

Abstract
A novel C17 resorcylic acid was synthesized by a structure-guided Vitis vinifera stilbene synthase (STS) mutant, in which threonine 197 was replaced with glycine (T197G). Altering the architecture of the coumaroyl binding and cyclization pocket of the enzyme led to the attachment of an extra acetyl unit, derived from malonyl-CoA, to p-coumaroyl-CoA. The resulting novel pentaketide can be produced strictly by STS-like enzymes and not by Chalcone synthase-like type III polyketide synthases; due to the unique thioesterase like activity of STS-like enzymes. We utilized a liquid chromatography mass spectrometry-based data analysis approach to directly compare the reaction products of the mutant and wild type STS. The findings suggest an easy to employ platform for precursor-directed biosynthesis and identification of unnatural polyketides by structure-guided mutation of STS-like enzymes.
Keywords: resorcylic acid, novel non-natural polyphenols, structure-guided mutants, stilbene synthase, type III polyketides synthases
Introduction
Type III polyketide synthases (PKSs) are structurally simple homodimeric (∼42 kDa each) ketosynthase enzymes found in most plants that have also been more recently observed in bacteria and fungi.1–3 Like type II and many type I PKSs, type III PKSs are iterative enzymes, however their modulating mechanisms are much more complicated as they orchestrate acyltransfer, decarboxylation, condensation, cyclization, and aromatization reactions in two catalytically relevant cavities.2,4,5 They catalyze the decarboxylative Claisen condensation of one to several molecules of extender substrate onto a starter substrate. Three well characterized cavities, the CoA binding tunnel, the substrate binding pocket and the cyclization pocket are present in all type III PKSs.4 Different type III PKSs differ in their preference for starter substrate, number of iterative condensation of extender substrates, and the mechanism of cyclization of the poly-β-keto intermetidate formed through Claisen condensation, aldol condensation or lactonization (Scheme 1). Thus, these enzymes are capable of producing a wide array of compounds, such as chalcones, pyrones, acridones, phloroglucinols, stilbenes, and resorcinolic lipids.
Scheme 1.

Proposed mechanism for the formation of bisnoryangonin (BNY), p-coumaroyltriacetic acid lactone (CTAL) naringenin chalcone and resveratrol from p-coumaroyl-CoA and malonyl-CoA.
Stilbene synthase (STS), like chalcone synthase (CHS), catalyzes the iterative condensation of three acetyl units, derived from malonyl-CoA, onto p-coumaroyl-CoA to form a tetraketide intermediate. CHS cyclizes this intermediate through a C1-C6 Claisen condensation, automatically resulting in breaking of the thioester bond of the product to the enzyme. In contrast, STS preferentially cyclizes the polyketide through a C2-C7 aldol condensation (Scheme 1). Thus, STS requires an extra thioesterase-like (TE-like) activity to offload the product from the enzyme. Detailed structural studies of STS indicate the presence of a hydrogen-bonding network (HBN) between the threonine 132, serine 338 and glutamic acid 192 through a water molecule; this HBN is predicted to facilitate STS thioesterase activity.6 This HBN is sterically possible only in the case of STSs, as the amino acid residue at position 98 is replaced by a bulkier amino acid like methionine (as compared to a smaller residue like valine in CHS), bringing the threonine 132 into hydrogen-bonding distance with serine 338 and glutamic acid 192.6
Bisnoryangonin (BNY) and p-coumaroyltriacetic lactone (CTAL) are obtained as derailment products during the reaction.7 Several intuitive and structure-based mutations of CHS-like enzymes have been carried out to alter substrate specificity and size of the polyketide intermediate formed to produce novel, natural or unnatural products.8,9 Some of these novel compounds have also shown interesting biological activities.10–13
The coumaroyl binding pocket is composed of the following structurally important residues: serine 133, glutamic acid 192, threonine 194, threonine 197, and serine 338. The unique TE-like activity of STS-like enzymes has not yet been fully exploited for the biosynthesis of novel compounds. We attempted to exploit this TE-like activity of STS, with the aim of generating novel polyketides that have not been reported before. As a proof of concept, we first created a point mutation replacing serine 338 by valine 338 in the Vitis vinifera STS (VvSTS), and as expected an almost complete conversion of the STS into CHS was observed. A ∼7.9-fold increase in naringenin (1) production (Fig. 2) was noted, and a ∼6.0-fold decrease in resveratrol (2) production (data not shown) was seen upon carrying out a serine to valine mutation at amino acid 338. Serine 338 is a part of the HBN, replacing it with a hydrophobic valine 338 disrupted the HBN. Next, to retain the TE-like activity of the enzyme, we created a mutant with the HBN intact, replacing threonine 197 with glycine. The threonine to glycine mutation at amino acid 197 predictably results in an increase in the substrate binding pocket size. Upon carrying out homology modeling of the VvSTS based on the previously published Acharis hypogaea (AhSTS) crystal structure and inspecting the cavities for the wild type and mutant we found that the mutation results in connection of the substrate binding pocket to the cyclization pocket. In Figure 3 the residue at position 197 is shown lining the coumaroyl-binding pocket. The sterically important residues lining the CoA binding tunnel and cyclization pocket are also shown in Figure 3. The overall size of the coumaroyl binding pocket and cyclization pocket increased from 1029.2 Å3 to 1134.3 Å3 in the mutant as compared to the wild type.
Figure 1.

