

Abstract
Simvastatin (SIM), a widely used anti-lipidaemic drug, has been identified as a bone anabolic agent. Its poor water solubility and the lack of distribution to the skeleton, however, have limited its application in the treatment of bone metabolic diseases. In this study, an amphiphilic macromolecular prodrug of SIM was designed and synthesized to overcome these limitations. The polyethylene glycol (PEG)-based prodrug can spontaneously self-assemble to form micelles. The use of SIM trimer as the prodrug’s hydrophobic segment allows easy encapsulation of additional free SIM. The in vitro studies showed that SIM/SIM-mPEG micelles were internalized by MC3T3 cells via lysosomal trafficking and consistently induced expression of both BMP2 and DKK1 mRNA, suggesting that the prodrug micelle retains the biological functions of SIM. After systemic administration, optical imaging suggests that the micelles would passively target to bone fracture sites associated with hematoma and inflammation. Furthermore, flow cytometry study revealed that SIM/SIM-mPEG micelles had preferred cellular uptake by inflammatory and resident cells within the fracture callus tissue. The treatment study using a mouse osteotomy model validated the micelles’ therapeutic efficacy in promoting bone fracture healing as demonstrated by micro-CT and histological analyses. Collectively, these data suggest that the macromolecular prodrug-based micelle formulation of SIM may have great potential for clinical management of impaired fracture healing.
Keywords: Prodrug, Simvastatin, Bone Fracture, Micelle, ELVIS
1. INTRODUCTION
With a worldwide aging population, osteoporosis and osteoporosis-related bone fractures have become major public health challenges with a heavy economic burden [1]. Aging and the incipient risk of osteoporosis result in the loss of bone mass and deterioration of bone quality predisposing to fracture. In addition, the fracture repair process is delayed and less effective with aging, making the elderly population particularly susceptible to increased morbidity and mortality. Despite significant advances in the development of new drugs for osteoporosis and the intense effort to identify drugs that improve fracture healing, to date there has been no drug that has been approved by the US FDA for improved fracture healing.
Statins, which were developed as 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors, have been widely used to treat cardiovascular diseases for decades [2]. In 1999, Mundy et al. reported that two statins, simvastatin (SIM) and lovastatin, have strong bone anabolic effects that were attributed to induction of the bone inducing factor bone morphogenic protein-2 (BMP-2) [3, 4]. Major efforts have been invested since then, attempting to validate this finding [5-10]. The results remain controversial [11-14], however, in part due to the hepatotropic nature of the statins. Since 95% of orally administered SIM is retained by the liver [8, 11], only 5% of the drug is available for extra-hepatic activity, including effects on the skeleton system. Higher oral administration doses may increase the distribution of SIM to the bone [6, 15-17] but are impractical due to the associated systemic toxicities [8, 11].
Though transdermal application [18] and local delivery [3, 19-32] of statins to fracture sites have been explored, most fracture sites are not within sufficient proximity to produce a clinically relevant effect and direct delivery of statins have yielded variable results. Recognizing these issues associated with the delivery of statins for fracture repair, we hypothesized that a systemically administered statin formulation that selectively localizes and is retains at the injury site and gradually releases the statins locally would overcome their high hepatotropicity and low water solubility, and best facilitate their bone anabolic effects to promote fracture healing. The recently reported Extravasation through Leaky Vasculature and Inflammatory cell-mediated Sequestration (ELVIS) mechanism [33-36] provides a unique opportunity to systemically target therapeutic agents to inflammatory pathologies, including fractures. As hematoma and acute inflammation are key initial pathological features of bone fractures, we hypothesized that the leaky vasculature associated with the fracture would allow the extravasation of systemically administered colloids, where they could be sequestered by the inflammatory infiltrates and activated resident cells at the fracture site. The retained drug within the formulation could then be gradually released from cells at the fracture site to promote the healing process.
We here report the design and synthesis of a dendritic amphiphilic macromolecular prodrug of simvastatin (SIM-mPEG) based on ω-methoxypolyethylene glycol (mPEG) to address the poor water solubility and the lack of osteotropicity of the statins. When mixed with free SIM, the prodrug self-assembles to form stable micelles (SIM/SIM-mPEG), which can gradually release the encapsulated SIM. Our results demonstrate that the systemically delivered micelle formulation passively targets the bone fracture site and promotes fracture healing.
2. MATERIALS AND METHODS
2.1 Materials
Simvastatin was purchased from Zhejiang Ruibang Laboratories (Wenzhou, Zhejiang, China). 3-Butyn-1-ol was purchased from Matrix Scientific (Columbia, SC, USA). Polyethylene glycol monomethylether (mPEG, 1.9 kDa) was purchased from Alfa Aesar (Ward Hill, MA, USA). Heterofunctional PEG (NH2-PEG-N3, 3 kDa) was purchased from Rapp Polymere (Tübingen, Germany). Alexa Fluor® 488 NHS ester was purchased from Invitrogen (Camarillo, CA, USA). IRDye 800CW NHS ester was purchased from LI-COR Biosciences (Lincoln, NE, USA). All other reagents and solvents were purchased from Acros Organics (Morris Plains, NJ, USA) and used as received.
2.2 The synthesis of the amphiphilic macromolecular prodrug SIM-mPEG
2.2.1 Synthesis of α-methoxy-ω-iodo-PEG (compound 2)
mPEG (1.9 kDa, 3.8g, 2mmol) was dissolved in anhydrous dichloromethane (DCM, 40 mL) and cooled in an ice-water bath. Under the protection of argon (Ar), triphenyl phosphine (2.62g, 10mmol) was added at 0 °C, and the solution was stirred for 10 min. Iodine (2 g, 8mmol) was added in several portions at 0 °C, then the temperature of the solution was raised to room temperature and stirred for 24 h. A total of 20 mL of saturated Na2SO3 solution was added and stirred for 10 min. The organic phase was then separated and dried over anhydrous Na2SO4. The product was then loaded on a silica gel column and eluted with DCM, ethyl acetate (EtOAc) (1:1) and pure EtOAc to remove the byproduct. Lastly, DCM and methanol (1:1) were used as eluent to obtain 3.16 g compound 2, yield: 79.1%. For NMR analyses of all the new compounds synthesized, a Varian Inova Unity 500 NMR Spectrometer was used. 1H-NMR (500MHz, CDCl3): δ (ppm) = 3.75 (t, J = 6.8Hz, 2H), 3.65 (br, 164H), 3.38 (s, 3H), 3.26 (t, J = 6.9Hz, 2H). 13C-NMR (125MHz, CDCl3): δ (ppm) = 71.33, 69.97 (br), 69.62, 58.42, 2.57.
2.2.2 Synthesis of α-methoxy-ω-azido-PEG (compound 3)
Compound 2 (500mg, 0.25mmol) and sodium azide (325 mg, 5mmol) were dissolved in anhydrous dimethylformamide (DMF, 4 mL). The solution was stirred at 100 °C for 24 h under the protection of Ar. DCM (100 mL) was then added and washed with brine. The organic phase was dried and concentrated. The residue was loaded on a short silica gel column and eluted with DCM:MeOH = 1:1 to remove the salt. The solvent was evaporated and the residue was further purified by LH-20 column to give 450 mg compound 3. Yield: 93.4%. 1H-NMR (500MHz, CDCl3): δ (ppm) = 3.77 (t, J = 5.0Hz, 2H), 3.65 (br, 159H), 3.55 (t, J = 5.0Hz, 2H), 3.50 (t, J = 5.0Hz, 2H), 3.46 (t, J = 5.0Hz, 2H), 3.45 (s, 3H). 13C-NMR (125MHz, CDCl3): δ (ppm) = 71.68, 70.44 (br), 70.42, 70.32, 70.26, 69.78, 58.76, 50.42.
