

Abstract
Studies in humans and rodents support a role for muscarinic ACh receptor (mAChR) and nicotinic AChR in learning and memory, and both regulate hippocampal synaptic plasticity using complex and often times opposing mechanisms. Acetylcholinesterase (AChE) inhibitors are commonly prescribed to enhance cholinergic signaling in Alzheimer's disease in hopes of rescuing cognitive function, caused, in part, by degeneration of cholinergic innervation to the hippocampus and cortex. Unfortunately, therapeutic efficacy is moderate and inconsistent, perhaps due to unanticipated mechanisms. M1 mAChRs bidirectionally control synaptic strength at CA3-CA1 synapses; weak pharmacological activation using carbachol (CCh) facilitates potentiation, whereas strong agonism induces muscarinic long-term depression (mLTD) via an ERK-dependent mechanism. Here, we tested the prediction that accumulation of extracellular ACh via inhibition of AChE is sufficient to induce LTD at CA3-CA1 synapses in hippocampal slices from adult rats. Although AChE inhibition with eserine induces LTD, it unexpectedly does not share properties with mLTD induced by CCh, as reported previously. Eserine-LTD was prevented by the M3 mAChR-preferring antagonist 1,1-dimethyl-4-diphenylacetoxypiperidinium iodide (4-DAMP), and pharmacological inhibition of MEK was completely ineffective. Additionally, pharmacological inhibition of p38 MAPK prevents mLTD but has no effect on eserine-LTD. Finally, long-term expression of eserine-LTD is partially dependent on a decrease in presynaptic release probability, likely caused by tonic activation of mAChRs by the sustained increase in extracellular ACh. Thus these findings extend current literature by showing that pharmacological AChE inhibition causes a prolonged decrease in presynaptic glutamate release at CA3-CA1 synapses, in addition to inducing a likely postsynaptic form of LTD.
Keywords: acetylcholinesterase inhibitor, Alzheimer's disease, hippocampus, long-term depression, muscarinic AChR
cholinergic afferents to the hippocampus, projecting from the medial septum and diagonal band of Broca, modulate multiple synaptic properties that can be observed across excitatory and inhibitory circuits and encompass cell excitability, long-term plasticity, or network oscillations. Current understanding of how the cholinergic system impacts hippocampal synaptic function is derived from pharmacological manipulation of nicotinic ACh receptors (nAChRs) and muscarinic AChRs (mAChRs), lesions of cholinergic afferents, and genetic approaches. Whereas a prominent role for cholinergic receptors in hippocampal function is evident, knowledge of the exact mechanisms by which they exert their effects remains incomplete.
In humans, nonspecific cholinergic antagonists disrupt memory (Lasser et al. 1989; Little et al. 1998; Preston et al. 1989), and degeneration of cholinergic input from the basal forebrain to the hippocampus is a hallmark of Alzheimer's disease (AD) (Collerton 1986; Kasa et al. 1997; McKinney and Jacksonville 2005; Schliebs 2005). In rodent models, bilateral injection of scopolamine, a nonspecific muscarinic antagonist, into the dorsal hippocampus impairs spatial learning in rodents (Herrera-Morales et al. 2007). In accord with these observations, cholinergic lesion of the medial septum using 192-saporin has been shown to impair long-term potentiation (LTP), a putative cellular correlate of learning and memory, although some studies report no effect of septal lesion on LTP magnitude (Scheiderer et al. 2006). Also, 192-saporin-induced septal lesion reduces glutamatergic synaptic currents in CA1 pyramidal cells (Kanju et al. 2012), and other septohippocampal lesion studies in rodent brain produce memory and attentional deficits (Bartus et al. 1982; Berger-Sweeney et al. 2001; Callahan et al. 1993; Dekker et al. 1991; Drachman and Leavitt 1974; Fibiger 1991; Gold 2003; Nilsson et al. 1992; Perry et al. 1999; Sarter and Parikh 2005; Wrenn et al. 1999). In genetically engineered animals lacking M1 mAChRs, impaired LTP is evident (Seeger et al. 2004; Shinoe et al. 2005), as are impairments in spatial memory and consolidation, although deficits in these animals appear to encompass specific tasks rather than broadly disrupting all hippocampus-dependent learning (Anagnostaras et al. 2003; Hamilton et al. 2001; Miyakawa et al. 2001). This finding highlights the level of complexity in mAChR modulation of hippocampal function, and discrepancies within the learning and memory literature do exist. For instance, Sheffler and colleagues (2009) report no impairment in hippocampus-dependent learning, due to injection of a highly specific M1 mAChR antagonist, VU-022035, but allosteric M1 mAChR agonists enhance acquisition of hippocampus-dependent cognition in another report (Digby et al. 2012). Also, whereas disruption of the M2 gene causes clear behavioral deficits in spatial learning and hippocampal memory tasks (Bainbridge et al. 2008; Seeger et al. 2004), the injection of M2/M4-favoring antagonists actually improves cognitive performance in some studies (Aura et al. 1997; Baratti et al. 1993; Carey et al. 2001; Galli et al. 2000; Hamm et al. 1995; Kopf et al. 1998; Ohno et al. 1994; Packard et al. 1990; Vannucchi et al. 1997).
As suggested by the findings described above, the effects of activation of M1-M4 mAChRs within area CA1 are particularly complex and often times opposing, with the overall effect of their activation depending on several factors, including the dose of agonist used and the timing of activation. Presynaptically, it is known that the M3 mAChR-preferring antagonist, the 1,1-dimethyl-4-diphenylacetoxypiperidinium iodide (4-DAMP)-sensitive receptor, depresses excitatory synapses by reducing the probability of neurotransmitter release, and a postsynaptically expressed form of muscarinic long-term depression (mLTD) is induced with a high dose of carbachol (CCh), using a M1-dependent mechanism at CA3-CA1 synapses (Scheiderer et al. 2006, 2008; Volk et al. 2007) and in the visual cortex (McCoy and McMahon 2007). Conversely, an M2 mAChR-mediated potentiation of glutamatergic synapses, termed LTPm, can be induced by low-dose CCh application in vitro (Auerbach and Segal 1994). Interestingly, LTP has also been observed in vivo after an intracerebroventricular injection of the M2/M4 receptor-favoring antagonist methoctramine (Li et al. 2007).
In addition to evoking changes directly in synaptic strength, numerous studies show that mAChRs activated with an agonist or synaptically released ACh can modulate (in most cases, enhance) the magnitude of N-methyl-d-aspartate receptor (NMDAR)-mediated plasticity. The observed effects of mAChRs on NMDAR conductance include facilitation (Harvey et al. 1993; Markram and Segal 1990; Shinoe et al. 2005) and in one report, depression (Jo et al. 2010). In a M1- and NMDAR-dependent mechanism, likely involving Kv7/M potassium channels (Suzuki and Okada 2012), synaptically released ACh or a low dose of CCh increases the magnitude of tetanus-induced LTP in CA3-CA1 synapses (Shinoe et al. 2005). Gu and Yakel (2011) demonstrate that synaptically derived ACh, released 10 ms after Schaffer collateral stimulation, evokes LTP. Furthermore, an enhancement of spike timing-dependent LTP can occur due to repeated stimulation of stratum (s.) oriens or application of the acetylcholinesterase (AChE) inhibitor eserine (Sugisaki et al. 2011).
