

Abstract
We report on a strategy for using SABRE (signal amplification by reversible exchange) for polarizing 1H and 13C nuclei of weakly interacting ligands which possess biologically relevant and nonaromatic motifs. We first demonstrate this via the polarization of acetonitrile, using Ir(IMes)(COD)Cl as the catalyst precursor, and confirm that the route to hyperpolarization transfer is via the J-coupling network. We extend this work to the polarization of propionitrile, benzylnitrile, benzonitrile, and trans-3-hexenedinitrile in order to assess its generality. In the 1H NMR spectrum, the signal for acetonitrile is enhanced 8-fold over its thermal counterpart when [Ir(H)2(IMes)(MeCN)3]+ is the catalyst. Upon addition of pyridine or pyridine-d5, the active catalyst changes to [Ir(H)2(IMes)(py)2(MeCN)]+ and the resulting acetonitrile 1H signal enhancement increases to 20- and 60-fold, respectively. In 13C NMR studies, polarization transfers optimally to the quaternary 13C nucleus of MeCN while the methyl 13C is hardly polarized. Transfer to 13C is shown to occur first via the 1H–1H coupling between the hydrides and the methyl protons and then via either the 2J or 1J couplings to the respective 13Cs, of which the 2J route is more efficient. These experimental results are rationalized through a theoretical treatment which shows excellent agreement with experiment. In the case of MeCN, longitudinal two-spin orders between pairs of 1H nuclei in the three-spin methyl group are created. Two-spin order states, between the 1H and 13C nuclei, are also created, and their existence is confirmed for Me13CN in both the 1H and 13C NMR spectra using the Only Parahydrogen Spectroscopy protocol.
NMR is used extensively in molecular structure determination as well as in the elucidation of chemical reaction mechanisms. Its ubiquity as a method of choice in both areas is achieved in spite of the fact that NMR suffers from inherent low sensitivity due to the very small population differences that exist between probed nuclear spin states even when using the most expensive state-of-the-art NMR spectrometers. However, hyperpolarization techniques, such as DNP (dynamic nuclear polarization)1 and PHIP (para-hydrogen-induced polarization),2−4 have been used with great success to overcome this sensitivity issue and are now being used more widely in mainstream NMR5−7 and MRI.8−10
Both DNP and PHIP use different approaches to redistribute the nuclear spin state populations in target molecules, resulting in greatly enhanced sensitivity. Where DNP employs polarization transfer from an unpaired electron on a stable radical at low temperature,1 PHIP uses para-hydrogen (para-H2)2 to transfer polarization through its participation as a reactant in a hydrogenation reaction. para-H2, although existing in a nuclear singlet state which renders it invisible to NMR, can be made visible by breaking its symmetry in the context of a chemical reaction.6,11 Early investigations utilizing PHIP relied on the use of an inorganic catalyst to introduce the molecule into an unsaturated target such as an alkyne, thereby breaking its symmetry.3,7,12,13 In these types of reactions, the products are created with non-Boltzmann nuclear spin distributions and substantial signal enhancements are obtained when they are probed by NMR. Investigations employing this approach have been used to identify the involvement of molecules in catalytic reaction mechanisms that would otherwise have been NMR invisible.14−19
This approach has proved to be extremely effective,20−23 but as the target is chemically changed by the hydrogenation process, the reliance of PHIP on the existence of an unsaturated analogue to the target molecule has obvious consequences if the intention is to use PHIP as a means to hyperpolarize an agent for measurement in an MRI context. This shortcoming has been addressed in a refinement of the method which employs catalytic transfer of polarization rather than chemical by circumventing the hydrogenation step of PHIP. The approach is known as SABRE24 (signal amplification by reversible exchange) and it achieves hyperpolarization of a substrate by transferring polarization into the target molecule through the establishment of a J-coupled network during its temporary contact with a metal center that simultaneously binds with molecular para-H2.25 The result is a technique which offers a quick and relatively simple route to hyperpolarize a wide range of molecules capable of interacting with a metal center.6,26−28 It has proved to work in conjunction with several NMR methods that enable analyte characterization and is also capable of facilitating the detection of molecules that are present in very low concentrations.6,26,29 The employment of a catalyst in SABRE is unusual in that its role is to transfer magnetism rather than to provide a lower energy chemical pathway. We and others have developed a range of catalysts30−33 for this purpose that include the highly active species [Ir(H)2(IMes)(py)3]Cl (IMes =1,3-bis(2,4,6-trimethylphenyl)imidazole-2-ylidene). This complex is formed from the air-stable precursor Ir(IMes)(COD)Cl,341 (COD = cyclooctadiene), upon reaction with H2 and pyridine. When undertaken in low field, continuously polarized materials have been produced because SABRE is a reversible process that proceeds without chemical change of the analyte molecule.27
In this study, we illustrate how it is possible to use SABRE to hyperpolarize a series of weakly coordinating ligands including acetonitrile, the aromatic ligands, benzylcyanide and benzonitrile, as well as aliphatic ligands trans-3-hexenedinitrile and propionitrile.35 This work is extended to include all of these ligands which correspond to motifs that can be found in a range of biologically relevant materials, such as Anastrazole36−38 and key industrial reagents such as TCNE (tetracyanoethylene).39 Furthermore, nitriles are very important industrially as solvents (e.g., acetonitrile and benzonitrile), in hydrocyanation reactions (exemplified in the DuPont process for the formation of adiponitrile40) and in C–C bond activation.41,42 We link our experimental observations together with theoretical modeling to provide further insight into the array of magnetic states created by the SABRE process.25 Many of these states are absent during thermal equilibrium NMR experiments that rely solely on simple interactions of nuclei with the applied magnetic field. The importance of such nonequilibrium states is reflected in modern multiple-pulse 1-D and 2-D methods.43,44 Levitt, Bodenhausen, and Warren’s uses of thermally equilibrated magnetization in the creation of longer-lived pseudo singlet states45−47 illustrate further how nonequilibrium states are being accessed and utilized. Consistent with these other methods, the use of para-H2, which itself exists as a long-lived singlet state, in polarization transfer experiments opens up further opportunities for creation and use of nonequilibrium states.13,17
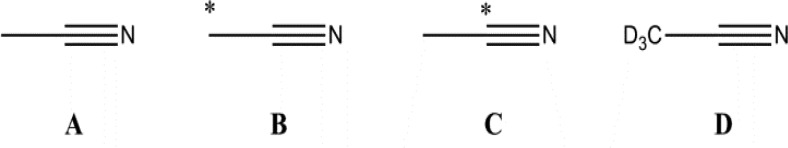
The investigations we describe here employ the four isotopomers of acetonitrile that are listed in Scheme 1. These smaller-sized spin systems offer a significant advantage over those used earlier for SABRE, insofar as they offer the possibility to match more closely the experimental and theoretical models. This situation is not simply, if at all realistically, achievable with more complex spin topologies. For example, even if all of the relevant chemical shift and J-coupling parameters were known, full theoretical treatment of a model containing all of the 17 1H-spins associated with the pyridine and hydride ligands of [Ir(H)2(IMes)(py)3]Cl under the strong coupling Hamiltonian is required. This is beyond the capabilities of normal desktop computation. Indeed, the challenges of fitting experimental and theoretical measurements to the SABRE effect have been commented upon,48 and we and others have recently described how careful consideration of the role of the catalyst is paramount.33,49−51 We also show here that high levels of hyperpolarization in the associated 1H and 13C nuclei of these cyano-containing derivatives can also be produced in order to illustrate the technique’s potential as a characterization aid and a possible route to chemical agents that are highly visible to MRI.
