

Abstract
The acute inflammatory response is a double-edged sword. On the one hand it plays a key role in initial host defense particularly against many infections. On the other hand its aim is imprecise and as a consequence, when it is drawn into battle, it can cause collateral damage in tissues. In situations where the inciting stimulus is sterile, the cost-benefit ratio may be high; because of this, sterile inflammation underlies the pathogenesis of a number of diseases. While there have been major advances in our understanding of how microbes trigger inflammation, much less has been learned about this process in sterile situations. This review focuses on a subset of the many sterile stimuli that can induce inflammation – specifically dead cells and a variety of irritant particles, including crystals, minerals, and protein aggregates. Although this subset of stimuli is structurally very diverse and might appear to be unrelated, there is accumulating evidence that the innate immune system may recognize them in similar ways and stimulate the sterile inflammatory response via common pathways. Here we review established and emerging data about these responses.
Keywords: Sterile particulates, inflammasome, danger signal, IL-1, NLRP3
Introduction
Inflammation is one of the oldest recorded medical conditions, presumably because in ancient times, as it is now, it was experienced by humans frequently and its signs and symptoms were not subtle. Written symbols for inflammation have been identified in Sumerian hieroglyphics dating back to 2700 BC and what we recognize today as the essential signs and symptoms of inflammation – redness, heat, swelling and pain – were described by the Roman academician Aulus Cornelius Celsus in the first century AD (1) (2) (3). Pus, which is another sign of inflammation primarily seen in certain infections, has also been described since ancient times (4).
We now know that most of the signs and symptoms of inflammation are caused by changes in the local vasculature of an affected tissue (5) (6, 7). The smooth muscle of arterioles relaxes, leading to vasodilation and the signs of erythema and heat. This also increases hydrostatic pressure across the vascular bed, which in combination with changes in the permeability of the vascular endothelial barrier, leads to the leakage of protein rich fluid into the tissue, thereby causing edema. Endothelial cells in venules express adhesion molecules that allow neutrophils and subsequently monocytes to adhere and migrate between endothelial cells into the tissue. If the recruitment of neutrophils into the tissue is sufficiently robust it becomes visible to the naked eye as pus (7).
Once triggered, the inflammatory response can develop very rapidly. Vasodilation can occur within seconds and fluid leakage and leukocyte extravasation within minutes to hours (8). The net result is rapid delivery of many of the body’s innate defenses to the offending site. This includes soluble defenses, such as antibody, complement, and collectins, and cellular ones, such as granulocytes and monocytes. Once in the tissue these various components attempt to neutralize, sequester and/or otherwise contain the inciting stimulus. If this is successful, then the innate defenses help clear debris and stimulate tissue repair. Once the inciting stimulus is gone, then the inflammatory response resolves.
One of the major triggers of inflammation is infection, with the inciting stimulus being certain proinflammatory molecules of the invading microbe (9–11). However, a potpourri of sterile stimuli including mechanical trauma, ischemia, toxins, minerals, crystals, chemicals and antigens also triggers inflammation. Most of these sterile stimuli can be broadly categorized into ones that are injurious, irritant or antigenic.
All of the various triggers of inflammation, whether they are infectious or sterile, ultimately lead to the same downstream vascular and cellular manifestations of inflammation. However, although the final inflammatory response is similar in all these situations, the initial events that elicit and control the response can be very different. This review focuses on inflammation that is triggered in sterile situations and in particular on a subset of sterile proinflammatory stimuli: Dead cells and irritant particles. The rationale for this focus is recent and emerging data that suggest that this diverse subset of proinflammatory stimuli may in fact stimulate responses through common mechanisms.
The medical significance of infectious and sterile inflammation
The inflammatory response plays an important role in host defense. It is one of the first lines of defense recruited to combat a potential threat. In the case of microbial invasion it often plays a critical role in clearing, containing and/or slowing the infection. Consequently animals or patients that have defects in mounting adequate inflammatory responses suffer from recurrent infections and, if the defects are profound, increased mortality (12). Because of this it is thought that infections have been one of the major forces driving the evolution of the inflammatory response.
While the inflammatory response is essential for host defense it is very much a double-edged sword. The effector mechanisms used by the innate immune system to kill microbes are extremely potent and can (and in fact do) also damage and kill mammalian cells. The recruited leukocytes kill microbes by producing highly reactive chemical species, such as reactive oxygen species, sodium hypochlorite (bleach), and other destructive molecules such as proteases. While these molecules are generally effective in destroying microbes, some of them leak from living and dying leukocytes and in so doing damage adjacent normal cells in the tissue. One of the major culprits in causing this collateral damage is the neutrophil, a cell type that is one of the focuses of this present review. In infections, tissue damage is a small price to pay to contain potentially life-threatening situations. Moreover, since infections are often rapidly cleared by immune mechanisms, the duration of the neutrophilic inflammatory response and (hence its attendant damage) are often limited.
However, the cost-benefit ratio may be very different in situations of sterile inflammation. Here the inciting stimulus may not be injurious to the host, and in any case, the innate immune mechanisms that are mobilized may do little or no good. As a result, the dominant effect of the inflammation in these situations may be collateral damage inflicted by the inflammatory response on otherwise healthy cells in the tissue. This process, if sufficiently robust, can cause acute disease and/or exacerbate damage from other etiologies. Moreover, if the sterile stimulus is not resolved this can lead to chronic inflammation and ongoing tissue damage that can also lead to and/or exacerbate disease. A number of these acute and chronic conditions will be described in the sections below.
Inflammation and disease caused by sterile particulates
A diverse set of sterile particles can stimulate inflammation, in some cases associated with prominent fibrosis. These particles include ones whose composition is inorganic (e.g. silica dioxide(13), iron oxide (14), calcium pyrophosphate (15), asbestos (13)) and organic (e.g. monosodium urate (16), amyloid-β (17), cholesterol (18)) and whose structure can be crystalline (e.g. silicates, monosodium urate) or amorphous (e.g. alum and iron oxide).
For many of these particles it is not actually clear whether the inflammatory response is subserving a useful role such as host defense. This is because the particles may not themselves be injurious and in any case the inflammatory response certainly fails to clear them, although it may help to collect and compartmentalize them. On the other hand, the damage caused by the sterile inflammatory response and its attendant fibrosis can lead to disease.
One of the classic crystal-based diseases, and the oldest to be recognized, is gout (16, 19). In this condition a fraction of patients that develop hyperuricemia nucleate crystals of monosodium urate (MSU) in their joints (20). The crystals incite an intense acute inflammatory response that can be exceedingly painful. These acute episodes wax and wane, but over time the recurrent inflammatory responses can damage the affected joint leading to chronic arthritis with associated dysfunction. Other crystals that deposit in joints can similarly lead to acute arthritis. This is seen for example with crystals of calcium pyrophosphate, which can form spontaneously in joints and cause the disease of pseudogout (21). In addition to affecting the joint, all of these particles can and do incite inflammation when they are deposited in other tissues, either spontaneously or experimentally by injection (22, 23).
Environmental and other exogenous particles can cause disease if they deposit in tissues. The situation where this most often occurs is when certain particles are inhaled during respiration and accumulate in sufficient amounts in the lung. This is typically seen in the setting of occupational exposure, the most common of which are with silicates (silica dioxide crystals and asbestos) (13). In the lung these particles cause inflammation and can lead to extensive pulmonary fibrosis and other complications. Particles that enter the body in other ways, e.g. through a wound or injection, can also cause inflammation. This is seen for example with injection of alum (an adjuvant composed of particles of aluminum salt) (24) or the introduction of talcum powder (25).
There are other conditions that are not traditionally thought of as particle-based diseases but in which particle-stimulated inflammation might play a role. Examples include the diseases of atherosclerosis and Alzheimer’s disease. In atherosclerosis chronic inflammation in arteries leads to intimal thickening that narrows the vascular lumen and compromises blood flow (18). In this condition cholesterol crystals develop in the artery wall and recent data suggests that this may be one of the stimulants of sterile inflammation (Duewell, et al. manuscript in preparation). In Alzheimer’s disease neurodegeneration occurs in the cortex of the brain and leads to dementia (17). This disease is associated with the deposition of amyloid peptide aggregates and recent data suggests that these aggregates may stimulate microglial cells to make proinflammatory mediators that may contribute to neural damage and disease pathogenesis (26).
Thus a wide range of sterile particulates can provoke inflammation and in so doing cause disease. Because of this, the sterile inflammatory response is medically important; consequently, this process and these particular stimuli are the focus of this review.
Inflammation to sterile cell death
Another kind of proinflammatory “particles”, albeit more complicated ones than those just discussed above, are necrotic cells and their debris. When cells die by necrosis they stimulate a robust acute inflammatory response in vivo (27, 28). In fact the occurrence and progression of the ensuing inflammation is so stereotypical that it can be used forensically to date the time of a tissue insult, e.g. in a heart attack (29). This inflammatory response is seen irrespective of the specific cause of cell injury and, of importance to this review, is seen in situations where cell death is caused by sterile insults e.g. from ischemia or toxins.
If cells are dying from infection, the acute inflammatory response may be beneficial in containing this process, as discussed above. However, in situations of sterile cell death, the inflammatory response and particularly the infiltration of tissues with neutrophils can increase the amount injury, similarly to what was discussed above for diseases caused by sterile particles. This has been shown in ischemic or toxic damage to the heart (30), lung (31), liver (32), brain (33), and kidney (34). In all these situations, depleting neutrophils with antibodies (30–32) or by blocking the signals that lead to their recruitment (33) (34) reduces the amount of tissue injury.
