar]
- 44.Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009;206:1457–1464. doi: 10.1084/jem.20090207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gu FM, Li QL, Gao Q, Jiang JH, Zhu K, Huang XY, Pan JF, Yan J, Hu JH, Wang Z, Dai Z, Fan J, Zhou J. IL-17 induces AKT-dependent IL-6/JAK2/STAT3 activation and tumor progression in hepatocellular carcinoma. Mol Cancer. 2011;10:150. doi: 10.1186/1476-4598-10-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsang JY, Chai JG, Lechler R. Antigen presentation by mouse CD4+ T cells involving acquired MHC class II:peptide complexes: another mechanism to limit clonal expansion? Blood. 2003;101:2704–2710. doi: 10.1182/blood-2002-04-1230. [DOI] [PubMed] [Google Scholar]
- 47.Davis DM. Intercellular transfer of cell-surface proteins is common and can affect many stages of an immune response. Nature reviews. Immunology. 2007;7:238–243. doi: 10.1038/nri2020. [DOI] [PubMed] [Google Scholar]
- 48.Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, Mantegazza AR, Ma HL, Crawford A, Angelosanto JM, Wherry EJ, Koni PA, Bushman FD, Elson CO, Eberl G, Artis D, Sonnenberg GF. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature. 2013;498:113–117. doi: 10.1038/nature12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wysocki CA, Panoskaltsis-Mortari A, Blazar BR, Serody JS. Leukocyte migration and graft-versus-host disease. Blood. 2005;105:4191–4199. doi: 10.1182/blood-2004-12-4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Castor MG, Pinho V, Teixeira MM. The role of chemokines in mediating graft versus host disease: opportunities for novel therapeutics. Front Pharmacol. 2012;3:23. doi: 10.3389/fphar.2012.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He S, Cao Q, Qiu Y, Mi J, Zhang JZ, Jin M, Ge H, Emerson SG, Zhang Y, Zhang Y. A new approach to the blocking of alloreactive T cell-mediated graft-versus-host disease by in vivo administration of anti-CXCR3 neutralizing antibody. Journal of immunology (Baltimore, Md. : 1950) 2008;181:7581–7592. doi: 10.4049/jimmunol.181.11.7581. [DOI] [PubMed] [Google Scholar]
- 52.Reshef R, Luger SM, Hexner EO, Loren AW, Frey NV, Nasta SD, Goldstein SC, Stadtmauer EA, Smith J, Bailey S, Mick R, Heitjan DF, Emerson SG, Hoxie JA, Vonderheide RH, Porter DL. Blockade of lymphocyte chemotaxis in visceral graft-versus-host disease. N Engl J Med. 2012;367:135–145. doi: 10.1056/NEJMoa1201248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rollins BJ. Chemokines. Blood. 1997;90:909–928. [PubMed] [Google Scholar]
- 54.Kawamura H, Yagita H, Nisizawa T, Izumi N, Miyaji C, Vance RE, Raulet DH, Okumura K, Abo T. Amelioration of acute graft-versus-host disease by NKG2A engagement on donor T cells. Eur J Immunol. 2005;35:2358–2366. doi: 10.1002/eji.200425933. [DOI] [PubMed] [Google Scholar]
- 55.Turman MA, Yabe T, McSherry C, Bach FH, Houchins JP. Characterization of a novel gene (NKG7) on human chromosome 19 that is expressed in natural killer cells and T cells. Hum Immunol. 1993;36:34–40. doi: 10.1016/0198-8859(93)90006-m. [DOI] [PubMed] [Google Scholar]
- 56.Zhu J, Jankovic D, Oler AJ, Wei G, Sharma S, Hu G, Guo L, Yagi R, Yamane H, Punkosdy G, Feigenbaum L, Zhao K, Paul WE. The transcription factor T-bet is induced by multiple pathways and prevents an endogenous Th2 cell program during Th1 cell responses. Immunity. 2012;37:660–673. doi: 10.1016/j.immuni.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sale GE, Anderson P, Browne M, Myerson D. Evidence of cytotoxic T-cell destruction of epidermal cells in human graft-vs-host disease. Immunohistology with monoclonal antibody TIA-1. Arch Pathol Lab Med. 1992;116:622–625. [PubMed] [Google Scholar]
- 58.Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). Journal of immunology (Baltimore, Md. : 1950) 1996;156:5–7. [PubMed] [Google Scholar]
- 59.Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. Journal of immunology (Baltimore, Md. : 1950) 1996;157:3223–3227. [PubMed] [Google Scholar]
- 60.Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Markiewicz MA, Carayannopoulos LN, Naidenko OV, Matsui K, Burack WR, Wise EL, Fremont DH, Allen PM, Yokoyama WM, Colonna M, Shaw AS. Costimulation through NKG2D enhances murine CD8+ CTL function: similarities and differences between NKG2D and CD28 costimulation. Journal of immunology (Baltimore, Md. : 1950) 2005;175:2825–2833. doi: 10.4049/jimmunol.175.5.2825. [DOI] [PubMed] [Google Scholar]
- 62.Karimi M, Leichner TM, Satake A, Raulet D, Kambayashi T. Transient NKG2D Blockade Attenuates Graft-Versus-Host Disease While Preserving Graft-Versus-Leukemia Effects. Blood. 2013;122:3242–3242. [Google Scholar]
- 63.Shono Y, Tuckett AZ, Ouk S, Liou HC, Altan-Bonnet G, Tsai JJ, Oyler JE, Smith OM, West ML, Singer NV, Doubrovina E, Pankov D, Undhad CV, Murphy GF, Lezcano C, Liu C, O'Reilly RJ, van den Brink MR, Zakrzewski JL. A small-molecule c-Rel inhibitor reduces alloactivation of T cells without compromising antitumor activity. Cancer discovery. 2014;4:578–591. doi: 10.1158/2159-8290.CD-13-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Skepner J, Ramesh R, Trocha M, Schmidt D, Baloglu E, Lobera M, Carlson T, Hill J, Orband-Miller LA, Barnes A, Boudjelal M, Sundrud M, Ghosh S, Yang J. Pharmacologic inhibition of RORgammat regulates Th17 signature gene expression and suppresses cutaneous inflammation in vivo. Journal of immunology (Baltimore, Md. : 1950) 2014;192:2564–2575. doi: 10.4049/jimmunol.1302190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.