

Abstract
RNA polymerase III (Pol III) synthesizes tRNAs and other small noncoding RNAs to regulate protein synthesis. Dysregulation of Pol III transcription has been linked to cancer, and germline mutations in genes encoding Pol III subunits or tRNA processing factors cause neurogenetic disorders in humans, such as hypomyelinating leukodystrophies and pontocerebellar hypoplasia. Here we describe an autosomal recessive disorder characterized by cerebellar hypoplasia and intellectual disability, as well as facial dysmorphic features, short stature, microcephaly, and dental anomalies. Whole-exome sequencing revealed biallelic missense alterations of BRF1 in three families. In support of the pathogenic potential of the discovered alleles, suppression or CRISPR-mediated deletion of brf1 in zebrafish embryos recapitulated key neurodevelopmental phenotypes; in vivo complementation showed all four candidate mutations to be pathogenic in an apparent isoform-specific context. BRF1 associates with BDP1 and TBP to form the transcription factor IIIB (TFIIIB), which recruits Pol III to target genes. We show that disease-causing mutations reduce Brf1 occupancy at tRNA target genes in Saccharomyces cerevisiae and impair cell growth. Moreover, BRF1 mutations reduce Pol III–related transcription activity in vitro. Taken together, our data show that BRF1 mutations that reduce protein activity cause neurodevelopmental anomalies, suggesting that BRF1-mediated Pol III transcription is required for normal cerebellar and cognitive development.
Three RNA polymerases synthesize the different classes of RNAs in eukaryotic cells. Pol I synthesizes most ribosomal RNAs; Pol II mRNAs and miRNAs; and Pol III a variety of noncoding RNAs with a structural and catalytic function, e.g., tRNAs, 7SK RNA, 5S rRNA, and U6 snRNA (Schramm and Hernandez 2002; White 2011; Dieci et al. 2013). Pol II–dependent transcription has attracted most attention because its mRNA products are protein coding, but recent years have seen an increasing interest in Pol III transcription, not least because of its implication in human disorders. Increased Pol III–dependent transcription has been linked to cell transformation and cancer (Marshall and White 2008; Cabarcas and Schramm 2011), and germline mutations in components of the Pol III or tRNA processing machinery have been associated with neurogenetic disorders, such as pontocerebellar hypoplasia (PCH), a heterogeneous group of severe, often lethal, progressive neurodegenerative conditions characterized by cerebellar hypoplasia, progressive microcephaly, seizures, and profound developmental impairment. At least 10 types of PCH are currently recognized, and mutations causing the most prevalent subtypes, PCH2 and PCH4, are predicted to lead to loss of function of tRNA splicing endonuclease (TSEN); other PCH types display anomalies of tRNA processing as well (Namavar et al. 2011; Akizu et al. 2013; Rudnik-Schöneborn et al. 2014; Schaffer et al. 2014). However, the pathomechanism leading to PCH is poorly understood in many subtypes.
More recently, mutations in the catalytic Pol III subunits POLR3A and POLR3B have been identified in syndromic hypomyelinating leukodystrophies, such as hypomyelination, hypodontia, and hypogonadotropic hypogonadism (4H syndrome) (Bernard et al. 2011; Saitsu et al. 2011; Tetreault et al. 2011). POLR3A mutations lead to a decrease in POLR3A levels and are predicted to interfere with the interaction with other Pol III subunits, which in turn would perturb Pol III–mediated transcription; a similar mechanism has been postulated for POLR3B-associated leukodystrophies. Although the molecular pathophysiology of these diseases is not fully understood, these findings suggest that an intact Pol III apparatus is essential for cognitive development and the structural and functional integrity of the cerebellum and white matter.
The basal Pol III transcription machinery requires the orchestrated interaction of Pol III, a multiprotein enzyme consisting of 17 polypeptide subunits, TFIIIB (transcription factor IIIB), and TFIIIC (White 2011; Vannini and Cramer 2012). These factors, as well as the underlying mechanisms of transcription initiation, are conserved in eukaryotes. TFIIIC recognizes and binds to specific sequence blocks in internal promoters (type 1 and 2 promoters) of target genes, such as tRNA genes. TFIIIC then recruits TFIIIB, which is usually composed of the B-related factor BRF1, B double prime 1 BDP1, and the TATA box-binding protein TBP, each of which is required for TFIIIB function in vitro (White 2011). TFIIIB recruits Pol III for transcription initiation. For the transcription of human RNA genes with external (type 3) promoters, such as U6 snRNA, BRF1 is replaced by BRF2 in TFIIIB, and TFIIIC is not required (Schramm and Hernandez 2002). Notably, TBP is a candidate gene for cognitive disorders in humans (Rooms et al. 2006), and TBP polyglutamine expansions cause spinocerebellar ataxia type 17 (Nakamura et al. 2001).
Here, we describe an autosomal recessive human syndrome characterized by intellectual disability and cerebellar, dental, and skeletal anomalies. We used whole-exome sequencing (WES) (Bamshad et al. 2011) and in vivo and in vitro assays to provide evidence that partial loss-of-function BRF1 mutations cause this recognizable disease by impairing Pol III–mediated transcription.
Results
Clinical delineation of a cerebellar-facial-dental syndrome
By comparing the clinical features of three pairs of siblings (three female and three male) (Fig. 1A–C) from three unrelated families, we observed a striking clinical overlap pointing to a previously undescribed disorder characterized by cerebellar hypoplasia, intellectual disability, characteristic facial dysmorphisms, and dental anomalies (Table 1; for the complete version of Table 1, see Supplemental Material). In each of the three families, two affected children were born to unaffected parents (Fig. 1C), suggesting autosomal recessive inheritance. In family 3 with first-cousin parents, Leber congenital amaurosis (LCA) segregated independently from cerebellar hypoplasia in two siblings (Fig. 1C). The six children affected by the cerebellar-facial-dental syndrome showed similar dysmorphic features that included sparse eyebrows, wave-shaped palpebral fissures, apparently low-set ears, malocclusion, and prominent upper incisors (Fig. 1A). They had proportionate short stature and microcephaly of prenatal onset, mild-to-severe intellectual disability with speech delay, scoliosis, and sparse hair (Table 1). Three children had laryngeal stridor or laryngomalacia. Cranial magnetic resonance imaging revealed a pattern that was reminiscent of TSEN-related PCH, with a very thin corpus callosum, a flattened brainstem, and cerebellar vermis hypoplasia; however, the pons was relatively well formed (Fig. 1B). On skull and orthopanoramic x-rays, we saw bialveolar protrusion with prominent alveolar processes as well as taurodontism, a specific malformation of the pulp of molar teeth. Array-CGH did not detect causative copy number variants. We refer to this syndrome as cerebellar-facial-dental syndrome.
Figure 1.

