

Abstract
We report on a therapeutic approach using thermo-responsive multi-fingered drug eluting devices. These therapeutic grippers referred to as theragrippers are shaped using photolithographic patterning and are composed of rigid poly(propylene fumarate) segments and stimuli responsive poly (N-isopropylacrylamide-co-acrylic acid) hinges. They close above 32°C allowing them to spontaneously grip onto tissue when introduced from a cold state into the body. Due to porosity in the grippers, theragrippers could also be loaded with fluorescent dyes and commercial drugs such as mesalamine and doxorubicin, which eluted from the grippers for up to seven days with first order release kinetics. In an in vitro model, theragrippers enhanced delivery of doxorubicin as compared to a control patch. We also released theragrippers into a live pig and visualized release of dye in the stomach. The design of such tissue gripping drug delivery devices offers an effective strategy for sustained release of drugs with immediate applicability in the gastrointestinal tract.
Keywords: Drug Delivery, Hydrogels, Actuators, Polymers, Diffusion, Soft-Robotics
Drug delivery mechanisms have an enormous impact on the efficacy and bioavailability of pharmaceuticals. There are many accepted routes of administration including but not limited to oral, rectal, buccal, nasal, ocular, vaginal, intravenous, and topical. Controlled release systems offer several advantages: better control over drug concentration, longer residence time, minimized side effects, drug protection from harsh conditions, and lower administration frequency.[1] Inflammatory bowel disease (IBD) and gastrointestinal (GI) cancer patients would benefit from such improvements over current treatments. In the case of IBD, achieving the therapeutic dose is difficult due to unpleasant delivery methods such as rectal suppositories or enemas; the wide range of pH, enzymatic activity, and pressure throughout the GI tract; low absorption and variable transit time through the GI tract.[2] Patients must often take a combination of up to 16 pills a day, rectal suppositories and enemas, reducing patient quality of life.[3, 4] Likewise, chemotherapy treatments for cancer are often delivered systemically, resulting in painful and unpleasant side effects for patients. An extended, site-specific delivery of drugs could potentially reduce these side effects, improve drug efficacy, and improve patient quality of life.
Many hydrogels have been developed for controlled release of pharmaceuticals, including micro or nanoparticles,[5-7] capsules or cylinders,[8, 9] and patches or discs,[10-14] made from a variety of materials and fabrication methods.[15, 16] In particular, stimuli responsive hydrogels are of interest to the drug delivery community due to their responsiveness to the unique range of pH and temperatures within the GI tract.[11, 17-19] In this paper, we describe a polymeric, biphasic drug eluting theragripper which actively grips into tissue in response to body temperature and releases drug from its layers and pores. Actuation of the theragrippers is derived from stimuli-responsive soft micro-origami paradigms.[18-23]
The theragrippers are composed of alternating rigid panels of biodegradable, photopatternable poly(propylene fumarate) (PPF)[24-25]and flexible hinges of biocompatible poly(N-isopropyl acrylamide-co-acrylic acid) (pNIPAM-AAc)(Figure 1a).[18] Our central hypothesis is that by combining, (a) the thermally responsive properties of pNIPAM, (b) the tissue gripping capabilities of a photolithographically shaped multi-fingered device with sharp tips, (c) the high stiffness of biodegradable PPF, and (d) the controlled release properties of porous polymers, that we can deliver sustained doses of drugs more effectively using a combined chemical and mechanical approach. Here, we characterize multiple controlled drug release profiles from the theragripper and demonstrate its uses in vitro and in vivo.
Figure 1.

Design and proof of principle of drug-eluting theragrippers. (a) Schematic of theragrippers with rigid PPF panels and flexible stimuli responsive pNIPAM-AAc hinges. Due to the thermal responsiveness of the pNIPAM-AAc, the grippers reversibly open and close around body temperature. (b) Theragrippers originally closed at 4°C, open as the solution temperature increases and finally close again in the opposite direction at 37°C. (c) Conceptual illustration of theragrippers attached to a colon wall, releasing a fluorescent drug to targeted areas of the colon.