Naringenin production by WtVvSTS and the single point mutant S338VVvSTS.
Figure 2.

Stereo view of malonyl-CoA bound to VvSTS. The sterically important residues from the (1) CoA binding pocket (Lys 55, Arg 58 and Lys 62) are shown in purple; (2) Coumaroyl binding pocket (Ser 133, Glu 192, Thr 194, Thr 197 and Ser 338) are shown in blue; (3) Cyclization pocket (Thr 132, Met 137, Phe 215, Ile 254, Glu 256, Phe 265 and Pro 375) are shown in brown. Malonyl-CoA is labeled as MLC 390.
We tested the catalytic activity of threonine to glycine 197 (T197G) with its natural substrates, malonyl-CoA and p-coumaroyl-CoA. We utilized online high resolution-liquid chromatography mass spectrometry (HR-LCMS) data comparison analysis software, made available through XCMS, to obtain the fold-changes in production of expected products and for the identification of peaks novel to this mutant. This software helps detect dysregulation between two or more sample groups. Because of the complexity of LC-MS data sets, direct manual comparison can be extremely cumbersome. Furthermore, minor fluctuations between two runs can result from column degradation, temperature changes, changes in pH, changes of the mobile phase, and so forth, leading to a variation in the retention time of a compound. XCMS overcomes these nonlinear deviations and helps align the retention times between two runs by utilizing a feature detection algorithm, centWave, for HR-LCMS data sets. It collects regions of interest in the raw-data and applies continuous wavelet transformation or Gauss-fitting in the chromatographic domain.14,15
The T197G mutant showed a twofold decrease in its ability to synthesize resveratrol, and a fourfold increase in naringenin production. Furthermore, the derailment products BNY and CTAL increased by sevenfold and fourfold, respectively (Supporting Information Fig. 2).
A novel parent-ion peak [M-H]− at m/z 313.0708 in the LC-ESIMS (± 5 PPM) indicated the formation of a coumaroyl-derived pentaketide, with the predicted molecular composition of C17H13O6 (∼2.8% yield). This peak was recorded as [M-H]− m/z 321.0976 (+5 PPM) when 13C3 labeled malonyl-CoA was used, indicating the presence of eight carbons derived from the acetyl units of malonyl-CoA, thus, confirming the formation of a pentaketide p-coumaroyl-CoA derived molecule. This molecule could possibly cyclize through a C2-C7 aldol condensation (resulting in compound 3) or C1-C6 Claisen condensation (resulting in compound 4) (Fig. 4). The MS/MS spectrum of [M-H]− m/z 313.0708 gave the fragment m/z 269.0819, corresponding to [M-H-CO2]− (Supporting Information Figs. 3 and 4), and that of [M-H]− m/z 321.0976 gave the fragment m/z 276.1050, corresponding to a loss of [M-H-13CO2] (Supporting Information Figs. 4 and 5) This indicates the loss of a carbon derived from malonyl-CoA, which is possible only for compound 3. Further comparison of the MS/MS spectrum of the stable-isotope labeled novel pentaketide to the unlabeled compound was completely consistent with the fragmentation pattern expected for compound 3 (Supporting Information Figs. 3–5).
Figure 3.