BRF1 mutations cause a cerebellar-dental-skeletal syndrome. (A) Patients 1 and 2 (family 1) at the ages of 12 and 10 yr, patients 3 and 4 (family 2) at the ages of 10 and 4 yr, and patients 5 and 6 (family 3). Note characteristic facial dysmorphism and dental anomalies. (B) Brain MRI (top, sagittal scans; bottom, coronal scans) of patients 1–4 (P1–P4) at the ages of 14 yr, 9 yr, 12 yr, and 18 mo, respectively, showing a thin corpus callosum (white filled arrows), flattened brainstem (white unfilled arrows), and cerebellar hypoplasia (black unfilled arrows). (C) Pedigrees of family 1 (left), family 2 (middle), and family 3 (right) with genotypes for BRF1 missense alterations. In family 2, the p.Pro292His mutation was likely transmitted by the unaffected father who did not participate in the study. In family 3, the two individuals denoted by an asterisk had Leber congenital amaurosis caused by a homozygous RDH12 mutation. (D) Multiple sequence alignment of BRF1 orthologs and human TFIIB showing evolutionary conservation of mutant BRF1 amino acid residues.
Table 1.
Clinical features of individuals with cerebellar-dental-skeletal syndrome

Identification of BRF1 mutations
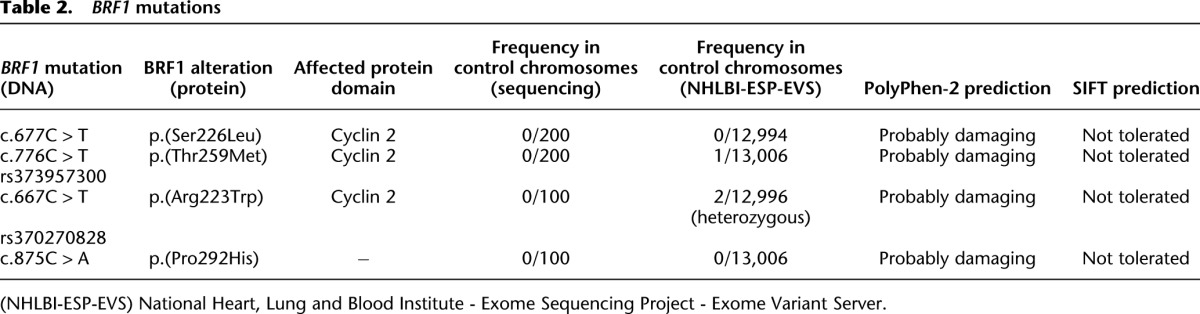
We performed WES in one or two affected individuals from each of the three families. Under the simplest assumption of genetic homogeneity for this rare autosomal recessive phenotypic constellation, we filtered for variants in the same gene with a predicted impact on protein function or RNA splicing that are rare in control populations of European descent. Because of parental consanguinity in family 3 and the lack of consanguinity in families 1 and 2, we searched for a gene harboring a rare homozygous variant in the affected individuals from family 3 and rare compound heterozygous (or homozygous) variants in the affected children in families 1 and 2. Subsequent to WES analysis and filtering, there was a single gene that contained biallelic mutations in all affected children: BRF1. Each affected child had two variants that predicted missense alterations, for a total of four distinct mutations. We confirmed all four BRF1 alleles, as well as cosegregation with the cerebellar-facial-dental syndrome, in each family by Sanger sequencing (Fig. 1C; Supplemental Fig. 1). In family 1, the affected siblings were compound heterozygous for a maternally inherited c.677C > T mutation and a paternally inherited c.776C > T mutation, predicting p.Ser226Leu (S226L) and p.Thr259Met (T259M), respectively. In family 2, the affected sisters had inherited a c.667C > T mutation from their mother and carried a c.875C > A mutation that was likely transmitted from their father, from whom no DNA was available (Fig. 1C). These mutations predict p.Arg223Trp (R223W) and p.Pro292His (P292H) missense alterations, respectively. The two affected boys in family 3 were homozygous for the c.677C > T (p.Ser226Leu) mutation that had been identified in family 1. This mutation cosegregated with the cerebellar-facial-dental syndrome in family 3, while LCA was caused by a homozygous mutation in RDH12 (c.784dupG; p.Ala262Glyfs*11), a gene previously shown to be mutated in LCA (Janecke et al. 2004). The four BRF1 mutations were absent from ethnically matched controls as shown by sequencing and were not present or very rare in public SNP databases (Table 2).
Table 2.
BRF1 mutations
The longest BRF1 isoform is a 677-amino-acid protein that, together with TBP and BDP1, forms the transcription factor IIIB (TFIIIB), which recruits Pol III to its templates and is involved in promoter opening (White 2011). The N-terminal region of BRF1 has a role in DNA binding and Pol III recruitment (Kassavetis et al. 1998), is homologous to the Pol II transcription factor TFIIB, and contains an N-terminal zinc ribbon domain and two cyclin domains (Khoo et al. 2014). The BRF1 gene on human chromosome 14q32.33 encodes several functional isoforms, some of which lack the zinc ribbon domain (McCulloch et al. 2000). The four identified missense alterations invariably affect amino acids that are evolutionarily conserved, including in orthologs from mouse and zebrafish (Fig. 1D), but are not conserved in BRF2. In Saccharomyces cerevisiae, Brf1 (Colbert and Hahn 1992), Arg223, Thr259, and Pro292 are conserved, whereas Ser226 is replaced conservatively by an alanine; for clarity, we will use the human amino acid numbering henceforth.
Functional analysis of brf1 in zebrafish
As a first test of the functional candidacy of BRF1, we sought an in vivo model with a credible phenotypic surrogate. We noted that all probands in our study have cerebellar hypoplasia and microcephaly, both of which have been modeled previously in zebrafish embryos for a variety of neurodevelopmental disorders (Golzio et al. 2012; Dauber et al. 2013; Margolin et al. 2013; Bernier et al. 2014). We therefore turned to developing zebrafish embryos and asked whether either suppression or overexpression of brf1 has an effect on neurogenesis, head size, and/or cerebellar morphology.
Given that brf1 has two copies in the zebrafish genome due to the teleost-specific genome duplication, we generated splice blocking morpholinos (sbMOs) for both copies (brf1a and brf1b), and we scored for neurogenesis by measuring the area of the optic tectum, the area between the eyes as a surrogate for brain size, and the cerebellar integrity. We note that while brf1a is known to be expressed at multiple stages of development as well as in multiple organs, little is known about brf1b in zebrafish. Our in-house RNA-seq data that interrogate the transcripts expressed specifically in the head of 5 d post fertilization (d.p.f.) zebrafish embryos indicated that brf1b has significantly increased expression when compared to brf1a. Indeed, brf1b was expressed, on average, 11.59 CPM (counts per million mapped reads), while brf1a expression was 4.32 CPM. In triplicate experiments scored by two investigators masked to the injection cocktails, suppression of brf1a gave no appreciable phenotypes (data not shown). In contrast, suppression of brf1b with a MO against the donor site of exon 8 (MO#1) (Supplemental Fig. 2) resulted in a significant reduction of both the size of the optic tectum (Fig. 2B′,D,F′,H) and the total area between the eyes (Fig. 2B,C,F,G). Furthermore, we observed cerebellar hypoplasia as determined by the lack of axons in the midline of the morphant zebrafish cerebellum (Fig. 2B′,F′,I). We did not observe further overt morphological abnormalities or developmental delay in the injected embryos as judged by the shape and position of the heart, the absence of edema, the number and shape of somites, and the presence of a normal swim bladder. The observed phenotypes were specific; coinjection of MO and wild-type (WT) human mRNA was able to rescue the phenotypes (Fig. 2B–D,F–I). To assess the functionality of the alleles, we coinjected MO with human mRNA bearing one of the variants of interest (P292H, R223W, S226L, and T259M) at a time. In the context of BRF1 isoform 2 (Fig. 2E), P292H and R223W scored as functionally nulls as they failed to rescue the reduction in head size and optic tecta as well as the cerebellar hypoplasia (Fig. 2F–I), while S226L and T259M scored as hypomorphic: The mutants were significantly more affected than MO + WT but not as severe as MO alone. Strikingly, testing of three of the alleles (R223W, S226L, T259M) in isoform 1 showed them all to be benign (rescue was indistinguishable from WT) (Fig. 2A–D), suggesting that the effect of the mutations is isoform specific. Overexpression of WT human BRF1 and human P292H, R223W, S226L, or T259M BRF1 showed no difference in head size, optic tecta, or cerebellar hypoplasia (Fig. 2G′,H′,I′).