How does a cell, which is not inflammatory when alive, become proinflammatory after death? This is incompletely understood and there may be multiple different mechanisms. One of the presently favored models is that after dying, cells release or expose proinflammatory molecules (also called proinflammatory damage-associated molecular patterns or “DAMPs”) that are normally intracellular and “hidden” by the plasma membrane (35, 36). Indeed one of the common events when cells undergo necrosis from whatever cause is a loss of integrity of the plasma membrane. Consistent with this idea, simply rupturing the plasma membrane by freeze-thawing or mechanical stress makes cells proinflammatory in vivo (28). Similarly, injecting cytoplasm from healthy cells into mice induces inflammation (37). Such findings have led to the hypothesis that the innate immune system has evolved mechanisms to sense cell injury by detecting the presence of a subset of molecules that are only exposed after death.
The identities of only a few proinflammatory DAMPs are known and it is likely that there are others yet to be discovered. Interestingly, there is emerging evidence that one of these is uric acid (Kono manuscript in preparation), the same molecule responsible for the inflammatory disease is gout (see above), and this may in part account for some of the similarities between inflammation to sterile particles and dead cells. Cells contain very high levels of uric acid from purine catabolism and can even continue to generate it after death (Kono et al., manuscript in preparation). In addition to uric acid there are other known DAMPs. DNA and probably more specifically unmethylated CpG rich DNA regions may also contribute to death-induced inflammation (38). In addition, a few intracellular molecules have also been found to have intrinsic proinflammatory activity including HMGB1 (39), SAP130 (40), IL-1α (37), IL-33 (41), DNA (42, 43), S100 proteins (44), heat shock proteins (45, 46) and others (47). HMGB1 is normally a chromatin-associated protein that is released from necrotic cells and sometimes from living cells by a non-classical secretion mechanism (48). SAP130 is a protein found in the U2 small nuclear ribonucleoprotein–associated protein complex, which is associated with the splicesome (49). Other intracellular proteins may not be proinflammatory directly but work by generating other bioactive mediators. This has been reported for non-muscle myosin heavy chain that when released into the extracellular fluids binds a natural (preexisting) anti-myosin IgM antibody leading to the activation of complement and the production of proinflammatory complement split products (50) (51). Similarly, released cellular proteases can cleave extracellular matrix components into bioactive fragments (52) . It is also possible that the particulate nature of dead cells contributes to triggering inflammation (see below). For a more complete discussion of proinflammatory DAMPs interested readers are referred to recent reviews (47) (53).
The role of IL-1 in the neutrophilic inflammatory response to sterile particulates and cell death
Until recently much more was known about how innate immunity senses infection than sterile particles and dead cells. In infections, the innate immune system uses Toll-like receptors (TLRs) and other receptors to sense many extracellular microbes and their products and trigger an inflammatory response. While TLRs typically recognize unique microbial (“non-self”) molecules (AKA pathogen-associated molecular patterns or PAMPs), there are also examples where these receptors recognize mammalian molecules. Given this and the fact that microbes and sterile particles/dead cells both trigger the same acute inflammatory response, it was investigated whether TLRs might be involved in detecting and responding to sterile proinflammatory particulates such as MSU and dead cells. While initial experiments suggested a potential role for TLR2 and TLR4 in the responses to MSU (54), subsequent studies found no role for these or other individual TLRs (22). Similarly, mice deficient in TLR2+4 showed only a minor reduction in the inflammatory response to sterile dead cells and animals deficient in other individual TLR had normal responses (although not all individual or combinations of TLRs were examined) (28). There is also some evidence for a role of TLR3 or TLR9 in sensing cell injury in gut ischemia (55) or in the liver toxicity (38), respectively.
Although, it currently appears that TLRs do not play a major role in triggering the inflammatory responses to at least some sterile particles and dead cells, the investigations into this issue led to an important insight into these responses. It was found that mice that lacked the adaptor protein MyD88, which is needed for signal transduction by most TLRs, generated almost no neutrophilic inflammation to either MSU or dead cells (22, 28). This led to an evaluation of the two other MyD88-dependent cellular receptors, the IL-1 and IL-18 receptors. These studies revealed that mice deficient in the IL-1 receptor (IL-1R) were similar to those lacking MyD88 and generated almost no neutrophilic inflammation to MSU (22) or dead cells (28). Thus investigations into the requirement for MyD88 led to the recognition of a key role for the IL-1 pathway in the neutrophilic inflammatory response to dead cells and MSU.
Subsequent studies have found a similar requirement for the IL-1 pathway in the generation of neutrophilic inflammation to a diverse set of sterile particulate stimuli. This has been seen in vivo for silica dioxide crystals (56, 57), alum (24, 58–60), asbestos (57), and cholesterol crystals (Duewell et al., manuscript in preparation). In contrast, IL-1 is not required for neutrophilic inflammation to at least some microbial stimuli. Thus IL-1R-deficient mice respond normally to the yeast cell wall preparation zymosan (28) or muramyl dipeptide (61). Interestingly, although IL-1R mutant mice had little neutrophilic inflammation to sterile stimuli, the subsequent recruitment of monocytes was less reduced or unaffected (28). This difference in IL-1 dependence suggests that different signals were involved in the recruitment of the two different leukocytes. Together these results demonstrated that the IL-1 pathway plays a key role in the neutrophilic inflammation to diverse sterile stimuli including a variety of irritant particles and dead cells.
A brief primer on the IL-1 pathway
Since the IL-1 pathway plays a critical role in the sterile neutrophilic inflammatory response and will be discussed further below, it is important to briefly review some of the essential features of this cytokine and its receptor. There are three major forms of IL-1: IL-1α, IL-1β, and IL-1ra, each of which is encoded by a separate but related gene. The mature forms of IL-1α and IL-1β both bind to and stimulate the same receptor, IL-1R1 (referred to here as the IL-1R), while IL-1ra is a competitive antagonist for this receptor. There are also other IL-1-related cytokine genes that utilize other receptors (e.g. IL-18, IL-33), but there are as of yet few studies to investigate whether they are involved in the sterile inflammatory response (41, 62, 63).
IL-1β is a potent multifunctional proinflammatory cytokine, whose activity is controlled at the transcriptional, translational, maturation and secretion levels (64). There are many cell stimuli (including TLR-activators, TNFα and IL-1 or IL18 (65, 66)), which activate the transcription of the IL-1β gene. While most inflammatory cytokines are mainly transcriptionally regulated and contain a leader sequence resulting in their release via the secretory pathway after translation and transport into the endoplasmic reticulum, the regulation and release of IL-1β is more complex. IL-1β is first produced in the cytosol as a biologically inactive pro-form (pro-IL-1β), which requires proteolytic cleavage for its activation and release from cells. The cleavage of pro-IL-1β to mature IL-1β is catalyzed by the cysteine protease caspase-1 (formerly known as IL-1β converting enzyme, ICE) (67). While intracellular cleavage of pro-IL-1β is under control of caspase-1, pro-IL-1β can also be released from cells and cleaved extracellularly by other proteases into mature IL-1 (68) (69).
In resting cells caspase-1 itself is present as a zymogen, pro-caspase-1, and its conversion to mature and active caspase-1 is under the control of members of the Nod-like receptor (NLR) family. In 2002, an NLR-containing multi-protein complex was isolated that controls the cleavage of pro-caspase-1 into the catalytically active form (70). In analogy to the apoptosome, which controls the activation of apoptosis-inducing caspases, the NLR protein complex that controls the activity of the inflammatory caspase-1 was termed “inflammasome” (70). Activation of inflammasomes cleaves caspase-1, which then hydrolyzes pro-IL-1β into its mature and active form.
Since the original description of the inflammasome, many more of these complexes have been identified and these all differ from one another in their NLR subunits. There are inflammasomes containing the NLRP1, NLRP3, NLRC4 and AIM-2 NLR proteins, and since there are many more NLR proteins there may be other complexes yet to be discovered (71). NLRP1, NLRC4 and AIM-2 inflammasomes are thought to be mainly involved in recognizing microbial products, while as it will be discussed below, the NLRP3 complex is the one implicated in responses to a number of sterile stimuli. The NLRP3 protein contains a C-terminal leucine-rich repeat (LRR)-rich domain, followed by a central nucleotide-binding NACHT oligomerization domain, and an N-terminal protein–protein interaction pyrin domain (PYD) (72). Upon activation NLRP3 interacts with the adaptor molecule called apoptosis-associated speck-like protein (ASC; also termed pycard or TMS1), and induces changes in its aggregation status (73). ASC serves as an adaptor protein, linking NLRP3 to caspase-1. After ASC interacts with NLRP3 via PYD domain interactions it associates via its C-terminal CARD domain with the CARD domain of pro-caspase-1. Induced close association of pro-caspase-1 is believed to provoke its self-cleavage into active caspase-1. Active caspase-1, in turn, can cleave pro-IL-1β into mature IL-1β (74). ASC also interacts with other NLR proteins and is similarly critical for caspase-1 activation in response to a broad range of stimuli, although there are some differences in the nature of the intermolecular interactions. NLRC4 (also called IPAF) recognizes bacterial flagellin and with ASC forms an inflammasome (75). In contrast to NRLP3, NLRC4 lacks a PYD domain and instead contains a N-terminal CARD domain, which can directly associate with the CARD domain of pro-caspase-1. NALP1 is the only NALP family member that contains both a PYD domain and a CARD domain, the latter of which can recruit caspase-5 (76). Another inflammasome-forming protein, AIM-2 is a bipartite protein consisting of a DNA binding domain (HIN200 domain) and a pyrin domain, which upon activation by intracellular double stranded DNA forms an inflammasome with ASC to activate pro-caspase-1 (77–79).