Considerable thought was given to the choice of materials for the theragripper design. We realize that pNIPAM and other hydrogels have been used extensively in active drug delivery devices due to their stimuli-responsiveness.[11,15-17] Below 32°C, pNIPAM-AAc is hydrophilic and swells in water; above 32°C it becomes hydrophobic and collapses as a result of the dehydration of its hydrophobic groups.[26] However while this mechanism makes pNIPAM an ideal hinge material, this hydrogel suffers from a low modulus that makes it weak as a gripping device. By combining pNIPAM with PPF, which has a modulus three orders of magnitude greater than that of most hydrogels and has been previously used in bone tissue engineering,[27-30] we are able to create a robust gripping device able to latch onto cells and tissue at body temperature (Figure 1a-b), and this distinguishes our work from prior uses of stimuli responsive polymers in drug delivery. Further, by developing a photolithographic approach, we could precisely shape this stiff, bone-like material to create sharp tips which can dig into tissue to secure the theragrippers in place, without damaging the tissue of the GI tract. These grippers are similar in shape to previously developed metallic microgrippers used for in vitro and in vivo tissue biopsies, but importantly they are all-polymeric.[31-33] Thus the theragrippers can store and elute drugs from the polymer networks in one or both layers. Consequently, the material composition and properties of the theragrippers make them ideal as drug delivery devices, as they could grip firmly onto tissue and elute a drug in close proximity to a targeted tissue such as in the GI tract (Figure 1c).
Drug-loaded theragrippers can be engineered using three different methods (Figure S1). In Method 1, we fabricate theragrippers and then soak them in a chemical solution overnight (TG1). We observed that the chemical was loaded primarily in the pNIPAM-AAc. We attribute this to the hydrophilicity of pNIPAMAAc at 4°C, porosity, and high capacity for swelling.[34]We increased the extent of loading by creating pores in the PPF via salt leaching during fabrication,[28,30] and then soaked the theragrippers in the chemical solution (Method 2, TG2). Thus, chemicalwas loaded both in the pNIPAM-AAc layer as well as in the porous PPF layer, leading to a larger total amount of dye released. In Method 3, chemical was loaded by in situ polymerization, wherein dry powder was mixed into the PPF pre-polymer prior to photo-crosslinking (TG3). This method provided extended release over longer periods of time due to the dense crosslinking and narrow interstitial spaces of the PPF polymer network.[27]
We quantified chemical elution from the three methods and plotted cumulative release over a period of 7 days at 37°C using fluorescein dye (Figure 2a-b, Figure S2). The cumulative release profile signifies the total amount of drug released over that period and plotted per gripper. Although all three samples were prepared using the same 1 mg/mL solution, we observed that the cumulative elution from each was very different. Cumulative elution from TG 2 was more than double that from TG1, due to high loading of dye into the salt-leached PPF pores.[28,30] TG3 showed the least cumulative elution (Figure 2a zoom), due to the dye being trapped in the interior of the PPF polymer network. The cumulative amount of each method could be increased or decreased as needed by altering the concentration of loading solution or the amount of dry chemical in the prepolymer. Further, the release rate from TG1 and TG2 is much higher than TG3; both these grippers eluted 90% of the plateau concentration in the first 6 hours, while TG3 eluted consistently over 7 days and did not plateau. These profiles can be combined for biphasic drug release (Figure S3).
Figure 2.

Quantified release profiles and kinetic analysis. (a) Graph of the cumulative fluorescein release profiles from the theragrippers. Zoomed inset details the slower TG3 release. (b) Zoomed in graph over a shorter time showing the cumulative release over the first 6 hours. (c) Graph of the fractional release of mesalamine and doxorubicin from the theragrippers. (d) First order kinetic model applied to each release profile.
Our processing methodology is compatible with real drugs, which are often formulated as powders. We highlight this applicability using mesalamine, a well-known anti-inflammatory drug for IBD. We fabricated mesalamine-TG3 grippers and measured the elution over 15 days at 37°C (Figure 2c). The drug eluted over 7 days and showed a slow and steady release profile consistent with TG3. Localized delivery over a full week is a significant improvement in quality of life for IBD patients currently administering rectal treatments on a nightly basis.