2.2.3 Synthesis of compound 4
Simvastatin (418 mg, 1 mmol) and TsOH monohydrate (19 mg, 0.1 mmol) were dissolved in 3-butyn-1-ol (420 mg, 6 mmol) and stirred at room temperature for 3 h. Ethyl acetate (50 mL) was added and then washed with saturated NaHCO3 (5 mL) and brine (20 mL). The aqueous phase was extracted 3 times with ethyl acetate (20 mL). The combined organic phase was dried by anhydrous sodium sulfate and then the solvent was evaporated. Toluene (30 mL) was added to the residue and then evaporated to remove the 3-butyn-1-ol. The residue was purified by flash chromatography (EtOAc:hexanes = 1:1 to 3:1), 148 mg of compound 4 was obtained and 252 mg of unreacted simvastatin was recovered. Yield: 30.3%. 1H-NMR (500MHz, CDCl3): δ (ppm) = 5.98 (d, J = 9.75Hz, 1H), 5.78 (dd, J = 9.75Hz, 6.34Hz, 1H), 5.49 (br, 1H), 5.39 (d, J = 2.92Hz, 1H), 4.27 (m, 1H), 4.23 (t, J = 6.83Hz, 2H), 3.98 (s, 1H), 3.78 (m, 1H), 3.68 (s, 1H), 2.55 (td, J = 6.83Hz, 2.44Hz, 2H), 2.53 (d, J = 2.93Hz, 1H), 2.51 (s, 1H), 2.44 (m, 1H), 2.37 (dd, J = 11.71Hz, 6.34Hz, 1H), 2.24 (dd, J = 11.71Hz, 2.44Hz, 1H), 2.02 (t, J = 2.44Hz, 1H), 1.94 (m, 1H), 1.94 (s, 1H), 1.50-1.64 (m, 8H), 1.21 (m, 1H), 1.12 (s, 3H), 1.11 (s, 3H), 1.09 (d, J = 7.31Hz, 3H), 0.87 (d, J = 7.32Hz, 3H), 0.82 (t, J = 7.32Hz, 3H). 13C-NMR (125MHz, CDCl3): δ (ppm) = 178.01, 171.80, 132.99, 131.50, 129.44, 128.20, 79.82, 72.08, 70.02, 68.87, 67.99, 62.17, 42.85, 42.24, 41.74, 37.62, 36.08, 34.70, 32.94, 32.88, 30.38, 27.18, 24.69, 24.58, 24.10, 22.99, 18.81, 13.79, 9.20. MS (ESI): m/z = 511 (M + Na+), calculated MW = 488.
2.2.4 Synthesis of compound 5
The diol compound 4 (300 mg, 0.6 mmol) and succinic anhydride (360 mg, 3.6 mmol) were dissolved in anhydrous DMF (10 mL). Triethylamine (TEA, 240mg, 2.4mmol) and 4-dimethylaminopyridine (DMAP, 29.28 mg, 0.24 mmol) were added. The solution was stirred at 45 °C for 20 h. Dilute hydrochloric acid (0.1 M, 30 mL) was added, followed by 100 mL of EtOAc. The solution was washed with brine and then dried. Flash chromatography separation gave 402 mg of compound 5. Yield: 94.6 %. 1H-NMR (500 MHz, CDCl3): δ (ppm) = 10.53 (br, 2H), 5.97 (d, J = 8.9Hz, 1H), 5.76 (dd, J = 9.27Hz, 6.34Hz, 1H), 5.48 (s, 1H), 5.36 (s, 1H), 5.25 (m, 1H), 4.90 (m, 1H), 4.18 (t, J = 6.83Hz, 2H), 2.67 (t, J = 6.34Hz, 4H), 2.64 (t, J = 6.34, 2H), 2.63 (d, J = 6.34, 2H), 2.59 (t, J = 6.34, 2H), 2.51 (td, J = 6.83Hz, 2.44Hz, 2H), 2.42 (m, 1H), 2.35 (q, J = 5.86, 1H), 2.22 (d, J = 12.19Hz, 2H), 2.02 (t, J = 2.44Hz, 1H), 1.94 (br, 2H), 1.89 (m, 2H), 1.60-1.70 (m, 2H), 1.40-1.60 (m, 3H), 1.33 (m, 1H), 1.11 (s, 6H), 1.07 (d, J = 7.31Hz, 3H), 0.86 (d, J = 6.83Hz, 3H), 0.82 (t, J = 7.31Hz, 3H). 13C-NMR (125MHz, CDCl3): δ (ppm) = 177.98, 177.53, 176.95, 171.65, 171.16, 169.68, 132.72, 131.36, 129.52, 128.20, 79.86, 71.50, 69.94, 67.99, 67.95, 62.29, 42.84, 38.22, 37.58, 37.37, 36.10, 32.79, 32.76, 31.06, 30.30, 28.92, 28.74, 28.70, 28.29, 27.10, 24.54, 24.51, 23.53, 22.90, 18.69, 13.62, 9.13. MS (ESI): m/z = 711 (M + Na+), calculated MW = 688.
2.2.5 Synthesis of simvastatin trimer (compound 6)
Compound 5 (495 mg, 0.719mmol) and N,N’-dicyclohexylcarbodiimide (DCC, 370.5 mg, 1.799 mmol) were dissolved in DCM (5 mL) at 0 °C. The solution was stirred for 5 min, DMAP (5.2 mg, 0.043 mmol) and simvastatin (750 mg, 1.799 mmol) were then added. The solution was stirred at 0 °C for about 1.5 h, then diluted with EtOAc and washed with brine. After drying with Na2SO4, the solvent was evaporated and the residue was purified by flash chromatography (EtOAc/hexanes = 1:3) to give 0.627g of the simvastatin trimer compound 6 with 287 mg of unreacted simvastatin recovered. Yield 58.6%. 1H-NMR (500 MHz, CDCl3): δ (ppm) = 5.98 (d, J = 9.75Hz, 2H), 5.97 (d, J = 9.27Hz, 1H), 5.76 (m, 3H), 5.51 (m, 3H), 5.36 (d, J = 2.44Hz, 2H), 5.33 (d, J = 2.93Hz, 1H), 5.27 (m, 2H), 5.22 (m, 1H), 4.87 (m, 1H), 4.50 (m, 2H), 4.19 (t, J = 6.24Hz, 2H), 2.79 (dd, J = 8.05Hz, 5.36Hz, 2H), 2.72 (m, 1H), 2.68 (m, 1H), 2.56-2.66 (m, 10H), 2.53 (td, J = 6.83Hz, 2.44Hz, 2H), 2.44 (m, 3H), 2.36 (m, 3H), 2.25 (d, J = 11.70Hz, 2H), 2.23 (d, J = 12.7Hz, 1H), 2.09 (m, 2H), 2.02 (t, J = 2.44Hz, 1H), 1.93-1.98 (m, 7H), 1.60-1.90 (m, 10H), 1.40-1.60 (m, 10H), 1.20-1.40 (m, 4H), 1.13 (s, 6H), 1.12 (s, 6H), 1.11 (s, 3H), 1.10 (s, 3H), 1.08 (d, J = 7.31Hz, 6H), 1.07 (d, J = 5.37Hz, 3H), 0.89 (d, J = 6.83Hz, 6H), 0.87 (d, J = 6.83Hz, 3H), 0.827 (t, J = 7.32Hz, 6H), 0.822(t, J = 7.32Hz, 3H). 13C-NMR (125MHz, CDCl3): δ (ppm) = 177.54, 177.40, 171.59, 171.35, 171.19, 171.01, 169.52, 168.65, 168.61, 132.73, 132.69, 132.68, 131.44, 131.39, 131.38, 129.65, 129.60, 128.31, 79.89, 76.48, 71.49, 69.97, 68.06, 67.83, 67.71, 65.78, 65.73, 62.29, 42.87, 42.81, 38.25, 37.68, 37.39, 36.55, 36.26, 35.22, 33.19, 33.16, 32.95, 32.88, 32.73, 31.11, 30.53, 30.40, 28.87, 27.18, 24.72. MS (ESI): m/z = 1511 (M + Na+), calculated MW = 1488.