The molecular mechanisms that control the inflammasome protein assembly are not fully understood. It is believed that the sensor NLR, for example NALP3, is held in an inactive conformation in the absence of stimuli. In this conformation self-interaction via the NACHT domains is prevented. However, stimulation of the NLR is thought to induce a change in its conformation, leading to homoassociation and inflammasome assembly with subsequent caspase-1 activation. Indeed, several activating mutations in NLRs have been identified as the cause for a number of auto-inflammatory hereditary periodic fever syndromes, which are diseases characterized by spontaneous attacks of systemic inflammation, severe local inflammation and episodes of fever (80). Many mutations within NALP3 have been described in patients with the auto-inflammatory diseases Muckle-Wells syndrome (MWS), familial cold autoinflammatory syndrome (FCAS) and chronic neurologic cutaneous and articular syndrome (CINCA) (80). These mutations can induce the spontaneous assembly of the NALP3 inflammasome with concomitant constitutive release of IL-1β or these mutations influence NALP3 to respond to stimuli with a lower threshold than the wild-type protein (81). The stimuli that lead to the activation of NLRP3 and potential mechanisms of how NLRP3 stimuli induce inflammasome assembly are discussed below.
The production of IL-1α has both similarities as well as differences from that of IL-1β. IL-1α can be produced by a broader range of cells and low levels may be expressed in some tissues without overt stimulation. Stimulation of these cells activates transcription of the IL-1α gene. Like IL-1β, the IL-1α protein is initially synthesized as a longer pro-form, which can be cleaved by intracellular proteases into the mature cytokine, and is also released from cells by a non-classical secretion mechanism. However, in contrast to IL-1β, this pro-form is biologically active and a portion of it is also expressed on the plasma membrane (82) (83). Furthermore, the proteases involved in cleaving pro- IL-1α into IL-1α may be different than for the processing of pro-IL-1β, although this is less well understood (84, 85). This step may not require the inflammasome, although reduced IL-1α production has been reported from caspase I-deficient cells (74) and physical interactions between pro- IL-1α and caspase-1 have been reported (86)
IL-1 has long been known to be a proinflammatory cytokine. In fact it was discovered based on its bioactivity in promoting fever (65, 87). It was soon appreciated that in addition to inducing fever, IL-1 had a spectrum of other proinflammatory effects that were largely similar to those induced by TNF. Injection of IL-1 by itself is sufficient to induce inflammation and patients with mutations in inflammasome components that lead to overproduction of IL-1β develop autoinflammatory diseases (80). However, in many settings of inflammation, IL-1 is just one of the many cytokines produced and probably often not the dominant one driving the responses; presumably because of this, neutralizing IL-1 (e.g. with IL-1ra), has incomplete or limited efficacy in reducing inflammation in many situations (often less effect than blocking other cytokines such as TNF). In contrast, it appears to play a much more central role in driving the neutrophilic inflammation to cell death and sterile irritant particles, and this had not been appreciated before the studies described above (28, 37).
The form and sources of IL-1 in sterile inflammation
While a role for the IL-1R has been shown in the sterile neutrophilic inflammatory response in vivo to a large group of sterile particulates and dead cells (see above), reciprocal experiments to examine the role of IL-1 in animals have been reported for only a few of these stimuli. Antibody neutralization of IL-1 inhibits the neutrophilic inflammatory response in mice to dead cells (28) and MSU (22). Similarly, the IL-1R antagonist IL-1ra was reported to ameliorate MSU-induced inflammatory symptoms in a small study of human patients with gout (88). Given these findings and the observations that other sterile particulates induce IL-1 (56, 57), it is almost certain IL-1 is driving the neutrophilic inflammation to all of the IL-1R-dependent, sterile proinflammatory particulates.
Information about the contribution of the different forms of IL-1 (i.e. IL-1α versus IL-1β) in these sterile inflammatory responses is even more limited. For dead cells, neutralizing anti- IL-1α antibodies significantly inhibited the neutrophilic inflammatory response, indicating an important role for IL-1α. In contrast, anti-IL-1β antibodies did not inhibit responses (28); however, this negative result does not rule out a role for IL-1β because complete blockade with antibodies is difficult to achieve and small amounts of residual IL-1β can be sufficient to cause biological effects. In fact, more recent studies using IL-1-mutant mice have shown a role for both IL-1α and IL-1β (HK, manuscript in preparation). Studies have not yet been performed to evaluate the relative roles of IL-1α and IL-1β in the inflammatory response to the other sterile particulates. However, in responses to MSU (23) cholesterol crystal (Duewell et. al. manuscript in preparation) silica (89) and amyloid β (26), mice lacking components of the inflammasome show decreased inflammation in vivo, which suggests a role for IL-1β (although these results do not rule out a contribution from IL-1α). There may also be caspase I-independent pathways of inflammation (90) (28) (91, 92) (93)(H.K. and K.L.R unpublished).
Since IL-1 is critical in these responses, where is it coming from (Fig. 1)? This is an important question because it gets at the fundamental issue of how cell death is sensed by the innate immune system. In the case of dead cells this cytokine could, on the one hand, be generated by host cells. Alternatively, since cells can contain an intracellular pool of IL-1 molecules, the IL-1 could come from the dead cell itself, released when the cells become necrotic. Supporting this latter idea, injection of necrotic dendritic cells (wild type) stimulated inflammation in vivo, while necrotic IL-1α -deficient dendritic cells did not (37). However, this is unlikely to be the whole story. Necrotic IL-1-deficient and wild type cells of other tissues (e.g. brain, liver) stimulate equivalent inflammation (H.K., D.K. and K.L.R unpublished). In addition, for sterile particles other than dead cells, the IL-1 must obviously come from cells in the host. Newly emerging data suggest that the innate immune system of the host also recognizes dead cells and in response produces IL-1. In these experiments (H.K. and K.L.R unpublished), inflammation to necrotic wild type cells is markedly decreased in IL-1-deficient mice. Therefore the contribution of IL-1 from the dead cell versus the host may depend on the particular kind or state of the cell that is dying or other factors.
Figure 1.
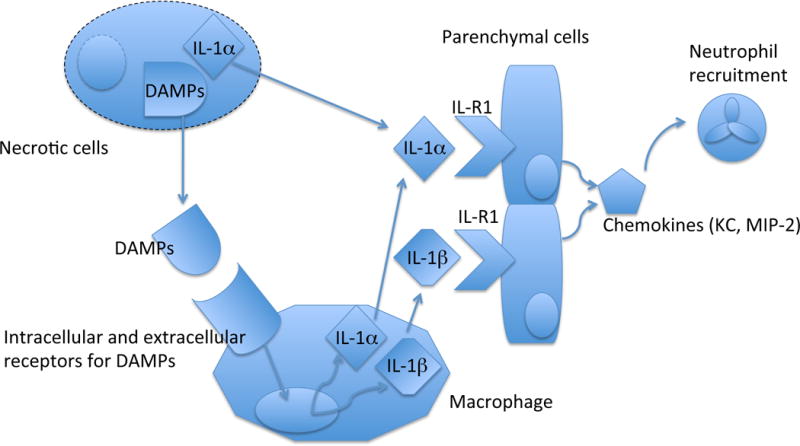
Source and role of IL-1 in recruiting neutrophils to sterile cell death.
IL-1α in necrotic cells can be released and directly activate parenchmal (radioresistant) cells. In addition, necrotic cells release DAMPs (damage associated molecular patterns) that are recognized by macrophages, which in turn produce IL-1α and IL-1β to stimulate parenchymal cells. IL-1-stimulated parenchymal cells secrete chemokines to recruit neutrophils.
In situations where the host is making IL-1, what is the cellular source of this cytokine? For dead cells, depletion of CD11b+ cells (which include macrophages) in vivo inhibits the inflammatory response and, importantly, the response can be reconstituted by transfer of wild type but not IL-1-deficient macrophages (H.K. and K.L.R unpublished). Therefore, macrophages are at least one important cell in producing IL-1 in response to dead cells. The source of IL-1 that drives the acute neutrophilic inflammatory response to sterile particulates in vivo has not been elucidated. However, in vitro LPS-primed macrophages have been shown to produce IL-1β in response to MSU, silica, alum, cholesterol crystals and asbestos (56) (Duewell et. al. manuscript in preparation). Another cell type of the macrophage lineage, microglia, produce IL-1β when stimulated with amyloid β aggregates (26). MSU has also been shown to stimulate IL-1 production from neutrophils (94, 95) synovial exudate cells and human monocytes (96, 97); therefore it is not unlikely that macrophages will be an important source of IL-1 that drives neutrophilic inflammation to many of the irritant particles.
The role of NLRP3 in the generation of IL-1β to sterile particles
How do the various sterile proinflammatory particles stimulate the production of IL-1 that drives the acute neutrophilic inflammatory response? A key observation in beginning to understand this problem was the discovery by Tschopp and his colleagues that the NOD-like receptor NLRP3 (NALP3) and its associated inflammasome components were required for LPS-primed macrophages to produce mature IL-1β when stimulated by MSU crystals (23) (Fig. 2). Subsequent studies by a number of groups found that NLRP3-deficient macrophages failed to produce mature IL-1β after stimulation with a variety of sterile particulate particles including silica (56, 57), alum (24, 56, 58, 59, 98), asbestos (57), and cholesterol crystals (Duewell et. al. manuscript in preparation). Similarly NLRP3 was required for microglia to produce mature IL-1β when stimulated with beta amyloid aggregates (26). Therefore, NLRP3 is a key component in the pathway by which IL-1 is generated in response to these various sterile particles.
Figure 2.