The observation that AD patients experience degeneration of cholinergic terminals in the hippocampus and cortex motivates the prescription of AChE inhibitors to AD patients with the goal of increasing the concentration of endogenously released ACh at the synaptic cleft (Blount et al. 2002; Imbimbo 2001). Whereas it is clear that NMDAR-dependent forms of synaptic potentiation of established importance to learning and memory can be enhanced in vitro by the application of AChE inhibitors (Sugisaki et al. 2011), their benefits in the clinical arena are moderate and inconsistent. The varied and often times competing nature of cholinergic regulatory mechanisms could account for the disappointing degree by which this therapy restores cognitive function in AD patients. Few studies have investigated the effects of AChE inhibitors on neurotransmission in the hippocampus, and further elucidation of their effects on synaptic physiology could better inform AChE use in the clinical arena. As described above, pharmacological activation of M1 mAChRs by a high concentration of an exogenous agonist can induce synaptic depression of CA3-CA1 synapses. Therefore, we hypothesized that accumulation of ACh due to AChE inhibitor application may be sufficient to trigger mLTD. To test this hypothesis, extracellular field recordings were performed at CA3-CA1 synapses in hippocampal slices in vitro from adult rats, and the effects of acute eserine treatment were assessed.
METHODS
Animals
Three- to 4-mo-old or 3- to 5-wk-old male Sprague-Dawley rats (The Jackson Laboratory, Bar Harbor, ME) were used in this study. The younger, 3- to 5-wk-old animals were used as a positive control for efficacy of the MEK inhibitor U0126. The rats were housed in a specific, pathogen-free facility under veterinary supervision at an ambient temperature of 22–23°C and under a 12:12-h light/dark cycle. Rats were allowed ad libitum access to food and water. All animal procedures used for this study were prospectively reviewed and approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
Slice Preparation and Electrophysiology
Hippocampal slices (400 μm) were prepared using methods described previously (Mans et al. 2010) with modifications. Briefly, rats were anesthetized with isoflurane, and brains were removed from the skull and immersed in ice-cold, high-sucrose artificial cerebrospinal fluid (aCSF) solution, composed of (in mM) the following: NaCl 85, KCl 2.5, MgSO4 4, CaCl2 0.5, NaH2PO4 1.25, NaHCO3 25, glucose 25, and sucrose 75 and saturated with 95% O2-5% CO2 (pH 7.4). This solution contains less Na+ and Ca2+ and higher Mg2+ than sodium-based aCSF and promotes neuronal survival during the slicing procedure by reducing excitotoxicity (Kuenzi et al. 2000). Coronal slices of dorsal hippocampi were cut on a vibratome (Leica Microsystems, Buffalo Grove, IL) and incubated at room temperature in kynurenic acid-supplemented, sodium-based aCSF, containing (in mM) the following: NaCl 119, KCl 2.5, CaCl2 2.5, MgSO4 1.3, NaH2PO4 1, NaHCO3 26, and glucose 10 and saturated with 95% O2-5% CO2 (pH 7.4). To record field excitatory postsynaptic potentials (fEPSPs), slices were placed in a submersion recording chamber and perfused continuously at ∼1.5–2.0 ml/min, with aCSF warmed to 26–28°C and recirculated via a peristaltic pump. Before placing electrodes, kynurenic acid was removed from slices during a 10-min washout period, in which recording solution was not recirculated. CA1 extracellular dendritic fEPSPs were recorded (Axopatch 200B; Molecular Devices, Sunnyvale, CA) using standard methods (Mans et al. 2010). Stimulus frequency was 0.1 Hz (100 μs duration; model DS3 stimulus isolator; Digitimer, Hertfordshire, UK), and stimulus intensity was adjusted to yield fEPSPs with amplitudes of 0.8–1.04 mV. Schaffer collaterals were stimulated with a bipolar tungsten stimulating electrode placed in CA1 s. radiatum, and fEPSPs were recorded using a glass microelectrode filled with aCSF, also placed in CA1 s. radiatum. If stable fEPSPs were maintained for at least 15 min, then mAChR-dependent LTD was induced with one of two treatments: 1) 10 min CCh (50 μM) bath application; 2) 10 min eserine (100 nM or 10 μM) bath application. When attempting to block LTD pharmacologically, inhibitors were present for the entire experiment, including during baseline stimulation, CCh or eserine application, and washout.
Preparation of Solutions for Electrophysiology Experiments
CCh.
CCh was purchased from Sigma (St. Louis, MO; cat. #C4382). A 50-mM stock solution was prepared in double-distilled (dd)H2O, aliquoted, and stored at −20°C. On the day of use, 100 μl stock solution was dissolved in 100 ml aCSF to reach a final concentration of 50 μM. As a vehicle control for experiments pairing U0126 with CCh, 100 μl DMSO was added to reach a final DMSO concentration of 0.1%.
Eserine.
Eserine was purchased from Sigma (cat. #E8375). A 20-mM stock solution was prepared in DMSO, aliquoted, and stored at −20°C. On the day of use, 12.5 μl stock solution was dissolved in 25 ml aCSF to reach a final concentration of 10 μM. As a vehicle control for experiments pairing U0126 with eserine, 12.5 μl DMSO was added to reach a final DMSO concentration of 0.1%. In the absence of eserine or U0126, DMSO concentration was maintained at 0.1% as a vehicle control.
U1026.
U0126, manufactured by Promega (Madison, WI), was purchased from Fisher Scientific (Pittsburgh, PA; cat. #V1121). A 20-mM stock solution was prepared in DMSO, aliquoted, and stored at −20°C. On the day of use, 75 μl stock solution was dissolved in 150 ml aCSF to reach a final concentration of 10 μM. In the absence of eserine, additional DMSO was added to bring the final DMSO concentration to 0.1%.
SB203580.
SB203580 was purchased from Tocris (Bristol, UK; cat. #1402). A 5-mM stock solution was prepared in ddH2O, aliquoted, and stored at −20°C. On the day of use, 75 μl stock solution was dissolved in 75 ml aCSF to reach a final concentration of 5 μM. As a vehicle control for experiments pairing SB203580 with eserine, additional DMSO was added to reach a final DMSO concentration of 0.1% in the absence of eserine.
Pirenzepine.
Pirenzepine was purchased from Tocris (cat. #1071). A 75-mM solution was prepared in ddH2O, aliquoted, and stored at −20°C. On the day of use, 10 μl stock solution was dissolved in 100 ml aCSF to reach a final concentration of 75 nM.
4-DAMP.
4-DAMP was purchased from Tocris (cat. #0482). A 10-mM stock solution was prepared in ddH2O, aliquoted, and stored at −20°C. On the day of use, stock solution was diluted in aCSF to reach a final concentration of 100 nM.
Picrotoxin.
Picrotoxin was purchased from Sigma (cat. #P7412). A 4-mM stock solution was prepared in aCSF and stored at 4°C. On the day of use, 25 ml stock solution was diluted to a final volume of 100 ml and a final concentration of 100 μM.
Atropine.
Atropine was purchased from Sigma (cat. #0257). A 10-mM stock solution was prepared in ddH2O, aliquoted, and stored at −20°C. On the day of use, stock solution was diluted in aCSF to a final concentration of 1 μM.
CGP 52432.
CGP 52432 (CGP) was purchased from Tocris (cat. #1246). A 1-mM stock solution was prepared in ddH2O, aliquoted, and stored at −20°C. On the day of use, stock solution was diluted in aCSF to a final concentration of 1 μM.
Data Analysis
Data were expressed as mean ± SE. Comparison of data from different treatment groups was performed by Student's t-test, and P < 0.05 was considered statistically significant. Data from electrophysiology experiments were filtered at 3 kHz, digitized at 10 kHz, and acquired using LabVIEW data acquisition software. The slope of the rising phase of fEPSP was measured and plotted vs. time. Each point represents the average of five raw data points. To determine the magnitude of LTD, the slopes of the rising phase of fEPSPs were normalized to baseline, and 5 min of raw fEPSPs was averaged. In the majority of experiments, the magnitude of LTD was measured 40 min postdrug (eserine or CCh) application. Exceptions occurred (see Fig. 1A; analysis at 65 min posteserine), because a lower concentration of eserine was used and (see Fig. 7A; analysis at 20 min posteserine application) because atropine was applied during the expression phase of the LTD.