GI cancers would also benefit from delivery of a low but consistent concentration of chemotherapeutic drugs directly to targeted tumor sites. To study this application, we quantified the elution profile of doxorubicin DOX-TG1 grippers at 37°C (Figure 2c). Although the DOX-TG1 were prepared in the same way as the fluorescein-TG1, they showed a more extended release, which we attribute to the higher hydrophobicity of DOX.
After comparing several kinetic models to the release data, we observe that our data best fits a first order kinetic model (Figure 2d).[1, 35-38] First order kinetics (ie. concentration dependent diffusion) is common with diffusion-controlled polymer matrix systems with the assumption that the dye or drug is uniformly distributed within the polymer layer. The widely varying rate constants (Table 1) explain the different rates of elution and are affected by the drug solubility in both the polymer and the solution, drug-polymer interactions, diffusivity and morphology of the polymers, and the method of loading.[34]
Table 1.
Rate constant and coefficient of determination (R2) values for the first order kinetic model.
| Gripper | k(hr-1) | R2 |
|---|---|---|
| Fluorescein TG1 | 0.128 | 0.9007 |
| Fluorescein TG2 | 0.138 | 0.8994 |
| Fluorescein TG3 | 0.007 | 0.9662 |
| Mesalamine TG3 | 0.003 | 0.9617 |
| DOX TG1 | 0.015 | 0.9093 |
An important hypothesis in this work is that the gripping action of multi-fingered theragrippers enhances drug delivery efficacy compared to a non-gripping patch under flow conditions, as would occur in the GI tract. We developed an in vitro model to study this hypothesis. The patch was designed with the same pNIPAM-AAc bilayer composition and volume as the grippers. However, patches were not patterned with sharp, segmented fingers, thus could not grip onto cells or tissue and curved only slightly in response to temperature. After loading both the theragrippers and patches with 0.75 μM DOX, we pipetted each onto a clump of MDA-MB-231 breast cancer cells at 37°C. The gripper closed around the cells, while the patch retained its slight curvature (Figure 3a, c). Under flow, the gripper remained attached to the cells for the whole experiment, while the patch drifted away from the cells in the direction of the flow after an average of 20 minutes. We then tested the efficacy of DOX in killing the cells using a Live/Dead assay and found that the DOX-patch killed fewer cells than the DOX-TG1 (Figure 3b, d; Figure S4). The results show that DOX was more efficiently delivered directly to the cells via the gripper than the patch, and that the gripping action played a critical role in achieving this effectiveness. Additionally, the effectiveness of the gripping action is shown by the retrieved theragrippers that contained cells within their grasp (Figure 3e-f). Thus, theragrippers could potentially grasp mucosal tissue and resist being carried away under flow in the GI tract. It is noteworthy that mucoadhesive coatings have previously enhanced hydrogel attachment to mucosa.[39] One limitation of this approach is that the initial application force and contact time significantly affect bonding strength,[39-40] and it is challenging to apply a patch with sufficient adhesion to the lower GI tract by oral or endoscopic delivery. The autonomous gripping action of the theragrippers reduces this requirement for application of high forces to enable attachment. Further, if needed, our approach facilitates inclusion of mucoadhesive coatings by dip-coating or spin coating.
Figure 3.

In vitro model of doxorubicin (DOX) elution from a non-folding control patch and a theragripper. (a) Optical image of the square non-gripping control patch on top of a cell clump (outlined with the dotted line). Scale bar is 2 mm. (b) The same cell pellet stained with ethidium homodimer after 2 hr of elution via the control patch. (c) The DOX-TG1 gripping into a cell clump (outlined with the dotted line). Scale bar is 1 mm. (d) The same cell pellet stained with ethidium homodimer after 2 hr of elution from DOX-TG1. (e-f) Optical and fluorescent images of the detached theragripper tightly closed around and gripping a clump of cells. Scale bars are 1 mm.