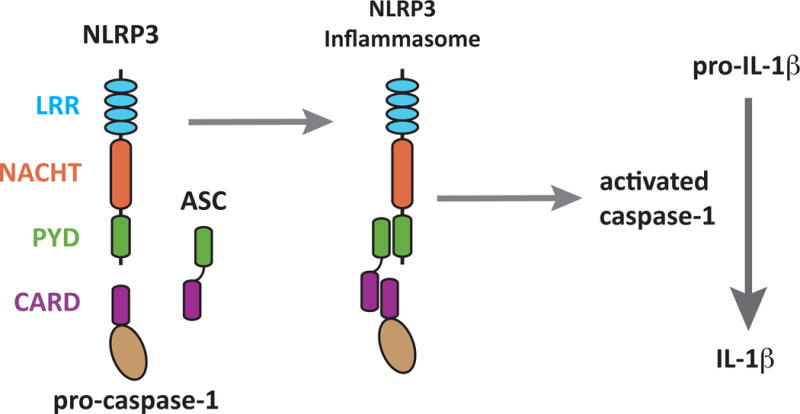
The NLRP3 Inflammasome.
The NLRP3 inflammasome is a heterotrimeric protein complex composed of NLRP3, the apoptosis associated speck-like protein (ASC) and pro-caspase I. Its NLRP3 subunit is composed of three distinct domains. One of these is a leucine-rich repeat region (LRR) that is thought to be involved in ligand recognition. The two other domains, NACHT and Pyrin-domain (PYD), are involved in protein-protein interactions. The PYD domain of NLRP3 binds to the PYD domain in ASC. Using its CARD domain, ASC recruits pro-caspase 1. This complex assembles in the cytosol of cells. Upon activation it cleaves and releases activated caspase-1, which then cleaves pro-IL-1β into its mature and active form.
These findings led to the question of how NLRP3 was actually working in this pathway. Since it is a leucine-rich repeat (LRR) protein and other LRR repeat proteins (such as TLRs) are involved in ligand recognition, it seemed likely that NLRP3 somehow sensed the presence of the sterile particulates, However, it was unclear how this might be occurring because NLRP3 and the sterile particles are present, at least initially, in distinct locations – the cytosol versus the extracellular fluids. Moreover, the particles that stimulated the NLRP3-dependent response were chemically quite diverse, making it unclear how they might all bind to the same receptor. Insights into this problem came from a series of studies that examined the interaction of particles with macrophages.
It had long been known that particles are avidly internalized into macrophages by phagocytosis and it was found that blocking this process with cytochalasin inhibited the NLRP3-dependent generation of mature IL-1β. Importantly, blocking phagocytosis inhibited IL-1β production stimulated by sterile particles but not by ATP, a soluble stimulator of the NLRP3 pathway (56). These findings did not themselves solve the problem of how NLRP3 might sense the particles, because particles in phagosomes are still segregated from NLRP3 in the cytosol. However, the results pointed to a potentially important role of internalization of particles in NLRP3 stimulation of macrophages and led to further studies of the events occurring in the particle-containing phagosomes.
One of the events that occurs after particles are internalized into phagosomes is the activation of NADPH-oxidase and the generation of reactive oxygen species (ROS). Tschopp found that NLRP3-dependent IL-1β production was blocked in cells that were unable to produce ROS or in which ROS were eliminated with chemical scavengers (57), although this was not seen in another system (56). This has raised the possibility that NLRP3 senses ROS or a ROS-dependent event. However, the precise connection between ROS and NLRP3 stimulation is not presently known.
Another study followed the fate of particles after internalization into phagosomes. It was found that surprisingly some of the internalized particles were not in membrane bound vesicles but instead free in the cytosol, presumably due to rupture of phagosomes. This finding raised the possibility that NLRP3 might be able to recognize the free particles directly. However, an alternative possibility was that NLRP3 might somehow sense the rupture of the phagosomes. In support of this latter mechanism it was found that experimentally rupturing endocytic vesicles that did not contain particles, e.g. by hypertonic lysis of pinosomes, stimulated the generation of mature IL-1β and importantly this was dependent on NLRP3 (56). Thus, sterile particles cause rupture of vesicles and such rupture can be sufficient to trigger the NLRP3 inflammasome. Whether the role of ROS generation in NLRP3 activation (see above) is through this mechanism (i.e. by damaging the phagosomal membrane and promoting rupture) or independent of it, remains to be determined. In any case these findings have led to the hypothesis that one of the things that NLRP3 senses is internal cell damage and specifically rupture of endocytic vesicles. This model is attractive because it can easily explain how structurally dissimilar particles can all lead stimulate NLRP3.
How might NLRP3 sense vesicular rupture? Another event that occurs after phagosomes form is that these vesicles acidify. It was found that blocking this acidification inhibited NLRP3 activation by particles but not by a soluble stimulus such as ATP (56, 99). Why might acidification be required for this process? One of the effects of vesicular acidification is to activate acid optimal proteases in the vacuole. Therefore one possibility was that activated vacuolar proteases might be involved in NLRP3 activation. Consistent with this hypothesis, macrophages in which cathepsin B was inhibited or genetically absent showed a partial but significant reduction in IL-1β production to particles but not to ATP(56, 99). There is emerging data that cathepsin L may also contribute to this response (Duewell et al. manuscript in preparation). These findings have suggested a model (Fig. 3) wherein activated cathepsins cleave a substrate, either in the vacuole or the cytosol, and in so doing generate a ligand for NLRP3.
Figure 3.
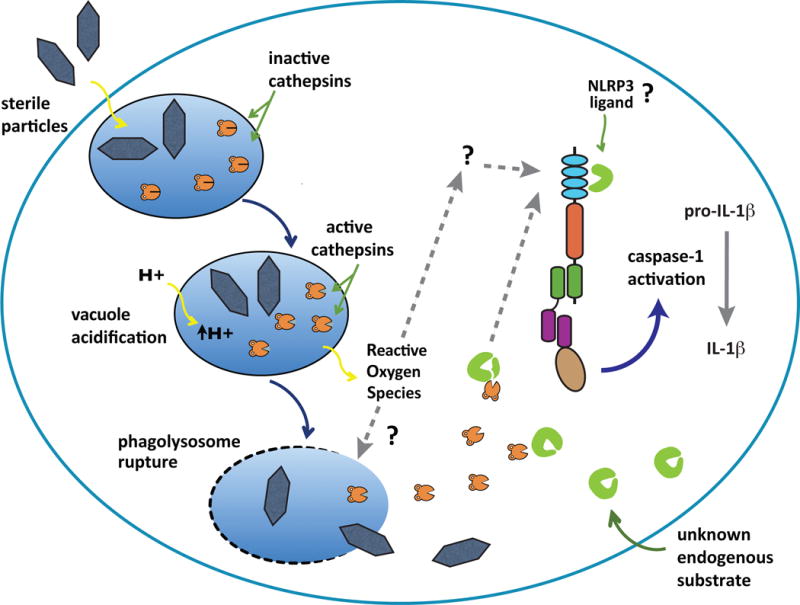
Phagolysosome disruption and activation of the inflammasome.
Phagosomes containing sterile particles acidify, generate reactive oxygen species (ROS) and then some of these vesicles rupture. The ROS that are produced have been implicated in the activation of NLRP3 inflammasomes through an unknown mechanism, possibly involving phagosomal destabilization or direct stimulation of NLRP3. The rupture of phagosomes releases activated cathepsins that trigger the NLRP3 inflammasome, possibly by cleaving an endogenous NLRP3 ligand into an active form. Dashed lines and question marks are hypothetical steps in this pathway.
The experiments described in this section have all been performed with macrophages in cell culture. Do these same mechanisms operate in animals during a sterile inflammatory response? There is limited data that sterile inflammation stimulated by MSU (23), silica (89), cholesterol (Duewell et al. manuscript in preparation) and dead cells (38) is reduced in NLRP3-mutant mice. However, in the NLRP3-negative animals, responses are often only partially reduced and in some cases seem largely independent of the inflammasome components (C.J. H.K. and K.L.R. unpublished). Other cellular processes such as autophagy may also be involved in IL-1 responses. Mice deficient in the autophagy protein Atg16L1 are susceptible to dextran sulphate sodium-induced colitis that is at least partially dependent on IL-1β. In this system, enhanced secretion of IL-1β is dependent on endotoxin priming. ((100)). Therefore it is possible that the mechanisms operating in vivo are more complicated and may involve redundant pathways.
Priming regulates the NLRP3 inflammasome activity
There are other interesting differences between the conditions needed to stimulate NLRP3-dependent responses in vivo versus in cell culture. In intact animals, NLRP3 stimuli (e.g. crystals, ATP or pore-forming toxins) will elicit a response by themselves but are insufficient to stimulate macrophages in culture unless these cells have first received a distinct “priming” stimulus. Of note, this priming is not only required for the induction of pro-IL-1 (as mentioned above), but also for the activation of caspase-1 (101). This indicates that priming induces factors upstream of caspase-1 that are limiting for NLRP3 activation in macrophages. One of these factors may be NLRP3 itself as a recent study revealed that NLRP3 is present at very low levels in resting macrophages and that “priming” stimuli leads to significant upregulation of NLRP3 in an NF-kB-dependent pathway (101). Importantly, heterologous expression of NLRP3 in macrophages was sufficient for caspase-1 activation in response to crystals, ATP or pore-forming toxins in the absence of prior priming (101).
