

Abstract
PTH, an 84-amino acid peptide hormone synthesized by the parathyroid glands, is essential for the maintenance of calcium homeostasis. While in its traditional metabolic role, PTH helps to maintain the serum calcium concentration within narrow, normal limits and participates as a determinant of bone remodeling, more specific actions, described as catabolic and anabolic are also well known. Clinically, the catabolic effect of PTH is best represented by primary hyperparathyroidism (PHPT), while the osteoanabolic effect of PTH is best seen when PTH or its biological aminoterminal fragment [PTH(1–34)] is used as a therapy for osteoporosis. These dual functions of PTH are unmasked under very specific pathological (PHPT) or therapeutic conditions. At the cellular level, PTH favors bone resorption, mostly by affecting the receptor activator of nuclear factor κ-B (RANK) ligand (RANKL)-osteoprotegerin-RANK system, leading to an increase in osteoclast formation and activity. Increased bone formation due to PTH therapy is explained best by its ability to enhance osteoblastogenesis and/or osteoblast survival. The PTH-induced bone formation is mediated, in part, by a decrease in SOST/sclerostin expression in osteocytes. This review focuses on the dual anabolic and catabolic actions of PTH on bone, situations where one is enhanced over the other, and the cellular and molecular mechanisms by which these actions are mediated.
Keywords: hyperparathyroidsm, osteoblastogenesis, osteoclastogenesis, PTH, PTH (1–84), teriparatide
INTRODUCTION
Parathyroid hormone (PTH), an 84-amino acid peptide hormone, is synthesized in the cells of the parathyroid glands. Release of PTH occurs both with circadian dynamics and in pulsatile fashion stochastically. Through its direct actions on bone and kidney, the principle target organs, and indirectly on the gastrointestinal tract (by facilitating the activation of vitamin D), PTH helps to maintain the serum calcium within narrow, normal limits. At the level of bone, it promotes calcium release; at the level of the kidney, it promotes tubular calcium reabsorption. The indirect effect on the gastrointestinal tract promotes calcium absorption (1). A hypocalcemic signal will lead to greater PTH release (and synthesis), thus leading to these organ-specific events and restoring the serum calcium to normal.
The direct actions of PTH are initiated by an interaction with its receptor (PTH1R), a G-protein-coupled receptor expressed in target cells, such as osteoblasts in bone and tubular cells in the kidney (2). Events following the binding of PTH to the PTH1R include stimulation of Gαs-mediated activation of adenyl cyclase, which in turn promotes cAMP production and subsequent activation of protein kinase A (PKA). The PTH1R is also linked to Gαq-mediated activation of phospholipase and protein kinase C (PKC) (3, 4). Regulation of these activation events occurs, in part, at the level of the PTH1R when it is internalized (5). Recently, PTH has been shown to downregulate sclerostin, an important regulator of bone formation. This effect is also mediated by cAMP signaling in osteocytes (6).
The catabolic effect of PTH is best represented by the classic disorder of PTH excess, primary hyperparathyroidism (PHPT). In this setting, in which patients are exposed to continuously high amounts of circulating PTH, bone loss is common. When PHPT was invariably a symptomatic disease, bone loss was often accompanied by fractures. With the more modern clinical profile of PHPT emerging at around the time that dual energy X-ray absorptiometry (DXA) became available in the 1980’s, discovery of PHPT was likely to be in asymptomatic individuals whose bone loss could be gleaned only by DXA. Insights into this phenotype revealed clues to the anabolic proclivity of this hormone (7), namely that cancellous bone microstructure is preserved in comparison to postmenopausal women without PHPT (8–10).
Further insight that exploited the idea that PTH could be primarily anabolic under certain circumstances came in the 1990’s when studies by Dobnig et al. (11) showed that the way in which PTH is administered dictates whether it will serve primarily an anabolic or catabolic role. In rats treated once daily (i.e., intermittently) with low doses of PTH, marked anabolic effects on the skeleton were observed while continuous, 24-h exposure was associated with the catabolic effects. This key observation was developed further as the foreshortened amino terminal fragment of PTH, teriparatide [PTH(1–34)] and, later, the full-length hormone [PTH(1–84)] were shown to be anabolic when administered once daily in low doses (12, 13).
At the cellular level, gene expression profiling of intermittent vs continuous PTH administration in vivo and in vitro suggests that the two modes of administration of PTH can regulate different set of genes, one favoring bone formation and the other favoring bone resorption (14, 15).
This review focuses on both the anabolic and catabolic skeletal effects of PTH, and discusses the cellular basis by which PTH exerts these effects.
PHPT
Historically, symptomatic PHPT is associated with a devastating, catabolic destruction of the skeleton with bone loss, brown tumors, bone cysts, and subperiosteal bone resorption of the phalanges (16, 17). Osteitis fibrosa cystica, the term given to this severe bone disease, is still seen in the developing world, but in most regions where biochemical screening is routine, asymptomatic PHPT predominates. Asymptomatic PHPT rarely is accompanied by these specific skeletal features (18–20). Rather, bone densitometry technology has permitted a different kind of insight into the skeleton of subjects with PHPT.
Bone density and skeletal microarchitecture
Silverberg et al. (7) evaluated the presence and extent of bone disease in patients with asymptomatic PHPT, by DXA and by histomorphometry of bone biopsies. The greatest reduction in bone mineral density (BMD) was found at the distal 1/3 radius, a site of predominantly cortical bone. The ability to perform 3-site DXA gave further information at the other 2 sites, the lumbar spine, a site primarily comprised of cancellous bone, and the hip, a site that is an even admixture of cortical and cancellous bone. BMD of the lumbar spine was within 5% of expected for non-hyperparathyroid, post-menopausal women. The hip regions showed values that were intermediate between the preferentially reduced cortical bone of the distal radius and the maintained BMD of the lumbar spine (Fig. 1).
Fig. 1.

The densitometric signature of primary hyperparathyroidism (PHPT) in the modern era. Bone densitometry at lumbar spine, femoral neck and radius in PHPT. Bone mineral density (BMD) is presented in comparison to expected values for normal controls. [Adapted from (7)].
The findings by DXA were followed up by an extensive series of histomorphometric studies by Dempster et al. (7, 9, 10, 21). Preferential involvement of cortical bone with preservation of cancellous areas was confirmed by histomorphometric analysis. The vast majority of patients with PHPT showed reductions in cortical width. In contrast, the cancellous compartment of the bone biopsy specimen showed greater than average values for trabecular bone volume. Other features of trabecular bone such as trabecular number, connectivity and separation indicated preservation of this compartment of bone in most patients with PHPT. Analysis of bone biopsy specimens by microcomputed tomography (μCT) also demonstrated in mild PHPT preserved cancellous bone architecture (Fig. 2) (8). Histomorphometric studies of bone biopsies in PHPT have confirmed that while the trabecular compartment is preserved, the cortical compartment is at risk with cortical thinning and increased cortical porosity commonly seen (9, 10, 21, 22).
Fig. 2.
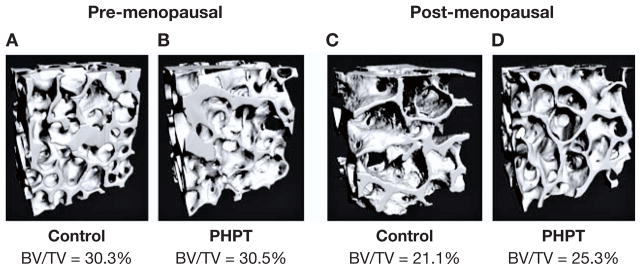
Microarchitectural features in pre-and post-menopausal women with primary hyperparathyroidism (PHPT). 3D micorcomputed tomography reconstructions of cancellous bone in pre- and post-menopausal women with PHPT (B and D), and normal controls (A and C). [Adapted from (8)].
The 10- and 15-yr natural history studies of Silverberg et al. (18, 20) showed that lumbar spine bone density remains stable for as long as subjects were followed, while the sites with more cortical bone, namely the distal 1/3 radius and the femoral neck, began to experience substantial declines after 10 yr of observation.