To demonstrate feasibility of drug delivery in clinical conditions, we performed an in vivo experiment using TG1 grippers loaded with a blue food dye in a porcine GI model. After an endoscope was introduced into the esophagus of a pig, cold theragrippers were delivered by injection over 5 seconds through a catheter in an endoscope port. Grippers closed on warming up from 4°C to body temperature, which typically occurred within 5 minutes. The theragrippers closed after sliding into the stomach due to the large quantity of fluid and the forceful contractions of the esophagus, and were scattered around the stomach with good coverage (Figure 4). The model drug was observed to be contained with the gripper (Figure 4a) and then eluting from the gripper (Figure 4b). While we did not recover the grippers from the stomach, we expect them to be completely shed through the natural mucus turnover in the GI tract, which occurs every 1-2 weeks (See supplemental note on biocompatibility).
Figure 4.
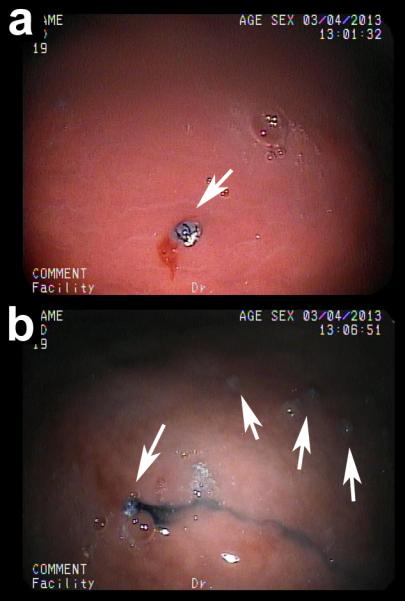
Endoscopic in vivo delivery of theragrippers to porcine stomach. Drug eluting grippers were loaded with a blue food dye by absorption and delivered to the stomach of a pig through an endoscope. (a,b) Representative grippers inside a porcine stomach containing (a) the dye and (b) eluting the dye. Arrows point to grippers spread throughout the stomach.
In summary, we have demonstrated a new approach for sustained drug release from therapeutic all-polymeric multi-fingered relatively stiff grippers with sharp tips for gripping into tissue. The theragrippers can be fabricated in a high throughput, parallel and cost-effective manner using lithographic processes. Due to the stimuli-responsive gripping action of these devices, they function autonomously and can be deployed en masse. They can absorb small molecule drugs, and their biphasic release can elute one or more drugs, rapidly or slowly up to 7 days, enabling a highly tunable device. Their extended release period of a week offers a significant improvement over the daily rectal drug delivery currently used in the treatment of IBD, yet is short enough to avoid concerns of the breakdown of pNIPAM. Their multi-fingered design shows improved site-specific delivery under flow compared to a patch. Thus, these devices can more effectively provide rapid or extended site-specific administration of one or more drugs by actively gripping into tissue with the combination of stiff, bone-like tips and thermoresponsive hinges. Future experiments investigating drug stability and bioavailability within the device and the pharmacokinetics of mesalamine when delivered via site-specific controlled release will need to be evaluated in vivo. These studies are limited by the lack an effective porcine IBD model which is still under development.
Experimental Section
PPF was synthesized using a previously published protocol; [41] the molecular weight was 952 Da with a polydispersity of 1.9. To make the PPF solution, we mixed three parts PPF stock solution with one part diethyl fumarate (DEF, Sigma Aldrich) at 65°C and added 5% Irgacure 2100 (Ciba) by weight. pNIPAM-AAc solution was synthesized using a previously published protocol.[20]
Grippers were fabricated on a silicon wafer with a spin coated 30% solution of polyvinyl alcohol (PVA, Sigma Aldrich, 9000 Da molecular weight, 80% hydrolyzed). We spin coated PPF solution at 3000 rpm and exposed at 650 mJ/cm2 on a mask aligner (Quintel). Without developing, we cast 1 mL of pNIPAM-AAc solution on top of the PPF and exposed at 100 mJ/cm2. We developed both layers in ethanol and deionized (DI) water. We released the theragrippers in DI water for 20 minutes. For TG2, we mixed 5% by weight ground sodium chloride (NaCl) into the PPF solution prior to patterning. After release, the NaCl dissolved in DI water overnight. For TG3, 1-5% powder was mixed into PPF prior to patterning.