Figure 2.

Functional annotation of variants in isoforms 1 and 2 of BRF1 and its effects on the head size, optic tectum size, and cerebellar formation in zebrafish embryos. (A,E) Schematic representation of the location of BRF1 variants examined in zebrafish within each of the two isoforms evaluated: isoform 1 (NP_001229717.1; 650 aa) and isoform 2 (NP_001229715.1; 584 aa). In purple are alleles that when tested are shown to be null, and in brown are alleles that score as hypomorphs based on zebrafish assays. (B,B′) Dorsal views of control zebrafish embryos and embryos injected with brf1b MO, brf1b MO + WT human BRF1, and brf1bMO + variant (R223W, S226L, or T259M) human BRF1 RNA in the context of isoform 1, respectively, at 3 d.p.f., stained with anti-α acetylated tubulin. (B) Head size measurements were taken using brightfield images (highlighted with a yellow outline in panel F). (B′) The area of the optic tecta was measured in the fluorescent images (highlighted with a cyan oval in panel F′). (C,D) Bar graphs showing the relative head size (C) and the optic tecta area (D). Data are presented as mean ± SE. Two-tailed t-tests were performed to assess statistical significance. The embryos coinjected with the MO and each of the variants were statistically different from MO alone but not statistically different when compared to embryos injected with MO + WT human BRF1, therefore scoring as benign. (F–I′) Functional assessment of the BRF1 missense variants in the context of isoform 2. Three d.p.f. embryos injected with brf1b MO, brf1b MO + WT human BRF1, brf1b MO + variant human BRF1 RNA (P292H, R223W, S226L, or T259M), or variant human BRF1 RNA alone in the context of isoform 2 were observed following staining with anti-α acetylated tubulin for head size (F) and for optic tecta area (F′), as well as for cerebellar defects (F′; illustrated with a red dashed box where maximum disorganization is observed). (G–I′) Bar graphs showing average head size, optic tecta area, as well as the percentage of embryos with cerebellar defects evaluated among each condition. To assess statistical significance among the evaluated conditions, two-tailed t-tests were performed for head size and optic tecta, and χ2 tests were performed for cerebellar disorganization to evaluate statistical significance across the conditions. In the context of isoform 2, functional analysis using zebrafish show that P292H and R223W are null, while S226L and T259M are hypomorphic alleles, showing an isoform-specific effect in which head size and optic tecta size are reduced and cerebellar disorganization occurs. No significant effects were observed for any of the phenotypes when the variants themselves were overexpressed. Each experiment was done at minimum in triplicate with at least 50 embryos per condition per replicate. (***) P-value ≤ 0.001 relative to MO + WT; (◊) P-value ≤ 0.001 relative to controls; (†) P-value ≤ 0.01 relative to MO.
To confirm the specificity of our assay, we designed a second sbMO against the donor site of brf1b exon 6 (MO#2). Injections of MO#2 recapitulated the results obtained with MO#1, showing a reduction of head size and optic tecta as well as cerebellar disorganization phenotypes in 3 d.p.f. embryos; again, the phenotypes could be at least partially rescued by coinjection of human WT BRF1 mRNA (Supplemental Fig. 3). Further supplementing these findings, we utilized the CRISPR system and induced aberrations in brf1b by CAS9/gRNA. In the F0 brf1bCas9/gRNA, we observed phenotypes that are fully concordant with the brf1b morphant phenotypes produced by both sbMOs (Supplemental Fig. 3). We conclude that suppression of brf1 leads to phenotypes that recapitulate key symptoms of the human pathology, arguing in favor of the candidacy of this gene for disease pathogenesis.
BRF1 mutations affect cell growth
We next turned to the question of the precise effect of the discovered missense alleles. Consistent with their predicted pathogenic nature, yeast lacking the chromosomal BRF1 gene and expressing BRF1 with mutations corresponding to R223W or P292H did not grow in spot dilution tests (Fig. 3A), consistent with loss of Brf1 function and similar to our zebrafish functional assay in which R223W and P292H were classified as nulls. Yeast cells transformed with plasmids containing BRF1 substitutions T259M or S226L (as found in families 1 and 3) or A226S (reflecting the human reference sequence) grew normally. We observed similar results in liquid cultures (Supplemental Fig. 4). To mimic the compound heterozygosity present in families 1 and 2, we transformed yeast with two distinct plasmids expressing the BRF1 variants R223W/P292H or S226L/T259M. Whereas S226L/T259M yeast showed essentially normal growth, the R223W/P292H combination was lethal (Fig. 3A). With respect to the mutations identified in family 2, these results further supported the candidacy of human biallelic BRF1 missense alterations causing the cerebellar-facial-dental syndrome.
Figure 3.

BRF1 mutations cause growth defects and reduced target promoter occupancy. (A) BRF1 mutations affect cell growth. Spot dilutions of the variants introduced into a BRF1 knockout strain grown at 30°C. Wild-type (WT) and variant Brf1 were encoded on plasmids. For the combination of two mutations, two plasmids were used, each harboring one mutation and a distinct marker. (5-FOA)5-fluoroorotic acid. (B) Three-dimensional modeling of human BRF1 missense alterations. The four identified amino acid substitutions were mapped to the structure of human TFIIB (green) in a complex with TBP (purple) and DNA (pdb code 1C9B) (Tsai and Sigler 2000). Amino acids affected by mutations in families 1 and 2 are shown in yellow and orange, respectively. (C–E) BRF1 variants show a decreased occupancy of tRNA promoters in yeast. Fold enrichments of ChIP experiments performed with tandem affinity purification (TAP)—tagged BRF1 variants in yeast. Data are presented as mean ± SD. (C) Mutations A226S to serine or S226L show no effect on fold enrichment compared to WT. (D) Mutation T259M leads to decreased fold enrichment on tRNA genes. (E) Combination of the two variants results in a much lower occupancy of the S226L and T259M variants. Summing up both signals results in less occupancy of the two mutated BRF1 than the WT BRF1.