Scheme 1. Acetonitrile Isotopomers A–D, Where ∗ Denotes Labeled 13C.
Results and Discussion
In this work, a MeOH-d4 solution of A and 1 was first exposed to 3 bar para-H2, to form [Ir(H)2(IMes)(MeCN)3]Cl, 2. When this reaction was completed in a magnetic field (referred to as the polarization transfer field or PTF) of 65 G, the resulting 1H NMR signal for free acetonitrile was observed 3 s later with an intensity 8-fold larger than its normal, fully relaxed level. This is due to SABRE and clearly confirms that 1 is able to catalyze the transfer of the spin order of para-H2 into A. However, when the same sample was reexamined 5 min later, after the introduction of fresh para-H2, the enhancement seen for the protons of A had fallen to 3-fold. While in both these measurements 2 was seen, it is clearly not itself an efficient SABRE catalyst for A. The fact that stronger SABRE enhancement was seen during the conversion of 1 into 2, and hence before the reaction reached completion, reveals that the introduction of para-H2 into 2 could be limiting. Hence we added phenylacetylene to the MeOH-d4 solution to see if it would increase the hyperpolarization level of A. This approach has been used successfully in related studies to increase the detectability of metal dihydride complexes by forcing H2 exchange.52 However, this treatment proved unsuccessful here, although weak PHIP was observed in the alkenic proton resonances of the product styrene which reflects the relatively poor hydrogenation activity of 2 (see Supporting Information).
In the light of this result, a small amount of pyridine was introduced as a cosubstrate into the reaction system such that the acetonitrile to pyridine ratio was 20:3. Now the methyl protons signal of A, seen in emission, was 9-fold enhanced and remained observable at this intensity for over an hour when the polarization transfer process was repeated. These investigations also revealed that 2 is converted into cis,cis-[Ir(H)2(IMes)(MeCN)(py)2]Cl, 3 (Scheme 2). This reactivity matched that predicted by density function theory (DFT) calculations which revealed 3 to be more thermodynamically stable than 2 (see Supporting Information). It also predicted that cis,cis-[Ir(H)2(IMes)(MeCN)2(py)]Cl is stable, although 3 is lower in energy. We conclude based on these observations, and our DFT data, that 3 is more efficient at hyperpolarizing A than 2.
Scheme 2. Reaction Pathways To Form 2 and 3 from 1 (Cl– and the Reaction Product Cyclooctane Omitted for Clarity).

The three 1H signals for free pyridine were also observed in this NMR spectrum in emission, with a collective 20-fold enhancement. Furthermore, the proton signals for equatorially bound pyridine of 3 were also enhanced. In contrast, the protons of the axially bound pyridine ligand of 3 failed to show any observable hyperpolarization. This tells us that the transfer of polarization in the plane trans to hydride is more efficient than that in the plane at right angles to it.
3 yielded 1H NMR signals for its two magnetically inequivalent hydride ligands at δ −20.56 and δ −22.12 where the former is trans to acetonitrile and the latter trans to pyridine (see Supporting Information). Both these hydride signals show PHIP in these measurements and when EXSY methods are used to examine 3, pyridine and A ligand exchange processes were readily observed. The experimentally determined rate constant for pyridine loss from 3 proved to be 1.72 and 0.33 s–1 for the equatorial and axial ligands, respectively, at 298 K. In contrast, the experimentally determined rate constant for the loss of A was 10.42 s–1 and, therefore, six times larger than that of equatorially bound pyridine. The experimentally determined rate constant for hydride interchange in 3 proved to be 5.80 s–1. No evidence of hydride exchange into H2 was apparent in these measurements, even though PHIP is visible in the hydride signals of 3. The mechanisms of ligand loss in related systems have recently been reported.31 The lifetime of 3 under these SABRE conditions was 0.096 s. Consequently, if polarization transfer into A and pyridine were to be equally efficient, then the hyperpolarization of A should dominate given its more rapid dissociation. Experimentally, the reverse is true thus polarization transfer into pyridine must be far more efficient than into acetonitrile.
In the corresponding 1H–1H COSY spectrum of 3, the hydride ligands showed evidence for spin–spin coupling to their respective trans-pyridine and trans-A ligands. These data concur with our suggestion that 3 hyperpolarizes A and pyridine by SABRE through its J-coupling network.25 However, polarization transfer in 3 is complicated by the fact that its hydride ligands are both magnetically- and chemically distinct from one another. This means that SABRE transfer in 3 could proceed not just in the equatorial plane but also into the axial ligands as they also see different hydride couplings.31 However, the slower exchange of the axial ligand allows us to critically demonstrate that it does not receive any direct polarization transfer. The small cis-hydride coupling, therefore, precludes the observation of polarization transfer into this ligand (confirmed experimentally as noted above). In stark contrast, for 2, transfer proceeds only into the ligands in the equatorial plane which are trans to hydride.
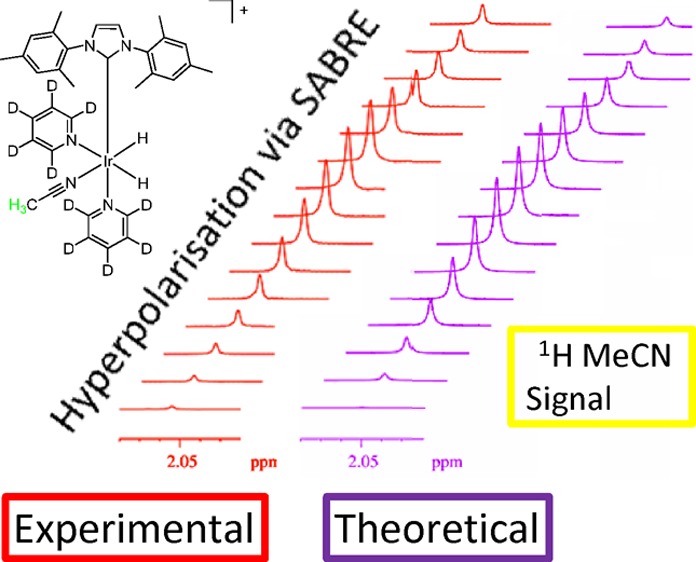
Nonetheless, while 3 achieves the 9-fold signal enhancement of A (in a PTF of 65 G), it can be readily optimized further. In order to do so, these measurements were conducted using a mixing chamber and flow probe apparatus30,49 and the results are illustrated in Figure 1a. It is important to note that zero PTFs are not achieved here experimentally. While the field-coil in the mixing chamber on a 0 G setting delivers a zero field, the resultant field experienced by the sample is better approximated by the Earth’s field. Therefore, an experimental setting of zero should be regarded as 0.5 G. This field value was used in producing the theoretically derived spectra which we compare to our experimental spectra.