A key role for NLRP3 levels in regulating the activity of the inflammasome pathway is also suggested by clinical data. Certain patients with autoinflammatory syndromes (cryopyrinopathies) have mutations in the coding sequence of NLRP3 and it is thought that these mutations can lead to overly active NLRP3, resulting in the inflammatory phenotype. However, a substantial fraction (~40%) of cryopyrinopathy patients have classical clinical symptoms in the absence of any mutations in the coding region of NLRP3 (80). A recent promoter analysis of NLRP3 identified various unique sequence variants in the promoter region of NLRP3 of cryopyrinopathy patients suggesting that promoter SNPs could lead to dysregulated NLRP3 expression (102). Indeed, a unique SNP (-1064T) in the NLRP3 promoter led to increased promoter activity and therefore dysregulation of NLRP3 expression levels could be the cause for the clinical inflammatory symptoms found in these patients.
These data suggest that, in normal individuals, macrophages only commit to NLRP3 inflammasome activation and concomitant caspase I-dependent (pyroptotic) cell death after they were primed by molecules present during infection (i.e., activator of PRRs), or by encountering potent pro-inflammatory cytokines such as TNFα (66). These findings further imply that many of the reported NLRP3 inflammasome activators (such as LPS, lipopeptides, or imiquimod/resiquimod, etc) that act together with ATP to induce NLRP3 inflammasome activation are not inflammasome activators per se. Instead these pro-inflammatory pattern recognition receptor ligands are necessary for priming and assembly of the NLRP3 inflammasome rather than being bona fide inflammasome activators.
The requirement of two or more independent signals for triggering cellular responses is a recurrent theme in immune cell activation. In macrophages, these mechanisms may operate to prevent uncontrolled NLRP3 activation. Unrestricted NLRP3 inflammasome activation could lead to the excessive production of pro-inflammatory IL-1β cytokine family members and could additionally induce pyroptotic cell death and the release of a range of additional caspase-1 regulated factors other than IL-1β (103). The regulation of the NLRP3 inflammasome via NF-kB activity provides a secure mechanism of control, because the activity of NF-kB itself is controlled by an elaborate set of molecules and by finely regulated feedback mechanisms (104).
How the inflammasome is primed in vivo remains to be fully determined. Recent reports (38, 105), however, support the hypothesis that priming via a pattern recognition receptor might also play a fundamental role for the recognition of danger signals in vivo. It was demonstrated that induction of sterile inflammation by pharmacologically induced liver damage was dependent on both TLR9 and NLRP3. Studies with TLR9 inhibitors as well as with TLR9 deficient mice suggested that TLR9 activation by excessively released host DNA provides a priming step for pro-IL-1β synthesis (38). Thus, it appears likely that TLR9 could also play a role in NLRP3 priming in vivo allowing NLRP3 inflammasome activation by released danger signals, at least in some situations. Similar mechanisms could operate during crystal induced NLRP3 activation in vivo, however, these hypotheses have yet to be tested.
Other signaling pathways stimulated by sterile stimuli
Particles and dead cells also stimulate macrophages in other ways besides the NLRP3 pathway described above, although the precise receptors and pathways are poorly understood overall. Most of the information on these processes is for MSU crystals because these have been the most studied sterile proinflammatory particles. The activation of a number of kinases has been described in different cell types. Stimulation of macrophages and neutrophils with MSU leads to the activation of ERK1/2 (106) and of TEC (107), respectively. Exactly how MSU leads to the activation of these kinases is not clear. This may be mediated by membrane receptors. MSU has been reported to stimulate TLR 2 and 4 ((54)), although the inflammatory response is not diminished in TLR 2+4 double mutant animals ((28)). It has also been suggested that blockade of the surface markers CD16 and CD11b (108) or deficiency in CD14 (109) can reduce or prevent MSU from inducing an inflammatory response. More recently, a receptor independent pathway of kinase activation has been described ((110)). MSU crystals were shown to bind with high affinity to cholesterol in the plasma membrane and in so doing cause aggregation of lipid rafts. This clustering of lipid rafts, which contain a number of kinases, led to the activation of the syk kinase.
The precise role of these additional signaling events in the generation of the sterile inflammatory response is not clear. It is possible that they contribute to some of the events reviewed above such as stimulating phagocytosis, triggering the oxidative burst, or priming of macrophages to produce pro-IL-1β. Alternatively, there might be many other inflammatory mediators that are generated in response to the sterile particulates and these involve signaling pathways distinct from the ones needed to generate IL-1. Unfortunately, at this point in time there is not sufficient information available to understand all the various signaling pathways and their role in the sterile inflammatory response.
The target of IL-1 in the neutrophilic inflammatory response to sterile particles and dead cells
Since IL-1 is required for the neutrophilic inflammatory response to sterile particulates and dead cells, where is it acting? The IL-1R is expressed broadly on many tissues (111 and therefore IL-1 could affect a large number of cell types. There is at present very limited information about which of these cells is the key target(s) in IL-1-stimulated neutrophil recruitment. Experiments have been performed with radiation bone marrow chimeras to address whether the IL-1R is needed on cells of hematopoietic origin, such as leukocytes, and/or on ones of non-hematopoietic origin (referred to herein as parenchymal cells). Chimeric mice in which the IL-1R is lacking from all bone marrow-derived elements, but not other cells showed no impairment in their neutrophilic inflammatory responses to either sterile particles (MSU) {Chen, 2006 #6) or dead cells ((28)). In contrast, chimeras that expressed the IL-1R on bone marrow elements, but lacked it on all other cells, showed reduced sterile neutrophilic inflammation to both particles ((22)) and dead cells ((28)). Therefore, a key target(s) of IL-1 in the sterile inflammatory response to both MSU and dead cells is a radioresistant parenchymal cell(s). Presumably these same mechanisms are also operative in the response to other sterile particulates, but this has not yet been formally tested.
The identity of the key parenchymal target of IL-1 in sterile inflammation in vivo is not known. However an in vitro study found that IL-1α stimulated cultured peritoneal mesothelial cells to produce the neutrophil chemotactic chemokines KC and MIP-2 (112)(37)). Based on these findings it was proposed that when dead cells are injected into the peritoneum, IL-1α is released and acts on peritoneal mesothelial cells to produce chemokines that then recruit neutrophils. Whether these mesothelial cell responses actually occur and/or are required in vivo and, if so, whether they are the only target of IL-1 that is required is not yet known. However, in other settings there must be other essential cellular targets because necrosis-induced neutrophilic inflammation is seen in tissue sites where mesothelium is not present, e.g. in the parenchyma of organs.
Therapeutic implications
Since the inflammation stimulated by sterile particles and dead cells contributes to the pathogenesis of a number of diseases it would be medically useful to develop therapeutics to inhibit this process. It would be ideal if this could be done in ways that would block the damaging component of the sterile inflammation without compromising host defense to e.g. infection. The findings reviewed in earlier sections raise the possibility that such selective inhibition could be obtained and point to some potential molecular targets that might allow this to be achieved.
Blocking IL-1 production or action would be potential therapeutic targets to inhibit sterile inflammation because IL-1 is required for the neutrophil recruitment in this setting and this cell type is the most damaging component of the response. One such inhibitor is already available clinically (IL-1ra) (113) and others are under development (64) There are at least two reasons why these targets might give some relative selectivity. First it is clear that animals that lack the IL-1 pathway can still mount neutrophilic inflammation to at least some microbial components (22, 28). Second, the monocytes-macrophage component of the acute inflammatory response is much less dependent on IL-1 ((28)). Therefore, important components of the innate immune response to infection may still be intact after the IL-1 pathway is blocked. Supporting this idea, patients that are treated with IL-1ra have only a very small increase in the incidence of infections (113)((88, 114); although it remains to be seen if this becomes a greater problem with agents that may block IL-1 responses more effectively than IL-1ra. However, blocking IL-1 itself will affect many other responses than just those stimulated by dead cells and sterile particulates.
Further selectivity might be achieved by blocking steps in the pathway that are more restricted or unique to the production of IL-1 to sterile particles and dead cells. NLRP3 might be one such target. Moreover, it is possible that the upstream mechanisms by which sterile particles trigger NLRP3 would be even more selective ones. Based on the emerging data about this process such targets might include phagosomal rupture, specific cathepsins and/or the putative endogenous ligand of NLRP3; as far as is known, these steps function uniquely in the sterile inflammatory response and not in host defense to microbes.
It is presently unclear whether the overall concept and the specific molecular targets considered above would truly be efficacious in treating many of the sterile inflammatory diseases. Similarly, it is not known whether and/or how much selectivity these targets would provide and whether they would be tractable for drug design. Nevertheless is intriguing that there is limited data that inhibition of the IL-1 pathway may be effective in blocking sterile inflammation in gout (88). If this holds up and is found to be true in other of the sterile inflammatory diseases then there would be a strong rationale to try and develop additional and selective inhibitors of this pathway.
Other sterile inflammation
This review has focused on a small subset of the sterile stimuli that can cause inflammation: cell death and irritant particles. Sterile inflammation also occurs in response to trauma, immunogenic antigens, and autoimmune conditions, to name just a few other examples. In these settings, whether and to what extent the inflammation is stimulated by similar IL-1-inflammasome-dependent mechanisms is mostly unknown at this time. However, in a number of situations where the IL-1 pathway is lacking genetically or has been neutralized, sterile inflammatory responses still occur (115) (116–118) (28, 119). It is also clear that molecules like zymosan that engage scavenger receptors and TLRs can stimulate acute neutrophilic inflammation in the absence of the IL-1 pathway. Therefore, while there may be a common molecular pathway underlying the inflammation to dead cells and particles, it seems likely that for many other sterile stimuli there will be additional and/or distinct molecular mechanisms that underlie the sterile inflammatory response.