The characteristic densitometric and histomorphometric pattern described above, with preferential reduction of the cortical compartment, is not always seen in PH-PT. The descriptions provided are the most common ones. Obviously, these features will vary with the extent of the disease, and predisposing factors that could favor losses in other skeletal compartments and thus other patterns. For example, Silverberg et al. described a minority of patients with PHPT whose lumbar spine bone density was preferentially reduced (23). This could reflect preferential loss of cancellous bone due to the menopause per se, prior to the development of PHPT. Other studies have demonstrated more universal loss of BMD in PHPT (24–26), a finding that would not be unexpected in patients with more severe disease. Recently, Hansen et al. (24), using a newer non-invasive technology, high-resolution peripheral CT (HR-pQCT), showed decreased bone mass in the radius in both the cortical and trabecular compartments, in 27 women with mild PHPT, as compared to a normal control group. In this study, subjects with PHPT had reduced BMD at the lumbar spine by DXA. It is not surprising, therefore, that the cancellous compartment would be abnormal by HR-pQCT in this cohort. It is likely that when more typical phenotype of PHPT is examined by HR-pQCT, microstructural analysis by HR-pQCT will be consistent with preserved cancellous bone.
Fracture risk
Given the catabolic skeletal actions of continuously elevated PTH levels, typically at cortical sites, one would expect increased non-vertebral fracture risk in patients with PHPT. The preserved cancellous skeleton would be expected to be associated with reduced fracture risk in the spine. Some studies, though, have reported an increase in overall fracture risk (27, 28), including vertebral (17, 28, 29), forearm, rib, and pelvic fractures (28) in PHPT. Increased risk of vertebral and hip fractures has not, however, been uniformly observed (30–32). These controversial findings may reflect reports that vary in the severity of the PHPT. Vignali et al. (29) assessed vertebral fracture risk in 150 subjects with PHPT according to the severity of the disease. Patients with symptomatic PHPT had a higher rate of vertebral fracture than patients with asymptomatic PHPT. When subjects with asymptomatic PHPT were classified according to whether they did or did not meet the criteria for surgery, established by the 2002 Workshop on asymptomatic PHPT, the rate of fracture was significant in those who met surgical guidelines but not in those who did not meet surgical guidelines.
It is hard to draw any conclusions from these studies because many of them are observational and cross-sectional. Some studies suffer by a surveillance bias in which the known PHPT state may have been more likely to be associated with x-rays for complaints of back pain, for example. It is still not clear, then, whether or not patients with mild, asymptomatic PHPT have increased fracture risk.
There are structural issues that may confound the simple expectations by BMD of increased fracture risk at cortical sites and reduced fracture risk at cancellous sites in PH-PT. PTH is known, for example, to have other effects on bone qualities beside BMD. As reported in many series, preserved cancellous microarchitecture in mild PHPT might counteract the cortical thinning at cortical sites. Moreover, PTH may increase periosteal apposition, leading to an increase in cross sectional diameter of the bone, favorably altering bone geometry (33, 34). The increase in the outer diameter of bone will increase bone strength independent of BMD (35). Thus, a number of other factors have to be taken into account that altogether defines fracture risk in PHPT.
PTH AS AN ANABOLIC SKELETAL THERAPY
The clues described earlier to the anabolic potential of PTH led to its successful development and that of its biologically active but foreshortened fragment, PTH(1–34) as a therapy for osteoporosis. PTH represents the only osteoanabolic class available at this time for the treatment of osteoporosis. These PTH forms have been shown to increase BMD, improve microarchitecture of the bone, and reduce vertebral fractures. For teriparatide [PTH(1–34)], a reduction in non-vertebral fractures has also been demonstrated (12, 13, 36–41).
Mode of action
Treatment with PTH leads to an increase in bone turnover, with an interesting bitemporal characteristic, in which there is an early stimulation of bone formation followed later by a stimulation of bone turnover (bone resorption and bone formation). The dichomatous kinetics between early effects on bone formation (a bone modeling effect) and a general increase in bone turnover (a bone remodeling effect) has led to the concept of an “anabolic window”, the period of time when PTH is maximally anabolic (Fig. 3) (13, 42–45). Even when bone remodeling is stimulated, for at least a limited period of time, bone formation exceeds bone resorption, continuing the anabolic property of PTH albeit perhaps less marked.
Fig. 3.

PTH as an anabolic agent for bone: a kinetic model. Treatment with PTH leads to increased bone turnover, with an early stimulation of bone formation followed later by a stimulation of bone resorption. It is thus formed an “anabolic window”, the period of time when PTH is maximally anabolic. When bone formation and resorption are stimulated, bone formation exceeds bone resorption, continuing the anabolic property of PTH. [Adapted from (45)]
The concept of the anabolic window is supported not only by studies of the kinetics of bone markers in the circulation but also by histomorphometric analysis of iliac crest bone biopsies. Using techniques of standard double-labeling and novel quadruple labeling techniques, it has been demonstrated by Lindsay et al. (46) that PTH initially stimulates bone formation without prior resorption. This suggests that the process of bone accrual is occurring on quiescent bone surfaces, which is classically a modeling-based event. Modeling is not usually observed in normal, adult human bone, but would appear to occur in this special situation of early PTH exposure (47). Lindsay et al. (46) evaluated bone biopsies of 10 post-menopausal women treated with teriparatide for 4 weeks and compared them to a matched control group. The authors relied on the appearance of the bone surface underneath the newly formed bone to classify the bone formation as modeling or remodeling-based. In the case of modeling-based bone formation, the surface underneath newly formed bone is smooth, and the collagen fibers have similar orientation to the adjacent bone tissue. In remodeling-based bone formation, that surface is irregular, with interrupted collagen fibers, indicating that bone resorption has occurred (47). In the analysis, modeling-based bone formation with teriparatide accounted for approximately 30% and 22% of the bone formation in cancellous and endocortical bone respectively. In control subjects, formation was exclusively remodeling-based (46). In agreement with the concept of the anabolic window, the ability of PTH to increase bone formation in the absence of prior resorption appears to be more pronounced in the early stages of the therapy, since the proportion of modeling-based bone formation decreases over the course of the treatment. When biopsies are carried out after 12–24 months after treatment with 20 or 40 μg daily of teriparatide treatment, only 2.8% and 7.7%, respectively, of bone formation in cancellous bone was a modeling process (38). The modeling-based bone formation induced by PTH can occur not only on quiescent bone surfaces, but also in areas of remodeling in which there is overfilling of bone resorption pits with extension of bone formation beyond the margins of the resorption cavity (46–48). Anabolic action also is appreciated when remodeling becomes the dominant profile of PTH action because there is more bone formation occurring than bone resorption (47).
Bone density and microarchitecture
Densitometric findings in men and women who are treated with PTH(1–84) or teriparatide demonstrate major increments in BMD at the lumbar spine (12, 13, 40, 44). By histomorphometry and μCT of paired iliac crest biopsy specimens from women treated with teriparatide for 11–24 months, improvements in cancellous bone volume, connectivity, and cancellous bone morphology with conversion from a more rod-like to a more normal plate-like appearance have been appreciated (39). Similar effects on cancellous bone were seen upon administration of PTH(1–84) for 18 or 24 months (Fig. 4) (37, 41).
Fig. 4.

Microcomputed tomography images of iliac crest biopsies of postmenopausal women treated with either PTH(1–84) or teriparatide. A) Osteoporotic postmenopausal women treated with placebo or PTH(1–84) for 18 months. B) Paired biopsy specimens from a 64-yr-old woman before and after treatment with teriparatide for 36 months. [Adapted from (36, 37)].
Smaller increases in BMD are appreciated at the hip sites (total hip and femoral neck). The 1/3 radial site typically is not increased and may actually show a small reduction in BMD (12, 13, 40, 44). The small or even negative effects of PTH on BMD at sites containing predominantly cortical bone were not confirmed when bone microarchitecture was assessed. Dempster et al. (36) have shown maintenance or an increase in cortical width in men and women treated with teriparatide for 18 and 36 months, respectively, without increases in cortical porosity. Images obtained during μCT analysis suggest that the increase in cortical thickness seen with teriparatide results from increased bone formation at both the periosteal and endosteal surfaces (39). The increase in cortical thickness, however, is not always seen (37, 41).