2.2.6 Synthesis of SIM-mPEG (compound 7)
The simvastatin trimer (compound 6, 772 mg, 0.520 mmol), mPEG-N3 (compound 3, 400 mg, 0.208 mmol), copper sulfate pentahydrate (52 mg, 0.208 mmol) were added to a solution of t-BuOH (3 mL) and water (2 mL). After the solution was bubbled with Ar for 2 min, L-ascorbic acid sodium salt (82 mg, 0.416 mmol) was added. The solution was stirred at room temperature for 60 h under the protection of Ar. DCM (100 mL) was added and washed with a solution (186 mg EDTA disodium and 42 mg NaHCO3 in 30 mL water) and brine (40 mL × 3) to remove the copper catalyst. The aqueous phase was extracted with DCM (50 mL). The combined organic phase was concentrated. The residue was loaded on a silica gel column and eluted with EtOAc to recover the unreacted simvastatin trimer (260 mg) and then eluted with a mixed solution (DCM:MeOH = 1:1) to yield the final product. After lyophilization, 548 mg white solid was obtained. Yield: 77.3%. 1H-NMR (500MHz, CDCl3): δ (ppm) = 7.55 (s, 1H), 5.98 (d, J = 9.75, 2H), 5.97 (d, J = 9.25, 1H), 5.76 (m, 3H), 5.50 (m, 3H), 5.37 (d, J = 2.93Hz, 2H), 5.33 (m, 1H), 5.27 (m, 2H), 5.22 (m, 1H), 4.87 (m, 1H), 4.50 (t, J = 5.37Hz, 2H), 4.48 (m, 2H), 4.36 (t, J = 6.83Hz, 2H), 3.88 (t, J = 5.37Hz, 2H), 3.77 (t, J = 4.87Hz, 2H), 3.62 (br, 164H), 3.54 (t, J = 4.88Hz, 2H), 3.37 (s, 3H), 3.05 (t, J = 7.31Hz, 2H), 2.78 (dd, J = 8.04Hz, 5.37Hz, 2H), 2.70 (m, 2H), 2.68 (m, 1H), 2.56-2.66 (m, 12H), 2.43 (m, 3H), 2.36 (m, 3H), 2.25 (d, J = 12.20Hz, 2H), 2.23 (d, J = 12.7Hz, 1H), 2.08 (m, 2H), 1.93-1.98 (m, 7H), 1.60-1.90 (m, 14H), 1.40-1.60 (m, 9H), 1.20-1.40 (m, 4H), 1.12 (s, 6H), 1.117 (s, 6H), 1.106 (s, 3H), 1.100 (s, 3H), 1.09 (d, J = 7.31Hz, 6H), 1.07 (d, J = 5.37Hz, 3H), 0.89 (d, J = 6.83Hz, 6H), 0.87 (d, J = 6.83Hz, 3H), 0.830 (t, J = 7.32Hz, 6H), 0.821 (t, J = 7.32Hz, 3H). 13C-NMR (125MHz, CDCl3): δ (ppm) = 177.56, 177.42, 171.61, 171.38, 171.22, 171.05,169.68, 168.70, 168.67, 143.45, 132.77, 132.70, 131.46, 131.40, 129.67, 129.66, 128.32, 122.59, 76.50, 71.85, 70.48 (br), 69.44, 68.07, 67.85, 67.74, 65.84, 65.47, 58.94, 50.05, 42.89, 42.83, 38.31, 37.73, 37.40, 36.56, 36.27, 35.23, 33.19, 32.96, 32.89, 32.74, 32.42, 31.07, 30.54, 30.40, 28.89, 27.19, 26.08, 25.31, 24.74, 24.66, 24.12, 23.51, 22.94, 13.83, 13.77, 9.26.
2.2.7 Synthesis of IRDye® 800CW-labeled SIM-mPEG (SIM-mPEG-IRDye)
The heterofunctional PEG (NH2-PEG-N3, 46.8 mg, 0.01562 mmol) and IRDye800CW NHS ester (1.214 mg, 0.00104 mmol) were dissolved in anhydrous DMF (1 mL) and bubbled with Ar for 1 min. After addition of N,N-diisopropylethylamine (DIPEA, 5.38 mg, 0.0416 mmol), the solution was stirred at RT for 20 h. Acetic anhydride (15.9mg, 0.156mmol) was then added and stirred for 12 h. The mixture was purified by LH-20 column to give 45.4 mg final product, which was a mixture of PEG labeled by IRDye 800CW and PEG capped by acetyl group. Yield: 97%. 1H-NMR (500MHz, CDCl3): δ (ppm) = 6.38 (s, 1H), 3.77 (t, J = 5.36Hz, 2H), 3.63 (br, 301H), 3.55 (t, J = 4.88Hz, 2H), 3.38 (t, J = 4.88Hz, 2H), 1.97 (s, 3H). 13C-NMR (125MHz, CDCl3): δ (ppm) = 170.19, 70.61, 70.58, 70.55, 70.47 (br), 70.08, 69.97, 69.80, 50.58, 39.20, 23.14.
Simvastatin trimer compound 6 (133.9 mg, 0.09 mmol), PEG mixture (45 mg, 0.015 mmol) and copper sulfate pentahydrate (7.5 mg, 0.03 mmol) were dissolved in Ar bubbled solution of t-BuOH (1 mL), H2O (0.5 mL), DMF (1 mL). The solution was bubbled by Ar for another 1 min and then L-ascorbic acid sodium (11.88 mg, 0.06 mmol) was added. The mixture was stirred at room temperature for 48 h. The product was purified by LH-20 column chromatography. After lyophilization, 35.7mg product was obtained. The IRDye 800CW content was determined as 1.34±0.053 μmol/g in the final product using a NanoDrop™ 2000 UV-Vis Spectrophotometer (Thermo Scientific, Wilmington, DE, USA). 1H-NMR (500MHz, CDCl3): δ (ppm) = 7.56 (s, 1H), 6.46 (s, 1H), 5.98 (d, J = 9.75, 2H), 5.97 (d, J = 9.25, 1H), 5.76 (m, 3H), 5.50 (m, 3H), 5.37 (d, J = 2.93Hz, 2H), 5.33 (m, 1H), 5.27 (m, 2H), 5.22 (m, 1H), 4.87 (m, 1H), 4.50 (t, J = 5.37Hz, 2H), 4.48 (m, 2H), 4.36 (t, J = 6.83Hz, 2H), 3.88 (t, J = 5.37Hz, 2H), 3.77 (t, J = 4.87Hz, 2H), 3.62 (br, 294H), 3.54 (t, J = 4.88Hz, 2H), 3.37 (s, 3H), 3.05 (t, J = 7.31Hz, 2H), 2.78 (dd, J = 8.04Hz, 5.37Hz, 2H), 2.70 (m, 2H), 2.68 (m, 1H), 2.56-2.66 (m, 12H), 2.43 (m, 3H), 2.36 (m, 3H), 2.25 (dd, J = 12.20Hz, 2H), 2.23 (d, J = 12.7Hz, 1H), 2.08 (m, 2H), 1.98 (s, 3H), 1.93-1.98 (m, 7H), 1.60-1.90 (m, 14H), 1.40-1.60 (m, 9H), 1.20-1.40 (m, 4H), 1.12 (s, 6H), 1.117 (s, 6H), 1.106 (s, 3H), 1.100 (s, 3H), 1.09 (d, J = 7.31Hz, 6H), 1.07 (d, J = 5.37Hz, 3H), 0.89 (d, J = 6.83Hz, 6H), 0.87 (d, J = 6.83Hz, 3H), 0.830 (t, J = 7.32Hz, 6H), 0.821 (t, J = 7.32Hz, 3H). 13C-NMR (125MHz, CDCl3): δ (ppm) = 177.56, 177.44, 171.62, 171.38, 171.22, 171.06, 170.15, 169.69, 168.68, 143.49, 132.71, 131.47, 131.41, 129.68, 128.33, 122.64, 76.51, 71.52, 70.49 (br), 70.12, 69.79, 69.44, 68.08, 67.86, 67.74, 65.80, 65.76, 63.47, 50.09, 42.90, 42.84, 39.23, 38.33, 37.75, 37.41, 36.57, 36.28, 35.25, 33.20, 32.97, 32.91, 32.76, 32.00, 30.55, 30.41, 28.90, 27.21, 25.31, 24.75, 24.67, 24.13, 23.52, 23.14, 22.96, 13.84, 13.78, 9.27.
2.2.8 Synthesis of Alexa Fluor® 488-labeled SIM-mPEG (SIM-mPEG-Alexa)
A procedure similar to the synthesis of SIM-mPEG-IRDye was utilized to obtain SIM-mPEG-Alexa.
2.3 Micelle formulation, characterization and free SIM loading
2.3.1 Micelle formulation
Two different methods were explored for the preparation of free simvastatin-loaded micelles (SIM/SIM-mPEG). For the film hydration method, SIM (10 mg) was dissolved in a methanol solution of SIM-mPEG (1 mL, 10 mg/mL). Methanol was then removed by rotor evaporation at 60 °C to form a film in the round bottom flask, which was placed in vacuum overnight to remove any residue solvent. The film was then hydrated by distilled water at room temperature for 30 min. For the direct dissolution method, SIM-mPEG was first dissolved in water (1 mL, 10 mg/mL) and placed at 4 °C overnight, and then the system was equilibrated at 24 °C for 24 h in order to allow micelle formation. SIM (10 mg) was then added to this solution, and dissolution of the drug proceeded under stirring for 24 h at room temperature. In both cases, the undissolved SIM was removed by centrifugation at 9,000 g for 0.5 min, followed by filtration of the supernatant through a 0.2 μm filter. The direct dissolution of SIM-mPEG alone in water was used as a control for the following characterization.
2.3.2 Characterization of the micelles and drug loading efficiency
Effective hydrodynamic diameters (Deff), polydispersity index (PDI) and ζ-potential of the micelles were measured by dynamic light scattering (DLS) using a Zetasizer Nano ZS90 (Malvern Instruments, Worcestershire, UK). To estimate loading efficiency, the undissolved SIM was collected and quantified by an Agilent 1100 HPLC system with a quaternary pump (with degasser), an autosampler and a diode-array based UV detector. The loading efficiency was calculated by subtracting the amount of undissolved SIM from the total amount of SIM used to prepare the micelle formulation. In order to ensure that all the undissolved SIM is accounted for, the weight of SIM pellet after centrifugation was combined with the weight of the undissolved the SIM collected from the 0.2 um filter paper after the filtration of the centrifuge supernatant. The dynamic light scattering (DLS) data confirmed the absence of the undissolved large SIM particles in the filtrate.