Figure 1.

(a) Plot of the 1H NMR signal intensity seen for free CH3CN as a function of the PTF (indicated); data produced by a CD3OD solution containing 1 (10 mg), CH3CN (0.1 M) and pyridine (0.02 M). (b) Matching theoretical plot of the predicted 1H NMR signals. The phase of the detected signals has been inverted for ease of viewing.
Experimentally, an emission signal is seen for the CH3 proton signal of A at all of the probed PTF values, with the maximum signal intensity, corresponding to a 20-fold signal enhancement, being seen at 80 G. Note the convention which we use here: the hyperpolarized signals appear in emission in contrast to the thermal signals which appear in absorption. Therefore, the enhancement in the above case is 20-fold with a sign change which technically could be stated as a −20-fold enhancement. This may appear somewhat contradictory to some readers and so we have stated enhancements as positive amounts throughout. Likewise, in some of the spectra presented, we have inverted the signal phases from those actually observed for ease of viewing.
While ≈10% pyridine polarization30 has previously been reported, a 20-fold signal enhancement still far exceeds the signal gain achieved using a cryoprobe.53,54 Here our aim was to explore the SABRE effect using nitrile-containing substrates. We did not seek to produce an optimized catalytic system, although we aimed to develop a strategy to aid in this development.
In order to gain insight into the characteristics of hyperpolarization transfer via SABRE, theoretical calculations were performed using the approach reported previously,25 the results of which are shown in Figure 1b. These calculations explore the evolutions of the resulting spin systems through a spin density operator treatment under the strong coupling Hamiltonian and were performed using Mathematica.55 The theoretical predictions accord well with the observations made in the experiment.
The level of hyperpolarization observed for the methyl protons of acetonitrile can be further increased when the pyridine-h5 used as a cosubstrate is replaced by pyridine-d5. This is a consequence of the fact that polarization is not transferred effectively into the 2H-nuclei of the deuterated ligands because of the greatly reduced hydride-2H J-couplings compared to the hydride-1H J-couplings and their frequency difference. Instead it is transferred into the polarization transfer receptors of 3 that remain, the three protons of A and potentially those in the IMes ligand. In this case a 60-fold increase in signal strength for A was observed when the PTF was 80 G rather than the 20-fold enhancement described earlier. Therefore, there is a net 3-fold increase in acetonitrile signal polarization upon changing from pyridine-h5 to pyridine-d5.
In order to test more comprehensively the potential role of the IMes ligand in the polarization transfer process, the analogous complex with a deuterated IMes ligand was prepared. Samples consisting of either 1 or Ir(IMes-d22)(COD)Cl31 in the presence of 20 equiv of pyridine and 4 equiv of A were then compared and found to give almost identical enhancements for A after being shaken in either a PTF of 0.5 or 80 G. A series of EXSY measurements on cis,cis-[Ir(H)2(IMes-d22)(MeCN)(py)2]Cl revealed that pyridine and A loss occurred on the same time scale to those observed for 1, within the limits of experimental uncertainty. Furthermore, vibrational analysis of the CO stretch in the two related Ir(CO)2(NHC)Cl complexes revealed a 4 cm–1 difference in their frequencies and thus is indicative that both the protio and deuterio NHCs have comparable electron-donating abilities (confirmed further by DFT calculations; see Supporting Information). The IMes ligand in these complexes, therefore, plays a minimal role as an acceptor of polarization, and deuteration of the coligand pyridine clearly provides for the best route to increase SABRE efficiency.
We note, however, that upon the addition of 1.1 equiv of PCy3 (Cy = cyclohexyl) to these samples, a significant difference is observed. The PCy3 ligand binds by displacing the axial pyridine and forms [Ir(IMes)(MeCN)(PCy3)(H)2(py)]+ (4). Thus, the PCy3 ligand in 4 is placed trans to the IMes ligand and cis to both the hydride ligands. 1 now proved to be 100% more effective at polarizing A than 4-h24. In contrast, when the 4-d22-derivative was examined identical polarization levels resulted. Hence when PCy3 is located in the axial position different SABRE behavior is seen and now deuteration of the axial ligand acts to improve efficiency. These observations confirm that polarization transfer into the axial groups leads to a reduction in SABRE activity with some catalysts.31
As indicated earlier in this paper, our theoretical calculations, in which we assumed a single MeCN ligand coming into contact with the metal center of the catalyst, reproduced the experimental behavior well for both the 1H NMR spectra. These calculations show that the key spin-system parameters in 3 are the 2-bond hydride–hydride coupling, the 5-bond trans hydride-A coupling, and the lifetime of the H–Ir–NCMe interaction. These were determined experimentally; the splitting 2JHH was measured at −7.6 Hz, the trans5JHH at 2.1 Hz, and the lifetime of 3 at 0.096 s. The lifetime was obtained from the rate of exchange of A with the metal center of 3 determined under saturation kinetics as necessary for a dissociative process. Close agreement between the theoretical and experimental spectrum resulted when the 2JHH value was set to −7.6 Hz, 5JHH to −2.1 Hz, and the complex lifetime set to 0.096 s (see Supporting Information for further details of the parameters used in the theoretical calculations). Furthermore, the sign of 2JHH controls the sign of the coefficient defining the observed hyperpolarization amplitude, and hence the phase of the signal, which in this case are both negative.
The 1H NMR signals that were detected in these measurements were encoded using a π/2 pulse. The observed peaks reflect the measurement of longitudinal magnetization as would be created in a normal NMR measurement, albeit with much higher absolute amplitude. Additionally, the formation of three two-spin order terms, such as 2I1zI2z, and the three-spin order term, 4I1zI2zI3z, are predicted according to our theoretical modeling to be formed under SABRE in A. However, such two- and three-spin order terms are not visible after a π/2 measurement pulse (or indeed any pulse angle) because the spins are completely magnetically equivalent.56 Normally, when at least one nucleus is magnetically inequivalent, OPSY49,57,58 (only parahydrogen spectroscopy) can be employed to view and differentiate these terms (see Supporting Information for description). In the case of A, this should not be possible because of the magnetic equivalence of the methyl protons. However, a small residual signal was observed for A in the 1H double-quantum filtered OPSY spectrum. Similar signals were also observed for B and C where their magnitude relative to the hydride peak at δ −22.12 was 0.05–0.1%, whereas it was ≈6% for A. Given the fact that we are dealing with the creation of hyperpolarized states, low levels of residual magnetization might be expected.