BRF1 mutations impair Pol III–dependent transcription
To investigate whether the altered BRF1 residues may be important for DNA binding, we mapped the mutated BRF1 residues on the known crystal structure of the homologous human TFIIB-TBP-DNA complex (Tsai and Sigler 2000). Residues Arg223 and Thr259 are conserved in human TFIIB (Fig. 1D) and are predicted to contact DNA (Fig. 3B). Residues Ser226 and Pro292 are not conserved in human TFIIB and are predicted to be in the proximity of the DNA, but not in direct contact (Fig. 3B). These modeling results suggested that the BRF1 mutations might affect BRF1 binding to DNA, impair TFIIIB-mediated Pol III recruitment, and reduce Pol III–mediated transcription.
To test whether the BRF1 mutations influence Pol III recruitment to gene promoters in vivo, we performed chromatin immunoprecipitation (ChIP) with tandem affinity purification (TAP)–tagged versions of BRF1 variants on Pol III target genes in yeast (Fig. 3C–E). Consistent with our predictions, the BRF1 variant T259M showed strongly decreased promoter occupancy at four tested tRNA loci and a significant decrease in occupancy of the U6 snRNA promoter (Fig. 3D). The ChIP signals for TAP-tagged A226S and S226L variants were not different from wild-type Brf1 (Fig. 3C). Although the R223W and P292H variants could not be tested due to their lethal effect, these results indicate that at least one BRF1 alteration can affect occupancy of different promoters in vivo.
Finally, to test whether the BRF1 mutations cause a transcriptional defect, we used an established in vitro assay (Hahn and Roberts 2000). This assay is based on a nuclear extract from a temperature-sensitive yeast strain carrying the BRF1 substitution p.Trp107Arg (W107R). Extracts from this strain are transcriptionally inactive, but addition of recombinant Brf1 protein restores activity on a template encoding the native SUP4 promoter. When we used purified recombinant Brf1 variants in this assay, we observed significantly less transcription for the BRF1 variant R223W in comparison with the same concentration of WT Brf1 (Fig. 4). Milder, but both significant and reproducible defects were observed for T259M, P292H, A226S, and S226L. BRF1 variants combining two mutations also showed transcriptional defects in this assay (Fig. 4). Taken together, these results show that BRF1 mutations identified in patients can impair transcription in a well-controlled, in vitro transcription assay.
Figure 4.

In vitro transcription defects caused by BRF1 mutations in yeast. (Top) A representative gel of the in vitro transcription reaction using a nuclear extract harboring a deficient Brf1 protein (p.Trp107Arg, W107R) that is impaired in Pol III–dependent transcription. Addition of WT Brf1 rescues activity whereas the different Brf1 variants and the combinations present in the affected children show a defect in transcription. Data are presented as mean ± SD. A quantification is shown in the middle panel. (*) P < 0.01; (**) P < 0.0001. The lower panel shows a twelve times excess of the Brf1 protein amount used for this assay separated on an SDS gel.
Discussion
Whereas intellectual disability can be due to mutations in synaptic genes (Pavlowsky et al. 2012; Zoghbi and Bear 2012), recent studies have highlighted impairments of basal cellular functions and pathways such as transcription and translation in the etiology of cognitive disorders and neurogenetic syndromes (Najmabadi et al. 2011). Some examples include pontocerebellar hypoplasias, Pol III–related leukodystrophies, X-linked intellectual disability, and leukoencephalopathy with vanishing white matter (Bugiani et al. 2010; Namavar et al. 2011; Borck et al. 2012; Daoud et al. 2013). Complete inactivation of processes such as translation initiation or tRNA transcription is not compatible with life, suggesting that most disease-causing mutations are hypomorphs.
Our identification and characterization of BRF1 mutations causing the syndromic cerebellar-facial-dental phenotype is in line with these observations. No mutant Brf1 mouse has been reported to our knowledge; in yeast, deletion of BRF1 is lethal (Colbert and Hahn 1992). Accordingly, one of the missense variants we have identified is recruited to a lesser extent than wild type to target genes. Several other mutants have reduced transcriptional activity, suggesting that in the context of this assay they behave as hypomorphs. BRF1 mutations likely confer a specific phenotype; we did not identify BRF1 mutations in three individuals with clinically overlapping but distinct malformation syndromes or in seven individuals with genetically unresolved forms of PCH (data not shown). However, there is compelling evidence for the pathogenicity of the identified variants in the cerebellar-facial-dental syndrome: BRF1 was the only gene exome-wide that harbored very rare or unique biallelic mutations in the three affected sibships. These missense variants affect conserved amino acids that lie within a predicted functional domain, and two of the affected amino acids likely interact directly with DNA. In vivo experiments in zebrafish embryos showed that suppression of brf1b phenocopied some of the key pathognomonic features of the syndrome. Of note, while we cannot exclude a possible effect of overexpression on the outcome of our zebrafish assays, the effects of the variants seemed to be restricted to isoform 2 of BRF1. Thus our results possibly illustrate an isoform-specific effect underlying the phenotype and offer a plausible hypothesis about how mutations in a ubiquitous, key component of the cellular machinery can induce specific, albeit severe, phenotypes. Similar splice isoform-specific pathogenic effects have been found in other disorders (Sarparanta et al. 2012; Schulte et al. 2014), suggesting that this mechanism might be a significant regulator of pleiotropy. Previous studies have shown that full-length human BRF1 is the canonical isoform in pol III transcription, and whether other isoforms, including isoforms lacking the evolutionary conserved Zn ribbon domain such as isoform 2, are biologically active remains unknown. Biochemical data indicate that overexpression of isoforms lacking the Zn ribbon domain can induce BRF1–TBP interactions in vitro and reconstitute transcription in crude systems (McCulloch et al. 2000; Schramm and Hernandez 2002). While the in vivo role of different BRF1 isoforms remains to be elucidated, our zebrafish data provide further evidence of the pathogenic nature of the four missense variants.
Further studies showed each of the missense variants to affect a variety of relevant activities in yeast, including target gene promoter occupancy, in vitro transcription, and cell growth. We do not know the precise mechanism by which BRF1 contributes to the phenotype in humans. We note, however, that mouse Brf1 is expressed in anatomical structures that are relevant to the observed human pathology: In situ hybridization of postnatal day 56 mouse brain had previously revealed prominent Brf1 expression in the olfactory bulb, hippocampus, and the Purkinje and granule cell layers of the cerebellum (Allen Brain Atlas) (Sunkin et al. 2013), underscoring the important role of BRF1 in brain development.