2.4 In vitro release profile
The film hydration method was used to prepare the SIM/SIM-mPEG micelles for evaluation of the in vitro SIM release. The micelles solutions were placed into the dialysis bag (MWCO=3500) suspended in a PBS solution (pH=7.4, containing 0.2% sodium dodecyl sulfate) as releasing medium. The total volume of each medium was 30 mL. The sealed vials were placed in a water bath with thermostatic oscillator (Thermo Scientific, USA) at 75 rpm and 37 °C. At predetermined time intervals, 1 mL of the solution was withdrawn from the release medium and replaced with fresh medium. The simvastatin concentration was analyzed by measuring the UV absorbance at 238 nm. The SIM released was calculated by adding up the amount of SIM left in the vial and those withdrawn at different time points.
2.5 Cell culture, RNA extraction and real time PCR
MC3T3 murine osteoblast lineage cells (1×105 cells per well in 12 well plates) were grown to confluence in DMEM (with 10% fetal calf serum) and then treated with free SIM (5 μM) or freshly prepared SIM/SIM-mPEG micelles (5 μM) for 1-3 days, without media changes. Following culture, cells were harvested and subjected to RNA isolation (RNeasy Mini kit, QIAGEN Inc., Valencia, CA) and assessed for concentration and purity by optical density measurement. Five hundred nano gram aliquots of total cellular RNA were reverse transcribed using oligo-dT primers and MMLV reverse transcriptase (First Strand cDNA Synthesis Kit, Fermentas) as recommended by the manufacturer. Real time quantitative PCR (qPCR) was carried out in duplicate using the Opticon 2 thermal cycler detection system (Bio-Rad Laboratories Inc., Hercules, CA). Reactions included iQ™SYBR® Green Supermix reagent (Bio-Rad Laboratories Inc., Hercules, CA), 10 nano gram cDNA, and forward and reverse primers each at a concentration of 250 nM in a total volume of 25 μl. mRNA amounts were normalized relative to the housekeeping gene GAPDH, and quantified using the DDCt method. Generation of only the correct amplification products was confirmed using melting point curve analysis of the products. The sequences of the oligonucleotide primers used were: GAPDH (GGTGCTGAGTATGTCGTGGA/GTGGTTCACACCCATCACAA) BMP2 (GGGACCCGCTGTCTTCTAGT/ TCAACTCAAATTCGCTGAGGAC) DKK1 (CTCATCAATTCCAACGCGATCA/ GCCCTCATAGAGAACTCCCG).
2.6 Intracellular localization studies
Freshly made SIM/SIM-mPEG micelles in which 20% of the SIM-mPEG was replaced with an equivalent amount of Alexa Fluor 488 labeled SIM-mPEG (SIM-mPEG-Alexa) were added to subconfluent cultures of MC3T3 cells grown on chamber slides to a concentration of 20 μM for 4 h, followed by an incubation of additional 12 h in the absence of micelles. Cells were then labeled with Lysotracker Red DND-99 (to identify lysosomes) and Hoechst 33342 (to label nuclei in accordance with the manufacturer’s recommendations (Image-IT LIVE Lysosomal and Nuclear Labeling Kit, Molecular Probes). Cells were then imaged using a Nikon Eclipse fluorescent microscope.
2.7 Mouse open fracture model (osteotomy)
Ten-week-old male Swiss Webster mice were purchased from Charles River Laboratories, and maintained under standard housing conditions. The animals were acclimated for at least 1 week before any experimental procedure. Under general anesthesia with isoflurane, the hips, thighs, and knees on the right side of the animal were prepared with Betadine (povidone-iodine) solution. A 1.0 cm right medial parapatellar incision was made on the right knee. The patella was dislocated laterally to expose the femoral condyles. A 25-gauge needle was introduced into the canal and driven in a retrograde intramedullary fashion to the level of the greater trochanter. Distally, the needle was cut flush with the cortex (length was about 1.4 cm) so as not to interfere with motion of the knee. Controlled unilateral transverse femoral shaft fractures were created with a #15 blade. The extensor mechanism was closed with interrupted absorbable sutures (size 4-0) in a standard fashion followed by closure of the skin with 4-0 sutures. Postoperatively, mice were placed on clean paper towels in their cages (to avoid inhalation/aspiration of the bedding material) and monitored until the animals were fully awake. The mice were monitored for signs of pain, with analgesia and antibiotics given according to established protocol. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of University of Nebraska Medical Center (UNMC).
2.8 In vivo imaging
Near-infrared (NIR) optical imaging was performed to observe the in vivo biodistribution of the SIM/SIM-mPEG micelles after its systemic administration. The IRDye® 800CW labeled and un-labeled prodrug mixture was used to prepare the IRDye-labeled SIM/SIM-mPEG micelles (SIM-PEG-IRDye, 1 mg/mouse, n = 3) and then given to fractured mice via tail vein injection 7 days post osteotomy. The mice were imaged prior to and then daily after the micelle administration using a LI-COR Pearl™ Impulse Small Animal Imaging System (Lincoln, NE, USA) to evaluate the distribution of the micelles continuously for 7 days. All of the mice were scanned under anesthesia with isoflurane (inhalational 1%~1.5%, Piramal Healthcare, Andhra Pradesh, India). The images were captured using channels 800 nm and white (optical) with the resolution of 170 μm. The NIR signal intensity was measured semi-quantitatively at a consistent region of interest (ROI) at the fracture and the contralateral site for all the mice.
2.9 Fluorescence-Activated Cell Scanning (FACS) Analysis
Alexa Fluor® 488 labeled SIM/SIM-mPEG micelles (SIM-PEG-Alexa Fluor® 488 1.5 mg/mice) were given to mice by tail vein injection on day 8 post osteotomy. At necropsy (24 h post injection), callus tissues were isolated from the fractured femurs and minced aseptically. The tissues were further digested with type II collagenase (1 mg/mL, Sigma-Aldrich, St. Louis, MO) at 37 °C for 2 h. After passing through a 70 μm cell strainer, a single cell suspension (1 × 106 cells/mL) was obtained. ACK Lysing Buffer (Quality Biological, Gaithersburg, MD) was then used to remove the red blood cells. For FACS evaluation of CD44, F4/80, Ly-6G (Gr-1, Gr1) and Ly-6B positive cells, the samples were incubated with antibodies {allophycocyanin (APC)-labeled rat anti-mouse F4/80 (eBioscience, San Diego, CA), APC-labeled rat anti-mouse Ly-6G (Gr-1, Gr1) (eBioscience, San Diego, CA), APC-labeled rat anti-mouse/human CD44 (eBioscience, San Diego, CA) and PE-labeled rat anti-mouse Ly-6B (AbD Serotec, Raleigh, NC)} for 30 min on ice. Isotype-matched APC-labeled rat IgG1 (eBioscience, San Diego, CA), APC-labeled rat IgG2a and IgG2b (eBioscience, San Diego, CA), and PE-labeled rat IgG2a (eBioscience, San Diego, CA) were used as negative controls, respectively. After the final wash, the cells were analyzed with Becton Dickinson FACSCalibur flow cytometer (BD Biosciences, Pharmingen). This experiment was repeated in triplicate.
2.10 Treatment of mouse femur fracture with SIM/SIM-mPEG micelles
Forty-five 10-week-old male Swiss Webster mice were fractured according to the procedure described in section 2.7 and randomly assigned into three groups (15 mice/group) (scheme 2). In order to minimize soft tissue inflammation caused by the surgery, the treatments were initiated 7 days post fracture. The mice were treated respectively with saline (vehicle control or CON) once daily, simvastatin acid (SIMA, equiv. SIM 6 mg/kg/day, intraperitoneal or i.p. injection) once daily or SIM/SIM-mPEG micelles (equiv. SIM 42 mg/kg/wk, intravenous or i.v. injection) once every 7 days by tail vein injection during the treatment period of two weeks. Simvastatin acid (SIMA) is the active form of SIM in vivo and was prepared as described before [29]. The overall SIM doses given to the SIMA group and SIM/SIM-mPEG group were equivalent while the drug injection times are different (14 times vs. twice). All mice were euthanized 21 days post fracture.
Scheme 2.

Experiment design of the therapeutic study of SIM/SIM-mPEG on osteotomized mice.