Fig. 1.
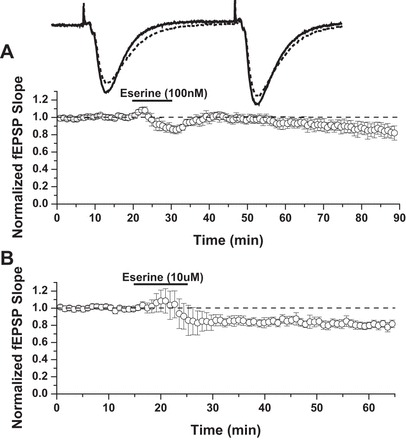
Acute eserine treatment induces long-term depression (LTD; eserine-LTD) at CA3-CA1 synapses. A: 10 min bath application of 100 nM eserine. LTD: 83 ± 8% of baseline field excitatory postsynaptic potentials (fEPSP) slope, 85–90 min (n = 4). B: 10 min bath application of 10 μM eserine. LTD: 80 ± 3% of baseline fEPSP slope, 60–65 min (n = 6).
Fig. 7.
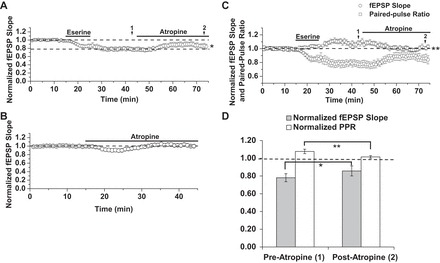
Atropine partially attenuates eserine-LTD but fully reverses an eserine-induced increase in PPR. A: atropine (1 μM), applied between 45 and 75 min of eserine-LTD (10 μM) expression. LTD: 78 ± 5% of baseline fEPSP slope preatropine (time point 1) vs. 86 ± 6% of baseline fEPSP slope postatropine (time point 2), n = 6; P = 0.02, Student's paired t-test. B: 30 min treatment with atropine alone. Postatropine slope: 1.01% of baseline (40–45 min, n = 7). C: paired-pulse facilitation ratios during atropine experiments shown in A, plotted with respect to fEPSP slope. Eserine-LTD and atropine-mediated attenuation of LTD are accompanied by an increase and normalization of PPR, respectively. PPR: 1.08 ± 3% of baseline PPR preatropine (time point 1) vs. 1.01 ± 2% of baseline PPR postatropine (time point 2), n = 6; P = 0.002, Student's paired t-test. D: LTD and PPR data from time points 1 and 2 (pre- and postatropine, respectively), presented in a bar chart. fEPSP slope remains significantly depressed, despite normalization of PPR by atropine. *P ≤ 0.05; **P ≤ 0.01.
RESULTS
Pharmacological Blockade of AChE Induces a Long-Lasting Synaptic Depression Requiring mAChR Activation
To test the effect of AChE inhibition on synaptic transmission, hippocampal slices from adult male rats (3–4 mo) were treated with eserine (100 nM) for 10 min during extracellular dendritic field potential recordings. We find this acute eserine treatment sufficient to induce a long-lasting depression, which we term eserine-LTD, at CA3-CA1 synapses (Fig. 1A; LTD: 83 ± 8% of fEPSP baseline slope; 85–90 min, n = 4). To test if a higher dose of eserine could accelerate the time course of LTD expression, we applied 10 μM eserine for 10 min. Compared with our initial experiments using 100 nM eserine, in which a clear depression of fEPSP slope was not observed consistently until 35–40 min after eserine washout, slices treated with 10 μM eserine displayed a stable depression more rapidly; a clear decrease in fEPSP slope consistently occurred as soon as 5 min after the start of eserine washout (Fig. 1B; LTD: 80 ± 3% of fEPSP baseline slope; 60–65 min, n = 6). To ensure the effects of eserine are indeed a consequence of AChE inhibition and accumulation of extracellular ACh, we used a second AChE inhibitor, donepezil (1 μM), and observed significant synaptic depression similar to eserine (data not shown; 77.9 ± 8.0% of fEPSP baseline slope; 60–65 min, n = 6; P = 0.03). The next series of experiments was aimed at elucidating the mechanism(s) underlying eserine-LTD.
In light of previous data from our lab, demonstrating a role for M1 mAChRs in mediating CCh-induced LTD at CA3-CA1 synapses (mLTD) (McCutchen et al. 2006; Scheiderer et al. 2006, 2008), and 4-DAMP-sensitive receptors, likely M3, mediating presynaptic depression during CCh application, we asked if eserine-LTD also requires M1 and/or M3 mAChR activation. To this end, the mAChR antagonist pirenzepine was bath applied at 75 nM, a dose highly selective for M1 mAChRs (Marino et al. 1998), in conjunction with 4-DAMP (100 nM) before the application of eserine. We found this combination of inhibitors capable of completely blocking eserine-LTD [Fig. 2A; LTD: 74 ± 5% fEPSP baseline slope in control solution (n = 5) vs. 1.02 ± 5% in pirenzepine + 4-DAMP (n = 5); P = 0.002, Student's t-test]. To elucidate further the receptors necessary for eserine-LTD, we treated slices with pirenzepine alone and found that the eserine-LTD magnitude was not affected [Fig. 2B; LTD: 77 ± 3% fEPSP baseline slope in control solution (n = 3) vs. 72 ± 4% in pirenzepine (n = 7); P > 0.05 between groups]. In contrast to pirenzepine treatment alone, we found 4-DAMP (100 nM) to be sufficient in blocking eserine-LTD [Fig. 2C; LTD: 80 ± 2% of fEPSP baseline slope in control solution (n = 5) vs. 1.05 ± 5% in 4-DAMP (n = 5); P = 0.001, Student's t-test]. This finding indicates a form of LTD induced by 4-DAMP-sensitive receptors.
Fig. 2.
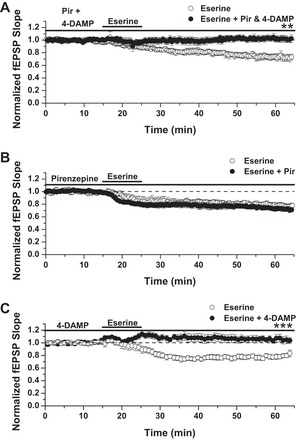
Eserine-LTD is blocked by inhibiting 1,1-dimethyl-4-diphenylacetoxypiperidinium iodide (4-DAMP)-sensitive receptors. A: eserine-LTD (100 nM) is blocked by combined treatment with pirenzepine (Pir) and 4-DAMP. LTD: 74 ± 5% of baseline fEPSP slope in control solution (n = 5) vs. 1.02 ± 5% in pirenzepine + 4-DAMP (n = 5). B: eserine-LTD (100 nM) is unaffected by pirenzepine alone. LTD: 77 ± 3% of baseline fEPSP slope in control solution (n = 3) vs. 72 ± 4% in pirenzepine (n = 7). C: eserine-LTD (10 μM), blocked by treatment with 4-DAMP alone. LTD: 80 ± 2% of baseline fEPSP slope in control solution (n = 5) vs. 1.05 ± 5% in 4-DAMP (n = 5); Student's t-test. **P ≤ 0.01; ***P ≤ 0.001.