We speculate that one plausible pathomechanism underlying the cerebellar-facial-dental syndrome is a reduced steady-state level of tRNAs that would be predicted to impair translation elongation; this may explain the growth defect in yeast and contribute to short stature in the affected individuals. Whether the major drivers of the phenotype are tRNAs or other Pol III–dependent transcripts remains to be determined. Finally, it is also unknown why partial disruption of Pol III transcription leads to organ- and tissue-specific phenotypes. There are ∼450 nuclear encoded tRNA genes in the human genome for only 61 anticodons. It has been shown previously that up to 26% of tRNA genes are active in one cell type versus another (Barski et al. 2010) and that tRNA expression varies by as much as tenfold among human tissues (Dittmar et al. 2006), suggesting the existence of active cell and tissue-specific regulation of Pol III transcription (Oler et al. 2010). Although the genome-wide architecture of Pol III–mediated transcription in neurons or specific brain regions is unknown, a pilot study using a microarray containing 42 probes for nuclear encoded tRNAs and assaying their relative expression among eight human tissues revealed a high overall tRNA expression level in brain (Dittmar et al. 2006). This observation may partially explain the vulnerability of brain regions toward impaired tRNA transcription or processing. Whether other affected tissues, such as the teeth, also show specific tRNA expression patterns remains to be investigated. We note that dental anomalies such as delayed dentition, hypodontia, oligodontia, and abnormally placed or shaped teeth are part of the Pol III–related leukodystrophy syndromes 4H; ataxia, delayed dentition, and hypomyelination (ADDH); and leukodystrophy with oligodontia (LO) (Daoud et al. 2013). An alternative explanation for the tissue-restricted phenotypes engendered by BRF1 dysfunction might be a partial functional redundancy with BRF2. It has been shown, however, that BRF1 and BRF2 are almost completely mutually exclusive as components of TFIIIB under physiological conditions (Moqtaderi et al. 2010), and whether BRF2 can partially compensate for defective BRF1 is unknown.
In conclusion, our study defines a syndrome with cerebellar hypoplasia, intellectual disability, and growth retardation that is caused by partial deficiency of the conserved Pol III transcription factor BRF1. These results add an example to the short list of genetic diseases caused by dysregulation of the Pol III machinery, most of which predominantly affect the central nervous system and suggest that improved understanding of the potential target specificity provided by BRF1 might offer pathomechanistic clues.
Methods
Subjects
The study was performed with the approval of the University of Ulm Ethics Committee. Affected individuals were evaluated at the genetics outpatient clinics of the Bambino Gesù Children’s Hospital, Rome (family 1); the Hospital S. Maria, Lisboa (family 2); and IRCCS Casa Sollievo Della Sofferenza Hospital, San Giovanni Rotondo, and Giovanni XIII Hospital, Bari, Italy (family 3). Patients were enrolled with written parental consent for participation in the study. The clinical evaluation included medical history interviews, a physical examination, and review of medical records. Clinical information of affected individuals from the first two families had been independently submitted to the web-based Dysmorphology Diagnostic System (DDS) of the DYSCERNE network (a European network of centers of expertise for dysmorphology) (Douzgou et al. 2014) by the respective attending geneticists (M.L.D. and B.D.; A.M.). After review of the clinical information and pictures, the DDS Expert Panel (including B.D.) concluded that the four affected individuals were likely affected by a previously unreported autosomal recessive disorder. Blood samples were obtained from each participating individual, and genomic DNA was extracted by standard procedures.
Whole exome and BRF1 sequencing
Families 1 and 2
Genomic DNA was enriched for exonic and adjacent splice site sequences with the SeqCap EZ Human Exome Library v2.0 kit, and libraries were run on an Illumina HiSeq 2000 Sequencer via a paired-end 100-bp protocol (Hussain et al. 2013). For data analysis, the Cologne Center for Genomics Varbank pipeline v2.6 and user interface was used. Primary data were filtered according to signal purity by the Illumina Realtime Analysis (RTA) software v1.8. Subsequently, the reads were mapped to the human genome reference build GRCh37/hg19 (http://www.genome.ucsc.edu/) using the BWA-SW alignment algorithm (Li and Durbin 2010). GATK v.1.6 (McKenna et al. 2010) was used to mark duplicated reads, perform a local realignment around short insertion and deletions (indels), recalibrate the base quality scores, and call SNPs and short indels. Mean coverage was >100× in both exomes and ∼90% of target bases were covered more than 30×. In the target regions, we detected 25,894 SNVs and 2004 short indels in the family 1 individual and 35,642 SNVs and 2795 short indels in the family 2 individual. Scripts developed in-house at the Cologne Center for Genomics were applied to detect protein changes, affected donor and acceptor splice sites, and overlaps with known variants. Splice site variants were analyzed with a maximum entropy model (Yeo and Burge 2004). We filtered for high-quality rare variants (MAF < 0.1%; based on the 1000 Genomes database, build 20110521 [The 1000 Genomes Project Consortium 2012] and the NHLBI Exome Sequencing Project Exome Variant Server [EVS], build ESP5400 [Tennessen et al. 2012]) with a predicted impact on protein sequence or splicing. We also filtered against an in-house database containing variants from 511 exomes from individuals with epilepsy to exclude pipeline-related artifacts.
Family 3
WES and data analysis were performed as previously described (Alfaiz et al. 2014). Briefly, exomes were captured using the Agilent SureSelect human all exon V4 enrichment kit and sequenced on an Illumina HiSeq platform. Variants were filtered based on adherence to an autosomal recessive inheritance pattern, prediction by SIFT (http://sift.jcvi.org/) (Kumar et al. 2009) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) (Adzhubei et al. 2010), and absence from dbSNP129 (http://www.ncbi.nlm.nih.gov/projects/SNP/).
PCR and Sanger sequencing were performed according to standard protocols for the validation of variants of interest and for cosegregation studies. BRF1 mutation nomenclature is based on transcript NM_001519.3 and RDH12 mutation nomenclature on transcript NM_152443.2.
In vivo modeling in zebrafish
Reciprocal BLAST of the human BRF1 protein sequence against the zebrafish genome identified two orthologs, suggesting that this gene is duplicated in the zebrafish genome: brf1a (NP_956192.1, 60% identity, 72% similarity) and brf1b (NP_956183.1, 60% identity, 72% similarity). We targeted each of these transcripts with a sbMO designed and obtained from Gene Tools. The MOs target the donor site of exon 6 for brf1a sbMO (TATTTGAAGCTTACCGATAGCTGGT), the donor site of exon 8 for brf1b sbMO (AGCTGTGACATTCACCTTTTTCTCA), and the donor site of exon 6 for brf1b sbMO#2 (GTCATTACAGGGAGACTTACCGACA). For rescue experiments, human WT mRNAs of isoform 1 (NP_001229717.1) and isoform 2 (NP_001229715.1) of BRF1 were cloned into pCS2+ vector and transcribed in vitro using the SP6 mMessage machine kit (Ambion). All variants were introduced with Phusion high-fidelity DNA polymerase (New England Biolabs) and custom designed mutagenesis primers and Sanger sequence confirmed on a 3730 ABI sequencer. We injected 1 nL of solution (12 ng MO and/or 200 pg of human mRNA, (WT, P292H, R223W, S226L, or T259M) into one- to four-cell-stage WT zebrafish embryos (Niederriter et al. 2013) and collected them at 3 d.p.f., fixed in Dent’s fixative overnight at 4°C. Standard immunohistochemistry was then performed using control embryos and embryos injected with brf1b MO, MO + WT human BRF1, and MO + variant human BRF1 (P292H, R223W, S226L, or T259M) stained with anti-α acetylated tubulin (T7451, mouse, Sigma-Aldrich, dilution 1:1000) as the primary antibody and Alexa Fluor goat anti-mouse IgG (A21207, Invitrogen, dilution 1:1000) as a secondary antibody. Stained zebrafish were observed using the Nikon AZ100 Microscope with a DS-Qi1MC digital camera head. All experiments were performed in triplicate (∼200 embryos), and significance was determined with a Student’s t-test.