As well as the 1H homonuclear longitudinal two-spin order terms, corresponding polarized coupled states between protons and the 13C nuclei are also predicted to be produced simultaneously via SABRE. This provides a route of polarization transfer to 13C. Indeed, it has been reported that 13C nuclei can be successfully polarized using SABRE.6,49 When a 13C NMR measurement was made on a sample of A, pyridine-d5 and 3 under 3 bar para-H2, at the same concentrations as those listed in Figure 1 and following evolution at a PTF of 0.5 G, a hyperpolarized signal for the quaternary 13C in natural abundance of free A at 116.7 ppm was observed with a S/N ratio of 11 in a single scan measurement. This appeared as an antiphase quartet with relative intensities −1:–1:1:1 and a splitting of 9.2 Hz. It is important to note that no signal was observed for the methyl 13C resonance in this measurement (see Supporting Information).
In order to probe polarization transfer to 13C more effectively, the 13C labeled materials B and C shown in Scheme 1 were employed together with 1 and pyridine-d5. First, for B, where the 13C label is located at the methyl site, its 1H signal was observed to hyperpolarize and yielded a 14-fold enhancement following polarization transfer in an optimal PTF of 80 G. The drop in 1H hyperpolarization level, relative to that seen for A, can be associated with the extra 13C nuclear spin, in 100% abundance, which causes a significant increase in the number of available magnetic states that can be populated (many of which are not directly observable). Transfer of hyperpolarization to the methyl 13C nucleus, albeit very weak, was also observed (see Figure 2a). This signal appeared as a 1:1:3:0.3 multiplet with a 136 Hz splitting, simulated theoretically in Figure 2b, instead of the expected 1:3:3:1 profile. The difference in intensity between the hyperpolarized and the thermal NMR spectra was actually very slight. The calculated relative contributions to the spectrum of the thermal and polarized components are 3:1. When we monitored the 13C spectra as a function of PTF we saw no change in the efficiency of polarization transfer even when the PTF of our apparatus was set to 0.5 G, in order to minimize the 1H–13C Zeeman frequency difference. As noted in the Supporting Information, this equipment setting does not achieve a resultant zero field, and the actual field produced by the apparatus was insufficiently low to produce an effectively enhanced signal. We note, however, that the methyl 13C resonance can also be observed as a weakly hyperpolarized signal under INEPT transfer from the CH3 protons. Our theoretical calculations agreed with this low level of polarization transfer and showed that a PTF of 5 mG would be required to effect efficient polarization transfer into the methyl 13C nucleus.
Figure 2.

13C NMR spectra of the methyl 13C of B obtained after hyperpolarization in a PTF of 5 G obtained using eight scans (a) and theoretical calculations (b).
In contrast, when C is studied, its quaternary 13C signal at δ 116.7 becomes strongly hyperpolarized at a PTF of 0.5 G yielding a signal for a 0.1 M sample in a single shot experiment with a S/N value of 105 as shown in Figure 3a. We obtained excellent agreement between the experimental and theoretical spectra of free C recorded and calculated following evolution in a PTF of 0.5 G as seen in Figure 3b. Figure 3c shows, the corresponding PTF plot which reveals enhancement maxima at 0.5 and 100 G are observed. The bound quaternary 13C signal for equatorial C of 3 is also readily observed experimentally at δ 117.8. Again no significant hyperpolarization of the methyl 13C signal was detected (Figure 3a). In the corresponding 1H NMR spectrum, the free methyl group signal enhancement for C was 12-fold at the optimum PTF of 90 G. Over the PTF range of 0–140 G, these signals generally appear as antiphase doublets. They are observed with unequal line amplitudes resulting from varying contributions of the theoretically predicted longitudinal magnetization and longitudinal two-spin order terms. Analysis of the experimental spectra revealed that the longitudinal two-spin order terms were the dominant terms at all fields except 90 and 140 G.
Figure 3.

(a) Single scan 13C NMR spectrum of free C (δ 116.7, 15.5 μL) in the presence of 1 (10 mg) and pyridine-d5 (3.75 μL). Hyperpolarization transfer was completed in a PTF of 0.5 G. The inset trace shows the corresponding methyl 13C-region, magnified 128 times relative to the main trace. The antiphase multiplet at 117.8 ppm is due to the quaternary 13C signal of bound C in 3. (b) Experimental (upper) and theoretically calculated (lower) 13C NMR spectra of the quaternary signal of C after hyperpolarization in a PTF of 0.5 G; (c) 13C field plot of C after hyperpolarization transfer using 1 over the PTF range of 0.5 to 140 G at intervals of 20 G collected experimentally (left) and theoretically calculated (right).
In the experimental intensity versus field profile for the quaternary 13C resonance shown in Figure 3(c), that is detected after a π/2 pulse, a complex intensity variation with increase in PTF is observed with a reversal of the signals sign being seen between 120 and 140 G. However, the theoretical model of these data only produced the signal reversal in this field range when the rate of loss of C was set to 55 s–1, corresponding to a residence time of the ligand on the catalyst of 0.018 s. The profile is also sensitive to small variations in the hydride–hydride 2JHH coupling. When the experimentally determined rate of exchange of C (10.42 s–1, corresponding to a residence time of 0.096 s) is employed in the calculation, the reversal occurs at around 70 G. We are working to understand this difference.
Our theoretical calculations involving C confirmed that SABRE creates longitudinal two-spin order terms, 2IkzSz (k = 1, 2, 3), where the Ik spins are proton nuclei of the three-spin methyl group and S is the quaternary carbon. The terms resulting from these states after a π/2 pulse dominate the spectra. No Sz longitudinal magnetization terms are produced with any meaningful amplitude at PTF’s other than pure (theoretical) 0 G. This is because of the large 1H–13C Zeeman frequency difference at anything other than the most minute PTFs. The initial magnetization created after a π/2(y) rf pulse to 13C is 2IkzSx and antiphase with respect to a single 2JCH coupling. Further calculation of the evolution of 2IkzSx within a four-spin system under a weak-coupling Hamiltonian during the FID acquisition obtained the following modulations of the real part of the observable terms: 90° out-of-phase oscillations with respect to I2 and I3, in-phase oscillation with respect to I1, and 90° out-of-phase oscillations with respect to the chemical shift of S, all of which are 180° phase shifted from original 2IkzSx term. The couplings between I1, I2, I3, and S are all equal and this results in a cos2[Jπτ] × sin[Jπτ] × sin[Ωτ] modulation. It is the combination of these effects that produces the unusual [−1:–1:1:1] multiplet seen in these measurements (Figure 3a). Three-spin order terms, 4IkzIlzSz, (k = 1, 2; m = 2, 3; k ≠ m) and four-spin order terms, 8IkzIlzImzSz (k = 1; l = 2, m = 3), are also produced but with amplitudes an order of magnitude lower than the two-spin order term. These do not appear to contribute significantly to the experimentally produced quaternary 13C spectra. In addition to their smaller amplitudes, the absence of their contributions could also be attributable to their relatively fast relaxation (T1 ∼1 s).49 In fact, only the quaternary 13C resonance was observed as an enhanced signal at a PTF of 0.5 G and upon proton decoupling the expected singlet appeared with diminished intensity. Refocusing of the antiphase coupling did, however, improve the resulting signal intensity (see Supporting Information).