Conclusion
There has been major progress in understanding the mechanisms underlying the generation of the neutrophilic inflammatory response in response to sterile irritant particles and dead cells. An important advance was the finding that all of these diverse stimuli elicit this response through the same key mediator, IL-1. Surprisingly, emerging data further suggested that there might be common mechanisms through which these diverse stimuli lead to IL-1 production. The sterile particulates all stimulate the NLRP3 inflammasome, which processes pro-IL-1β to the mature and bioactive form. Moreover these particles may all stimulate NLRP3 via the same mechanisms that operate in the phagosomes of macrophages. New potential players that function in this process have been identified, including ROS and cathepsins (56, 57, 99). These findings are opening up the possibility of developing new therapies and therapeutics to treat diseases that are caused by sterile inflammation.
Nevertheless, many of these new insights need further validation, particularly in vivo, and there are many missing pieces to the puzzle. For example, in the IL-1 pathway it is unclear how phagocytes are “primed” to make pro-IL-1β, how ROS and cathepsins function, what causes phagosomal rupture, what are the roles and regulation of IL-1α versus IL-1β, what is the potential redundancy of mechanisms, and what are the key targets of IL-1, to name but a few of the unresolved issues. Beyond this we don’t understand what controls other aspects of the sterile inflammatory response, such as monocyte recruitment, and the role of other receptors and signaling pathways. Given the medical importance of the sterile inflammatory response, these are key issues that need to be addressed. Solving these issues and developing a comprehensive understanding of the mechanisms controlling the sterile inflammatory responses will be important to understanding disease pathogenesis and in devising novel rationale methods to for treatment.
Table 1.
Sterile stimulus and consequences
| Sterile stimulus | Diseases | Organs | Symptoms Or consequences | IL-1 dependence | Inflammaosme dependence | References |
|---|---|---|---|---|---|---|
| Cell death | Many (Ischemia, toxic damage, etc.) | Many | Increased damage Fibrosis | Yes (In vivo and vitro) | Yes (In vivo and vitro) | (27, 28) (30),(31), (32), (33),(34) |
| Monosodium urate crystal | Gout | Joint | Inflammation Chronic arthritis | Yes (In vivo and vitro) | Yes (In vivo and vitro) | (16, 19, 20, 22, 23)Kono, manuscript in preparation |
| Calcium pyrophosphate | Psuedogout | Joint | Inflammation Chronic arthritis | Yes (In vivo and vitro) | Yes (In vivo and vitro) | (15, 21, 23) |
| Silica | Silicosis | Lung | Inflammation Fibrosis | Yes (In vivo and vitro) | Yes (In vivo and vitro) | (13, 56, 89) |
| Iron oxide | Siderosis | Lung | Inflammation Fibrosis | ND | ND | (120) |
| Metal wear particulates | Joint loosening | Joint (implanted) | periprosthetic osteolysis, revision surgery | Yes (In vivo and vitro) | Yes (In vitro) | (121, 122) |
| Asbestos | Asbestosis | Lung | Inflammaiton Fibrosis Mesothelioma Lung Cancer | Yes (In vivo and vitro) | Yes (In vivo and vitro) | (13, 57) |
| Cholesterol crystal | Atherosclerosis | Arteries | Inflammation Occulusion of arteries to ischemia | Yes (In vivo and vitro) | Yes (In vivo and vitro) | (18) Duewell, manuscript in preparation |
| b-amyloid | Arzheimer’s disease | Brain | Neuronal degeneration | Yes (In vivo and vitro) | Yes (In vivo and vitro) | (17, 26) |
| Aluminium salts | Skin Peritoneal cavity | Inflammation Adjuvant activity | Yes (In vivo and vitro) | Yes (In vivo and vitro) | (24, 58, 59, 98, 123) | |
| talcum powder | Pleural cavity | Inflammation pleurodesis | ND | ND | (25) |
Acknowledgments
This work was supported by grants to EL and KLR from the NIH, and to KLR from the American Asthma Foundation (AAF). Core resources supported by the Diabetes Endocrinology Research Center grant DK32520 were also used.
References
- 1.Celsus AC. circa 30 A.D. De Medicina [Google Scholar]
- 2.Rocha e Silva M. A brief survey of the history of inflammation. Agents Actions. 1978;8:45–9. doi: 10.1007/BF01972401. [DOI] [PubMed] [Google Scholar]
- 3.Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 4.Majno G. The Healing Hand : Man and Wound in the Ancient World. Cambridge, Massachusetts: Harvard University Press; 1991. [Google Scholar]
- 5.Waller A. Microscopical observation on the perforation of capillaries by the corpuscles of the blood and on the origin of mucus- and pus globules. (3rd Series).Philosophical Magazine. 1846;29:397–405. [Google Scholar]
- 6.Cohnheim J. Ueber Entzündung und Eiterung. Virchows Arch (Path Anat) 1867;40:1–79. [Google Scholar]
- 7.Majno G, Joris I. Cells, Tissues and Diseases: Principles of general pathology. Cambridge, MA: Oxford University Press; 2004. [Google Scholar]
- 8.Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, Lawyer P, Fay MP, Germain RN, Sacks D. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science. 2008;321:970–4. doi: 10.1126/science.1159194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–35. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 10.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 11.Inohara C, McDonald C, Nunez G. NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu Rev Biochem. 2005;74:355–83. doi: 10.1146/annurev.biochem.74.082803.133347. [DOI] [PubMed] [Google Scholar]
- 12.Boxer L, Dale DC. Neutropenia: causes and consequences. Semin Hematol. 2002;39:75–81. doi: 10.1053/shem.2002.31911. [DOI] [PubMed] [Google Scholar]
- 13.Mossman BT, Churg A. Mechanisms in the pathogenesis of asbestosis and silicosis. Am J Respir Crit Care Med. 1998;157:1666–80. doi: 10.1164/ajrccm.157.5.9707141. [DOI] [PubMed] [Google Scholar]
- 14.Billings CG, Howard P. Occupational siderosis and welders’ lung: a review. Monaldi Arch Chest Dis. 1993;48:304–14. [PubMed] [Google Scholar]
- 15.Ea HK, Liote F. Calcium pyrophosphate dihydrate and basic calcium phosphate crystal-induced arthropathies: update on pathogenesis, clinical features, and therapy. Curr Rheumatol Rep. 2004;6:221–7. doi: 10.1007/s11926-004-0072-6. [DOI] [PubMed] [Google Scholar]
- 16.Nuki G, Simkin PA. A concise history of gout and hyperuricemia and their treatment. Arthritis Res Ther. 2006;8(Suppl 1):S1. doi: 10.1186/ar1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weiner HL, Frenkel D. Immunology and immunotherapy of Alzheimer’s disease. Nat Rev Immunol. 2006;6:404–16. doi: 10.1038/nri1843. [DOI] [PubMed] [Google Scholar]
- 18.Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol. 2009;27:165–97. doi: 10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hippocrates . The Genuine Works of Hippocrates. Volume I and II. New York: William Wood and Company; 1886. [Google Scholar]
- 20.McCarty DJ, Hollander JL. Identification of urate crystals in gouty synovial fluid. Ann Int Med. 1961;54:452. doi: 10.7326/0003-4819-54-3-452. [DOI] [PubMed] [Google Scholar]
- 21.Beck C, Morbach H, Richl P, Stenzel M, Girschick HJ. How can calcium pyrophosphate crystals induce inflammation in hypophosphatasia or chronic inflammatory joint diseases? Rheumatol Int. 2009;29:229–38. doi: 10.1007/s00296-008-0710-9. [DOI] [PubMed] [Google Scholar]
- 22.Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, Akira S, Rock KL. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest. 2006;116:2262–71. doi: 10.1172/JCI28075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 24.Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–6. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van den Heuvel MM, Smit HJ, Barbierato SB, Havenith CE, Beelen RH, Postmus PE. Talc-induced inflammation in the pleural cavity. Eur Respir J. 1998;12:1419–23. doi: 10.1183/09031936.98.12061419. [DOI] [PubMed] [Google Scholar]
- 26.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–65. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Majno G, La Gattuta M, Thompson TE. Cellular death and necrosis: chemical, physical and morphologic changes in rat liver. Virchows Arch Pathol Anat Physiol Klin Med. 1960;333:421–65. doi: 10.1007/BF00955327. [DOI] [PubMed] [Google Scholar]
- 28.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–6. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 29.B E, ZD P, L P, editors. Heart Disease: A Textbook of Cardiovascular Medicine. 6. Philadelphia, PA: WB Saunders; 2001. Acute myocardial infarction; pp. 1114–231. [Google Scholar]
- 30.Romson JL, Hook BG, Kunkel SL, Abrams GD, Schork MA, Lucchesi BR. Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation. 1983;67:1016–23. doi: 10.1161/01.cir.67.5.1016. [DOI] [PubMed] [Google Scholar]
- 31.Kishi M, Richard LF, Webster RO, Dahms TE. Role of neutrophils in xanthine/xanthine oxidase-induced oxidant injury in isolated rabbit lungs. J Appl Physiol. 1999;87:2319–25. doi: 10.1152/jappl.1999.87.6.2319. [DOI] [PubMed] [Google Scholar]
- 32.Liu ZX, Han D, Gunawan B, Kaplowitz N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology. 2006;43:1220–30. doi: 10.1002/hep.21175. [DOI] [PubMed] [Google Scholar]
- 33.Abulafia DP, de Rivero Vaccari JP, Lozano JD, Lotocki G, Keane RW, Dietrich WD. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J Cereb Blood Flow Metab. 2009;29:534–44. doi: 10.1038/jcbfm.2008.143. [DOI] [PubMed] [Google Scholar]