CRISPR target sequences (brf1b: CATGCATGAATTCCGGCGCA) were identified using crispr.mit.edu and cloned into pT7-gRNA (Addgene) following the protocol described at http://www.addgene.org/crispr/Chen/. gRNAs and mRNAs were synthesized by a MEGAshortscript T7 kit (Ambion), and Cas9 mRNAs were synthesized by mMESSAGE mMACHINE SP6 (Ambion). One hundred twenty pg gRNA + 150 pg Cas9 mRNA were injected into one-cell stage zebrafish and collected at 3 d.p.f. in Dent’s fixative overnight at 4°C and stained as mentioned above.
For RNA sequencing (RNA-seq), total RNA from 5 d.p.f. zebrafish heads was extracted using a TRIzol/chloroform protocol. Libraries were constructed using a customized version of strand-specific dUTP and run on an Illumina HiSeq 2000 platform (Sugathan et al. 2014).
Yeast strains and growth assays
To introduce BRF1 mutations in yeast, the shuffle vector system developed by Sikorski and Hieter was used (Sikorski and Hieter 1989). BRF1 mutations were introduced in the vector pRS315, which contains yeast BRF1 expressed under the control of its own promoter. Mutations were introduced using site-directed mutagenesis and confirmed by DNA sequencing. The identified mutations from humans (h) were transferred to yeast (y). The corresponding amino acid positions are as follows: hR223 = yR218, hS226 = yA221, hT259 = yT254, and hP292 = yP288. For combinations of two mutations, BRF1 variants T259M and S226L were cloned into pRS314 vector and cotransformed with the S226L and T259M mutations in the pRS315 vector, respectively. Plasmids were transformed into the yeast strain SHY285 (gift from S. Hahn, MATα, ade2Δ∷hisG, his3Δ200, leu2Δ0, lys2Δ0, met15Δ0, trp1Δ63, ura3Δ0, brf1Δ∷HIS3/pSH524), and WT BRF1 was replaced with the mutant gene by plasmid shuffle at 30°C. The rescue plasmid pSH524 contains a URA3 marker (Ars Cen URA3, BRF1) (Hahn and Roberts 2000). For the spotting assays, strains were grown overnight in selective media, normalized to equal densities (107 cells/200 μL), serially diluted (1:5), and spotted on YPD media or selective media (Leu-, Leu/Trp-) supplemented with 5-fluoroorotic acid (5-FOA; Carl Roth). Plates were incubated for 3 d at 30°C. For the liquid assays, cultures were inoculated in selective media, grown overnight and normalized to equal densities (OD600nm = 0.33). We used 3.5 μL of these dilutions to inoculate a 96-well plate with 200 μL of liquid media (YPD and selective media supplemented with 5-FOA). Plates were incubated for 3 d at 30°C. As a control strain we used WT BY4741 yeast.
Protein purification
Protein purification was performed essentially as previously described (Alexander et al. 2004), with minor modifications. Proteins were expressed in Escherichia coli BL21-Codon plus (DE3) RIL cells in LB medium. Protein expression was induced in 2 l cultures with 1 mM IPTG at an OD600 of 0.7. Cells were grown for 3 h at 37°C and harvested (20 min, 4000 rpm, 4°C). The pellet was washed with 40 mL LB medium and flash-frozen in liquid nitrogen. The cells were thawed on ice and resuspended in 50 mL lysis buffer (25 mM Tris/HCl at pH 7.5 and 25°C; 200 mM KCl; 12.5 mM MgCl2; 10% [v/v] glycerol; 1 × PI). Subsequently, 0.8 mg lysozyme (Roth) per 1 mL lysate was added and incubated rotating in the cold room. After 10 min, 0.2% IGEPAL CA-630 (MP Biomedicals) was added for further 5 min. For cell disruption, lysate was sonicated for 15 min, at 25% duty cycle and 40 output value. Brf1 in inclusion bodies was washed twice in H.35 buffer (20 mM HEPES at pH 8.0 and 25°C; 350 mM KCl; 2 mM MgCl2; 1% NaOH; 20% [v/v] glycerol; 0.1% [v/v] IGEPAL CA-630; 1 × PI). Inclusion bodies were extracted with 5 mL G6 buffer (100 mM Tris/HCl at pH 7.5 and 4°C; 6 M GdmCl; 2 mM β-mercaptoethanol) and rotation for 1 h at 4°C. The solution was bound in batch to 2.5 mL Ni-NTA bead volume in G6 buffer overnight at 4°C. Beads were washed with 12.5 mL G6 buffer and protein eluted in batch with three times 2.5 mL Ni-NTA elution buffer (100 mM Tris/HCl at pH 7.5 and 4°C; 6 M GdmCl; 10 mM β-mercaptoethanol; 500 mM imidazole; 1 × PI). To the combined eluate, ZnSO4 and DTT were added to final concentrations of 10 μM and 5 mM, respectively. Finally, the denatured Brf1 was refolded via rapid dilution and dialysis. For dilution, 100 μL of the eluate was placed in the lid of a 1.5-mL Eppendorf tube containing 1 mL dialysis buffer (20 mM Tris acetate 80% cation at pH 7.5 and 4°C; 200 mM KCl; 2 mM MgCl2; 20% [v/v] glycerol; 0.1% [v/v], IGEPAL CA-630; 1 × PI), and the lid was closed quickly. For every tube this could be repeated twice. After flash dilution the protein was dialyzed in dialysis buffer for 2 h, frozen in liquid nitrogen, and stored at −80°C. Usually the purification resulted in 30 mL of 0.4–0.8 mg/mL protein solution.
ChIP assay
For the ChIP assay, TAP-tagged BRF1 and its mutant variants were introduced in the pRS315 vector, which contains BRF1 expressed under the control of its own promoter. For combinations of the mutations, S226L and T259M were cloned into pRS314 vector and cotransformed with the TAP-tagged BRF1 variants T259M-TAP and S226L-TAP in the pRS315 vector, respectively. ChIP experiments were performed as previously described (Aparicio et al. 2005).
In vitro transcription assay
The Pol III–dependent in vitro transcription assay was performed as described by Hahn and Roberts (2000), with minor modifications. The nuclear extract was prepared as described by Seizl et al. (2011). The template used contained the SUP4 gene from [−159] − [+244] with respect to the transcription start site of the tRNA. SUP4 encodes a tyrosine tRNA. The 25 μL transcription reaction contained 100 mM KOAc (pH 7.6), 20 mM HEPES (pH 7.6), 1 mM EDTA, 5 mM MgOAc, 2.5 mM DTT, 150 ng template, 192 μg phosphocreatine, 0.2 μg creatine phosphokinase, 10 U RiboLock RNase inhibitor (Fermentas), 50 μg temperature-sensitive nuclear extract harboring a temperature-sensitive W107R mutation in BRF1, 40 μM α-amanitin, and 100 ng of recombinant protein where indicated. Proteins were added to the transcription reaction mix for 5 min before addition of 0.1 mM NTPs. The transcription reaction was carried out for 60 min at 30°C. The RNA was isolated, and for primer extension, 0.125 pmol primer was annealed for 45 min at 60°C in 20 μL 5 mM Tris (pH 8.3), 75 mM KCl, 1 mM EDTA (pH 8.0). The sequence of the 5′-Cy5 labeled primer was TCTCCCGGGGGCGAGTCGAACGCCC. Two micrograms of Actinomycin D was added before 0.25 units MuLV reverse transcriptase in 60 μL 5 mM Tris (pH 8.3), 75 mM KCl, 4.5 mM MgCl2, 15 mM DTT, and 0.1 mM dNTPs extended the primer. The resulting cDNA was isolated by ethanol precipitation and resuspended in 4 μL 0.04 mg/mL RNase A and 4 μL formamide buffer (80% formamide, 25 mM EDTA, 1.5% bromphenolblue). Before loading, samples were boiled for 2 min at 95°C and put directly on ice. Samples were run on a 7 M urea, 8% polyacrylamide (35:1) gel in 1× TBE for 45 min at 180 V. The gel was pre-run for 5–10 min, and the pockets were rinsed before loading. Gels were analyzed and quantified with a typhoon scanner FLA9400 and ImageQuant Software (GE Healthcare).