2.11 Evaluation of fracture healing
At necropsy, the fractured femurs were collected, fixed in 10% formalin and processed for further micro-CT imaging and analysis (Bruker SkyScan1172, Kontich, Belgium). Each femur was scanned and reconstructed into a 3D-structure with a voxel size of 5.5 μm. The X-ray tube voltage was 70 kV and the current was 141 μA, with a 0.5 mm thick aluminum filter. Exposure time was 530 ms. The X-ray projections were obtained at 0.7° intervals with a scanning angular rotation of 180° and eight frames were averaged for each rotation. 3D reconstructions were performed using NRecon software.
For the quantitative analysis, we specifically focused on the site of fracture callus formation. The position of each bone was corrected with DataViewer and then saved on a volume of interest (VOI) of 400 slices (resized by 2). This was followed by careful placement of a constant square region of interest (ROI) in the callus with 200 slices above and below the fracture line. To standardize analysis within the animals, special care was taken so as to keep the constant ROI in the center for each sample, with the fracture line and metal rod (fixation needle) serving as horizontal and vertical points of reference, respectively. A custom analysis process was performed with series of specific threshold settings in CTan (MicroPhotonic) and these were kept constant for all samples. The following parameters were measured and calculated: callus bone volume to tissue volume (BV/TV), callus bone surface to volume (BS/TV), trabecular number (Tb.N) and trabecular separation (Tb.Sp). For nonmetric indices, connectivity density (Conn.D) and mean polar moment of inertia (MMI) were also analyzed. While the total callus volume and the total mineralized callus tissue volumes are of great interests, the above analysis protocol was selected due to the large variation of the callus shape/volume.
After micro-CT analysis, the fractured femurs were decalcified using 14% EDTA at room temperature for 30 days. The decalcification solution was changed daily. The specimens were then paraffin embedded, sectioned (5 μm), stained with Safranin O and histologically evaluated.
2.12 Statistical analysis
For the quantitative analysis of micro-CT, data is expressed as mean ± SD. Statistical analysis was done by one-way analysis of variance (ANOVA) with LSD/Tamhane’s T2 test as post-hoc test. A value of P < 0.05 was considered statistically significant.
3. RESULTS
3.1 Characterization of the micelle formulation
Due to their amphiphilic nature, SIM-mPEG can spontaneously self-assemble into micelles and incorporate additional free SIM molecules into the hydrophobic micellar cores. As shown in Table 1, compared with the control, the particle size of the SIM-mPEG micelles increases with the free SIM incorporation into their hydrophobic cores. According to turbidity test (data not shown), for the film hydration method, a loading capacity of close to 100% (Wd/Wp) was achieved (i.e. 1 mg/mL SIM-mPEG can dissolve 1 mg of free SIM into the micelle solution) with micelle diameter at approximately 30 nm, a PDI value of 0.053 and a ζ-potential of −3.3 mV. Since the film hydration method provided higher drug loading efficiency, smaller particle size and more consistent PDI than the direct dissolution method, it was selected to prepare SIM/SIM-mPEG micelles for all studies. Using the pyrene-based fluorescence method, the critical micelle concentration (CMC) of SIM-mPEG was determined as ~ 10−6 M.
Table1.
Characterization of SIM/SIM-mPEG micelles
| Preparation Method |
Drug Loading Efficiency (%) |
Deff (nm)a | PDIa | ζ-Potential (mV) |
|---|---|---|---|---|
| Film Hydration | 99.76±0.04 | 30.09±0.22 | 0.053±0.015 | −3.29±0.29 |
| Direct Dissolution | 34.27±0.78 | 252.03±1.50 | 0.247±0.006 | −10.7±0.3 |
| Control | N/A | 18.46±1.50 | 0.396±0.013 | −5.96±0.35 |
Effective diameter (Deff) and polydispersity indices (PDI) were determined by DLS at 25 °C (n = 3)
During in vitro release study, SIM/SIM-mPEG micelles were found to quickly release 8-10 % of the encapsulated free SIM within 6 h (Fig. 1) followed by a sustained release profile. The ratio of SIM vs SIM-mPEG in the micelle formulation was found to affect drug release kinetics. Micelles with a lower SIM content produce relatively slower release characteristics. Due to the absence of esterases and the neutral pH of the releasing medium, the free SIM released is likely from the encapsulated free SIM rather than SIM cleaved from SIM-mPEG. In the in vivo setting, the local conditions in the micro-environment of the fracture site, including the low pH during the early stage of healing process (hematoma and inflammation phase) and the presence of enzymes (e.g. esterase) will lead to drug cleavage and release from the SIM-mPEG. To ensure a sustained presence of SIM at the fracture site, the micelle formulation containing 75% SIM equivalent in SIM-mPEG and 25% encapsulated free SIM was selected for the in vivo study.
Figure 1.
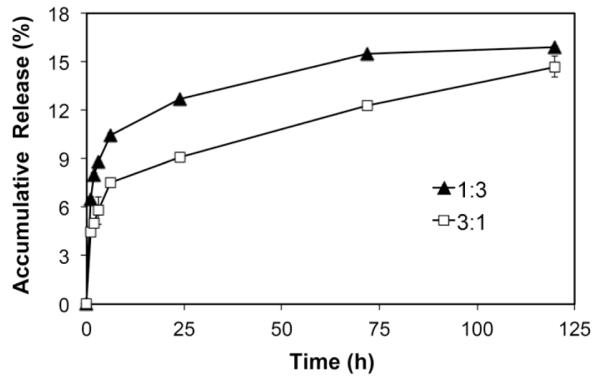
In vitro release of SIM from SIM/SIM-mPEG in PBS. Two micellar formulations of SIM/SIM-mPEG were prepared using different molar ratios of simvastatin distributed in prodrug portion and free drug portion (1:3 or 3:1). Data is expressed as mean ± SD, n=3.
3.2 SIM/SIM-mPEG micelle osteoinductivity is mediated in part through BMP-2
As shown in Figure 2, fluorescence microscopic analysis revealed that MC3T3 cells in culture were able to internalize Alexa Fluor 488 labeled SIM/SIM-mPEG micelles, and that the micelles subsequently trafficked to an endosomal/lysosomal compartment within the cells, as evidenced by co-localization of the Alexa Fluor 488 signal with the lysotropic marker dye DND-99. This is consistent with establishment within the cells of an intracellular depot of micelles that may provide a mechanism for sustained therapeutic action.
Figure 2.

Micelle internalization, lysosomal uptake in MC3T3 pre-osteoblast cell line. Hoechst 33342 (blue, nuclear), LysoTracker DND-99 (red, lysosomes), SIM-mPEG-Alexa Fluor 488 (green, micelles). Bar size = 100 μm.
To evaluate the ability of SIM/SIM-mPEG micelles to modulate the osteogenic phenotype, MC3T3 cells, an osteoblast lineage cell line, were treated with free SIM or SIM/SIM-mPEG micelles and examined for expression levels of the osteoinductive statin target gene BMP2 and the BMP2 regulated gene Dickkopf-related protein 1 (DKK1) using real time PCR. As shown in Figure 3, while less potent than an equivalent dose of free SIM, SIM/SIM-mPEG induced expression of both BMP2 and DKK1 mRNA levels. In contrast the treatments did not affect expression levels of BMP4, which is not a statin target gene. These results suggest that free SIM encapsulated in the SIM/SIM-mPEG micelles may be released to regulate the expression of BMP2.
Figure 3.

Induction of BMP2 expression and BMP2 target genes (qPCR)
3.3 Systemically administered SIM/SIM-mPEG micelles passively target to the fracture site
To explore the in vivo distribution of the systemically administered SIM/SIM-mPEG micelles, SIM/SIM-mPEG micelles were labeled with IRDye® 800CW by incorporation of SIM-mPEG-IRDye. As shown in Figure 4A, at one day post administration, the micelles were found to passively accumulate at the fractured femur, with intense and sustained NIR signals, which persisted for more than 7 days. Due to the renal clearance of the polymeric micelles, the NIR signal was found to distribute to the bladder at day 1 and 2 post-micelle administration. Weak signals were also observed at an anatomical location corresponding to the liver. After selection of the region of interest (ROI), the NIR signal intensity was analyzed semi-quantitatively (Fig. 4B). The signal intensity differences between the fracture site and the intact site were statistically significant (P < 0.05) up to 7 days, which further confirmed the direct visual observation.
Figure 4.