Bone geometry and fracture risk
Iliac crest bone biopsies from post-menopausal women treated with teriparatide for 1 month confirm a 4- to 5-fold increase in bone formation rate on the cancellous, endocortical, and periosteal surfaces when compared to a control a group. The increase in bone formation rate on the periosteal surface suggests that PTH has also the potential to increase bone diameter (48).
Bone geometry was assessed by peripheral quantitative CT (pQCT) in a subgroup of 101 women enrolled in the teriparatide fracture prevention trial, at the forearm, the site at which cortical bone predominates (49). There were significant increases in cortical bone area, total bone mineral content and total bone area in teriparatide-treated subjects (20 or 40 μg daily) as compared to patients receiving placebo. Cortical thickness was not changed. Periosteal circumference was significantly higher in both teriparatide groups, as well as the polar cross-sectional moment of inertia. These changes in bone geometry are known to be associated with increased bone strength and improved resistance to fracture (49).
Although PTH can increase cortical porosity and bone resorption at the inner endocortical surface of the bone, it can stimulate periosteal bone apposition, and consequently, increase the outer diameter of the bone. The periosteal apposition partly offsets the loss of compressive and bending strength produced by cortical thinning and porosity, and the resultant change in the ratio of the outer to inner diameter of bone, leads, ultimately, to increased bone strength (45, 50). This favorable change in bone geometry observed with PTH could well account for the reduction in non-vertebral fracture risk (12), even though BMD is not changed or somewhat reduced at cortical sites. The paradox of reduced cortical BMD and reduced fracture risk at sites of cortical bone is thus explained by these affects of PTH on bone size and microstructure.
CELLULAR ACTIONS OF PTH ON THE SKELETON
Cellular actions of PTH contributing to increased bone resorption: Catabolic
Increased bone resorption is the most recognized catabolic action of PTH. It is one of the essential mechanisms by which PTH maintains calcium homeostasis, particularly in the face of a hypocalcemic stimulus. In vivo, PTH enhances bone resorption by increasing osteoclastic activity. However, in vitro studies demonstrate that osteoclast-like cells in culture do not show increased activity in response to PTH, unless osteoclasts are co-cultured with stromal or osteoblast-like cells or conditioned medium from osteoblasts previously treated with PTH (51–53). It is likely therefore, that PTH induces bone resorption by activating osteoclasts indirectly, through its actions on osteoblasts.
In osteoblasts, PTH regulates the expression of the receptor activator of nuclear factor-κB (RANK) ligand (RANKL), and its soluble decoy receptor osteoprotegerin (OPG), which both play a dominant role in osteoclastogenesis (54–56). RANKL, a tumor necrosis factor (TNF) family member, binds to the RANK on the surface of hematopoietic precursors of osteoclast, promoting their differentiation and survival. RANKL also stimulates fully formed osteoclasts. The catabolic effects of RANKL are inhibited by OPG, a TNF receptor family member that binds to RANKL and thereby prevents access of RANKL to its receptor RANK. The balance between amounts of RANKL and OPG is a determinant of osteoclastogenesis (57). Continuous infusion of PTH increases RANKL and inhibits OPG mRNA expression in primary murine osteoblasts and in bone from rats (54, 55, 58). In vitro studies conducted by Fu et al. (59) showed that PTH directly increases RANKL expression by activation of cAMP/PKA-CREB pathway, and inhibits OPG expression via a PKA-CREB-AP-1 pathway. These PTH actions lead to an increase in the RANKL/OPG ratio, which is believed to be the main mechanism by which PTH influences osteoclastogenesis and bone resorption (Fig. 5).
Fig. 5.

Anabolic and catabolic pathways of PTH on the skeleton. A) PTH decreases SOST/sclerostin expression in osteocytes. Sclerostin functions as a negative regulator of bone formation, and its downregulation by PTH contributes for the PTH-induced osteoanabolism. B) PTH favors bone resorption, mostly by increasing receptor activator of nuclear factor kappa-B ligand (RANKL) and decreasing osteoprotegerin (OPG) expression in osteoblasts, which ultimately leads to an increase in osteoclast formation and activity.
Clinical studies also argue for RANKL as a key intermediate in the catabolic actions of PTH. Circulating levels of RANKL were elevated in 29 patients with mild PHPT, correlating positively with bone resorption markers and with rates of bone loss at the total femur (60). The RANKL/OPG ratio, as determined by mRNA analysis of bone biopsies, significantly declines after parathyroid surgery (61). The pre-operative RANKL/OPG ratio correlated positively with 1-yr post-operative increases in bone mass. In addition to the RANKL/OPG system, it has been reported that the monocyte chemoattractant protein-1 (MCP-1) can mediate the action of PTH on osteoclastogenesis. PTH increases the expression of MCP-1 in rat osteoblastic cells and in the femur of rats treated with intermittent or continuous infusion of PTH via the PKA pathway (62). MCP-1 promotes chemoattraction of pre-osteoclasts, and enhances RANKL-induced osteoclastogenesis and fusion, contributing to the increase in bone resorption (62). Although the increase in MCP-1 expression was more pronounced in rats treated with intermittent doses of PTH than in rats treated with continuous infusion of this hormone, the latter mode of administration led to moderated but sustained up-regulation of MCP-1 mRNA levels, explaining the catabolic action of PTH observed upon continuous infusion of PTH.
Although many studies have failed to demonstrate consistently a direct effect of PTH on osteoclasts, Langub et al. (63) showed that the PTH receptor PTH1R is expressed in osteoclasts from patients with the secondary hyperparathyroidism of chronic kidney disease. More recently, Dempster et al. (64) have also demonstrated that the PTH1R is expressed in human osteoclasts derived from peripheral blood mononuclear cells at the mRNA and protein level. It is still unclear, though, if PTH can directly activate osteoclasts independent of its actions on osteoblasts.
Cellular actions of PTH contributing to increased bone formation: Osteoanabolic
Pre-clinical and clinical studies have demonstrated that intermittent administration of PTH promotes bone formation (12, 13, 65–67). The anabolic actions of PTH on bone mass depend on its direct action on cells of osteoblastic lineage. Following the interaction PTH-PTH1R (6), Gαs and Gαq are activated, with subsequent activation of PKA and PKC. It has been demonstrated that cAMP/PKA signaling is a dominant mechanism by which PTH increases bone anabolism, and that PKC is not required for the osteoanabolic action of PTH (68). In fact, a recent study showed that the Gαq-PKC signal in osteoblasts is inhibitory to the anabolic actions of PTH on bone mass (69).
The osteoanabolic action of PTH is due to its ability to increase osteoblast number, which can be achieved by an increase in osteoblastogenesis, decrease in osteoblast apoptosis or a combination of the two events (70, 71). The increase in osteoblastogenesis is explained mostly by an increase in osteoblast differentiation rather than increased proliferation. It is generally accepted that differentiation requires exit from the cell cycle and, as a result, proliferation is attenuated as differentiation proceeds (71). Thus, it has been suggested that one of the mechanisms by which PTH promotes its anabolic actions is to arrest the cell cycle progression of osteoblasts, enhancing their commitment to a differentiated osteogenic fate (72). Indeed, anti-proliferative effects have been reported in osteoblastic cell lines, cultures of primary cells, and in rodents treated with PTH, which is explained by both an attenuation of the expression of cyclin D1, a protein required for cell cycle progression, and an increase in the expression of the cell cycle inhibitors p27Kip1 and p21Cip1 (73–75). However, the effect of PTH on osteoblast proliferation may be specific to the differentiation/developmental stage of the osteoblastic cell. Although PTH suppresses proliferation of committed osteoprogenitor cells, there is evidence that suggests that it does not affect or increase the replication of uncommitted osteoblast progenitors (71, 73, 75, 76). In agreement with these data, in vitro and in vivo studies in rodents conducted by Ogita et al. (77), showed that cyclic PTH treatment promotes osteoblast differentiation from periosteum-derived mesenchymal progenitors, and has a biphasic effect to enhance, then suppress proliferation of periosteal osteoblast progenitors.