Kinetic studies: 6 theragrippers of each type with 1 mg/mL fluorescein were rinsed three times with 25°C DI water and added to 20 mL of 37°C DI water. 300 μL samples of the solution were taken at each time point, and were excited and analyzed at 485 nm and 538 nm on a plate reader (Spectramax Gemini XPS, Molecular Devices). Mesalamine was removed from Pentasa® capsules (Shire) and ground into a fine powder. 5% by weight was added to the PPF solution prior to patterning (TG3). 60 theragrippers were rinsed with DI water and added to 15 mL of 37°C DI water. 300 μL samples of the solution were taken at each time point and analyzed using liquid chromatography mass spectroscopy (Waters Acquity H Class UPLC).DOX-TG1 grippers were soaked in 400 μg/mL doxorubicin hydrochloride (Sigma Aldrich) overnight. 6 theragrippers were rinsed three times with 25°C DI water, and added to 20 mL of 37°C DI water. 300 μL samples of the solution were taken at each time point, and were excited and analyzed at 485 nm and 590 nm on a plate reader.
In vitro model: MDA-MB-231 cells were cultured, centrifuged into a clump, and pipetted into MDA-MB-231 media at 37°C. A square PPF/pNIPAM-AAc patch with the same polymer volume as the theragrippers was fabricated using Method 1. TG1 and the patch were soaked in a 0.75 μM doxorubicin (DOX) solution at 4°C for 1 hour. Both were rinsed twice in DI water before addition to the 6-well plate. Using a syringe pump (Harvard Apparatus), warm MDA-MB-231media was flowed over the cells at a rate of 5 mL/min. After 2 hours of DOX exposure, the theragripper and patch were removed. The cells were rinsed in 5 mL/min PBS for 20 minutes to remove any remaining DOX and media. The cells were stained with calcein AM (Life Technologies) and ethidium homodimer (Life Technologies) and imaged using fluorescent microscopy.
In vivo porcine experiment: TG1 were soaked in blue dye for 24 hrs. 20 theragrippers were suspended in 10 mL of 4°C DI water and endoscopically delivered to the esophagus and stomach of a pig through a catheter in an endoscope (EG-3830TK PENTAX, Tokyo, Japan). Experimental protocols were approved by the Johns Hopkins University IACUC and meet guidelines of the National Institutes of Health guide for the Care and Use of Laboratory Animals.
Supplementary Material
Footnotes
This research is supported by the NSF grant NSF CBET-1066898 (to D.H. G.), the NIH Director's New Innovator Award Program through grant DP2-OD004346-01 (to D.G.), NIH R01 AR061460 (to J.P.F.), Broad Medical Research Institute grant (IBD-0336 to F.M.S.), and Northrop Grumman (to K.M.).
Supporting information for this article is available on the WWWunder http://dx.doi.org/10.1002/anie.201xxxxxx.
Contributor Information
Kate Malachowski, Department of Chemical and Biomolecular Engineering The Johns Hopkins University 3400 N. Charles St., Baltimore, MD 21218 (USA).
Joyce Breger, Department of Chemical and Biomolecular Engineering The Johns Hopkins University 3400 N. Charles St., Baltimore, MD 21218 (USA).
Hye Rin Kwag, Department of Chemical and Biomolecular Engineering The Johns Hopkins University 3400 N. Charles St., Baltimore, MD 21218 (USA).
Martha O. Wang, Fischell Department of Bioengineering University of Maryland, College Park, MD 20742 (USA)
John P. Fisher, Fischell Department of Bioengineering University of Maryland, College Park, MD 20742 (USA)
Florin M. Selaru, Department of Medicine, The Johns Hopkins University, Baltimore, MD21218 (USA)
David H. Gracias, Department of Chemical and Biomolecular Engineering The Johns Hopkins University 3400 N. Charles St., Baltimore, MD 21218 (USA).