Data access
The zebrafish RNA-seq data have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE63191. BRF1 mutation data have been submitted to the NCBI ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar/) under accession numbers SCV000192004, SCV000192005, SCV000192006, and SCV000195766.
Supplementary Material
Acknowledgments
We thank the families for their collaboration. The clinical information and the patient pictures were uploaded to the web-based Dysmorphology Diagnostic System (DDS) developed by DYSCERNE—a European Network of Centres of Expertise for Dysmorphology (EU DG Sanco)—and discussed among the DDS expert panel. We thank Sarah Sainsbury for mapping the BRF1 mutations on the hTFIIB structure; Karl Bertram and Ali A. Alfaiz for help with cloning and bioinformatic analyses; Bernd Lapatki and Leopoldo Zelante for discussions on clinical (including dental) aspects of the disorder; Birgit Budde, Dagmar Wieczorek, and Hilde van Esch for providing DNA; and Christelle Golzio and Michael Talkowski for zebrafish RNA-seq data. F.H. was supported by the Elite Network Bavaria program “Protein Dynamics in Health and Disease”; P.C. by the Deutsche Forschungsgemeinschaft (SFB646, GraKo1721, SFB960, SFB1064, CIPSM, NIM), an Advanced Grant of the European Research Council, the Jung-Stiftung, and the Vallee Foundation; G.B. by the Deutsche Forschungsgemeinschaft; G.M. by a grant from the Italian Ministry of Health (Ricerca Corrente 2012–14); and A.R. by the Lithuanian-Swiss Cooperation Program.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.176925.114.
References
- The 1000 Genomes Project Consortium 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akizu N, Cantagrel V, Schroth J, Cai N, Vaux K, McCloskey D, Naviaux RK, Van Vleet J, Fenstermaker AG, Silhavy JL, et al. . 2013. AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder. Cell 154: 505–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander DE, Kaczorowski DJ, Jackson-Fisher AJ, Lowery DM, Zanton SJ, Pugh BF. 2004. Inhibition of TATA binding protein dimerization by RNA polymerase III transcription initiation factor Brf1. J Biol Chem 279: 32401–32406. [DOI] [PubMed] [Google Scholar]
- Alfaiz AA, Micale L, Mandriani B, Augello B, Pellico MT, Chrast J, Xenarios I, Zelante L, Merla G, Reymond A. 2014. TBC1D7 mutations are associated with intellectual disability, macrocrania, patellar dislocation, and celiac disease. Hum Mutat 35: 447–451. [DOI] [PubMed] [Google Scholar]
- Aparicio O, Geisberg JV, Sekinger E, Yang A, Moqtaderi Z, Struhl K. 2005. Chromatin immunoprecipitation for determining the association of proteins with specific genomic sequences in vivo. Curr Protoc Mol Biol 69: 21.3.1–21.3.33. [DOI] [PubMed] [Google Scholar]
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. 2011. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 12: 745–755. [DOI] [PubMed] [Google Scholar]
- Barski A, Chepelev I, Liko D, Cuddapah S, Fleming AB, Birch J, Cui K, White RJ, Zhao K. 2010. Pol II and its associated epigenetic marks are present at Pol III-transcribed noncoding RNA genes. Nat Struct Mol Biol 17: 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard G, Chouery E, Putorti ML, Tetreault M, Takanohashi A, Carosso G, Clement I, Boespflug-Tanguy O, Rodriguez D, Delague V, et al. . 2011. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am J Hum Genet 89: 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O, Witherspoon K, Gerdts J, Baker C, Vulto-van Silfhout AT, et al. . 2014. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158: 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borck G, Shin BS, Stiller B, Mimouni-Bloch A, Thiele H, Kim JR, Thakur M, Skinner C, Aschenbach L, Smirin-Yosef P, et al. . 2012. eIF2γ mutation that disrupts eIF2 complex integrity links intellectual disability to impaired translation initiation. Mol Cell 48: 641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugiani M, Boor I, Powers JM, Scheper GC, van der Knaap MS. 2010. Leukoencephalopathy with vanishing white matter: a review. J Neuropathol Exp Neurol 69: 987–996. [DOI] [PubMed] [Google Scholar]
- Cabarcas S, Schramm L. 2011. RNA polymerase III transcription in cancer: the BRF2 connection. Mol Cancer 10: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbert T, Hahn S. 1992. A yeast TFIIB-related factor involved in RNA polymerase III transcription. Genes Dev 6: 1940–1949. [DOI] [PubMed] [Google Scholar]
- Daoud H, Tetreault M, Gibson W, Guerrero K, Cohen A, Gburek-Augustat J, Synofzik M, Brais B, Stevens CA, Sanchez-Carpintero R, et al. . 2013. Mutations in POLR3A and POLR3B are a major cause of hypomyelinating leukodystrophies with or without dental abnormalities and/or hypogonadotropic hypogonadism. J Med Genet 50: 194–197. [DOI] [PubMed] [Google Scholar]
- Dauber A, Golzio C, Guenot C, Jodelka FM, Kibaek M, Kjaergaard S, Leheup B, Martinet D, Nowaczyk MJ, Rosenfeld JA, et al. . 2013. SCRIB and PUF60 are primary drivers of the multisystemic phenotypes of the 8q24.3 copy-number variant. Am J Hum Genet 93: 798–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieci G, Conti A, Pagano A, Carnevali D. 2013. Identification of RNA polymerase III-transcribed genes in eukaryotic genomes. Biochim Biophys Acta 1829: 296–305. [DOI] [PubMed] [Google Scholar]
- Dittmar KA, Goodenbour JM, Pan T. 2006. Tissue-specific differences in human transfer RNA expression. PLoS Genet 2: e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douzgou S, Clayton-Smith J, Gardner S, Day R, Griffiths P, Strong K, DYSCERNE expert panel . 2014. Dysmorphology at a distance: results of a web-based diagnostic service. Eur J Hum Genet 22: 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golzio C, Willer J, Talkowski ME, Oh EC, Taniguchi Y, Jacquemont S, Reymond A, Sun M, Sawa A, Gusella JF, et al. . 2012. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 485: 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn S, Roberts S. 2000. The zinc ribbon domains of the general transcription factors TFIIB and Brf: conserved functional surfaces but different roles in transcription initiation. Genes Dev 14: 719–730. [PMC free article] [PubMed] [Google Scholar]
- Hussain MS, Baig SM, Neumann S, Peche VS, Szczepanski S, Nurnberg G, Tariq M, Jameel M, Khan TN, Fatima A, et al. . 2013. CDK6 associates with the centrosome during mitosis and is mutated in a large Pakistani family with primary microcephaly. Hum Mol Genet 22: 5199–5214. [DOI] [PubMed] [Google Scholar]
- Janecke AR, Thompson DA, Utermann G, Becker C, Hübner CA, Schmid E, McHenry CL, Nair AR, Rüschendorf F, Heckenlively J, et al. . 2004. Mutations in RDH12 encoding a photoreceptor cell retinol dehydrogenase cause childhood-onset severe retinal dystrophy. Nat Genet 36: 850–854. [DOI] [PubMed] [Google Scholar]