Beyond the effects on the cell cycle itself, PTH enhances osteoblast differentiation. It stimulates osteoblast differentiation and osteoblastic lineage commitment in primary calvarial cells, bone marrow-derived, and in periosteal cells (15, 77, 78). Evidence from in vitro and in vivo studies suggests that PTH increases the expression of genes that typically signal bone formation, such as the osteoblast-specific transcription factor Runx2, as well as alkaline phosphatase (79), collagen type I alpha 1 (COL1A1), and osteocalcin (14, 15, 78, 80). Recently, a novel bone formation-related factor, Tmem119, was shown to be rapidly stimulated in mouse osteoblastic cell lines by PTH (81). Consistent with a PTH-induced increase in the number of differentiating osteoblasts, intermittent PTH enhances ossicle development from bone marrow derived stromal cells implanted into immunocompromised mice (82).
Alternatively, PTH can increase osteoblast number by decreasing osteoblast apoptosis. Daily injections of PTH to adult mice showed an anti-apoptotic effect of PTH on osteoblasts in femoral and vertebral sections (83). The prevalence of osteoblast apoptosis was inversely correlated with serum osteocalcin, bone formation rate, and osteoblast number, suggesting that the prosurvival effect of PTH on osteoblasts accounts, at least in part, for its anabolic effect on bone (83). In vitro, PTH treatment also inhibits pro-apoptotic effects of dexamethasone, etoposide, hydrogen peroxide induced oxidative stress, UV irradiation, serum withdrawal and nutrient deprivation in a variety of osteoblastic cells (79, 83, 84). Mechanistically, PTH phosphorylates and inactivates the pro-apoptotic protein Bad, and increases the expression of survival genes like Bcl-2. These actions are mediated by activation of PKA (83). The increased expression of Runx2 is also required for the anti-apoptotic effect of intermittent PTH (83). Moreover, it was recently shown that PTH treatment of cultured osteoblasts augments DNA repair, and enhances cell survival under extreme metabolic stress and direct DNA damage, which was proposed as another mechanism by which PTH can suppress osteoblast apoptosis (84).
MEDIATORS OF PTH ACTIONS ON BONE FORMATION
Sclerostin
Sclerostin, a product of the SOST gene expressed primarily by osteocytes, is a secreted glycoprotein that functions as a key negative regulator of bone formation (85). In the human genetic diseases van Buchem’s disease and sclerosteosis, reduced sclerostin concentration and/or activity are translated into generalized and progressive overgrowth of bone and sclerosis of the skeleton (86–88). Likewise, Sost knockout mice have high bone mass (89). Preclinical studies show that an antisclerostin antibody has osteoanabolic effects in rodents (90). Early studies in human subjects are also confirming the anabolic effects of an antisclerostin antibody (91).
The finding that osteocytes secrete sclerostin, and express the PTH1R (92) raised the hypothesis that the osteocyte-derived sclerostin could be a mediator of the anabolic action of PTH on the skeleton. Indeed, downregulation of SOST/sclerostin by PTH has been demonstrated in vitro, in animals, and in humans, and the regulation of SOST by this hormone is currently recognized as having a key role in PTH-induced bone formation (Fig. 5) (93–98). PTH decreases Sost mRNA levels in vitro, and in rodents treated with either continuous or intermittent administration of PTH (93, 96). Similarly, transgenic mice overexpressing a constitutively active PTH1R specifically in osteocytes, have increased bone mass, and decreased Sost expression (94). In these mice, concomitant overexpression of Sost, also in osteocytes, abolishes the increase in cortical bone area, periosteal bone formation rate, and cancellous bone volume, supporting the hypothesis that the anabolic effect of PTH requires downregulation of sclerostin in osteocytes (94). In human subjects, serum sclerostin levels are reported to be lower in patients with PHPT than in controls, and a negative correlation between circulating sclerostin and PTH is observed (97–99). Moreover, intermittent PTH therapy in post-menopausal women decreases circulating sclerostin levels, which sustains the idea that the downregulation of sclerostin accounts for the osteoan-abolic action of PTH also in humans (95).
The mechanism by which sclerostin inhibits bone formation is not completely understood. Until recently, it was assumed that sclerostin would pass through the osteocytic canaliculi to access the bone surface, where it would bind to the LDL-receptor related protein (LRP) 5 and 6, inhibiting the osteoblastic canonical Wnt/β-catenin signaling (6, 100, 101). In this way, sclerostin would antagonize the pro-differentiating and survival actions of Wnts on osteoblasts. However, an effect through LRP5 in the Wnt signaling pathway has recently been questioned by studies suggesting that LRP5 does not function as a Wnt coreceptor (102). Thus, the definitive mechanism(s) by which sclerostin inhibits bone formation is still unknown.
IGF
IGF-I is a regulator of cell growth and function, and, in osteoblasts, acts as a prodifferentiating and a prosurvival factor. PTH can induce skeletal expression of IGF-I, which would act as an autocrine/paracrine factor to mediate the PTH effects on osteoblasts (103, 104). Indeed, animal studies have shown that IGF-I plays a key role in the osteoanabolic effects of PTH. IGF-I-deficient mice had an attenuated effect of the anabolic actions of PTH on cortical bone (105), and deletion of the insulin receptor substrate adapting molecule-1 (which transmits IGF-I receptor signaling) blunted the anabolic response to intermittent PTH administration (106). The anabolic actions of PTH on bone were also altered in mice with deletion of the IGF-I receptor specifically in mature osteoblasts (103). Increased cancellous bone volume at the primary spongiosa, and enhanced bone formation, mineralization, and cortical thickness at the cortical bone observed upon PTH administration to the control mice were blunted in the IGF-I receptor knockout mice (103). At the cellular level, IGF-I receptor deletion decreased the ability of PTH to stimulate osteoprogenitor cell proliferation and differentiation, as measured by the number of ALP-expressing colonies and mineralized nodules in cultures of bone marrow-derived stromal cells treated with PTH.
The role of T cells
T cells that express PTH receptors may be involved in both the anabolic and catabolic actions of PTH through CD40 Ligand, a surface molecule of activated T cells that induces CD40 signaling in stromal cells (107, 108). The work of Pacifici et al. has shown that deletion of T cells or T cell-expressed CD40 Ligand blunts the catabolic activity of PTH in bone by decreasing bone marrow stromal cell number, RANK/OPG production and osteoclastogenic activity (109). Silencing of the PTH receptor in T cells also blocks the bone loss and the osteoclastic expansion induced by continuous PTH, thus demonstrating that PTH signaling in T cells may also be central to PTH-induced bone loss. T cells also play a permissive role in the anabolic effect of intermittent PTH, which is reduced in T cell-deficient mice. The mechanism involves activation of Wnt signaling by T cells in pre-osteoblasts.
CONCLUSION
The human skeleton is being renewed constantly by the dynamic process of bone remodeling, which consists of two normally balanced phases of bone resorption and bone formation. When these two processes are balanced, bone is neither gained nor lost. An imbalance between these two, favoring bone resorption, results in a catabolic net effect on bone mass, while an anabolic net effect ensues if bone formation exceeds bone resorption. Although the increase in bone resorption was the most recognized action of PTH in the skeleton, this hormone can, indeed, increase bone formation, and its final effect on bone mass, either catabolic or anabolic, will depend on which process or processes are being favored.
Continuous exposure to high levels of PTH is associated with catabolic effects on bone, while intermittent exposure to low doses of PTH has anabolic actions. The former is exemplified by the chronic disorder of PTH excess, PH-PT, and the second is best represented by the use of PTH to treat osteoporosis. However, it is now recognized that PTH can have anabolic effects even in states of continuously elevated PTH, as evidenced by a preserved cancellous bone and a normal or greater trabecular connectivity observed in patients with PHPT. Similarly, decrease in BMD at cortical sites can be appreciated in subjects being treated with PTH, which, however, does not lead to negative effects on bone strength. In fact, decrease in facture risk at vertebral and non-vertebral sites was demonstrated in patients treated with PTH.