- 34.Kelly KJ, Williams, C Rb, M Sm, S Ta, G-R Jc, B Jv. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. J Clin Invest. 1996;97:1056–63. doi: 10.1172/JCI118498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 36.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–5. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 37.Eigenbrod T, Park JH, Harder J, Iwakura Y, Nunez G. Cutting edge: critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1alpha released from dying cells. J Immunol. 2008;181:8194–8. doi: 10.4049/jimmunol.181.12.8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, Flavell RA, Mehal WZ. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest. 2009 doi: 10.1172/JCI35958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 40.Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, Saito T. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol. 2008;9:1179–88. doi: 10.1038/ni.1651. [DOI] [PubMed] [Google Scholar]
- 41.Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS ONE. 2008;3:e3331. doi: 10.1371/journal.pone.0003331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishii KJ, Suzuki K, Coban C, Takeshita F, Itoh Y, Matoba H, Kohn LD, Klinman DM. Genomic DNA released by dying cells induces the maturation of APCs. J Immunol. 2001;167:2602–7. doi: 10.4049/jimmunol.167.5.2602. [DOI] [PubMed] [Google Scholar]
- 43.Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, Chang B, Duramad O, Coffman RL. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. 2005;202:1131–9. doi: 10.1084/jem.20050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Foell D, Wittkowski H, Vogl T, Roth J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol. 2007;81:28–37. doi: 10.1189/jlb.0306170. [DOI] [PubMed] [Google Scholar]
- 45.Osterloh A, Veit A, Gessner A, Fleischer B, Breloer M. Hsp60-mediated T cell stimulation is independent of TLR4 and IL-12. Int Immunol. 2008;20:433–43. doi: 10.1093/intimm/dxn003. [DOI] [PubMed] [Google Scholar]
- 46.Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001;14:303–13. doi: 10.1016/s1074-7613(01)00111-x. [DOI] [PubMed] [Google Scholar]
- 47.Rock KL, Kono H. The inflammatory response to cell death. Annu Rev Pathol. 2008;3:99–126. doi: 10.1146/annurev.pathmechdis.3.121806.151456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Landsman D, Bustin M. A signature for the HMG-1 box DNA-binding proteins. Bioessays. 1993;15:539–46. doi: 10.1002/bies.950150807. [DOI] [PubMed] [Google Scholar]
- 49.Das BK, Xia L, Palandjian L, Gozani O, Chyung Y, Reed R. Characterization of a protein complex containing spliceosomal proteins SAPs 49, 130, 145, and 155. Mol Cell Biol. 1999;19:6796–802. doi: 10.1128/mcb.19.10.6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weiser MR, Williams JP, Moore, K L, M M, H Hb, C Mc. Reperfusion injury of ischemic skeletal muscle is mediated by natural antibody and complement. J Exp Med. 1996;183:2343–8. doi: 10.1084/jem.183.5.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang M, Alicot EM, Chiu I, Li J, Verna N, Vorup-Jensen T, Kessler B, Shimaoka M, Chan R, Friend D, Mahmood U, Weissleder R, Moore FD, Carroll MC. Identification of the target self-antigens in reperfusion injury. J Exp Med. 2006;203:141–52. doi: 10.1084/jem.20050390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–9. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 53.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–89. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu-Bryan R, Scott P, Sydlaske A, Rose DM, Terkeltaub R. Innate immunity conferred by Toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum. 2005;52:2936–46. doi: 10.1002/art.21238. [DOI] [PubMed] [Google Scholar]
- 55.Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, Hogaboam CM, Kunkel SL. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med. 2008;205:2609–21. doi: 10.1084/jem.20081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–56. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–7. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kool M, Petrilli V, De Smedt T, Rolaz A, Hammad H, van Nimwegen M, Bergen IM, Castillo R, Lambrecht BN, Tschopp J. Cutting edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J Immunol. 2008;181:3755–9. doi: 10.4049/jimmunol.181.6.3755. [DOI] [PubMed] [Google Scholar]
- 59.Franchi L, Nunez G. The Nlrp3 inflammasome is critical for aluminium hydroxide-mediated IL-1beta secretion but dispensable for adjuvant activity. Eur J Immunol. 2008;38:2085–9. doi: 10.1002/eji.200838549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li H, Nookala S, Re F. Aluminum hydroxide adjuvants activate caspase-1 and induce IL-1beta and IL-18 release. J Immunol. 2007;178:5271–6. doi: 10.4049/jimmunol.178.8.5271. [DOI] [PubMed] [Google Scholar]
- 61.Rosenzweig HL, Martin TM, Planck SR, Galster K, Jann MM, Davey MP, Kobayashi K, Flavell RA, Rosenbaum JT. Activation of NOD2 in vivo induces IL-1beta production in the eye via caspase-1 but results in ocular inflammation independently of IL-1 signaling. J Leukoc Biol. 2008;84:529–36. doi: 10.1189/jlb.0108015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leung BP, Culshaw S, Gracie JA, Hunter D, Canetti CA, Campbell C, Cunha F, Liew FY, McInnes IB. A role for IL-18 in neutrophil activation. J Immunol. 2001;167:2879–86. doi: 10.4049/jimmunol.167.5.2879. [DOI] [PubMed] [Google Scholar]
- 63.Li X, Kovacs EJ, Schwacha MG, Chaudry IH, Choudhry MA. Acute alcohol intoxication increases interleukin-18-mediated neutrophil infiltration and lung inflammation following burn injury in rats. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1193–201. doi: 10.1152/ajplung.00408.2006. [DOI] [PubMed] [Google Scholar]
- 64.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 65.Dinarello CA, Ikejima T, Warner SJ, Orencole SF, Lonnemann G, Cannon JG, Libby P. Interleukin 1 induces interleukin 1. I. Induction of circulating interleukin 1 in rabbits in vivo and in human mononuclear cells in vitro. J Immunol. 1987;139:1902–10. [PubMed] [Google Scholar]
- 66.Franchi L, Eigenbrod T, Nunez G. TNF-alpha Mediates Sensitization to ATP and Silica via the NLRP3 Inflammasome in the Absence of Microbial Stimulation. The Journal of Immunology. 2009;183:0000. doi: 10.4049/jimmunol.0900173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356:768–74. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 68.Fantuzzi G, Ku G, Harding MW, Livingston DJ, Sipe JD, Kuida K, Flavell RA, Dinarello CA. Response to local inflammation of IL-1 beta-converting enzyme- deficient mice. J Immunol. 1997;158:1818–24. [PubMed] [Google Scholar]
- 69.Coeshott C, Ohnemus C, Pilyavskaya A, Ross S, Wieczorek M, Kroona H, Leimer AH, Cheronis J. Converting enzyme-independent release of tumor necrosis factor alpha and IL-1beta from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc Natl Acad Sci U S A. 1999;96:6261–6. doi: 10.1073/pnas.96.11.6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 71.Franchi L, Warner N, Viani K, Nunez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev. 2009;227:106–28. doi: 10.1111/j.1600-065X.2008.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29:301–5. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Masumoto J, Taniguchi S, Ayukawa K, Sarvotham H, Kishino T, Niikawa N, Hidaka E, Katsuyama T, Higuchi T, Sagara J. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem. 1999;274:33835–8. doi: 10.1074/jbc.274.48.33835. [DOI] [PubMed] [Google Scholar]
- 74.Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–3. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 75.Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Oz√∂ren N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A, Grant EP, N√∫√±ez G. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol. 2006;7:576–82. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 76.Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–65. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 77.Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL, Superti-Furga G. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009 doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- 78.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009 doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009 doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*) Annu Rev Immunol. 2009;27:621–68. doi: 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20:319–25. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 82.Kurt-Jones EA, Beller DI, Mizel SB, Unanue ER. Identification of a membrane-associated interleukin 1 in macrophages. Proc Natl Acad Sci U S A. 1985;82:1204–8. doi: 10.1073/pnas.82.4.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brody DT, Durum SK. Membrane IL-1: IL-1 alpha precursor binds to the plasma membrane via a lectin-like interaction. J Immunol. 1989;143:1183–7. [PubMed] [Google Scholar]
- 84.Black RA, Kronheim SR, Cantrell M, Deeley MC, March CJ, Prickett KS, Wignall J, Conlon PJ, Cosman D, Hopp TP. Generation of biologically active interleukin-1 beta by proteolytic cleavage of the inactive precursor. J Biol Chem. 1988;263:9437–42. [PubMed] [Google Scholar]
- 85.Hazuda DJ, Strickler J, Kueppers F, Simon PL, Young PR. Processing of precursor interleukin 1 beta and inflammatory disease. J Biol Chem. 1990;265:6318–22. [PubMed] [Google Scholar]