- Kassavetis GA, Kumar A, Ramirez E, Geiduschek EP. 1998. Functional and structural organization of Brf, the TFIIB-related component of the RNA polymerase III transcription initiation complex. Mol Cell Biol 18: 5587–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo SK, Wu CC, Lin YC, Lee JC, Chen HT. 2014. Mapping the protein interaction network for TFIIB-related factor Brf1 in the RNA polymerase III preinitiation complex. Mol Cell Biol 34: 551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4: 1073–1081. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26: 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin DH, Kousi M, Chan YM, Lim ET, Schmahmann JD, Hadjivassiliou M, Hall JE, Adam I, Dwyer A, Plummer L, et al. . 2013. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med 368: 1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall L, White RJ. 2008. Non-coding RNA production by RNA polymerase III is implicated in cancer. Nat Rev Cancer 8: 911–914. [DOI] [PubMed] [Google Scholar]
- McCulloch V, Hardin P, Peng W, Ruppert JM, Lobo-Ruppert SM. 2000. Alternatively spliced hBRF variants function at different RNA polymerase III promoters. EMBO J 19: 4134–4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. . 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moqtaderi Z, Wang J, Raha D, White RJ, Snyder M, Weng Z, Struhl K. 2010. Genomic binding profiles of functionally distinct RNA polymerase III transcription complexes in human cells. Nat Struct Mol Biol 17: 635–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, Hosseini M, Behjati F, Haas S, Jamali P, et al. . 2011. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 478: 57–63. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I. 2001. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 10: 1441–1448. [DOI] [PubMed] [Google Scholar]
- Namavar Y, Barth PG, Poll-The BT, Baas F. 2011. Classification, diagnosis and potential mechanisms in pontocerebellar hypoplasia. Orphanet J Rare Dis 6: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederriter AR, Davis EE, Golzio C, Oh EC, Tsai IC, Katsanis N. 2013. In vivo modeling of the morbid human genome using Danio rerio. J Vis Exp 78: e50338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oler AJ, Alla RK, Roberts DN, Wong A, Hollenhorst PC, Chandler KJ, Cassiday PA, Nelson CA, Hagedorn CH, Graves BJ, et al. . 2010. Human RNA polymerase III transcriptomes and relationships to Pol II promoter chromatin and enhancer-binding factors. Nat Struct Mol Biol 17: 620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlowsky A, Chelly J, Billuart P. 2012. Emerging major synaptic signaling pathways involved in intellectual disability. Mol Psychiatry 17: 682–693. [DOI] [PubMed] [Google Scholar]
- Rooms L, Reyniers E, Scheers S, van Luijk R, Wauters J, Van Aerschot L, Callaerts-Vegh Z, D’Hooge R, Mengus G, Davidson I, et al. . 2006. TBP as a candidate gene for mental retardation in patients with subtelomeric 6q deletions. Eur J Hum Genet 14: 1090–1096. [DOI] [PubMed] [Google Scholar]
- Rudnik-Schöneborn S, Barth PG, Zerres K. 2014. Pontocerebellar hypoplasia. Am J Med Genet C Semin Med Genet 166C: 173–183. [DOI] [PubMed] [Google Scholar]
- Saitsu H, Osaka H, Sasaki M, Takanashi J, Hamada K, Yamashita A, Shibayama H, Shiina M, Kondo Y, Nishiyama K, et al. . 2011. Mutations in POLR3A and POLR3B encoding RNA Polymerase III subunits cause an autosomal-recessive hypomyelinating leukoencephalopathy. Am J Hum Genet 89: 644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarparanta J, Jonson PH, Golzio C, Sandell S, Luque H, Screen M, McDonald K, Stajich JM, Mahjneh I, Vihola A, et al. . 2012. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat Genet 44: 450–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer AE, Eggens VR, Caglayan AO, Reuter MS, Scott E, Coufal NG, Silhavy JL, Xue Y, Kayserili H, Yasuno K, et al. . 2014. CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell 157: 651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm L, Hernandez N. 2002. Recruitment of RNA polymerase III to its target promoters. Genes Dev 16: 2593–2620. [DOI] [PubMed] [Google Scholar]
- Schulte EC, Kousi M, Tan PL, Tilch E, Knauf F, Lichtner P, Trenkwalder C, Hogl B, Frauscher B, Berger K, et al. . 2014. Targeted resequencing and systematic in vivo functional testing identifies rare variants in MEIS1 as significant contributors to restless legs syndrome. Am J Hum Genet 95: 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seizl M, Lariviere L, Pfaffeneder T, Wenzeck L, Cramer P. 2011. Mediator head subcomplex Med11/22 contains a common helix bundle building block with a specific function in transcription initiation complex stabilization. Nucleic Acids Res 39: 6291–6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugathan A, Biagioli M, Golzio C, Erdin S, Blumenthal I, Manavalan P, Ragavendran A, Brand H, Lucente D, Miles J, et al. . 2014. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc Natl Acad Sci 111: E4468–E4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkin SM, Ng L, Lau C, Dolbeare T, Gilbert TL, Thompson CL, Hawrylycz M, Dang C. 2013. Allen Brain Atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res 41: D996–D1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, et al. . 2012. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337: 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetreault M, Choquet K, Orcesi S, Tonduti D, Balottin U, Teichmann M, Fribourg S, Schiffmann R, Brais B, Vanderver A, et al. . 2011. Recessive mutations in POLR3B, encoding the second largest subunit of Pol III, cause a rare hypomyelinating leukodystrophy. Am J Hum Genet 89: 652–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai FT, Sigler PB. 2000. Structural basis of preinitiation complex assembly on human pol II promoters. EMBO J 19: 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini A, Cramer P. 2012. Conservation between the RNA polymerase I, II, and III transcription initiation machineries. Mol Cell 45: 439–446. [DOI] [PubMed] [Google Scholar]
- White RJ. 2011. Transcription by RNA polymerase III: more complex than we thought. Nat Rev Genet 12: 459–463. [DOI] [PubMed] [Google Scholar]
- Yeo G, Burge CB. 2004. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11: 377–394. [DOI] [PubMed] [Google Scholar]
- Zoghbi HY, Bear MF. 2012. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol 4. doi: 10.1101/cshperspect.a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.