Mechanistically, although PTH can regulate different sets of genes when it is administered intermittently or continuously, favoring bone formation or resorption, respectively, some genes are regulated in the same way upon continuous or intermittent endogenous exposure to PTH. Decreased circulating sclerostin levels, for example, are observed in patients with PHPT, as well as in patients treated with recombinant PTH. In the same way, Sost mRNA levels decrease in rodents treated with either continuous or intermittent administration of PTH.
Despite advances in elucidating the mechanism of action of PTH on bone, the different skeletal responses to PTH are still incompletely understood. Although the response to PTH can be clearly catabolic or anabolic depending on the mode of exposure to this hormone, increases and decreases in bone mass at different skeletal sites can actually coexist in the same subject or condition. Future studies may elucidate a means to perturb molecular pathways that are regulated by PTH so that the anabolic response to this hormone is primarily expressed. Then these new insights could be applied into a medical therapy for PHPT.
References
- 1.Hanley DA, Watson PH, Hodsman AB, Dempster DW. Pharmacological Mechanisms of Therapeutics: Parathyroid Hormone. In: Bilezikian J, Raisz LG, Martin TJ, editors. Principles of Bone Biology. Amsterdam: Elsevier; 2008. pp. 1661–95. [Google Scholar]
- 2.Datta NS, Abou-Samra AB. PTH and PTHrP signaling in osteoblasts. Cell Signal. 2009;21:1245–54. doi: 10.1016/j.cellsig.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kousteni S, Bilezikian JP. The cell biology of parathyroid hormone in osteoblasts. Curr Osteoporos Rep. 2008;6:72–6. doi: 10.1007/s11914-008-0013-9. [DOI] [PubMed] [Google Scholar]
- 4.Potts JT. Parathyroid hormone: past and present. J Endocrinol. 2005;187:311–25. doi: 10.1677/joe.1.06057. [DOI] [PubMed] [Google Scholar]
- 5.Datta NS, Samra TA, Mahalingam CD, Datta T, Abou-Samra AB. Role of PTH1R internalization in osteoblasts and bone mass using a phosphorylation-deficient knock-in mouse model. J Endocrinol. 2010;207:355–65. doi: 10.1677/JOE-10-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kramer I, Keller H, Leupin O, Kneissel M. Does osteocytic SOST suppression mediate PTH bone anabolism? Trends Endocrinol Metab. 2010;21:237–44. doi: 10.1016/j.tem.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 7.Silverberg SJ, Shane E, de la Cruz L, et al. Skeletal disease in primary hyperparathyroidism. J Bone Miner Res. 1989;4:283–91. doi: 10.1002/jbmr.5650040302. [DOI] [PubMed] [Google Scholar]
- 8.Dempster DW, Muller R, Zhou H, et al. Preserved three-dimensional cancellous bone structure in mild primary hyperparathyroidism. Bone. 2007;41:19–24. doi: 10.1016/j.bone.2007.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parisien M, Mellish RW, Silverberg SJ, et al. Maintenance of cancellous bone connectivity in primary hyperparathyroidism: trabecular strut analysis. J Bone Miner Res. 1992;7:913–9. doi: 10.1002/jbmr.5650070808. [DOI] [PubMed] [Google Scholar]
- 10.Parisien M, Silverberg SJ, Shane E, et al. The histomorphometry of bone in primary hyperparathyroidism: preservation of cancellous bone structure. J Clin Endocrinol Metab. 1990;70:930–8. doi: 10.1210/jcem-70-4-930. [DOI] [PubMed] [Google Scholar]
- 11.Dobnig H, Turner RT. The effects of programmed administration of human parathyroid hormone fragment (1–34) on bone histomorphometry and serum chemistry in rats. Endocrinology. 1997;138:4607–12. doi: 10.1210/endo.138.11.5505. [DOI] [PubMed] [Google Scholar]
- 12.Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in post-menopausal women with osteoporosis. N Engl J Med. 2001;344:1434–41. doi: 10.1056/NEJM200105103441904. [DOI] [PubMed] [Google Scholar]
- 13.Greenspan SL, Bone HG, Ettinger MP, et al. Treatment of Osteoporosis with Parathyroid Hormone Study Group. Effect of recombinant human parathyroid hormone (1–84) on vertebral fracture and bone mineral density in postmenopausal women with osteoporosis: a randomized trial. Ann Intern Med. 2007;146:326–39. doi: 10.7326/0003-4819-146-5-200703060-00005. [DOI] [PubMed] [Google Scholar]
- 14.Onyia JE, Helvering LM, Gelbert L, et al. Molecular profile of catabolic versus anabolic treatment regimens of parathyroid hormone (PTH) in rat bone: an analysis by DNA microarray. J Cell Biochem. 2005;95:403–18. doi: 10.1002/jcb.20438. [DOI] [PubMed] [Google Scholar]
- 15.Locklin RM, Khosla S, Turner RT, Riggs BL. Mediators of the biphasic responses of bone to intermittent and continuously administered parathyroid hormone. J Cell Biochem. 2003;89:180–90. doi: 10.1002/jcb.10490. [DOI] [PubMed] [Google Scholar]
- 16.Cope O. The study of hyperparathyroidism at the Massachusetts General Hospital. N Engl J Med. 1966;274:1174–82. doi: 10.1056/NEJM196605262742105. [DOI] [PubMed] [Google Scholar]
- 17.Dauphine RT, Riggs BL, Scholz DA. Back pain and vertebral crush fractures: an unemphasized mode of presentation for primary hyperparathyroidism. Ann Intern Med. 1975;83:365–7. doi: 10.7326/0003-4819-83-3-365. [DOI] [PubMed] [Google Scholar]
- 18.Rubin MR, Bilezikian JP, McMahon DJ, et al. The natural history of nprimary hyperparathyroidism with or without parathyroid surgery after 15 years. J Clin Endocrinol Metab. 2008;93:3462–70. doi: 10.1210/jc.2007-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heath H, 3rd, Hodgson SF, Kennedy MA. Primary hyperparathyroidism. Incidence, morbidity, and potential economic impact in a community. N Engl J Med. 1980;302:189–93. doi: 10.1056/NEJM198001243020402. [DOI] [PubMed] [Google Scholar]
- 20.Silverberg SJ, Shane E, Jacobs TP, Siris E, Bilezikian JP. A 10-year prospective study of primary hyperparathyroidism with or without parathyroid surgery. N Engl J Med. 1999;341:1249–55. doi: 10.1056/NEJM199910213411701. [DOI] [PubMed] [Google Scholar]
- 21.Parisien M, Cosman F, Mellish RW, et al. Bone structure in post-menopausal hyperparathyroid, osteoporotic, and normal women. J Bone Miner Res. 1995;10:1393–9. doi: 10.1002/jbmr.5650100917. [DOI] [PubMed] [Google Scholar]
- 22.Uchiyama T, Tanizawa T, Ito A, Endo N, Takahashi HE. Microstructure of the trabecula and cortex of iliac bone in primary hyperparathyroidism patients determined using histomorphometry and node-strut analysis. J Bone Miner Metab. 1999;17:283–8. doi: 10.1007/s007740050096. [DOI] [PubMed] [Google Scholar]
- 23.Silverberg SJ, Locker FG, Bilezikian JP. Vertebral osteopenia: a new indication for surgery in primary hyperparathyroidism. J Clin Endocrinol Metab. 1996;81:4007–12. doi: 10.1210/jcem.81.11.8923852. [DOI] [PubMed] [Google Scholar]
- 24.Hansen S, Beck Jensen JE, Rasmussen L, Hauge EM, Brixen K. Effects on bone geometry, density, and microarchitecture in the distal radius but not the tibia in women with primary hyperparathyroidism: A case-control study using HR-pQCT. J Bone Miner Res. 2010;25:1941–7. doi: 10.1002/jbmr.98. [DOI] [PubMed] [Google Scholar]