The standard homonuclear OPSY57,58 sequence was modified to create a heteronuclear experiment by the application of simultaneous π/2 pulses to the 1H and 13C nuclei in conjunction with coherence selection gradients in the ratio of 64:80 for 1H observation and 16:80 for 13C. This facilitated the selective detection of the theoretically predicted 2IkzSz longitudinal two-spin order term through either its Ik or S nuclei. Typical NMR spectra are shown in parts a and b of Figure 4 that illustrate these observables.
Figure 4.

1H (a) and 13C (b) NMR spectra of a CD3OD solution (0.6 mL) consisting of 1 (2 mg), C (3.1 μL), and pyridine-d5 (0.75 μL) obtained using a modified OPSY sequence in which the double quantum coherence originating from the 2IkzSz starting state (Ik are 1H and S is 13C) was selected.
It should be noted, that the addition of the 13C spin allows the creation of the 4IkzIlzSz, term which can also be detected using the same heteronuclear OPSY pulse sequence but with a gradient ratio of 45.7:80. However, the resulting 1H NMR signal had a very small amplitude (S/N ratio 19) compared with the 1H signal arising from the two-spin order term (S/N ratio 995) as described and shown in Figure 4a. This corresponds with the theoretical calculations which show the amplitudes of these terms to be 75× smaller than the two-spin terms.
In the case of D, the absence of any 1H–1H coupling between the substrate and the hydride ligands of 3 results in no visible 1H or 13C-hyperpolarization transfer within the experimental range of PTFs explored here. In this instance, not even the quaternary 13C could be observed after 32 scans with repolarization between scans. Our theoretical calculations show no polarization transfer and agree completely with this result. This situation does not change with time thereby confirming 1 is very inefficient at driving H–D exchange in A–D. Deuteration effects have recently been reported to complicate the SABRE process with pyridine.50
We extended our experimental investigations to longer chain molecules containing cyano groups. In the Supporting Information, we detail experimental 1H and 13C data that were collected when propionitrile, benzylnitrile, benzonitrile, and trans-3-hexenedinitrile were examined. We note that for propionitrile, both the CH2 and CH3 protons were enhanced by 10- and 4-fold, respectively, in the 1H NMR spectrum. The spectrum can be simplified by the use of pyridine-d5 instead of pyridine-h5; the resulting hyperpolarized 1H NMR spectrum is shown in Figure 5. In the 13C NMR spectrum, recorded after exposure to 3 bar para-H2 in a PTF of 0.5 G, an antiphase multiplet was detected at δ 9.4 which is indicative of polarization transfer to the methyl 13C nucleus. For benzylnitrile, enhancements of 2- and 18-fold were seen for the benzyl and CH2 proton signals, respectively, after polarization transfer at a PTF of 65 G. Conversely, benzonitrile displayed no enhancement upon exposure to 3 bar para-H2 at a PTF of 65 G. These important observations support the hypothesis that polarization transfer must occur in the first instance via a suitable JHH coupling between the hydride and substrate protons. Such a coupling is effectively zero for benzonitrile due to the remote (six bonds) location of the substrate proton nearest to the hydride nucleus in this molecule. trans-3-hexenedinitrile also proved to polarize via SABRE and, interestingly, its alkene functionality proved resistant to hydrogenation. The CH2 and alkenic protons were enhanced by 4- and 10-fold, respectively. In this last case, the preference for either the nitrile or alkene ligating to the metal was probed using DFT. Structures were optimized using the PBE0 functional from Adamo and the basis sets from Aldrich described by the def2-SVP label. These calculations showed that, in terms of enthalpy, the dominant form of interaction with the iridium center was via the nitrile group of trans-3-hexenedinitrile, by 105.9 kJ mol–1 compared to binding through the alkenic component, hence reducing the propensity for alkene hydrogenation. It has been noted that in the absence of diisopropylethylamine (DIPEA) Ir/PHOX catalysts are unable to hydrogenate the alkene bond of α,β-unsaturated nitriles due to coordination of two substrate molecules bound through the nitrile group leading to deactivation of the catalyst.59 Even in the presence of DIPEA, 50–100 bar of H2 was required to achieve full conversion. This is despite α,β-unsaturated nitriles being easier to hydrogenate than β,γ-unsaturated nitriles due to activation by the nitrile group and increased conjugation.60 It is, therefore, not surprising that trans-3-hexenedinitrile, a β,γ-unsaturated nitrile, does not hydrogenate under the conditions we employ here.
Figure 5.

1H NMR spectrum of propionitrile (4.6 μL), pyridine-d5 (0.75 μL), and 1 (2 mg) in MeOH-d4 after being shaken in a PTF of 65 G.
In summary, the small 3JHH couplings that exist between hydride nuclei and the CH2/CH3 protons located directly next to the cyano group in A, propionitrile, benzylnitrile, and trans-3-hexenedinitrile are adequate to propagate polarization transfer. This contrasts to the 6JHH coupling to the ortho proton in benzonitrile which results in no detectable polarization transfer.
Conclusions
In this paper, we have proved that hyperpolarization transfer from the hydride ligands of the polarization transfer catalyst into the weakly coordinating acetonitrile ligand occurs in a homonuclear fashion via their spin–spin coupling network to the methyl protons when the polarization transfer field is >0.5 G. We have also demonstrated that a range of aromatic and nonaromatic nitrile-containing substrates can be polarized via SABRE, provided that there is a suitable homonuclear coupling pathway. This is a departure from the aromatic systems that have been more commonly polarized by SABRE.
Our theoretical calculations details how the transfer of polarization to the quaternary 13C nucleus occurs via the “long-range” coupling to the methyl protons rather than directly from the former para-H2 derived hydride nuclei in the catalyst. According to the theoretical study, it is the fact that this heteronuclear coupling is of the same order of magnitude as the hydride–hydride coupling that facilitates this process in fields >0.5 G. In sharp contrast, the “short-range” coupling between the methyl protons and the methyl 13C nucleus which is an order of magnitude larger than the hydride–hydride coupling enables very little hyperpolarization transfer to it. The inefficiency of transfer in this case was predicted in our calculations and observed as a result experimentally. Theoretically, the population of both homonuclear and heteronuclear two spin order terms within A-C was predicted. A modified OPSY sequence, was used to detect the longitudinal two-spin order existing between 1H and 13C to confirm this experimentally. The complete magnetic equivalence of the methyl protons precludes detection of this homonuclear state. This situation lowers the efficiency of polarization transfer into visible states by a theoretically predicted value of 20% at 70.5 G and increases to a maximum of 33% at 90.5 G.