Eserine-LTD Does Not Require pERK or p38 MAPK
Because mLTD induced by CCh requires activation of the ERK1/2 signaling pathway (Scheiderer et al. 2008; Volk et al. 2007), we next tested whether eserine-LTD shares this mechanism. We first performed positive control experiments in young rats, aged 3–5 wk, an age at which a 20-min bath application of the MEK inhibitor U0126 (20 μM) reliably blocks mLTD (Scheiderer et al. 2008). As shown in Fig. 3A, U0126 completely blocked CCh-induced mLTD [86 ± 3% of fEPSP baseline slope in control solution (n = 8) vs. 99 ± 5% of fEPSP slope in U0126 (n = 9); P < 0.05 between groups]. However, to block mLTD in adult rats, U0126 pretreatment, lasting at least 1 h, preceding CCh application, was required [Fig. 3B, LTD: 76 ± 3% of baseline fEPSP slope in control solution (n = 6) vs. 76 ± 4% of fEPSP slope in U0126 < 1 h (n = 5); Fig. 3C, LTD: 76 ± 7% of fEPSP slope in control solution vs. 1.05 ± 4% of fEPSP slope in U0126 > 1 h (n = 6); P = 0.002, Student's t-test]. With the use of a 1-h preincubation protocol, we found no effect of MEK inhibition on the magnitude of eserine-LTD [Fig. 3D; LTD: 77 ± 6% of baseline fEPSP slope in control solution (n = 5) vs. 75 ± 2% of baseline fEPSP slope in U0126 > 1 h (n = 4); P > 0.05 between groups], indicating that the cellular mechanisms of CCh mLTD and eserine-LTD do not share the requirement of ERK1/2 activation. We next tested for involvement of p38 MAPK, a signaling enzyme activated by metabotropic glutamate receptors (mGluRs) that share Gαq coupling with M1 mAChRs and induce mGluR-LTD (Moult et al. 2008) in the mechanism of eserine-LTD. Similar to mGluR-LTD, we find that blocking p38 MAPK, using SB203580 (5 μM; for at least 1 h before agonist application), completely blocks mLTD induced by CCh in adult rats [Fig. 4A; LTD: 86 ± 2% of fEPSP slope in control solution (n = 6) vs. 97 ± 3% of fEPSP slope in SB203580 (n = 7); P = 0.003 between groups, Student's t-test]. However, eserine-LTD remained intact, despite p38 MAPK blockade [Fig. 4B; LTD: 83 ± 3% of baseline fEPSP slope in control solution (n = 4) vs. 85 ± 10% of baseline fEPSP slope in SB203580 (n = 5); P > 0.05 between groups]. Taken as a whole, these data demonstrate that induction of eserine-LTD is independent of ERK1/2 or p38 MAPK signaling.
Fig. 3.
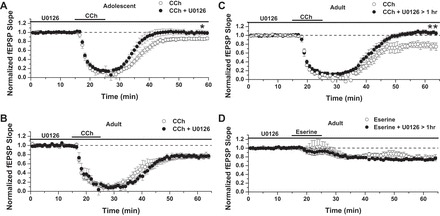
Eserine-LTD does not depend on ERK1/2 activity. A: U0126 treatment for 20 min before carbachol (CCh) application completely blocks CCh-induced muscarinic LTD (mLTD) in 3- to 5-wk-old rats. LTD: 86 ± 3% of baseline fEPSP slope in control solution (n = 8) vs. 99 ± 5% of fEPSP slope in U0126 (n = 9). B: the blocking of mLTD in adult rats (age 3–4 mo) requires pretreatment with U0126, lasting at least 1 h. LTD: 76 ± 3% of baseline fEPSP slope in control solution (n = 6) vs. 76 ± 4% of fEPSP slope in U0126 < 1 h (n = 5). C: LTD: 76 ± 7% of baseline fEPSP slope in control solution vs. 1.05 ± 4% of baseline fEPSP slope in U0126 > 1 h (n = 6); P = 0.002, Student's t-test. D: pretreatment with U0126 ≥ 1 h does not block eserine-LTD (10 μM): 77 ± 6% of baseline fEPSP slope in control solution (n = 5) vs. 75 ± 2% of baseline fEPSP slope in U0126 > 1 h (n = 4). *P ≤ 0.05; **P ≤ 0.01.
Fig. 4.
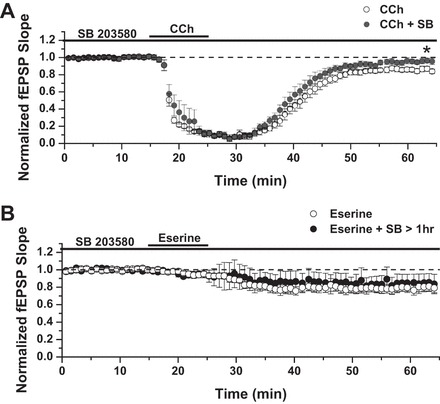
The inhibition of p38 MAPK prevents CCh-LTD but not eserine-LTD. A: pretreatment with SB203580 (SB; 5 μM) for at least 1 h before agonist application completely blocks LTD induced by CCh in adult rats; LTD: 86 ± 2% of baseline fEPSP slope in control solution (n = 6) vs. 97 ± 3% of baseline fEPSP slope in SB203580 (n = 7); P = 0.003, Student's t-test. B: eserine-LTD (10 μM) remained intact, despite p38 MAPK blockade; LTD: 83 ± 3% of baseline fEPSP slope in control solution (n = 4) vs. 85 ± 10% of baseline fEPSP slope in SB203580 (n = 5). *P ≤ 0.05.
The expression profile of mAChRs within the hippocampus spans both excitatory and inhibitory neurons, and published findings show that an AChE inhibitor can selectively enhance the activity of GABAergic interneurons expressing M3 mAChRs (Cea-del Rio et al. 2010). To eliminate the possibility that eserine induces LTD due to a potentiation of inhibitory GABAAR transmission, experiments were performed in the presence of 100 μM picrotoxin. Whereas it was evident from the observed population spikes that picrotoxin effectively blocked GABAAR transmission, the ability of eserine to induce LTD was unaffected [Fig. 5A; LTD: 86 ± 7% of baseline fEPSP slope (n = 6)]. We also tested for a role of GABABRs in eserine-LTD by performing experiments in the presence of CGP. Similar to results in picrotoxin, CGP did not affect the magnitude of LTD induced by eserine [Fig. 5B; LTD: 86 ± 3% of baseline fEPSP slope in control solution (n = 4) vs. 89 ± 6% of fEPSP slope in CGP (n = 5); P > 0.05 between groups, Student's t-test].
Fig. 5.
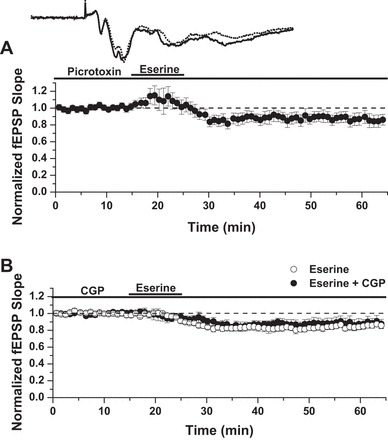
Eserine-LTD occurs independent of GABA channel conductance. A: picrotoxin, a GABAAR channel blocker, does not prevent LTD induced by 10 μM eserine. LTD: 86 ± 7% of baseline fEPSP slope (n = 6). B: CGP 52432 (CGP), a GABABR-selective inhibitor, does not prevent eserine-LTD (10 μM). LTD: 86 ± 3% of baseline fEPSP slope in control solution (n = 4) vs. 89 ± 6% in CGP (n = 5); P > 0.05, Student's t-test.