- 25.Bilezikian JP, Brandi ML, Rubin M, Silverberg SJ. Primary hyperparathyroidism: new concepts in clinical, densitometric and biochemical features. J Intern Med. 2005;257:6–17. doi: 10.1111/j.1365-2796.2004.01422.x. [DOI] [PubMed] [Google Scholar]
- 26.Kulak CA, Bandeira C, Voss D, et al. Marked improvement in bone mass after parathyroidectomy in osteitis fibrosa cystica. J Clin Endocrinol Metab. 1998;83:732–5. doi: 10.1210/jcem.83.3.4655. [DOI] [PubMed] [Google Scholar]
- 27.Yu N, Donnan PT, Flynn RW, et al. Increased mortality and morbidity in mild primary hyperparathyroid patients. The Parathyroid Epidemiology and Audit Research Study (PEARS) Clin Endocrinol (Oxf) 2010;73:30–4. doi: 10.1111/j.1365-2265.2009.03766.x. [DOI] [PubMed] [Google Scholar]
- 28.Khosla S, Melton LJ, 3rd, Wermers RA, Crowson CS, O’Fallon W, Riggs B. Primary hyperparathyroidism and the risk of fracture: a population-based study. J Bone Miner Res. 1999;14:1700–7. doi: 10.1359/jbmr.1999.14.10.1700. [DOI] [PubMed] [Google Scholar]
- 29.Vignali E, Viccica G, Diacinti D, et al. Morphometric vertebral fractures in postmenopausal women with primary hyperparathyroidism. J Clin Endocrinol Metab. 2009;94:2306–12. doi: 10.1210/jc.2008-2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson RJ, Rao S, Ellis B, Kleerekoper M, Parfitt AM. Mild asymptomatic primary hyperparathyroidism is not a risk factor for vertebral fractures. Ann Intern Med. 1988;109:959–62. doi: 10.7326/0003-4819-109-12-959. [DOI] [PubMed] [Google Scholar]
- 31.Melton LJ, 3rd, Atkinson EJ, O’Fallon WM, Heath H., 3rd Risk of age-related fractures in patients with primary hyperparathyroidism. Arch Intern Med. 1992;152:2269–73. [PubMed] [Google Scholar]
- 32.Larsson K, Ljunghall S, Krusemo UB, Naessén T, Lindh E, Persson I. The risk of hip fractures in patients with primary hyperparathyroidism: a population-based cohort study with a follow-up of 19 years. J Intern Med. 1993;234:585–93. doi: 10.1111/j.1365-2796.1993.tb01017.x. [DOI] [PubMed] [Google Scholar]
- 33.Parfitt AM. Parathyroid hormone and periosteal bone expansion. J Bone Miner Res. 2002;17:1741–3. doi: 10.1359/jbmr.2002.17.10.1741. [DOI] [PubMed] [Google Scholar]
- 34.Bilezikian JP. Bone strength in primary hyperparathyroidism. Osteoporos Int. 2003;14 (Suppl 5):S113–5. doi: 10.1007/s00198-003-1482-4. [DOI] [PubMed] [Google Scholar]
- 35.Burr DB, Hirano T, Turner CH, Hotchkiss C, Brommage R, Hock JM. Intermittently administered human parathyroid hormone(1–34) treatment increases intracortical bone turnover and porosity without reducing bone strength in the humerus of ovariectomized cynomolgus monkeys. J Bone Miner Res. 2001;16:157–65. doi: 10.1359/jbmr.2001.16.1.157. [DOI] [PubMed] [Google Scholar]
- 36.Dempster DW, Cosman F, Kurland ES, et al. Effects of daily treatment with parathyroid hormone on bone microarchitecture and turnover in patients with osteoporosis: a paired biopsy study. J Bone Miner Res. 2001;16:1846–53. doi: 10.1359/jbmr.2001.16.10.1846. [DOI] [PubMed] [Google Scholar]
- 37.Recker RR, Bare SP, Smith SY, et al. Cancellous and cortical bone architecture and turnover at the iliac crest of postmenopausal osteoporotic women treated with parathyroid hormone 1–84. Bone. 2009;44:113–9. doi: 10.1016/j.bone.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 38.Ma YL, Zeng Q, Donley DW, et al. Teriparatide increases bone formation in modeling and remodeling osteons and enhances IGF-II immunoreactivity in postmenopausal women with osteoporosis. J Bone Miner Res. 2006;21:855–64. doi: 10.1359/jbmr.060314. [DOI] [PubMed] [Google Scholar]
- 39.Jiang Y, Zhao JJ, Mitlak BH, Wang O, Genant HK, Eriksen EF. Recombinant human parathyroid hormone (1–34) [teriparatide] improves both cortical and cancellous bone structure. J Bone Miner Res. 2003;18:1932–41. doi: 10.1359/jbmr.2003.18.11.1932. [DOI] [PubMed] [Google Scholar]
- 40.Orwoll ES, Scheele WH, Paul S, et al. The effect of teriparatide [human parathyroid hormone (1–34)] therapy on bone density in men with osteoporosis. J Bone Miner Res. 2003;18:9–17. doi: 10.1359/jbmr.2003.18.1.9. [DOI] [PubMed] [Google Scholar]
- 41.Fox J, Miller MA, Recker RR, Bare SP, Smith SY, Moreau I. Treatment of postmenopausal osteoporotic women with parathyroid hormone 1–84 for 18 months increases cancellous bone formation and improves cancellous architecture: a study of iliac crest biopsies using histomorphometry and micro computed tomography. J Musculoskelet Neuronal Interact. 2005;5:356–7. [PubMed] [Google Scholar]
- 42.Dobnig H, Sipos A, Jiang Y, et al. Early changes in biochemical markers of bone formation correlate with improvements in bone structure during teriparatide therapy. J Clin Endocrinol Metab. 2005;90:3970–7. doi: 10.1210/jc.2003-1703. [DOI] [PubMed] [Google Scholar]
- 43.Lane NE, Sanchez S, Genant HK, Jenkins DK, Arnaud CD. Short-term increases in bone turnover markers predict parathyroid hormone-induced spinal bone mineral density gains in post-menopausal women with glucocorticoid-induced osteoporosis. Osteoporos Int. 2000;11:434–42. doi: 10.1007/s001980070111. [DOI] [PubMed] [Google Scholar]
- 44.Kurland ES, Cosman F, McMahon DJ, Rosen CJ, Lindsay R, Bilezikian JP. Parathyroid hormone as a therapy for idiopathic osteoporosis in men: effects on bone mineral density and bone markers. J Clin Endocrinol Metab. 2000;85:3069–76. doi: 10.1210/jcem.85.9.6818. [DOI] [PubMed] [Google Scholar]
- 45.Rubin MR, Bilezikian JP. Parathyroid hormone as an anabolic skeletal therapy. Drugs. 2005;65:2481–98. doi: 10.2165/00003495-200565170-00005. [DOI] [PubMed] [Google Scholar]
- 46.Lindsay R, Cosman F, Zhou H, et al. A novel tetracycline labeling schedule for longitudinal evaluation of the short-term effects of anabolic therapy with a single iliac crest bone biopsy: early actions of teriparatide. J Bone Miner Res. 2006;21:366–73. doi: 10.1359/JBMR.051109. [DOI] [PubMed] [Google Scholar]
- 47.Compston JE. Skeletal actions of intermittent parathyroid hormone: effects on bone remodelling and structure. Bone. 2007;40:1447–52. doi: 10.1016/j.bone.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 48.Lindsay R, Zhou H, Cosman F, Nieves J, Dempster DW, Hodsman AB. Effects of a one-month treatment with PTH(1–34) on bone formation on cancellous, endocortical, and periosteal surfaces of the human ilium. J Bone Miner Res. 2007;22:495–502. doi: 10.1359/jbmr.070104. [DOI] [PubMed] [Google Scholar]
- 49.Zanchetta JR, Bogado CE, Ferretti JL, et al. Effects of teriparatide [recombinant human parathyroid hormone (1–34)] on cortical bone in postmenopausal women with osteoporosis. J Bone Miner Res. 2003;18:539–43. doi: 10.1359/jbmr.2003.18.3.539. [DOI] [PubMed] [Google Scholar]
- 50.Seeman E, Delmas PD. Bone quality--the material and structural basis of bone strength and fragility. N Engl J Med. 2006;354:2250–61. doi: 10.1056/NEJMra053077. [DOI] [PubMed] [Google Scholar]