This series of observations have significant implications on how best to complete any 13C labeling if SABRE is to be used in vivo. For such nitrile containing systems, placing the label into a site that exhibits a long-range coupling to the SABRE polarized protons is suggested. The theoretical calculations provide excellent agreement with the experimental observations of the 1H spectrum and the variations of the spectral amplitudes with PTF. Also the theoretical 13C spectrum of the methyl 13C nucleus and the quaternary 13C nucleus at low field both provide excellent agreement with experiment. The only departure from this otherwise excellent agreement is the quaternary 13C PTF plot profile. Here the inversion of the phase of the spectrum predicted by our models does not occur at the same PTF as the experiment shows, when the experimentally measured parameters are used in the model. We are continuing our research to improve this fit.
We have further demonstrated that the precise identity of the ligands and their conformation within the complex determines the propagation of polarization to ligands that are cis to the hydride ligands. In the complex [Ir(H)2(IMes)(MeCN)(py)2]+, no detectable polarization is transferred into the IMes ligand. Crucially, the same PTF plot and enhancement levels are observed regardless of whether or not the IMes ligand is deuterated. However, addition of PCy3 to this complex and subsequent elimination of pyridine, forms Ir(IMes)(H)2(MeCN)(Py)(PCy3), 4, in which PCy3 is trans to IMes and cis to the hydrides. The resulting enhancement of the protons in A is half of that for the former complex. In stark contrast, the use of IMes-d22 instead of IMes, resulted in the enhancement level being maintained in the presence of PCy3. It is, therefore, possible to control transfer into the axial site by ligand design.
We believe that these methods illustrate a viable strategy to develop SABRE for weakly interacting, nonpyridine-based systems. We are currently exploring alternative catalysts to improve further SABRE’s polarization transfer efficiency and signal enhancement levels. Such increases in catalyst efficiency would improve the likelihood that drug molecules possessing weakly interacting groups, such as nitriles, could be polarized by this method. Once successfully hyperpolarized such molecules could potentially feature as contrast agents, or reagents that enable the study of reaction mechanisms and the detection of reaction intermediates. An assessment of the steric bulk and ligand exchange rates will have to be made before the polarization of an unknown nitrile by SABRE can be contemplated.
Acknowledgments
S.B.D. and G.G.R.G. thank the EPSRC (Grant No. EP/G009546/1) and the Wellcome Trust and Wolfson Foundation (092506 and 098335) for their generous funding. R.A.G. thanks Oxford Instruments for funding. R.O.J. thanks the University of York for a DTA scholarship. We are grateful to Cambridge Isotope Laboratories for providing the labeled materials used in this study.
Supporting Information Available
Experimental information including synthesis, methods, NMR characterization for 3, hyperpolarized 1H and 13C spectra for A–C, propionitrile, benzylnitrile, benzonitrile, and trans-3-hexenedinitrile, parameters used in theoretical models, DFT calculations, and details about the theoretical approach. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Abragam A.; Goldman M. Principles of Dynamic Nuclear-Polarization. Rep. Prog. Phys. 1978, 41, 395–467. [Google Scholar]
- Bowers C. R.; Weitekamp D. P. Para-Hydrogen and Synthesis Allow Dramitically Enhanced Nuclear Alignment. J. Am. Chem. Soc. 1987, 109, 5541–5542. [Google Scholar]
- Eisenschmid T. C.; Kirss R. U.; Deutsch P. P.; Hommeltoft S. I.; Eisenberg R.; Bargon J.; Lawler R. G.; Balch A. L. Para-Hydrogen Induced Polarization in Hydrogenation Reactions. J. Am. Chem. Soc. 1987, 109, 8089–8091. [Google Scholar]
- Pravica M. G.; Weitekamp D. P. Net NMR Alignment By Adibatic Transport of Para-Hydrogen Addition-Products to High Magnetic-Field. Chem. Phys. Lett. 1988, 145, 255–258. [Google Scholar]
- Natterer J.; Bargon J. Parahydrogen induced polarization. Prog. Nucl. Magn. Reson. Spectrosc. 1997, 31, 293–315. [Google Scholar]
- Lloyd L. S.; Adams R. W.; Bernstein M.; Coombes S.; Duckett S. B.; Green G. G. R.; Lewis R. J.; Mewis R. E.; Sleigh C. J. Utilization of SABRE-Derived Hyperpolarization To Detect Low-Concentration Analytes via 1D and 2D NMR Methods. J. Am. Chem. Soc. 2012, 134, 12904–12907. [DOI] [PubMed] [Google Scholar]
- Green R. A.; Adams R. W.; Duckett S. B.; Mewis R. E.; Williamson D. C.; Green G. G. R. The theory and practice of hyperpolarization in magnetic resonance using parahydrogen. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 67, 1–48. [DOI] [PubMed] [Google Scholar]
- Golman K.; Axelsson O.; Johannesson H.; Mansson S.; Olofsson C.; Petersson J. S. Parahydrogen-induced polarization in imaging: Subsecond C-13 angiography. Magn. Reson. Med. 2001, 46, 1–5. [DOI] [PubMed] [Google Scholar]
- Johansson E.; Mansson S.; Wirestam R.; Svesson J.; Petersson S.; Golman K.; Stahlberg F. Cerebral perfusion assessment by bolus tracking using hyperpolarized C-13. Magn. Reson. Med. 2004, 51, 464–472. [DOI] [PubMed] [Google Scholar]
- Magnusson P.; Johansson E.; Mansson S.; Petersson J. S.; Chai C. M.; Hansson G.; Axelsson O.; Golman K. Passive catheter tracking during interventional MRI using hyperpolarized C-13. Magn. Reson. Med. 2007, 57, 1140–1147. [DOI] [PubMed] [Google Scholar]
- Duckett S. B.; Mewis R. E.. Improving NMR and MRI Sensitivity with Parahydrogen. In Hyperpolarization Methods in NMR Spectroscopy; Kuhn L. T., Ed.; Topics in Current Chemistry 338; Springer: Berlin, 2013; pp 75–103. [DOI] [PubMed] [Google Scholar]