Targeting of SIM/SIM-mPEG micelles to the fracture site. (A) Near infrared optical imaging demonstrated the targeting of IRDye® 800CW-labeled SIM/SIM-mPEG to the fracture site in the right femur of the osteotomized Swiss-Webster mice (Supine position). (B) Semi-quantitative analysis of NIR signal intensity (from IRDye® 800CW-labeled SIM/SIM-mPEG micelles) at the selected region of interest. Data is presented as the mean ± SD (n = 3). The signal intensity differences between the fracture site and the intact site were statistically significant (t-test, P < 0.05)
3.4 Characterization of cell types in the facture callus demonstrating uptake and retention of SIM/SIM-mPEG micelles
To dissect the micelles’ targeting/retention mechanism at the fracture site, cells isolated from the callus tissues were analyzed by FACS. As shown in Figure 5, the most highly enriched cell populations showing micelle internalization were identified as Ly-6G (36.8 %) and F4/80 (30.8 %) positive cells, markers characteristic of monocytes and macrophages. Micelle internalization also was observed in Ly-6B+ cells (15.2 %) and CD44+ cells (14.2%), suggesting that neutrophils and chondrocytes also participated in the sequestration and retention of the Alexa Fluor 488-labeled SIM/SIM-mPEG at the fracture site.
Figure 5.
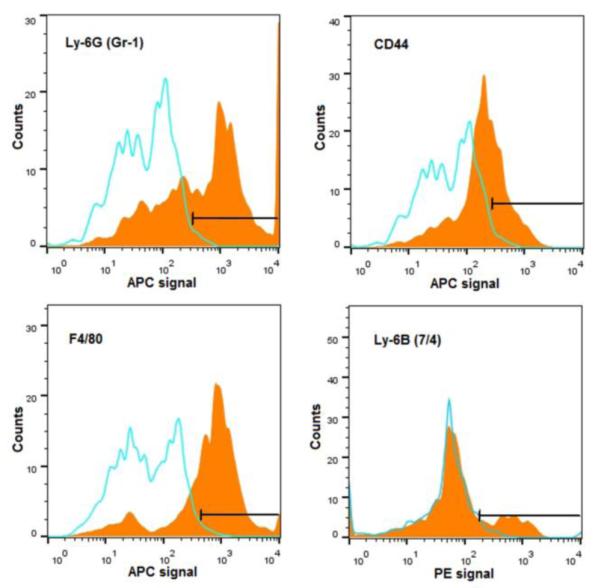
Representative data from Fluorescence Activated Cell Sorting (FACS) analysis of cells isolated from the fracture callus 24 h after systemic administration of Alexa Fluor® 488 labeled SIM/SIM-mPEG micelles. The histogram plots show the intensity of staining with the specific antibodies designated on the x-axis (orange fills) with isotype control antibodies (blue lines) on the same plots. The percentages represent the respective antibody positive cells among Alexa Fluor® 488 labeled SIM/SIM-mPEG micelles: 36.8 % were Ly-6G positive, 30.8 % were F4/80 positive, 15.2 % were Ly-6B positive and 14.2 % were CD44 positive. All data presented were isotype-control corrected.
3.5 Therapeutic efficacy of SIM/SIM-mPEG micelles on fracture healing
Callus mineralization is an essential feature of fracture repair. As shown in Figure 6, after 14 days of treatment, the SIM/SIM-mPEG treated group exhibited greater mineralization of the fracture callus. In contrast, the CON and SIMA treated groups demonstrated less mineralized callus formation at the fracture site (shown as dark areas or cavities in Fig. 6). Quantitative analysis of the micro-CT data further validates this observation. As shown in Figure 7, comparison of the SIM/SIM-mPEG treated and control animals reveals significant improvement in bone volume density (BV/TV, 6.07 % vs. 4.24 %), bone surface density (BS/TV, 8.61 mm−1 vs. 5.81 mm−1), connectivity density (Conn.D, 1691.00 mm−3 vs. 1136.12 mm−3), mean polar moment of inertia (MMI, 6.54 mm4 vs. 3.95 mm4), trabecular number (Tb.N, 2.13 mm−1 vs. 1.48 mm−1) and trabecular separation (Tb.Sp, 1.01 mm vs. 1.30 mm) for the SIM/SIM-PEG treated animals than CON (P < 0.05). When comparing equivalent doses of SIM to CON, none of the parameters demonstrated statistically significant differences except Tb.Sp (1.00 mm vs. 1.30 mm, P < 0.05).
Figure 6.
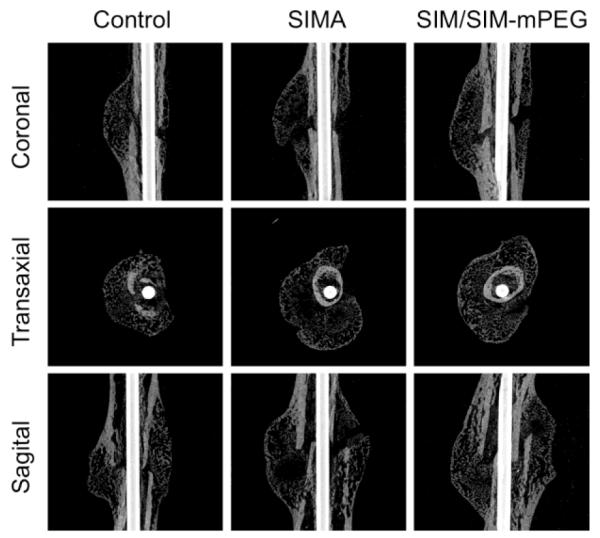
Representative micro-CT images of the healing of fractured femurs 21 days post osteotomy. The images for each animal group were selected from the animals showing the average values of callus Bone Volume/Tissue Volume (BV/TV) during quantitative analysis of the micro-CT data. SIM/SIM-PEG treated group exhibited extensively consolidated and calcified callus formation, as compared to CON and SIMA treated groups.
Figure 7.

Micro-CT morphometric analysis showing the quantitative indices of callus formation. Data is expressed as mean ± SD. * P < 0.05 when compared to CON.
Histological analysis of the callus tissue confirms the enhanced fracture healing in the SIM/SIM-mPEG micelle treated group compared to CON. As shown in Figure 8, at 21 days post fracture, the callus sections from the CON group were populated primarily by cells with morphologic features of chondrocytes surrounded by a proteoglycan rich ECM (stained red) consistent with a relatively early phase of fracture repair. In the callus sections from the animals treated with SIM/SIM-mPEG micelles, however, the fracture site has been replaced with mineralized tissues organized into woven bone, consistent with a more advanced stage of fracture repair. The histological features of SIM group are consistent with an intermediate stage between CON and SIM/SIM-mPEG groups [37].
Figure 8.

Representative images of Safranin O stained histological sections of callus from different treatment groups. The three panels on the right are an enlarged presentation (× 10) of the outlined region in the left panels (× 2). The callus sections of the SIM/SIM-mPEG group were devoid of chondrocytes and cartilage matrix, indicating advanced ossification. Bar = 1 mm (left panels) or 0.2 mm (right panels).
4. DISCUSSION
With an ever-growing aging population, delayed fracture healing and nonunion have placed a significant burden on the public healthcare system. Although several bone anabolic agents have been developed, the US FDA has not approved a systemic therapy that would improve fracture healing and nonunion. As an anti-lipidaemic drug class, statins have been shown to exhibit a bone anabolic effect [3, 4]; however their poor water solubility and lack of distribution to the skeleton [8, 11] have prevented their clinical use to modulate and accelerate the fracture-healing process.
The biologic events associated with fracture repair are well established and include the sequential development of a hematoma followed by local inflammation, granulation tissue formation, soft callus formation and eventual hard callus formation and remodeling [38]. We hypothesized that the early physiological processes associated with a fracture, especially the initial hematoma formation and subsequent inflammation, represent a window of opportunity for the delivery of bone anabolic agents to the fracture site for therapeutic intervention. We speculated that the inflammation associated with fracture repair creates an environment that is permissive for passive extravasation and retention of a systemically administered colloidal drug delivery system. We further hypothesized that the inflammatory and resident cells involved in the dynamic repair and remodeling process represent target cell populations that could sequester, retain and gradually release the drug loaded in the delivery vesicles to regulate the healing process.
To capitalize on the unique pathology of the fracture site for delivery of a statin with osteogenic capacity, we designed a dendritic amphiphilic simvastatin (SIM) prodrug by conjugating a cluster of three SIMs covalently to the chain terminus of mPEG. In designing the hydrophobic SIM trimer block, the ester bond was selected as the chemical linkage as it can not only hold the three SIMs together, but also can be hydrolyzed in vivo by esterases to release the conjugated SIM. Of the 3 SIMs conjugated to mPEG, two are in the prodrug form with an intact lactone ring and the other SIM is in the form of simvastatin acid. When exposed to in vivo environment factors (esterases, water, acidity, elevated temperature, etc.), the lactone ring will open to produce the active form of the drug simvastatin acid. Therefore, all of the drug released will be in the bioactive form. Click chemistry, one of the most efficient coupling methods [39], was then used to conjugate the SIM trimer to the mPEG terminus to form the amphiphilic macromolecule, which self-assembles into micelles. Because of the structural similarity between SIM and the SIM trimer, free SIM can be incorporated into the hydrophobic core to form SIM/SIM-mPEG micelles, which permits additional drug-loading capacity. This novel prodrug design of SIM not only solubilizes the highly hydrophobic drug, but also by self-assembly into micelles enables its selective extravasation at the fracture site associated with the initial phase of hematoma formation and inflammation. This preferential distribution of the SIM/SIM-mPEG micelles to the fracture site via the ELVIS mechanism [33-36] will increases the local SIM concentration to promote fracture healing.