We next assessed the effect of eserine on presynaptic activity by analyzing the paired-pulse facilitation ratios before and during expression of eserine-LTD. We combined control experiments from three data sets (eserine ± U0126, SB203580, or CGP; Figs. 2C, 3, and 4) to increase the statistical power of our paired-pulse ratio (PPR) analysis. In the event that eserine-LTD is due to a decrease in the probability of neurotransmitter release, we expected any decrease in fEPSP slope after eserine application to be accompanied by an increase in PPR (slope of fEPSP 2/slope of fEPSP 1). By normalizing the PPRs to the baseline PPRs of each experiment, we were able to overlay a plot of PPR on the same time course as the fEPSP slope. Consistent with a presynaptic mechanism of expression, changes in fEPSP slope during eserine-LTD experiments are accompanied by temporally matched fluctuations in PPR (Fig. 6A; n = 16). To test the strength of this correlation, a linear regression was performed, plotting fEPSP slope as a function of PPR over samples collected between 25 and 65 min of the LTD experiments. The results of this analysis indicate a very strongly negative correlation [Fig. 6B; linear regression: slope = −0.97, R2 = 0.46, P = 0.0 × 10−30 (n = 4,231 sweeps)] between PPR and fEPSP slope during eserine-LTD expression and strongly support a presynaptic component to the expression mechanisms of eserine-LTD.
Fig. 6.
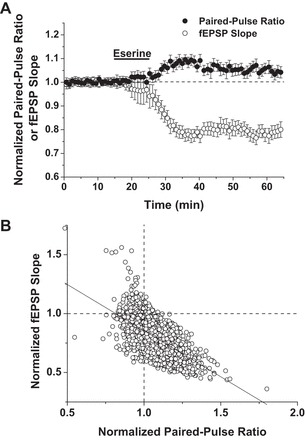
An increase in paired-pulse ratio (PPR) strongly correlates with eserine-LTD expression. A: control eserine-LTD (10 μM) experiments, presented in Figs. 2C, 3, and 4, averaged and overlayed with normalized PPRs. Consistent with a presynaptic mechanism of expression, changes in fEPSP slope during eserine-LTD experiments are accompanied by temporally matched fluctuations in PPR (n = 16). B: linear regression plotting fEPSP slope as a function of PPR over the time course of eserine-LTD expression (25–65 min). A very strong, negative correlation exists between the normalized fEPSP slope and PPR. Linear regression: slope = −0.97, R2 = 0.46, P = 0.0 × 10−30 (n = 4,231 sweeps).
Because AChE inhibition likely outlasts the 10-min bath application of eserine, we also asked if continued accumulation of extracellular ACh and constant activation of presynaptic mAChRs, which are known to depress synaptic transmission at CA3-CA1 synapses, account for the depression observed. To this end, we attempted to reverse eserine-LTD expression using a nonselective muscarinic receptor antagonist, atropine (1 μM), during 45–75 min of LTD expression. As shown in Fig. 7A, the synaptic depression is partially but significantly reversed by atropine [LTD: 78 ± 5% of baseline fEPSP slope preatropine (40–45 min, n = 6) vs. 86 ± 6% of baseline fEPSP slope postatropine (70–75 min, n = 6); P = 0.02, Student's paired t-test]. Importantly, control experiments show atropine treatment alone does not increase fEPSP slope (Fig. 7B; postatropine slope: 1.01% of baseline (40–45 min, n = 7, P > 0.05)]. We also analyzed PPRs during the atropine experiments and plotted them with respect to fEPSP slope (Fig. 7C). Consistent with results shown in Fig. 6A, eserine-LTD was accompanied by an increase in PPR. Strikingly, application of atropine resulted in PPR returning to the level of baseline on the same time course as the attenuation of LTD magnitude (Fig. 7C). It should be noted that fEPSP slope remained significantly depressed after atropine treatment (between 70 and 75 min; time point “2”), despite the return of PPR to baseline levels [Fig. 7, C and D; PPR: 1.08 ± 3% of baseline PPR preatropine (time point “1”) vs. 1.01 ± 2% of baseline PPR postatropine (time point 2), n = 6; P = 0.002, Student's paired t-test]. We interpret these results as evidence for a dual mechanism underlying eserine-LTD: a presynaptic mechanism dependent on constant activation of mAChRs and a mechanism independent of changes in release probability that likely occur at the postsynaptic site.
DISCUSSION
M1 mAChRs are required for normal hippocampal-dependent learning and memory and bidirectionally modulate synaptic efficacy at hippocampal CA3-CA1 synapses (Anagnostaras et al. 2003; Hamilton et al. 2001; Miyakawa et al. 2001; Scheiderer et al. 2006, 2008; Seeger et al. 2004; Shinoe et al. 2005; Suzuki and Okada 2012). Previous work from our lab shows that pharmacologically activating M1 mAChRs, using CCh, induce a Src kinase and phosphorylated ERK (pERK)-dependent form of LTD (mLTD) at CA3-CA1 synapses that are independent of the canonical M1-Gαq-PLC signaling pathway (Scheiderer et al. 2006, 2008). Because inhibitors of AChE are used clinically to increase extracellular ACh levels in the treatment of dementia in AD, we tested the hypothesis that LTD is induced as a consequence of AChE inhibition and that it is mechanistically similar to that induced by CCh. Here, we report that a 10-min application of eserine induces LTD (eserine-LTD) but with properties not shared by mLTD. Inhibition of M3 mAChRs, but not M1 mAChRs, was required to prevent eserine-LTD, and pharmacological inhibition of MEK, which prevents the increase in pERK, was completely ineffective. Furthermore, we find that mLTD requires p38 MAPK activation, a signaling enzyme previously implicated in mGluR-LTD, but eserine-LTD does not share this property. Finally, whereas previous reports suggest that mLTD expression requires internalization of AMPA receptors (Volk et al. 2007), long-term expression of eserine-LTD is partially dependent on a decrease in presynaptic release probability, caused by tonic activation of presynaptic mAChRs by the sustained increase in extracellular ACh.
Given published literature showing that CCh-induced LTD at synapses in the hippocampus and various areas of cortex requires M1 mAChRs and an increase in pERK (McCoy and McMahon 2007; Scheiderer et al. 2008), it was surprising that eserine-LTD was not prevented by the M1-favoring antagonist, pirenzepine alone, but instead was prevented by the M3 mAChR-preferring antagonist 4-DAMP. It was also surprising that eserine-LTD and CCh-induced mLTD are mechanistically different. This prompted us to consider that synaptic depression induced by eserine might be a consequence of sustained activation of mAChRs caused by continued elevation of ACh from persistent AChE inhibition, even after washout of eserine from the chamber. This concept is supported by the partial reversal of eserine-LTD by the nonselective mAChR antagonist atropine. Persistent antagonism of AChE would require a slowly dissociating eserine-AChE complex. Thorough characterizations of eserine pharmacology support this notion; the dissociation constant has been determined to be 7.1 μM [reviewed in Triggle et al. (1998)], and careful kinetic analysis of the three-step carbamylenzyme mechanism shows a notably slow dissociation (Stojan and Zorko 1997). It is likely that eserine remains bound to AChE for the duration of the expression phase of eserine-LTD, causing persistent elevation of synaptic ACh and concomitant activation of presynaptic mAChRs that are well known to decrease release probability at hippocampal synapses (Fernandez de Sevilla and Buno 2003; Fernandez de Sevilla et al. 2002; Valentino and Dingledine 1981). Unfortunately, the identity of the presynaptic mAChRs remains controversial. With the use of mAChR knockout mice, Dasari and Gulledge (2011) suggest that it is M4, not M1 or M2, mAChRs; however, the sensitivity of eserine-induced presynaptic depression to 4-DAMP suggests a role for M3 mAChRs.