- Kating P.; Wandelt A.; Selke R.; Bargon J. Nuclear Singlet-Triplet Mixing During Hydrogenations with Parahydrogen - an in-situ NMR Method to Investigate Catalytic Reaction-Mechanisms and their Kinetics 0.2. Homogeneous Hydogenation of 1,4-Dihydro-1,4-Epoxynaphthalene Using Different Rhodium Catalysts. J. Phys. Chem. 1993, 97, 13313–13317. [Google Scholar]
- Hommeltoft S. I.; Berry D. H.; Eisenberg R. A Metal-Centered Radical-Pair Mechanism for Alkyne Hydrogenation with a Binuclear Rhodium Hydride Complex - CIDNP without Organic Radicals. J. Am. Chem. Soc. 1986, 108, 5345–5347. [Google Scholar]
- Permin A.; Eisenberg R. Parahydrogen induced polarization and the oxidative addition of hydrogen to iridium tribromostannyl carbonylate anions. Inorg. Chem. 2002, 41, 2451–2458. [DOI] [PubMed] [Google Scholar]
- Deibele C.; Permin A. B.; Petrosyan V. S.; Bargon J. A study of the mechanism of platinum(II)/tin(II) dichloride mediated hydrogenation of alkynes and alkenes employing parahydrogen-induced polarization. Eur. J. Inorg. Chem. 1998, 1915–1923. [Google Scholar]
- Blazina D.; Duckett S. B.; Dyson P. J.; Lohmann J. A. B. Catalytic hydrogenation by triruthenium clusters: A mechanistic study with parahydrogen-induced polarization. Chem.—Eur. J. 2003, 9, 1046–1061. [DOI] [PubMed] [Google Scholar]
- Colebrooke S. A.; Duckett S. B.; Lohman J. A. B.; Eisenberg R. Hydrogenation studies involving halobis(phosphine)-rhodium(I) dimers: Use of parahydrogen induced polarisation to detect species present at low concentration. Chem.—Eur. J. 2004, 10, 2459–2474. [DOI] [PubMed] [Google Scholar]
- Giernoth R.; Heinrich H.; Adams N. J.; Deeth R. J.; Bargon J.; Brown J. M. PHIP detection of a transient rhodium dihydride intermediate in the homogeneous hydrogenation of dehydroamino acids. J. Am. Chem. Soc. 2000, 122, 12381–12382. [Google Scholar]
- Prestwich T. G.; Blazina D.; Duckett S. B.; Dyson P. J. A parahydrogen study of catalytic hydrogenation by diphosphane substituted triruthenium clusters. Eur. J. Inorg. Chem. 2004, 4381–4387. [Google Scholar]
- Feng B.; Coffey A. M.; Colon R. D.; Chekmenev E. Y.; Waddell K. W. A pulsed injection parahydrogen generator and techniques for quantifying enrichment. J. Magn. Reson. 2012, 214, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey A. M.; Shchepin R. V.; Wilkens K.; Waddell K. W.; Chekmenev E. Y. A large volume double channel H-1-X RF probe for hyperpolarized magnetic resonance at 0.0475 T. J. Magn. Reson. 2012, 220, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoevener J.-B.; Chekmenev E. Y.; Harris K. C.; Perman W. H.; Robertson L. W.; Ross B. D.; Bhattacharya P. PASADENA hyperpolarization of C-13 biomolecules: equipment design and installation. Magn. Reson. Mater. Phys., Biol. Med. 2009, 22, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell K. W.; Coffey A. M.; Chekmenev E. Y. In Situ Detection of PHIP at 48 mT: Demonstration Using a Centrally Controlled Polarizer. J. Am. Chem. Soc. 2011, 133, 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams R. W.; Aguilar J. A.; Atkinson K. D.; Cowley M. J.; Elliott P. I. P.; Duckett S. B.; Green G. G. R.; Khazal I. G.; Lopez-Serrano J.; Williamson D. C. Reversible Interactions with para-Hydrogen Enhance NMR Sensitivity by Polarization Transfer. Science 2009, 323, 1708–1711. [DOI] [PubMed] [Google Scholar]
- Adams R. W.; Duckett S. B.; Green R. A.; Williamson D. C.; Green G. G. R.. A theoretical basis for spontaneous polarization transfer in non-hydrogenative parahydrogen-induced polarization. J. Chem. Phys. 2009, 131. [DOI] [PubMed] [Google Scholar]
- Gong Q.; Gordji-Nejad A.; Blümich B.; Appelt S. Trace Analysis by Low-Field NMR: Breaking the Sensitivity Limit. Anal. Chem. 2010, 82, 7078–7082. [DOI] [PubMed] [Google Scholar]
- Hoevener J.-B.; Schwaderlapp N.; Borowiak R.; Lickert T.; Duckett S. B.; Mewis R. E.; Adams R. W.; Burns M. J.; Highton L. A. R.; Green G. G. R.; et al. Toward Biocompatible Nuclear Hyperpolarization Using Signal Amplification by Reversible Exchange: Quantitative in Situ Spectroscopy and High-Field Imaging. Anal. Chem. 2014, 86, 1767–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H.; Xu J.; Gillen J.; McMahon M. T.; Artemov D.; Tyburn J.-M.; Lohman J. A. B.; Mewis R. E.; Atkinson K. D.; Green G. G. R.; et al. Optimization of SABRE for polarization of the tuberculosis drugs pyrazinamide and isoniazid. J. Magn. Reson. 2013, 237, 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshuis N.; Hermkens N.; van Weerdenburg B. J. A.; Feiters M. C.; Rutjes F. P. J. T.; Wijmenga S. S.; Tessari M. Toward Nanomolar Detection by NMR Through SABRE Hyperpolarization. J. Am. Chem. Soc. 2014, 136, 2695–2698. [DOI] [PubMed] [Google Scholar]
- Cowley M. J.; Adams R. W.; Atkinson K. D.; Cockett M. C. R.; Duckett S. B.; Green G. G. R.; Lohman J. A. B.; Kerssebaum R.; Kilgour D.; Mewis R. E. Iridium N-Heterocyclic Carbene Complexes as Efficient Catalysts for Magnetization Transfer from para-Hydrogen. J. Am. Chem. Soc. 2011, 133, 6134–6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekete M.; Bayfield O.; Duckett S. B.; Hart S.; Mewis R. E.; Pridmore N.; Rayner P. J.; Whitwood A. Iridium(III) Hydrido N-Heterocyclic Carbene-Phosphine Complexes as Catalysts in Magnetization Transfer Reactions. Inorg. Chem. 2013, 52, 13453–13461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Weerdenburg B. J. A.; Gloggler S.; Eshuis N.; Engwerda A. H. J.; Smits J. M. M.; de Gelder R.; Appelt S.; Wymenga S. S.; Tessari M.; Feiters M. C.; et al. Ligand effects of NHC-iridium catalysts for signal amplification by reversible exchange (SABRE). Chem. Commun. 2013, 49, 7388–7390. [DOI] [PubMed] [Google Scholar]
- Lloyd L. S.; Asghar A.; Burns M. J.; Charlton A.; Coombes S.; Cowley M. J.; Dear G. J.; Duckett S. B.; Genov G. R.; Green G. G. R.; et al. Hyperpolarisation through reversible interactions with parahydrogen. Catal. Sci. Technol. 2014, 4, 3544–3554. [Google Scholar]