More importantly, from the Chemistry, Manufacturing, and Controls (CMC) and clinical translational perspective, the SIM-mPEG prodrug design and its micelle formulation has significant advantages. In comparison to conventional polymeric micelle design, the SIM-mPEG’s hydrophobic SIM trimer segment was synthesized by an organic chemistry methodology that generates a prodrug with a definitive structure and molecular mass. Furthermore, a single molecular weight PEG (i.e. dPEG®) is already commercially available and “click” coupling of dPEG® with the SIM trimer would yield a SIM-mPEG prodrug with a definitive molecular weight, which will maintain the batch-to-batch reproducibility in chemical manufacturing. When administered in vivo, its pharmacokinetics behavior will also be easier to assess in comparison to polymeric prodrugs with polydispersity in molecular weight. A limited number of previous studies have reported the design, synthesis and self-assembly behavior of single molecular weight prodrugs into nanomedicines, primarily involving cancer chemotherapeutic agents [40, 41]. Our study is the first to extrapolate this design concept beyond oncology and validate its utility in a clinically relevant fracture model.
The CMC of SIM-mPEG was ~ 10−6 M, suggesting that the micelles are stable and therefore may have the capacity to retain their size/aggregation in the circulation after systemic administration [42]. This allows the micelles’ passive targeting to the fracture site as evidenced by the imaging data. Fluorescently tagged micelles were readily internalized by cultured murine preosteoblast MC3T3 cells and localize to the subcellular endosomal/lysosomal compartment. This pattern of cellular uptake is consistent with establishment within cells of an intracellular depot that permits protracted release of an active therapeutic agent and thus provide sustained therapeutic efficacy. In line with these observations, SIM/SIM-mPEG micelles retained the SIM-mediated osteoinductive activity, as evidenced by enhanced expression of BMP2 mRNA within micelle treated cells. The induction of DKK1, which is a BMP2 target gene, provides evidence that the induced BMP2 is biologically active.
In vivo imaging data support the passive targeting of the SIM/SIM-mPEG micelles to the fracture site. Flow cytometric analysis confirms that the micelles retention at the fracture site is due to cell-mediated sequestration. Specifically, the data indicate that the micelles were primarily internalized by F4/80 and Gr-1 positive myeloid cell populations and CD44 positive mesenchymal cells, consistent with sequestration in infiltrating macrophage/monocytes, as well as mesenchymal cells within the callus. The local uptake and retention of the micelles are consistent with our proposed model of an ELVIS mechanism, in which altered vasculature permeability results in extravasation of the colloids, which subsequently are internalized and retained by inflammatory cells present at the fracture site.
The data from micro-CT analysis is consistent with accelerated callus formation and enhanced mineralization in the SIM/SIM-mPEG treated group compared to the CON. This contrasts with the effects of the free SIM treatment, which were not statistically different from the CON. Histological analysis of the SIM/SIM-mPEG treated group revealed enhanced mineralization and consolidation of the fracture callus when compared to the CON, confirming the findings from the micro-CT analysis. Our findings with the systemic delivery of the statin prodrug differ from results obtained with local drug delivery approaches, which require utilization of a matrix for drug delivery and sustained drug release [3, 19-32]. Unlike the local matrix delivery systems, direct surgical access to the fracture site is not required in this case. In addition, the uptake and retention by cells at the fracture site provide a unique mechanism for drug retention and sustained release.
While the in vivo treatment study validates the therapeutic efficacy of SIM/SIM-mPEG, additional studies are necessary to further optimize this new simvastatin micelle formulation for clinical translation. Though the animal group size selected allowed us to detect significant differences between SIM/SIM-mPEG treated animals and CONs in several of the histomorphometric parameters, the comparison between SIM/SIM-mPEG and free SIM was not statistically significant. This may be partially due to the variability in the pattern of fracture healing among the animals and potentially can be overcome by further increasing the group size. The SIM dosing level may be an additional contributing factor to be considered. In both the imaging and the treatment studies, SIM/SIM-mPEG was administered at 7 days post osteotomy to allow initial healing of the soft tissue injury created during the model generation. While this practice may greatly increase the distribution of the micelles to the site of direct bone injury, further studies are needed to determine the optimal time frame for drug delivery and to define the potential differential pattern of cell uptake during the progressive stages of callus formation and healing. The therapeutic benefits observed in this study were demonstrated in relatively young mice. Further studies need to be done in animals with known difficulties in fracture healing, such as aged mice or mice chronically exposed to glucocorticoids.
5. CONCLUSIONS
In this study, we report the successful synthesis of a novel amphiphilic dendritic simvastatin-methoxy polyethylene glycol conjugate (SIM-mPEG), which spontaneously self-assembles into stable micelles. Upon formulation with free simvastatin (SIM), the SIM/SIM-mPEG micelles demonstrated passive targeting to the fracture site in a femur osteotomy mouse model. As a direct result of the alteration of SIM’s pharmacokinetics and biodistribution profile, the targeting and retention of SIM at the fracture site led to accelerated fracture healing, as evident in the micro-CT and histological evaluation. Upon further optimization, this statin prodrug micelle system may have the potential to be translated into clinical applications in managing impaired fracture healing.
The success of this formulation also corroborates the novel macromolecular prodrug design as a viable strategy to solubilize hydrophobic compounds. Our results confirm that the colloidal nature of the formulation imparts tropism for the payloads, facilitating their in vivo passive targeting to sites of tissue injury and associated inflammation. We believe that this design principle can be extrapolated to the formulations of additional therapeutic agents to improve the treatment of many other inflammatory diseases.
Scheme 1.

Synthesis of the amphiphilic macromolecular prodrug SIM-mPEG. a) PPh3 (5 eq), I2 (4 eq), DCM, RT, 24 h, 79.1%; b) NaN3 (20 eq), DMF, 100°C, 24h, 93.4%; c) 3-butyn-1-ol (6 eq), TsOH·H2O (0.1 eq), RT, 3h, 30.3%; d) succinic anhydride (6eq), Et3N (4 eq), DMAP (0.4 eq), 45°C, 20 h, 94.6%; e) DCC (2.5 eq), SIM (2.5 eq), DMAP (0.06 eq), 0°C, 1.5 h, 58.6%; f) 6 (2.5 eq), 3 (1 eq), CuSO4·5H2O (1 eq), L-ascorbic acid sodium salt (2 eq), t-BuOH/H2O, 60 h, 77.3%.