A presynaptic expression mechanism for eserine-LTD should be revealed by analysis of the PPR, an accepted indicator of alterations in presynaptic release probability (Dobrunz 2002; Kamiya and Zucker 1994). Indeed, our analysis of PPR during the time course of eserine-LTD shows a very strongly negative correlation between fEPSP slope and PPR, supporting the interpretation that a decrease in presynaptic neurotransmitter release may partially contribute to eserine-LTD, and a body of literature supports this notion. Namely, agonism of presynaptic mAChRs with CCh triggers a rapid synaptic depression at CA3-CA1 synapses that is associated with an increase in PPR but does not persist after CCh washout (Fernandez de Sevilla et al. 2002; Scheiderer et al. 2006; Valentino and Dingledine 1981). Importantly, the increase in PPR induced by eserine is reversed completely by atropine treatment, supporting the concept that a presynaptic mechanism underlies eserine-LTD expression that is dependent on sustained elevation of synaptic ACh, likely due to slow dissociation of eserine from AChE. Importantly, the lack of effect of CGP on eserine LTD indicates that this presynaptic depression is independent of GABABRs, consistent with a previous report by Kremin et al. (2006), showing that presynaptic mAChR suppression is not dependent on these receptors.
A significant reduction in fEPSP slope was still evident, despite normalization of PPR by atropine to baseline, an observation that we interpret as evidence for a second, likely postsynaptic, mechanism of eserine-LTD expression. Although an increase in pERK signaling is required for other forms of LTD, including CCh-induced mLTD (Scheiderer et al. 2008), eserine-LTD persisted, despite blockade of MEK, the kinase that phosphorylates and activates ERK1/2, using U0126. In pursuing the signaling mechanism underlying eserine-LTD, downstream of M3 mAChR activation, we also tested for a role of p38 MAPK, an enzyme known to mediate hippocampal mGluR-LTD, which requires activation of Gαq-coupled mGluR5 during induction (Moult et al. 2008). We report that CCh-induced mLTD requires p38 MAPK activation, a new but anticipated finding, as both mGluR5 and M1/M3 are similarly coupled to Gαq signaling pathways. However, eserine-LTD was insensitive to p38 MAPK inhibition. Further experimentation will be necessary to determine the intracellular signaling cascades initiated during eserine-LTD induction.
A multitude of complex and sometimes competing mechanisms of synaptic modulation has been described in the context of cholinergic innervation in the hippocampus (Auerbach and Segal 1996). It is generally supported that AChE inhibitors facilitate synaptic function and plastic mechanisms required for learning and memory by elevating extracellular synaptic ACh concentration. In the present study, we demonstrate that AChE inhibition and the consequent increase in extracellular ACh modulate synaptic efficacy at CA3-CA1 synapses through several complicated mechanisms, likely involving M3 mAChRs that could be located at pre- and postsynaptic sites. Future studies are needed to identify the location of the M3 mAChRs and the downstream signaling mechanisms invoked, which could include postsynaptically released endocannabinoids that retrogradely depress presynaptic glutamate release via activation of presynaptic CB1 receptors (Ohno-Shosaku et al. 2003). Our findings add to the body of literature on cholinergic modulation of synaptic function in the hippocampus by showing that AChE inhibition causes a prolonged decrease in presynaptic glutamate release, in addition to inducing a likely postsynaptic form of LTD. The prolonged decrease in presynaptic glutamate release in the presence of AChE inhibitors, which display tight binding affinity to AChE and slow dissociation, may have the unintended consequence of decreasing synaptic flexibility or induction of other forms of activity-dependent plasticity, potentially explaining why this class of drugs is not always beneficial for treating cognitive deficits in AD. Clearly more work is needed to understand fully the complex role of AChE inhibitors and the cholinergic system in modulating hippocampal function.
GRANTS
Support for this work was provided by the National Institute on Aging (Grant 5R01.AG2161201, Muscarinic receptor-induced LTD in rat hippocampus; to L. L. McMahon, principal investigator/project leader) and the National Institute of General Medical Science [Grant 5k12GM088010, Mentored Experiences in Research, Instruction, and Teaching (MERIT) fellowship award to Lisa Schweibert, principal investigator/project leader, University of Alabama at Birmingham].
DISCLOSURES
The authors declare that there are no conflicts of interest, financial or otherwise, associated with this project or publication.
AUTHOR CONTRIBUTIONS
Author contributions: R.A.M., B.A.W., C.C.S., and L.L.M. conception and design of research; R.A.M., B.A.W., and C.C.S. performed experiments; R.A.M., B.A.W., and C.C.S. analyzed data; R.A.M., B.A.W., C.C.S., and L.L.M. interpreted results of experiments; R.A.M. prepared figures; R.A.M. drafted manuscript; R.A.M. and L.L.M. edited and revised manuscript; R.A.M. and L.L.M. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge the contributions of Dr. Teruko M. Bredemann for manuscript preparation and submission.
Present address of R. A. Mans: Dept. of Biology, Armstrong State Univ., Savannah, GA (e-mail: robert.mans@armstrong.edu).
REFERENCES
- Anagnostaras SG, Murphy GG, Hamilton SE, Mitchell SL, Rahnama NP, Nathanson NM, Silva AJ. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat Neurosci 6: 51–58, 2003. [DOI] [PubMed] [Google Scholar]
- Auerbach JM, Segal M. A novel cholinergic induction of long-term potentiation in rat hippocampus. J Neurophysiol 72: 2034–2040, 1994. [DOI] [PubMed] [Google Scholar]
- Auerbach JM, Segal M. Muscarinic receptors mediating depression and long-term potentiation in rat hippocampus. J Physiol 492: 479–493, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aura J, Sirvio J, Riekkinen P., Jr Methoctramine moderately improves memory but pirenzepine disrupts performance in delayed non-matching to position test. Eur J Pharmacol 333: 129–134, 1997. [DOI] [PubMed] [Google Scholar]
- Bainbridge NK, Koselke LR, Jeon J, Bailey KR, Wess J, Crawley JN, Wrenn CC. Learning and memory impairments in a congenic C57BL/6 strain of mice that lacks the M2 muscarinic acetylcholine receptor subtype. Behav Brain Res 190: 50–58, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baratti CM, Opezzo JW, Kopf SR. Facilitation of memory storage by the acetylcholine M2 muscarinic receptor antagonist AF-DX 116. Behav Neural Biol 60: 69–74, 1993. [DOI] [PubMed] [Google Scholar]
- Bartus RT, Dean RL, 3rd, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science 217: 408–414, 1982. [DOI] [PubMed] [Google Scholar]