References
- 1.Siegel R, Rathbone M. In: Fundamentals and Applications of Controlled Release Drug Delivery. Siepmann J, Siegel R, Rathbone M, editors. Controlled Release Society; 2012. [Google Scholar]
- 2.Basit AW. Drugs. 2005;65:1991. doi: 10.2165/00003495-200565140-00006. [DOI] [PubMed] [Google Scholar]
- 3.Lissner D, Siegmund B. Digestive Diseases. 2013;31:91–94. doi: 10.1159/000347194. [DOI] [PubMed] [Google Scholar]
- 4.Cheifetz AS. Journal of the American Medical Association. 2013;309:2150–2158. [Google Scholar]
- 5.Lee PI, Kim C-J. Journal of Controlled Release. 1991;16:229–236. [Google Scholar]
- 6.Tao SL, Desai TA. Advanced Drug Delivery Reviews. 2003;55:315–328. doi: 10.1016/s0169-409x(02)00227-2. [DOI] [PubMed] [Google Scholar]
- 7.Guan JJ, Ferrell N, Lee LJ, Hansford DJ. Biomaterials. 2006;27:4034–4041. doi: 10.1016/j.biomaterials.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 8.J de Leede LG, de Boer AG, Portzgen E. Journal of Controlled Release. 1986;4:17–24. [Google Scholar]
- 9.Guan JJ, He HY, Lee LJ, Hansford DJ. Small. 2007;3:412–418. doi: 10.1002/smll.200600240. [DOI] [PubMed] [Google Scholar]
- 10.Tao SL, Desai TA. Drug Discovery Today. 2005;10:909–915. doi: 10.1016/S1359-6446(05)03489-6. [DOI] [PubMed] [Google Scholar]
- 11.He HY, Guan JJ, Lee LJ. Journal of Controlled Release. 2006;110:339–346. doi: 10.1016/j.jconrel.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 12.Dadsetan M, Liu Z, Pumberger M, Giraldo CV, Ruesink T, Lu L, Yaszemski MJ. Biomaterials. 2010;31:8051–8062. doi: 10.1016/j.biomaterials.2010.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chirra HD, Desai TA. Small. 2012;8:3839–3846. doi: 10.1002/smll.201201367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernards DA, Lance KD, Ciaccio NA, Desai TA. Nanoletters. 2012;12:5355–5361. doi: 10.1021/nl302747y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta P, Vermani K, Garg S. Drug Discovery Today. 2002;7:569–579. doi: 10.1016/s1359-6446(02)02255-9. [DOI] [PubMed] [Google Scholar]
- 16.Chirra HD, Desai TA. Advanced Drug Delivery Reviews. 2012;64:1569–1578. doi: 10.1016/j.addr.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guan J, He H, Hansford DJ, Lee LJ. Journal of Physical Chemistry B. 2005;109:23134–23137. doi: 10.1021/jp054341g. [DOI] [PubMed] [Google Scholar]
- 18.Bassik N, Abebe B, Laflin K, Gracias DH. Polymer. 2010;51:6093–6098. [Google Scholar]
- 19.Fusco S, Sakar MS, Kennedy S, Peters C, Bottani R, Starsich F, Mao A, Sotiriou GA, Pané S, Pratsinis SE, Mooney D, Nelson B. Advanced Materials. 2014;26:952–957. doi: 10.1002/adma.201304098. [DOI] [PubMed] [Google Scholar]
- 20.Ionov L. Soft Matter. 2011;7:6786–6791. [Google Scholar]
- 21.Shim TS, Kim S-H, Heo C-J, Jeon HC, Yang S-M. AngewandteChemie. 2011;51:1420–1423. doi: 10.1002/anie.201106723. [DOI] [PubMed] [Google Scholar]
- 22.Gracias DH. Current Opinion in Chemical Engineering. 2013;2:112–119. [Google Scholar]
- 23.Wu ZL, Moshe M, Greener J, Therien-Aubin H, Nie Z, Sharon E, Kumacheva E. Nature Communications. 2012;4:1586. doi: 10.1038/ncomms2549. [DOI] [PubMed] [Google Scholar]