- Torres O.; Martín M.; Sola E. Labile N-Heterocyclic Carbene Complexes of Iridium. Organometallics 2009, 28, 863–870. [Google Scholar]
- Michelin R. A.; Mozzon M.; Bertani R. Reactions of transition metal-coordinated nitriles. Coord. Chem. Rev. 1996, 147, 299–338. [Google Scholar]
- Hillner B. E. Cost-effectiveness projections of anastrazole (AN) vs. tamoxifen (T) as initial adjuvant therapy in ER-positive early breast cancer using data from the ATAC trial. Breast 2003, 12Supplement 1S44–S45. [Google Scholar]
- Amsterdam L. L.; Gentry W.; Jobanputra S.; Wolf M.; Rubin S. D.; Bulun S. E. Anastrazole and oral contraceptives: a novel treatment for endometriosis. Fertil. Steril. 2005, 84, 300–304. [DOI] [PubMed] [Google Scholar]
- Fleming F. F.; Yao L.; Ravikumar P. C.; Funk L.; Shook B. C. Nitrile-Containing Pharmaceuticals: Efficacious Roles of the Nitrile Pharmacophore. J. Med. Chem. 2010, 53, 7902–7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer F. A.; Ottiger P.; Pfaffen C.; Leutwyler S. Structure and Intermolecular Vibrations of Perylene·trans-1,2-Dichloroethene, a Weak Charge-Transfer Complex. J. Phys. Chem. A 2013, 117, 10702–10713. [DOI] [PubMed] [Google Scholar]
- Bini L.; Muller C.; Vogt D. Mechanistic Studies on Hydrocyanation Reactions. ChemCatChem. 2010, 2, 590–608. [Google Scholar]
- Evans M. E.; Jones W. D. Controlling the Selectivity for C–H and C–CN Bond Activation at Rhodium: A DFT Examination of Ligand Effects. Organometallics 2011, 30, 3371–3377. [Google Scholar]
- Evans M. E.; Li T.; Vetter A. J.; Rieth R. D.; Jones W. D. Thermodynamic Trends in Carbon–Hydrogen Bond Activation in Nitriles and Chloroalkanes at Rhodium. J. Org. Chem. 2009, 74, 6907–6914. [DOI] [PubMed] [Google Scholar]
- Serrano P.; Pedrini B.; Mohanty B.; Geralt M.; Herrmann T.; Wuethrich K. The J-UNIO protocol for automated protein structure determination by NMR in solution. J. Biomol. NMR 2012, 53, 341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wüthrich K. NMR Studies of Structure and Function of Biological Macromolecules (Nobel Lecture). Angew. Chem., Int. Ed. 2003, 42, 3340–3363. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Theis T.; Liang X.; Wang Q.; Zhou P.; Warren W. S. Storage of Hydrogen Spin Polarization in Long-Lived C-13(2) Singlet Order and Implications for Hyperpolarized Magnetic Resonance Imaging. J. Am. Chem. Soc. 2013, 135, 9632–9635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvi N.; Buratto R.; Bornet A.; Ulzega S.; Rebollo I. R.; Angelini A.; Heinis C.; Bodenhausen G. Boosting the Sensitivity of Ligand-Protein Screening by NMR of Long-Lived States. J. Am. Chem. Soc. 2012, 134, 11076–11079. [DOI] [PubMed] [Google Scholar]
- Tayler M. C. D.; Levitt M. H. Accessing Long-Lived Nuclear Spin Order by Isotope-Induced Symmetry Breaking. J. Am. Chem. Soc. 2013, 135, 2120–2123. [DOI] [PubMed] [Google Scholar]
- Pravdivtsev A. N.; Yurkovskaya A. V.; Vieth H.-M.; Ivanov K. L.; Kaptein R. Level Anti-Crossings are a Key Factor for Understanding para-Hydrogen-Induced Hyperpolarization in SABRE Experiments. ChemPhysChem 2013, 14, 3327–3331. [DOI] [PubMed] [Google Scholar]
- Mewis R. E.; Atkinson K. D.; Cowley M. J.; Duckett S. B.; Green G. G. R.; Green R. A.; Highton L. A. R.; Kilgour D.; Lloyd L. S.; Lohman J. A. B.; et al. Probing signal amplification by reversible exchange using an NMR flow system. Magn. Reson. Chem. 2014, 52, 358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barskiy D. A.; Kovtunov K. V.; Koptyug I. V.; He P.; Groome K. A.; Best Q. A.; Shi F.; Goodson B. M.; Shchepin R. V.; Coffey A. M.; et al. The Feasibility of Formation and Kinetics of NMR Signal Amplification by Reversible Exchange (SABRE) at High Magnetic Field (9.4 T). J. Am. Chem. Soc. 2014, 136, 3322–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenstein O.; Crabtree R. H. Outer sphere hydrogenation catalysis. New J. Chem. 2013, 37, 21–27. [Google Scholar]
- Zhou R. R.; Aguilar J. A.; Charlton A.; Duckett S. B.; Elliott P. I. P.; Kandiah R. Parahydrogen derived illumination of pyridine based coordination products obtained from reactions involving rhodium phosphine complexes. Dalton Trans. 2005, 3773–3779. [DOI] [PubMed] [Google Scholar]
- Ratering D.; Baltes C.; Nordmeyer-Massner J.; Marek D.; Rudin M. Performance of a 200-MHz cryogenic RF probe designed for MRI and MRS of the murine brain. Magn. Reson. Med. 2008, 59, 1440–1447. [DOI] [PubMed] [Google Scholar]
- Sack M.; Wetterling F.; Sartorius A.; Ende G.; Weber-Fahr W. Signal-to-noise ratio of a mouse brain (13) C CryoProbe system in comparison with room temperature coils: spectroscopic phantom and in vivo results. NMR Biomed. 2014, 27, 709–15. [DOI] [PubMed] [Google Scholar]
- Mathematica Version 8.0; Wolfram Research, Inc.: Champaign, IL, 2010. [Google Scholar]
- Harris R. K.Nuclear Magnetic Resonance Spectrsocopy: A Physiochemical View; Longman Scientific and Technical: Harlow, England, 1986. [Google Scholar]
- Aguilar J. A.; Adams R. W.; Duckett S. B.; Green G. G. R.; Kandiah R. Selective detection of hyperpolarized NMR signals derived from para-hydrogen using the Only Para-hydrogen SpectroscopY (OPSY) approach. J. Magn. Reson. 2011, 208, 49–57. [DOI] [PubMed] [Google Scholar]
- Aguilar J. A.; Elliott P. I. P.; Lopez-Serrano J.; Adams R. W.; Duckett S. B. Only para-hydrogen spectroscopy (OPSY), a technique for the selective observation of para-hydrogen enhanced NMR signals. Chem. Commun. 2007, 1183–1185. [DOI] [PubMed] [Google Scholar]
- Mueller M.-A.; Pfaltz A. Asymmetric Hydrogenation of alpha,beta-Unsaturated Nitriles with Base-Activated Iridium N,P Ligand Complexes. Angew. Chem., Int. Ed. 2014, 53, 8668–8671. [DOI] [PubMed] [Google Scholar]
- Kukula P.; Koprivova K. Structure-selectivity relationship in the chemoselective hydrogenation of unsaturated nitriles. J. Catal. 2005, 234, 161–171. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.