- Berger-Sweeney J, Stearns NA, Murg SL, Floerke-Nashner LR, Lappi DA, Baxter MG. Selective immunolesions of cholinergic neurons in mice: effects on neuroanatomy, neurochemistry, and behavior. J Neurosci 21: 8164–8173, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount PJ, Nguyen CD, McDeavitt JT. Clinical use of cholinomimetic agents: a review. J Head Trauma Rehabil 17: 314–321, 2002. [DOI] [PubMed] [Google Scholar]
- Callahan MJ, Kinsora JJ, Harbaugh RE, Reeder TM, Davis RE. Continuous ICV infusion of scopolamine impairs sustained attention of rhesus monkeys. Neurobiol Aging 14: 147–151, 1993. [DOI] [PubMed] [Google Scholar]
- Carey GJ, Billard W, Binch H, 3rd, Cohen-Williams M, Crosby G, Grzelak M, Guzik H, Kozlowski JA, Lowe DB, Pond AJ, Tedesco RP, Watkins RW, Coffin VL. SCH 57790, a selective muscarinic M(2) receptor antagonist, releases acetylcholine and produces cognitive enhancement in laboratory animals. Eur J Pharmacol 431: 189–200, 2001. [DOI] [PubMed] [Google Scholar]
- Cea-del Rio CA, Lawrence JJ, Tricoire L, Erdelyi F, Szabo G, McBain CJ. M3 muscarinic acetylcholine receptor expression confers differential cholinergic modulation to neurochemically distinct hippocampal basket cell subtypes. J Neurosci 30: 6011–6024, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collerton D. Cholinergic function and intellectual decline in Alzheimer's disease. Neuroscience 19: 1–28, 1986. [DOI] [PubMed] [Google Scholar]
- Dasari S, Gulledge AT. M1 and M4 receptors modulate hippocampal pyramidal neurons. J Neurophysiol 105: 779–792, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker AJ, Connor DJ, Thal LJ. The role of cholinergic projections from the nucleus basalis in memory. Neurosci Biobehav Rev 15: 299–317, 1991. [DOI] [PubMed] [Google Scholar]
- Digby GJ, Noetzel MJ, Bubser M, Utley TJ, Walker AG, Byun NE, Lebois EP, Xiang Z, Sheffler DJ, Cho HP, Davis AA, Nemirovsky NE, Mennenga SE, Camp BW, Bimonte-Nelson HA, Bode J, Italiano K, Morrison R, Daniels JS, Niswender CM, Olive MF, Lindsley CW, Jones CK, Conn PJ. Novel allosteric agonists of M1 muscarinic acetylcholine receptors induce brain region-specific responses that correspond with behavioral effects in animal models. J Neurosci 32: 8532–8544, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE. Release probability is regulated by the size of the readily releasable vesicle pool at excitatory synapses in hippocampus. Int J Dev Neurosci 20: 225–236, 2002. [DOI] [PubMed] [Google Scholar]
- Drachman DA, Leavitt J. Human memory and the cholinergic system. A relationship to aging? Arch Neurol 30: 113–121, 1974. [DOI] [PubMed] [Google Scholar]
- Fernandez de Sevilla D, Buno W. Presynaptic inhibition of Schaffer collateral synapses by stimulation of hippocampal cholinergic afferent fibres. Eur J Neurosci 17: 555–558, 2003. [DOI] [PubMed] [Google Scholar]
- Fernandez de Sevilla D, Cabezas C, de Prada AN, Sanchez-Jimenez A, Buno W. Selective muscarinic regulation of functional glutamatergic Schaffer collateral synapses in rat CA1 pyramidal neurons. J Physiol 545: 51–63, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fibiger HC. Cholinergic mechanisms in learning, memory and dementia: a review of recent evidence. Trends Neurosci 14: 220–223, 1991. [DOI] [PubMed] [Google Scholar]
- Galli RL, Fine RE, Thorpe BC, Hale BS, Lieberman HR. Antisense oligonucleotide sequences targeting the muscarinic type 2 acetylcholine receptor enhance performance in the Morris water maze. Int J Neurosci 103: 53–68, 2000. [DOI] [PubMed] [Google Scholar]
- Gold PE. Acetylcholine modulation of neural systems involved in learning and memory. Neurobiol Learn Mem 80: 194–210, 2003. [DOI] [PubMed] [Google Scholar]
- Gu Z, Yakel JL. Timing-dependent septal cholinergic induction of dynamic hippocampal synaptic plasticity. Neuron 71: 155–165, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton SE, Hardouin SN, Anagnostaras SG, Murphy GG, Richmond KN, Silva AJ, Feigl EO, Nathanson NM. Alteration of cardiovascular and neuronal function in M1 knockout mice. Life Sci 68: 2489–2493, 2001. [DOI] [PubMed] [Google Scholar]
- Hamm RJ, Pike BR, Temple MD, O'Dell DM, Lyeth BG. The effect of postinjury kindled seizures on cognitive performance of traumatically brain-injured rats. Exp Neurol 136: 143–148, 1995. [DOI] [PubMed] [Google Scholar]
- Harvey J, Balasubramaniam R, Collingridge GL. Carbachol can potentiate N-methyl-d-aspartate responses in the rat hippocampus by a staurosporine and thapsigargin-insensitive mechanism. Neurosci Lett 162: 165–168, 1993. [DOI] [PubMed] [Google Scholar]
- Herrera-Morales W, Mar I, Serrano B, Bermudez-Rattoni F. Activation of hippocampal postsynaptic muscarinic receptors is involved in long-term spatial memory formation. Eur J Neurosci 25: 1581–1588, 2007. [DOI] [PubMed] [Google Scholar]
- Imbimbo BP. Pharmacodynamic-tolerability relationships of cholinesterase inhibitors for Alzheimer's disease. CNS Drugs 15: 375–390, 2001. [DOI] [PubMed] [Google Scholar]
- Jo J, Son GH, Winters BL, Kim MJ, Whitcomb DJ, Dickinson BA, Lee YB, Futai K, Amici M, Sheng M, Collingridge GL, Cho K. Muscarinic receptors induce LTD of NMDAR EPSCs via a mechanism involving hippocalcin, AP2 and PSD-95. Nat Neurosci 13: 1216–1224, 2010. [DOI] [PubMed] [Google Scholar]
- Kamiya H, Zucker RS. Residual Ca2+ and short-term synaptic plasticity. Nature 371: 603–606, 1994. [DOI] [PubMed] [Google Scholar]
- Kanju PM, Parameshwaran K, Sims-Robinson C, Uthayathas S, Josephson EM, Rajakumar N, Dhanasekaran M, Suppiramaniam V. Selective cholinergic depletion in medial septum leads to impaired long term potentiation and glutamatergic synaptic currents in the hippocampus. PLoS One 7: e31073, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasa P, Rakonczay Z, Gulya K. The cholinergic system in Alzheimer's disease. Prog Neurobiol 52: 511–535, 1997. [DOI] [PubMed] [Google Scholar]
- Kopf SR, Boccia MM, Baratti CM. AF-DX 116, a presynaptic muscarinic receptor antagonist, potentiates the effects of glucose and reverses the effects of insulin on memory. Neurobiol Learn Mem 70: 305–313, 1998. [DOI] [PubMed] [Google Scholar]
- Kremin T, Gerber D, Giocomo LM, Huang SY, Tonegawa S, Hasselmo ME. Muscarinic suppression in stratum radiatum of CA1 shows dependence on presynaptic M1 receptors and is not dependent on effects at GABA(B) receptors. Neurobiol Learn Mem 85: 153–163, 2006. [DOI] [PubMed] [Google Scholar]
- Kuenzi FM, Fitzjohn SM, Morton RA, Collingridge GL, Seabrook GR. Reduced long-term potentiation in hippocampal slices prepared using sucrose-based artificial cerebrospinal fluid. J Neurosci Methods 100: 117–122, 2000. [DOI] [PubMed] [Google Scholar]
- Lasser NL, Nash J, Lasser VI, Hamill SJ, Batey DM. Effects of antihypertensive therapy on blood pressure control, cognition, and reactivity. A placebo-controlled comparison of prazosin, propranolol, and hydrochlorothiazide. Am J Med 86: 98–103, 1989. [DOI] [PubMed] [Google Scholar]
- Li S, Cullen WK, Anwyl R, Rowan MJ. Muscarinic acetylcholine receptor-dependent induction of persistent synaptic enhancement in rat hippocampus in vivo. Neuroscience 144: 754–761, 2007. [DOI] [PubMed] [Google Scholar]