- 51.Chambers TJ, Fuller K, McSheehy PM, Pringle JA. The effects of calcium regulating hormones on bone resorption by isolated human osteoclastoma cells. J Pathol. 1985;145:297–305. doi: 10.1002/path.1711450403. [DOI] [PubMed] [Google Scholar]
- 52.McSheehy PM, Chambers TJ. Osteoblastic cells mediate osteoclastic responsiveness to parathyroid hormone. Endocrinology. 1986;118:824–8. doi: 10.1210/endo-118-2-824. [DOI] [PubMed] [Google Scholar]
- 53.Kaji H, Sugimoto T, Kanatani M, Fukase M, Chihara K. Involvement of dual signal transduction systems in the stimulation of osteoclast-like cell formation by parathyroid hormone and parathyroid hormone-related peptide. Biochem Biophys Res Commun. 1993;194:157–62. doi: 10.1006/bbrc.1993.1798. [DOI] [PubMed] [Google Scholar]
- 54.Lee SK, Lorenzo JA. Parathyroid hormone stimulates TRANCE and inhibits osteoprotegerin messenger ribonucleic acid expression in murine bone marrow cultures: correlation with osteoclast-like cell formation. Endocrinology. 1999;140:3552–61. doi: 10.1210/endo.140.8.6887. [DOI] [PubMed] [Google Scholar]
- 55.Ma YL, Cain RL, Halladay DL, et al. Catabolic effects of continuous human PTH (1--38) in vivo is associated with sustained stimulation of RANKL and inhibition of osteoprotegerin and gene-associated bone formation. Endocrinology. 2001;142:4047–54. doi: 10.1210/endo.142.9.8356. [DOI] [PubMed] [Google Scholar]
- 56.Kanzawa M, Sugimoto T, Kanatani M, Chihara K. Involvement of osteoprotegerin/osteoclastogenesis inhibitory factor in the stimulation of osteoclast formation by parathyroid hormone in mouse bone cells. Eur J Endocrinol. 2000;142:661–4. doi: 10.1530/eje.0.1420661. [DOI] [PubMed] [Google Scholar]
- 57.Kearns AE, Khosla S, Kostenuik PJ. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocr Rev. 2008;29:155–92. doi: 10.1210/er.2007-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang JC, Sakata T, Pfleger LL, et al. PTH differentially regulates expression of RANKL and OPG. J Bone Miner Res. 2004;19:235–44. doi: 10.1359/JBMR.0301226. [DOI] [PubMed] [Google Scholar]
- 59.Fu Q, Jilka RL, Manolagas SC, O’Brien CA. Parathyroid hormone stimulates receptor activator of NFkappa B ligand and inhibits osteoprotegerin expression via protein kinase A activation of cAMP-response element-binding protein. J Biol Chem. 2002;277:48868–75. doi: 10.1074/jbc.M208494200. [DOI] [PubMed] [Google Scholar]
- 60.Nakchbandi IA, Lang R, Kinder B, Insogna KL. The role of the receptor activator of nuclear factor-kappaB ligand/osteoprotegerin cytokine system in primary hyperparathyroidism. J Clin Endocrinol Metab. 2008;93:967–73. doi: 10.1210/jc.2007-1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stilgren LS, Rettmer E, Eriksen EF, Hegedüs L, Beck-Nielsen H, Abrahamsen B. Skeletal changes in osteoprotegerin and receptor activator of nuclear factor-kappab ligand mRNA levels in primary hyperparathyroidism: effect of parathyroidectomy and association with bone metabolism. Bone. 2004;35:256–65. doi: 10.1016/j.bone.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 62.Li X, Qin L, Bergenstock M, Bevelock LM, Novack DV, Partridge NC. Parathyroid hormone stimulates osteoblastic expression of MCP-1 to recruit and increase the fusion of pre/osteoclasts. J Biol Chem. 2007;282:33098–106. doi: 10.1074/jbc.M611781200. [DOI] [PubMed] [Google Scholar]
- 63.Langub MC, Monier-Faugere MC, Qi Q, Geng Z, Koszewski NJ, Malluche HH. Parathyroid hormone/parathyroid hormone-related peptide type 1 receptor in human bone. J Bone Miner Res. 2001;16:448–56. doi: 10.1359/jbmr.2001.16.3.448. [DOI] [PubMed] [Google Scholar]
- 64.Dempster DW, Hughes-Begos CE, Plavetic-Chee K, et al. Normal human osteoclasts formed from peripheral blood monocytes express PTH type 1 receptors and are stimulated by PTH in the absence of osteoblasts. J Cell Biochem. 2005;95:139–48. doi: 10.1002/jcb.20388. [DOI] [PubMed] [Google Scholar]
- 65.Shen V, Dempster DW, Birchman R, Xu R, Lindsay R. Loss of cancellous bone mass and connectivity in ovariectomized rats can be restored by combined treatment with parathyroid hormone and estradiol. J Clin Invest. 1993;91:2479–87. doi: 10.1172/JCI116483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wronski TJ, Yen CF, Qi H, Dann LM. Parathyroid hormone is more effective than estrogen or bisphosphonates for restoration of lost bone mass in ovariectomized rats. Endocrinology. 1993;132:823–31. doi: 10.1210/endo.132.2.8425497. [DOI] [PubMed] [Google Scholar]
- 67.Iwaniec UT, Moore K, Rivera MF, Myers SE, Vanegas SM, Wronski TJ. A comparative study of the bone-restorative efficacy of anabolic agents in aged ovariectomized rats. Osteoporos Int. 2007;18:351–62. doi: 10.1007/s00198-006-0240-9. [DOI] [PubMed] [Google Scholar]
- 68.Yang D, Singh R, Divieti P, Guo J, Bouxsein ML, Bringhurst FR. Contributions of parathyroid hormone (PTH)/PTH-related peptide receptor signaling pathways to the anabolic effect of PTH on bone. Bone. 2007;40:1453–61. doi: 10.1016/j.bone.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ogata N, Shinoda Y, Wettschureck N, et al. G alpha(q) signal in osteoblasts is inhibitory to the osteoanabolic action of parathyroid hormone. J Biol Chem. 2011;286:13733–40. doi: 10.1074/jbc.M110.200196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jilka RL. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone. 2007;40:1434–46. doi: 10.1016/j.bone.2007.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kousteni S, Bilezikian J. Cellular Actions of Parathyroid Hormone. In: Bilezikian J, Raisz LG, Martin TJ, editors. Principles of Bone Biology. Amsterdam: Elsevier; 2008. pp. 639–56. [Google Scholar]
- 72.Wang YH, Liu Y, Rowe DW. Effects of transient PTH on early proliferation, apoptosis, and subsequent differentiation of osteoblast in primary osteoblast cultures. Am J Physiol Endocrinol Metab. 2007;292:E594–603. doi: 10.1152/ajpendo.00216.2006. [DOI] [PubMed] [Google Scholar]
- 73.Qin L, Li X, Ko JK, Partridge NC. Parathyroid hormone uses multiple mechanisms to arrest the cell cycle progression of osteoblastic cells from G1 to S phase. J Biol Chem. 2005;280:3104–11. doi: 10.1074/jbc.M409846200. [DOI] [PubMed] [Google Scholar]
- 74.Onishi T, Hruska K. Expression of p27Kip1 in osteoblast-like cells during differentiation with parathyroid hormone. Endocrinology. 1997;138:1995–2004. doi: 10.1210/endo.138.5.5146. [DOI] [PubMed] [Google Scholar]
- 75.Datta NS, Kolailat R, Fite A, Pettway G, Abou-Samra AB. Distinct roles for mitogen-activated protein kinase phosphatase-1 (MKP-1) and ERK-MAPK in PTH1R signaling during osteoblast proliferation and differentiation. Cell Signal. 2010;22:457–66. doi: 10.1016/j.cellsig.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Datta NS, Pettway GJ, Chen C, Koh AJ, McCauley LK. Cyclin D1 as a target for the proliferative effects of PTH and PTHrP in early osteoblastic cells. J Bone Miner Res. 2007;22:951–64. doi: 10.1359/jbmr.070328. [DOI] [PubMed] [Google Scholar]