- 86.Keller M, Ruegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–31. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 87.King MK, Wood WB., Jr Studies on the pathogenesis of fever. IV. The site of action of leucocytic and circulating endogenous pyrogen. J Exp Med. 1958;107:291–303. doi: 10.1084/jem.107.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther. 2007;9:R28. doi: 10.1186/ar2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, Carter AB, Rothman PB, Flavell RA, Sutterwala FS. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A. 2008;105:9035–40. doi: 10.1073/pnas.0803933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miwa K, Asano M, Horai R, Iwakura Y, Nagata S, Suda T. Caspase 1-independent IL-1beta release and inflammation induced by the apoptosis inducer Fas ligand. Nat Med. 1998;4:1287–92. doi: 10.1038/3276. [DOI] [PubMed] [Google Scholar]
- 91.Maelfait J, Vercammen E, Janssens S, Schotte P, Haegman M, Magez S, Beyaert R. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1beta maturation by caspase-8. J Exp Med. 2008;205:1967–73. doi: 10.1084/jem.20071632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schonbeck U, Mach F, Libby P. Generation of biologically active IL-1 beta by matrix metalloproteinases: a novel caspase-1-independent pathway of IL-1 beta processing. J Immunol. 1998;161:3340–6. [PubMed] [Google Scholar]
- 93.Mehta VB, Hart J, Wewers MD. ATP-stimulated release of interleukin (IL)-1beta and IL-18 requires priming by lipopolysaccharide and is independent of caspase-1 cleavage. J Biol Chem. 2001;276:3820–6. doi: 10.1074/jbc.M006814200. [DOI] [PubMed] [Google Scholar]
- 94.Roberge CJ, Grassi J, De Medicis R, Frobert Y, Lussier A, Naccache PH, Poubelle PE. Crystal-neutrophil interactions lead to interleukin-1 synthesis. Agents Actions. 1991;34:38–41. doi: 10.1007/BF01993232. [DOI] [PubMed] [Google Scholar]
- 95.Roberge CJ, de Médicis R, Dayer JM, Rola-Pleszczynski M, Naccache PH, Poubelle PE. Crystal-induced neutrophil activation. V. Differential production of biologically active IL-1 and IL-1 receptor antagonist. J Immunol. 1994;152:5485–94. [PubMed] [Google Scholar]
- 96.Di Giovine FS, Malawista SE, Nuki G, Duff GW. Interleukin 1 (IL 1) as a mediator of crystal arthritis. Stimulation of T cell and synovial fibroblast mitogenesis by urate crystal-induced IL 1. J Immunol. 1987;138:3213–8. [PubMed] [Google Scholar]
- 97.di Giovine FS, Malawista SE, Thornton E, Duff GW. Urate crystals stimulate production of tumor necrosis factor alpha from human blood monocytes and synovial cells. Cytokine mRNA and protein kinetics, and cellular distribution. J Clin Invest. 1991;87:1375–81. doi: 10.1172/JCI115142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li H, Willingham SB, Ting JP, Re F. Cutting edge: inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J Immunol. 2008;181:17–21. doi: 10.4049/jimmunol.181.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sharp FA, Ruane D, Claass B, Creagh E, Harris J, Malyala P, Singh M, O’Hagan DT, Petrilli V, Tschopp J, O’Neill LA, Lavelle EC. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Natl Acad Sci U S A. 2009;106:870–5. doi: 10.1073/pnas.0804897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–8. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 101.Bauernfeind F, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemr T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E. NF-kB Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. The Journal of Immunology. 2009;183 doi: 10.4049/jimmunol.0901363. in print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Anderson JP, Mueller JL, Misaghi A, Anderson S, Sivagnanam M, Kolodner RD, Hoffman HM. Initial description of the human NLRP3 promoter. Genes Immun. 2008;9:721–6. doi: 10.1038/gene.2008.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gurcel L, Abrami L, Girardin S, Tschopp J, van der Goot FG. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell. 2006;126:1135–45. doi: 10.1016/j.cell.2006.07.033. [DOI] [PubMed] [Google Scholar]
- 104.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–62. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 105.Giamarellos-Bourboulis EJ, Mouktaroudi M, Bodar E, van der Ven J, Kullberg BJ, Netea MG, van der Meer JW. Crystals of monosodium urate monohydrate enhance lipopolysaccharide-induced release of interleukin 1 beta by mononuclear cells through a caspase 1-mediated process. Ann Rheum Dis. 2009;68:273–8. doi: 10.1136/ard.2007.082222. [DOI] [PubMed] [Google Scholar]
- 106.Jaramillo M, Godbout M, Naccache PH, Olivier M. Signaling events involved in macrophage chemokine expression in response to monosodium urate crystals. J Biol Chem. 2004;279:52797–805. doi: 10.1074/jbc.M403823200. [DOI] [PubMed] [Google Scholar]
- 107.Popa-Nita O, Marois L, Pare G, Naccache PH. Crystal-induced neutrophil activation: X. Proinflammatory role of the tyrosine kinase Tec. Arthritis Rheum. 2008;58:1866–76. doi: 10.1002/art.23801. [DOI] [PubMed] [Google Scholar]
- 108.Barabe F, Gilbert C, Liao N, Bourgoin SG, Naccache PH. Crystal-induced neutrophil activation VI. Involvment of FcgammaRIIIB (CD16) and CD11b in response to inflammatory microcrystals. FASEB J. 1998;12:209–20. doi: 10.1096/fasebj.12.2.209. [DOI] [PubMed] [Google Scholar]
- 109.Scott P, Ma H, Viriyakosol S, Terkeltaub R, Liu-Bryan R. Engagement of CD14 mediates the inflammatory potential of monosodium urate crystals. J Immunol. 2006;177:6370–8. doi: 10.4049/jimmunol.177.9.6370. [DOI] [PubMed] [Google Scholar]
- 110.Ng G, Sharma K, Ward SM, Desrosiers MD, Stephens LA, Schoel WM, Li T, Lowell CA, Ling CC, Amrein MW, Shi Y. Receptor-independent, direct membrane binding leads to cell-surface lipid sorting and Syk kinase activation in dendritic cells. Immunity. 2008;29:807–18. doi: 10.1016/j.immuni.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Deyerle KL, Sims JE, Dower SK, Bothwell MA. Pattern of IL-1 receptor gene expression suggests role in noninflammatory processes. J Immunol. 1992;149:1657–65. [PubMed] [Google Scholar]
- 112.Armstrong DA, Major JA, Chudyk A, Hamilton TA. Neutrophil chemoattractant genes KC and MIP-2 are expressed in different cell populations at sites of surgical injury. J Leukoc Biol. 2004;75:641–8. doi: 10.1189/jlb.0803370. [DOI] [PubMed] [Google Scholar]
- 113.Fleischmann R, Stern R, Iqbal I. Anakinra: an inhibitor of IL-1 for the treatment of rheumatoid arthritis. Expert Opin Biol Ther. 2004;4:1333–44. doi: 10.1517/14712598.4.8.1333. [DOI] [PubMed] [Google Scholar]
- 114.Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, Kim HJ, Brewer C, Zalewski C, Wiggs E, Hill S, Turner ML, Karp BI, Aksentijevich I, Pucino F, Penzak SR, Haverkamp MH, Stein L, Adams BS, Moore TL, Fuhlbrigge RC, Shaham B, Jarvis JN, O’Neil K, Vehe RK, Beitz LO, Gardner G, Hannan WP, Warren RW, Horn W, Cole JL, Paul SM, Hawkins PN, Pham TH, Snyder C, Wesley RA, Hoffmann SC, Holland SM, Butman JA, Kastner DL. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. 2006;355:581–92. doi: 10.1056/NEJMoa055137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kokkola R, Andersson A, Mullins G, Ostberg T, Treutiger CJ, Arnold B, Nawroth P, Andersson U, Harris RA, Harris HE. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol. 2005;61:1–9. doi: 10.1111/j.0300-9475.2005.01534.x. [DOI] [PubMed] [Google Scholar]
- 116.Lubberts E, Joosten LA, Oppers B, van den Bersselaar L, Coenen-de Roo CJ, Kolls JK, Schwarzenberger P, van de Loo FA, van den Berg WB. IL-1-independent role of IL-17 in synovial inflammation and joint destruction during collagen-induced arthritis. J Immunol. 2001;167:1004–13. doi: 10.4049/jimmunol.167.2.1004. [DOI] [PubMed] [Google Scholar]
- 117.Lin HW, Basu A, Druckman C, Cicchese M, Krady JK, Levison SW. Astrogliosis is delayed in type 1 interleukin-1 receptor-null mice following a penetrating brain injury. J Neuroinflammation. 2006;3:15. doi: 10.1186/1742-2094-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bertini R, Howard OM, Dong HF, Oppenheim JJ, Bizzarri C, Sergi R, Caselli G, Pagliei S, Romines B, Wilshire JA, Mengozzi M, Nakamura H, Yodoi J, Pekkari K, Gurunath R, Holmgren A, Herzenberg LA, Herzenberg LA, Ghezzi P. Thioredoxin, a redox enzyme released in infection and inflammation, is a unique chemoattractant for neutrophils, monocytes, and T cells. J Exp Med. 1999;189:1783–9. doi: 10.1084/jem.189.11.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Altemeier WA, Zhu X, Berrington WR, Harlan JM, Liles WC. Fas (CD95) induces macrophage proinflammatory chemokine production via a MyD88-dependent, caspase-independent pathway. J Leukoc Biol. 2007;82:721–8. doi: 10.1189/jlb.1006652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pauluhn J. Retrospective analysis of 4-week inhalation studies in rats with focus on fate and pulmonary toxicity of two nanosized aluminum oxyhydroxides (boehmite) and pigment-grade iron oxide (magnetite): the key metric of dose is particle mass and not particle surface area. Toxicology. 2009;259:140–8. doi: 10.1016/j.tox.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 121.Epstein NJ, Warme BA, Spanogle J, Ma T, Bragg B, Smith RL, Goodman SB. Interleukin-1 modulates periprosthetic tissue formation in an intramedullary model of particle-induced inflammation. J Orthop Res. 2005;23:501–10. doi: 10.1016/j.orthres.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 122.Caicedo MS, Desai R, McAllister K, Reddy A, Jacobs JJ, Hallab NJ. Soluble and particulate Co-Cr-Mo alloy implant metals activate the inflammasome danger signaling pathway in human macrophages: a novel mechanism for implant debris reactivity. J Orthop Res. 2009;27:847–54. doi: 10.1002/jor.20826. [DOI] [PubMed] [Google Scholar]
- 123.Kool M, Soullie T, van Nimwegen M, Willart MA, Muskens F, Jung S, Hoogsteden HC, Hammad H, Lambrecht BN. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J Exp Med. 2008;205:869–82. doi: 10.1084/jem.20071087. [DOI] [PMC free article] [PubMed] [Google Scholar]