- Little JT, Johnson DN, Minichiello M, Weingartner H, Sunderland T. Combined nicotinic and muscarinic blockade in elderly normal volunteers: cognitive, behavioral, and physiologic responses. Neuropsychopharmacology 19: 60–69, 1998. [DOI] [PubMed] [Google Scholar]
- Mans RA, Chowdhury N, Cao D, McMahon LL, Li L. Simvastatin enhances hippocampal long-term potentiation in C57BL/6 mice. Neuroscience 166: 435–444, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino MJ, Rouse ST, Levey AI, Potter LT, Conn PJ. Activation of the genetically defined m1 muscarinic receptor potentiates N-methyl-D-aspartate (NMDA) receptor currents in hippocampal pyramidal cells. Proc Natl Acad Sci USA 95: 11465–11470, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Segal M. Acetylcholine potentiates responses to N-methyl-D-aspartate in the rat hippocampus. Neurosci Lett 113: 62–65, 1990. [DOI] [PubMed] [Google Scholar]
- McCoy PA, McMahon LL. Muscarinic receptor dependent long-term depression in rat visual cortex is PKC independent but requires ERK1/2 activation and protein synthesis. J Neurophysiol 98: 1862–1870, 2007. [DOI] [PubMed] [Google Scholar]
- McCutchen E, Scheiderer CL, Dobrunz LE, McMahon LL. Coexistence of muscarinic long-term depression with electrically induced long-term potentiation and depression at CA3-CA1 synapses. J Neurophysiol 96: 3114–3121, 2006. [DOI] [PubMed] [Google Scholar]
- McKinney M, Jacksonville MC. Brain cholinergic vulnerability: relevance to behavior and disease. Biochem Pharmacol 70: 1115–1124, 2005. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Yamada M, Duttaroy A, Wess J. Hyperactivity and intact hippocampus-dependent learning in mice lacking the M1 muscarinic acetylcholine receptor. J Neurosci 21: 5239–5250, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moult PR, Correa SA, Collingridge GL, Fitzjohn SM, Bashir ZI. Co-activation of p38 mitogen-activated protein kinase and protein tyrosine phosphatase underlies metabotropic glutamate receptor-dependent long-term depression. J Physiol 586: 2499–2510, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson OG, Leanza G, Rosenblad C, Lappi DA, Wiley RG, Bjorklund A. Spatial learning impairments in rats with selective immunolesion of the forebrain cholinergic system. Neuroreport 3: 1005–1008, 1992. [DOI] [PubMed] [Google Scholar]
- Ohno M, Yamamoto T, Watanabe S. Blockade of hippocampal M1 muscarinic receptors impairs working memory performance of rats. Brain Res 650: 260–266, 1994. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Matsui M, Fukudome Y, Shosaku J, Tsubokawa H, Taketo MM, Manabe T, Kano M. Postsynaptic M1 and M3 receptors are responsible for the muscarinic enhancement of retrograde endocannabinoid signalling in the hippocampus. Eur J Neurosci 18: 109–116, 2003. [DOI] [PubMed] [Google Scholar]
- Packard MG, Regenold W, Quirion R, White NM. Post-training injection of the acetylcholine M2 receptor antagonist AF-DX 116 improves memory. Brain Res 524: 72–76, 1990. [DOI] [PubMed] [Google Scholar]
- Perry E, Walker M, Grace J, Perry R. Acetylcholine in mind: a neurotransmitter correlate of consciousness? Trends Neurosci 22: 273–280, 1999. [DOI] [PubMed] [Google Scholar]
- Preston GC, Ward C, Lines CR, Poppleton P, Haigh JR, Traub M. Scopolamine and benzodiazepine models of dementia: cross-reversals by Ro 15–1788 and physostigmine. Psychopharmacology (Berl) 98: 487–494, 1989. [DOI] [PubMed] [Google Scholar]
- Sarter M, Parikh V. Choline transporters, cholinergic transmission and cognition. Nat Rev Neurosci 6: 48–56, 2005. [DOI] [PubMed] [Google Scholar]
- Scheiderer CL, McCutchen E, Thacker EE, Kolasa K, Ward MK, Parsons D, Harrell LE, Dobrunz LE, McMahon LL. Sympathetic sprouting drives hippocampal cholinergic reinnervation that prevents loss of a muscarinic receptor-dependent long-term depression at CA3-CA1 synapses. J Neurosci 26: 3745–3756, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiderer CL, Smith CC, McCutchen E, McCoy PA, Thacker EE, Kolasa K, Dobrunz LE, McMahon LL. Coactivation of M(1) muscarinic and alpha1 adrenergic receptors stimulates extracellular signal-regulated protein kinase and induces long-term depression at CA3-CA1 synapses in rat hippocampus. J Neurosci 28: 5350–5358, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliebs R. Basal forebrain cholinergic dysfunction in Alzheimer's disease—interrelationship with beta-amyloid, inflammation and neurotrophin signaling. Neurochem Res 30: 895–908, 2005. [DOI] [PubMed] [Google Scholar]
- Seeger T, Fedorova I, Zheng F, Miyakawa T, Koustova E, Gomeza J, Basile AS, Alzheimer C, Wess J. M2 muscarinic acetylcholine receptor knock-out mice show deficits in behavioral flexibility, working memory, and hippocampal plasticity. J Neurosci 24: 10117–10127, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffler DJ, Williams R, Bridges TM, Xiang Z, Kane AS, Byun NE, Jadhav S, Mock MM, Zheng F, Lewis LM, Jones CK, Niswender CM, Weaver CD, Lindsley CW, Conn PJ. A novel selective muscarinic acetylcholine receptor subtype 1 antagonist reduces seizures without impairing hippocampus-dependent learning. Mol Pharmacol 76: 356–368, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoe T, Matsui M, Taketo MM, Manabe T. Modulation of synaptic plasticity by physiological activation of M1 muscarinic acetylcholine receptors in the mouse hippocampus. J Neurosci 25: 11194–11200, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojan J, Zorko M. Kinetic characterization of all steps of the interaction between acetylcholinesterase and eserine. Biochim Biophys Acta 1337: 75–84, 1997. [DOI] [PubMed] [Google Scholar]
- Sugisaki E, Fukushima Y, Tsukada M, Aihara T. Cholinergic modulation on spike timing-dependent plasticity in hippocampal CA1 network. Neuroscience 192: 91–101, 2011. [DOI] [PubMed] [Google Scholar]
- Suzuki E, Okada T. Stratum oriens stimulation-evoked modulation of hippocampal long-term potentiation involves the activation of muscarinic acetylcholine receptors and the inhibition of Kv7/M potassium ion channels. Eur J Neurosci 36: 1984–1992, 2012. [DOI] [PubMed] [Google Scholar]
- Triggle DJ, Mitchell JM, Filler R. The pharmacology of physostigmine. CNS Drug Rev 4: 87–136, 1998. [Google Scholar]
- Valentino RJ, Dingledine R. Presynaptic inhibitory effect of acetylcholine in the hippocampus. J Neurosci 1: 784–792, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannucchi MG, Scali C, Kopf SR, Pepeu G, Casamenti F. Selective muscarinic antagonists differentially affect in vivo acetylcholine release and memory performances of young and aged rats. Neuroscience 79: 837–846, 1997. [DOI] [PubMed] [Google Scholar]
- Volk LJ, Pfeiffer BE, Gibson JR, Huber KM. Multiple Gq-coupled receptors converge on a common protein synthesis-dependent long-term depression that is affected in fragile X syndrome mental retardation. J Neurosci 27: 11624–11634, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrenn CC, Lappi DA, Wiley RG. Threshold relationship between lesion extent of the cholinergic basal forebrain in the rat and working memory impairment in the radial maze. Brain Res 847: 284–298, 1999. [DOI] [PubMed] [Google Scholar]