- 24.Peter SJ, Miller ST, Zhu G, Yasko AW, Mikos AG. Journal of Biomedical Materials Research. 1998;41:1–7. doi: 10.1002/(sici)1097-4636(199807)41:1<1::aid-jbm1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 25.Fisher JP, Timmer MD, Holland TA, Dean D, Engel PS, Mikos AG. Biomacromolecules. 2003;4:1327–1334. doi: 10.1021/bm030028d. [DOI] [PubMed] [Google Scholar]
- 26.Oh KS, Yuk SH. In: Biomedical Applications of Hydrogels Handbook. Ottenbrite R, Park K, Okano T, editors. Springer; 2010. [Google Scholar]
- 27.Haesslein A, Ueda H, Hacker MC, Jo S, Ammon DM, Borazjani RN, Kunzler JF, Salamone JC, Mikos AG. Journal of Controlled Release. 2006;114:251–260. doi: 10.1016/j.jconrel.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 28.Fisher JP, Holland TA, Dean D, Engel PS, Mikos AG. Journal of Biomaterials Science, Polymer Edition. 2001;12:673–687. doi: 10.1163/156856201316883476. [DOI] [PubMed] [Google Scholar]
- 29.Fisher JP, Dean D, Mikos AG. Biomaterials. 2002;23:4333–4343. doi: 10.1016/s0142-9612(02)00178-3. [DOI] [PubMed] [Google Scholar]
- 30.Choi J, Kim K, Kim T, Liu G, Bar-Shir A, Hyeon T, McMahon MT, Bulte JWM, Fisher JP, Gilad AA. Journal of Controlled Release. 2011;156:239–245. doi: 10.1016/j.jconrel.2011.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leong TG, Randall CL, Benson BR, Bassik N, Stern GM, Gracias DH. Proceedings of the National Academy of Sciences. 2009;106:703–708. doi: 10.1073/pnas.0807698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gultepe E, Randhawa JS, Kadam S, Yamanaka S, Selaru FM, Shin EJ, Kalloo AN, Gracias DH. Advanced Materials. 2013;25:514–519. doi: 10.1002/adma.201203348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gultepe E, Yamanaka S, Laflin KE, Kadam S, Shim Y, Olaru AV, Limetkai B, Khashab MA, Kalloo AN, Gracias DH, Selaru FM. Gastroenterology. 2013;144:691–693. doi: 10.1053/j.gastro.2013.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vilar G, Tulla-Puche J, Albericio F. Current Drug Delivery. 2012;9:367–394. doi: 10.2174/156720112801323053. [DOI] [PubMed] [Google Scholar]
- 35.Langer RS, Peppas NA. Biomaterials. 1981;2:201–214. doi: 10.1016/0142-9612(81)90059-4. [DOI] [PubMed] [Google Scholar]
- 36.Gurny R, Doelker E, Peppas NA. Biomaterials. 1982;3:27–32. doi: 10.1016/0142-9612(82)90057-6. [DOI] [PubMed] [Google Scholar]
- 37.Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. International Journal of Pharmaceutics. 1983;15:25–35. [Google Scholar]
- 38.Siepmann J, Siepmann F. International Journal of Pharmaceutics. 2008;364:328–343. doi: 10.1016/j.ijpharm.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 39.Shaikh R, Raghu RST, James GM, Woolfson AD, Donnelly RF. Journal of Pharmacy and Bioallied Sciences. 2011;3:89–100. doi: 10.4103/0975-7406.76478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kremser C, Albrecht K, Greindl M, Wolf C, Debbage P, Bernkop-Schnurch A. Magnetic Resonance Imaging. 2008;26:638–643. doi: 10.1016/j.mri.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 41.Kasper FK, Tanahashi K, Fisher JP, Mikos AG. Nature Protocols. 2009;4:518–525. doi: 10.1038/nprot.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.