ACKNOWLEDGMENTS
This study was supported in part by NIH (R01 AR053325, AR062680) and Nebraska Arthritis Outcome Research Center (NAORC). We thank Mr. Arun Tatiparthi of Micro Photonics for his assistance with the micro-CT analysis. Y.Z. was supported by the Bukey Fellowship from University of Nebraska Medical Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
7. REFERENCES
- 1.Burge R, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22(3):465–75. doi: 10.1359/jbmr.061113. [DOI] [PubMed] [Google Scholar]
- 2.Laufs U, Liao JK. Direct vascular effects of HMG-CoA reductase inhibitors. Trends Cardiovasc Med. 2000;10(4):143–8. doi: 10.1016/s1050-1738(00)00044-x. [DOI] [PubMed] [Google Scholar]
- 3.Mundy G, et al. Stimulation of bone formation in vitro and in rodents by statins. Science. 1999;286(5446):1946–9. doi: 10.1126/science.286.5446.1946. [DOI] [PubMed] [Google Scholar]
- 4.Garrett IR, Mundy GR. The role of statins as potential targets for bone formation. Arthritis Res. 2002;4(4):237–40. doi: 10.1186/ar413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Staal A, et al. The ability of statins to inhibit bone resorption is directly related to their inhibitory effect on HMG-CoA reductase activity. J Bone Miner Res. 2003;18(1):88–96. doi: 10.1359/jbmr.2003.18.1.88. [DOI] [PubMed] [Google Scholar]
- 6.Oxlund H, Dalstra M, Andreassen TT. Statin given perorally to adult rats increases cancellous bone mass and compressive strength. Calcif Tissue Int. 2001;69(5):299–304. doi: 10.1007/s00223-001-2027-5. [DOI] [PubMed] [Google Scholar]
- 7.Yaturu S. Skeletal effects of statins. Endocr Pract. 2003;9(4):315–20. doi: 10.4158/EP.9.4.315. [DOI] [PubMed] [Google Scholar]
- 8.Jadhav SB, Jain GK. Statins and osteoporosis: new role for old drugs. J Pharm Pharmacol. 2006;58(1):3–18. doi: 10.1211/jpp.58.1.0002. [DOI] [PubMed] [Google Scholar]
- 9.Maeda T, et al. Induction of osteoblast differentiation indices by statins in MC3T3-E1 cells. J Cell Biochem. 2004;92(3):458–71. doi: 10.1002/jcb.20074. [DOI] [PubMed] [Google Scholar]
- 10.Garrett IR, Gutierrez G, Mundy GR. Statins and bone formation. Curr Pharm Des. 2001;7(8):715–36. doi: 10.2174/1381612013397762. [DOI] [PubMed] [Google Scholar]
- 11.Park JB. The use of simvastatin in bone regeneration. Med Oral Patol Oral Cir Bucal. 2009;14(9):e485–8. [PubMed] [Google Scholar]
- 12.Yao W, et al. Simvastatin did not prevent nor restore ovariectomy-induced bone loss in adult rats. J Musculoskelet Neuronal Interact. 2006;6(3):277–83. [PubMed] [Google Scholar]
- 13.von Stechow D, et al. Does simvastatin stimulate bone formation in vivo? BMC Musculoskelet Disord. 2003;4:8. doi: 10.1186/1471-2474-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rejnmark L, et al. Effects of simvastatin on bone turnover and BMD: a 1-year randomized controlled trial in postmenopausal osteopenic women. J Bone Miner Res. 2004;19(5):737–44. doi: 10.1359/JBMR.040209. [DOI] [PubMed] [Google Scholar]
- 15.von Knoch F, et al. Promotion of bone formation by simvastatin in polyethylene particle-induced osteolysis. Biomaterials. 2005;26(29):5783–9. doi: 10.1016/j.biomaterials.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 16.Ho ML, et al. Simvastatin increases osteoblasts and osteogenic proteins in ovariectomized rats. Eur J Clin Invest. 2009;39(4):296–303. doi: 10.1111/j.1365-2362.2009.02092.x. [DOI] [PubMed] [Google Scholar]
- 17.Uzzan B, et al. Effects of statins on bone mineral density: a meta-analysis of clinical studies. Bone. 2007;40(6):1581–7. doi: 10.1016/j.bone.2007.02.019. [DOI] [PubMed] [Google Scholar]
- 18.Gutierrez GE, et al. Transdermal application of lovastatin to rats causes profound increases in bone formation and plasma concentrations. Osteoporos Int. 2006;17(7):1033–42. doi: 10.1007/s00198-006-0079-0. [DOI] [PubMed] [Google Scholar]
- 19.Ayukawa Y, et al. Local application of statin promotes bone repair through the suppression of osteoclasts and the enhancement of osteoblasts at bone-healing sites in rats. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;107(3):336–42. doi: 10.1016/j.tripleo.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 20.Skoglund B, Aspenberg P. Locally applied Simvastatin improves fracture healing in mice. BMC Musculoskelet Disord. 2007;8:98. doi: 10.1186/1471-2474-8-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seto H, et al. Topical administration of simvastatin recovers alveolar bone loss in rats. J Periodontal Res. 2008;43(3):261–7. doi: 10.1111/j.1600-0765.2007.01024.x. [DOI] [PubMed] [Google Scholar]
- 22.Lee Y, et al. The effect of local simvastatin delivery strategies on mandibular bone formation in vivo. Biomaterials. 2008;29(12):1940–9. doi: 10.1016/j.biomaterials.2007.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanigo T, Takaoka R, Tabata Y. Sustained release of water-insoluble simvastatin from biodegradable hydrogel augments bone regeneration. J Control Release. 2010;143(2):201–6. doi: 10.1016/j.jconrel.2009.12.027. [DOI] [PubMed] [Google Scholar]
- 24.Fukui T, et al. Therapeutic effect of local administration of low-dose simvastatinconjugated gelatin hydrogel for fracture healing. J Bone Miner Res. 2012;27(5):1118–31. doi: 10.1002/jbmr.1558. [DOI] [PubMed] [Google Scholar]
- 25.Pradeep AR, et al. Clinical efficacy of subgingivally delivered 1.2-mg simvastatin in the treatment of individuals with Class II furcation defects: a randomized controlled clinical trial. J Periodontol. 2012;83(12):1472–9. doi: 10.1902/jop.2012.110716. [DOI] [PubMed] [Google Scholar]
- 26.Morris MS, et al. Injectable simvastatin in periodontal defects and alveolar ridges: pilot studies. J Periodontol. 2008;79(8):1465–73. doi: 10.1902/jop.2008.070659. [DOI] [PubMed] [Google Scholar]
- 27.Ho MH, et al. Highly efficient release of lovastatin from poly(lactic-co-glycolic acid) nanoparticles enhances bone repair in rats. J Orthop Res. 2011;29(10):1504–10. doi: 10.1002/jor.21421. [DOI] [PubMed] [Google Scholar]
- 28.Garrett IR, et al. Locally delivered lovastatin nanoparticles enhance fracture healing in rats. J Orthop Res. 2007;25(10):1351–7. doi: 10.1002/jor.20391. [DOI] [PubMed] [Google Scholar]
- 29.Yoshinari M, et al. Controlled release of simvastatin acid using cyclodextrin inclusion system. Dent Mater J. 2007;26(3):451–6. doi: 10.4012/dmj.26.451. [DOI] [PubMed] [Google Scholar]
- 30.Killeen AC, et al. Impact of local and systemic alendronate on simvastatin-induced new bone around periodontal defects. J Periodontol. 2012;83(12):1463–71. doi: 10.1902/jop.2012.110683. [DOI] [PubMed] [Google Scholar]
- 31.Lee Y, et al. Role of prostaglandin pathway and alendronate-based carriers to enhance statin-induced bone. Mol Pharm. 2011;8(4):1035–42. doi: 10.1021/mp200045p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chou J, et al. Controlled release of simvastatin from biomimetic beta-TCP drug delivery system. PLoS One. 2013;8(1):e54676. doi: 10.1371/journal.pone.0054676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang D, Goldring SR. The bone, the joints and the Balm of Gilead. Mol Pharm. 2011;8(4):991–3. doi: 10.1021/mp200328t. [DOI] [PubMed] [Google Scholar]
- 34.Ren K, et al. Early detection and treatment of wear particle-induced inflammation and bone loss in a mouse calvarial osteolysis model using HPMA copolymer conjugates. Mol Pharm. 2011;8(4):1043–51. doi: 10.1021/mp2000555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan F, et al. Dexamethasone prodrug treatment prevents nephritis in lupus-prone (NZB × NZW)F1 mice without causing systemic side effects. Arthritis Rheum. 2012;64(12):4029–39. doi: 10.1002/art.34667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan F, et al. Development of macromolecular prodrug for rheumatoid arthritis. Adv Drug Deliv Rev. 2012;64(12):1205–19. doi: 10.1016/j.addr.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosen CJ, American Society for Bone and Mineral Research . Primer on the metabolic bone diseases and disorders of mineral metabolism. 8th ed xxvi. Wiley-Blackwell; Ames, Iowa: 2013. p. 1078. [Google Scholar]
- 38.Marsh DR, Li G. The biology of fracture healing: optimising outcome. Br Med Bull. 1999;55(4):856–69. doi: 10.1258/0007142991902673. [DOI] [PubMed] [Google Scholar]
- 39.Hein CD, Liu XM, Wang D. Click chemistry, a powerful tool for pharmaceutical sciences. Pharm Res. 2008;25(10):2216–30. doi: 10.1007/s11095-008-9616-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cui H, et al. Amino Acid sequence in constitutionally isomeric tetrapeptide amphiphiles dictates architecture of one-dimensional nanostructures. J Am Chem Soc. 2014;136(35):12461–8. doi: 10.1021/ja507051w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen Y, et al. Prodrugs forming high drug loading multifunctional nanocapsules for intracellular cancer drug delivery. J Am Chem Soc. 2010;132(12):4259–65. doi: 10.1021/ja909475m. [DOI] [PubMed] [Google Scholar]
- 42.Lukyanov AN, et al. Polyethylene glycol-diacyllipid micelles demonstrate increased acculumation in subcutaneous tumors in mice. Pharm Res. 2002;19(10):1424–9. doi: 10.1023/a:1020488012264. [DOI] [PubMed] [Google Scholar]