- 77.Ogita M, Rached MT, Dworakowski E, Bilezikian JP, Kousteni S. Differentiation and proliferation of periosteal osteoblast progenitors are differentially regulated by estrogens and intermittent parathyroid hormone administration. Endocrinology. 2008;149:5713–23. doi: 10.1210/en.2008-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ishizuya T, Yokose S, Hori M, et al. Parathyroid hormone exerts disparate effects on osteoblast differentiation depending on exposure time in rat osteoblastic cells. J Clin Invest. 1997;99:2961–70. doi: 10.1172/JCI119491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen HL, Demiralp B, Schneider A, et al. Parathyroid hormone and parathyroid hormone-related protein exert both pro- and anti-apoptotic effects in mesenchymal cells. J Biol Chem. 2002;277:19374–81. doi: 10.1074/jbc.M108913200. [DOI] [PubMed] [Google Scholar]
- 80.Krishnan V, Moore TL, Ma YL, et al. Parathyroid hormone bone anabolic action requires Cbfa1/Runx2-dependent signaling. Mol Endocrinol. 2003;17:423–35. doi: 10.1210/me.2002-0225. [DOI] [PubMed] [Google Scholar]
- 81.Hisa I, Inoue Y, Hendy GN, et al. Parathyroid hormone-responsive Smad3-related factor, Tmem119, promotes osteoblast differentiation and interacts with the bone morphogenetic protein-Runx2 pathway. J Biol Chem. 2011;286:9787–96. doi: 10.1074/jbc.M110.179127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pettway GJ, Schneider A, Koh AJ, et al. Anabolic actions of PTH (1–34): use of a novel tissue engineering model to investigate temporal effects on bone. Bone. 2005;36:959–70. doi: 10.1016/j.bone.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 83.Bellido T, Ali AA, Plotkin LI, et al. Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts. A putative explanation for why intermittent administration is needed for bone anabolism. J Biol Chem. 2003;278:50259–72. doi: 10.1074/jbc.M307444200. [DOI] [PubMed] [Google Scholar]
- 84.Schnoke M, Midura SB, Midura RJ. Parathyroid hormone suppresses osteoblast apoptosis by augmenting DNA repair. Bone. 2009;45:590–602. doi: 10.1016/j.bone.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Poole KE, van Bezooijen RL, Loveridge N, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19:1842–4. doi: 10.1096/fj.05-4221fje. [DOI] [PubMed] [Google Scholar]
- 86.Balemans W, Ebeling M, Patel N, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Hum Mol Genet. 2001;10:537–43. doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- 87.Balemans W, Patel N, Ebeling M, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39:91–7. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Loots GG, Kneissel M, Keller H, et al. Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 2005;15:928–35. doi: 10.1101/gr.3437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li X, Ominsky MS, Niu QT, et al. Targeted deletion of the scle-rostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23:860–9. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- 90.Li X, Ominsky MS, Warmington KS, et al. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res. 2009;24:578–88. doi: 10.1359/jbmr.081206. [DOI] [PubMed] [Google Scholar]
- 91.Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26:19–26. doi: 10.1002/jbmr.173. [DOI] [PubMed] [Google Scholar]
- 92.Fermor B, Skerry TM. PTH/PTHrP receptor expression on osteoblasts and osteocytes but not resorbing bone surfaces in growing rats. J Bone Miner Res. 1995;10:1935–43. doi: 10.1002/jbmr.5650101213. [DOI] [PubMed] [Google Scholar]
- 93.Bellido T, Ali AA, Gubrij I, et al. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology. 2005;146:4577–83. doi: 10.1210/en.2005-0239. [DOI] [PubMed] [Google Scholar]
- 94.Rhee Y, Allen MR, Condon K, et al. PTH receptor signaling in osteocytes governs periosteal bone formation and intracortical remodeling. J Bone Miner Res. 2011;26:1035–46. doi: 10.1002/jbmr.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Drake MT, Srinivasan B, Mödder UI, et al. Effects of parathyroid hormone treatment on circulating sclerostin levels in post-menopausal women. J Clin Endocrinol Metab. 2010;95:5056–62. doi: 10.1210/jc.2010-0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Keller H, Kneissel M. SOST is a target gene for PTH in bone. Bone. 2005;37:148–58. doi: 10.1016/j.bone.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 97.Mirza FS, Padhi ID, Raisz LG, Lorenzo JA. Serum sclerostin levels negatively correlate with parathyroid hormone levels and free estrogen index in postmenopausal women. J Clin Endocrinol Metab. 2010;95:1991–7. doi: 10.1210/jc.2009-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.van Lierop AH, Witteveen JE, Hamdy NA, Papapoulos SE. Patients with primary hyperparathyroidism have lower circulating sclerostin levels than euparathyroid controls. Eur J Endocrinol. 2010;163:833–7. doi: 10.1530/EJE-10-0699. [DOI] [PubMed] [Google Scholar]
- 99.Costa A, Cremers S, Rubin M, et al. Circulating Sclerostin Levels in Disorders of Parathyroid Function: Primary Hyperparathyroidism (PHPT) and Hypoparathyroidism (HypoPT). ASBMR 2010 Annual Meeting; 2010. p. S158. (abstract) [Google Scholar]
- 100.Semënov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005;280:26770–5. doi: 10.1074/jbc.M504308200. [DOI] [PubMed] [Google Scholar]
- 101.Li X, Zhang Y, Kang H, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883–7. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 102.Yadav VK, Ryu JH, Suda N, et al. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell. 2008;135:825–37. doi: 10.1016/j.cell.2008.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang Y, Nishida S, Boudignon BM, et al. IGF-I receptor is required for the anabolic actions of parathyroid hormone on bone. J Bone Miner Res. 2007;22:1329–37. doi: 10.1359/jbmr.070517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lombardi G, Di Somma C, Vuolo L, Guerra E, Scarano E, Colao A. Role of IGF-I on PTH effects on bone. J Endocrinol Invest. 2010;33 (7 Suppl):22–6. [PubMed] [Google Scholar]
- 105.Yakar S, Bouxsein ML, Canalis E, et al. The ternary IGF complex influences postnatal bone acquisition and the skeletal response to intermittent parathyroid hormone. J Endocrinol. 2006;189:289–99. doi: 10.1677/joe.1.06657. [DOI] [PubMed] [Google Scholar]
- 106.Yamaguchi M, Ogata N, Shinoda Y, et al. Insulin receptor substrate-1 is required for bone anabolic function of parathyroid hormone in mice. Endocrinology. 2005;146:2620–8. doi: 10.1210/en.2004-1511. [DOI] [PubMed] [Google Scholar]
- 107.Pacifici R. T cells: critical bone regulators in health and disease. Bone. 2010;47:461–71. doi: 10.1016/j.bone.2010.04.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gao Y, Wu X, Terauchi M, et al. T cells potentiate PTH-induced cortical bone loss through CD40L signaling. Cell Metab. 2008;8:132–45. doi: 10.1016/j.cmet.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tawfeek H, Bedi B, Li JY, et al. Disruption of PTH receptor 1 in T cells protects against PTH-induced bone loss. PLoS One. 2010;5:e12290. doi: 10.1371/journal.pone.0012290. [DOI] [PMC free article] [PubMed] [Google Scholar]