Possible structures formed by 4 molecules of malonyl-CoA and one molecule of p-coumaroyl-CoA. Compound 3 is a resorcylic acid derivative. Compound 4 is a phlorophenone derivative.
We also carried out a complete methylation of the reaction mixture and carried out LC-MS analysis of the resulting products.16 A peak unique to the T197G reaction mixture was obtained with parent-ion peak [M+H]+ at m/z 357.0770 (±5 PPM) (6 Supporting Information Fig. 6). This correlates to the methylated lactone of 3. The fragmentation pattern corresponded to methylated lactone of 3 precisely (Supporting Information Fig. 6).
We propose a possible mechanism of reaction for the production of resorcylic acid by the T197G mutant.2,17 There are three possible scenarios for the cyclization of the polyketide intermediate through aldol condensation (Scheme 5). Scenario (1) indicates occurrence of aldol condensation and aromatization before hydrolysis, resulting in producing the resorcylic acid scaffold (Scheme 5a). If the hydrolysis precedes the aldol condensation, two cases for synthesis of the resorcinol scaffold are possible: the first (Scheme 5b), aldol condensation, which does not accompany decarboxylation, and the second (Scheme 5c) decarboxylative aldol condensation. In the second case (Scheme 5c), retention or detachment of the carboxylic acid depends on whether or not decarboxylation is concerted with aromatization. We observed that the T197G mutant produced the pentaketide resorcylic acid 3 exclusively when the reaction period was shortened. The prolonged reaction resulted in a decrease of 3, and an increase in 5. Therefore, we conclude that the resorcinol produced by the T197G mutant STS is a result of non-enzymatic decarboxylation of 3. This was further confirmed by heating the reaction products at 44°C, and observing that all of 3 was converted into 5 (Supporting Information Figs. 7 and 8). Because of this instability of carboxylic acids, a property of that has also been discussed by Gorham et al.,18 we were unable to obtain purified 3 and corresponding NMR data.
Scheme 2.
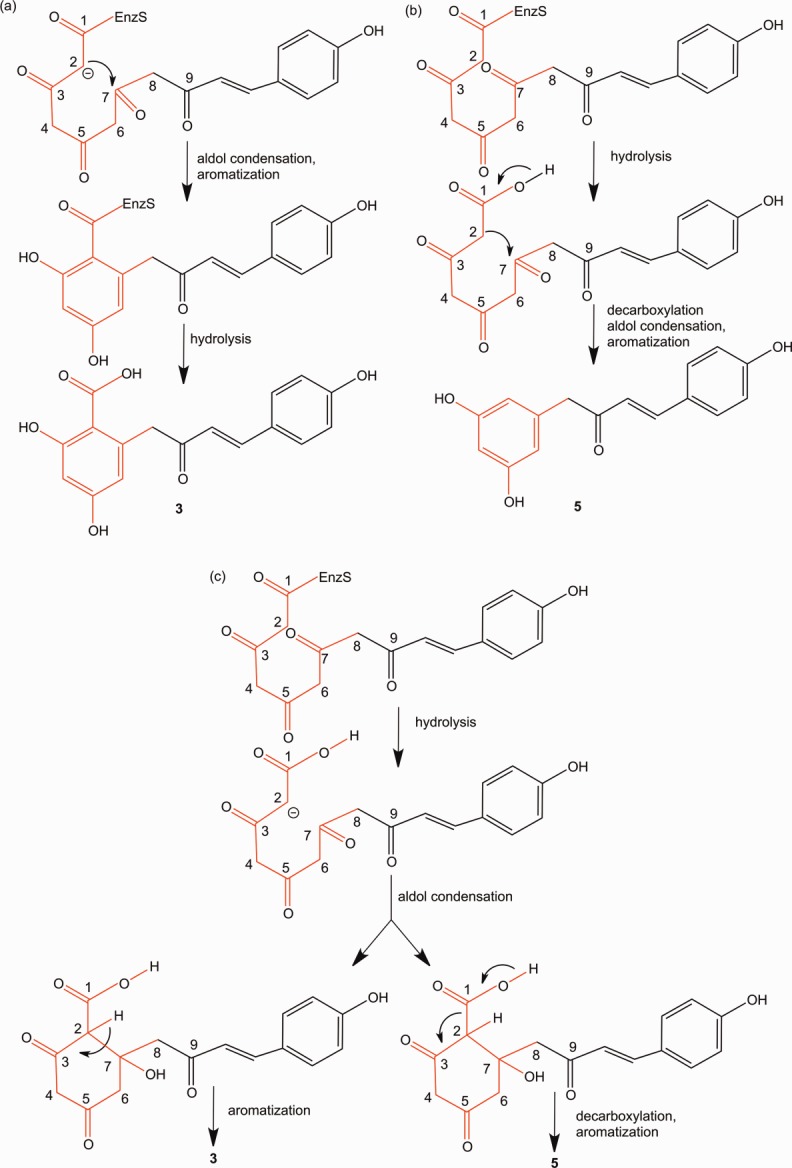
Possible scenarios for mechanism of formation of recorcylic acid: (a) Hydrolysis of the thioester takes place after aldol condensation and aromatization, and therefore the carboxyl group remains. (b) Resorcinol synthesis by hydrolysis followed by decarboxylative aldol condensation and aromatization. (c) Synchronous synthesis of alkylresorcylic acid and resorcinol by hydrolysis followed by aldol condensation and aromatization.
Our results suggested that the rational engineering of STS-like enzymes is a promising approach for the biosynthesis of chemically and structurally divergent non-natural polyketides. Because we can now manage to control the number of malonyl-CoA condensations, as well as manipulate the mechanism of the cyclization, the next step will be to carry out reactions to generate polyketides with desired ring systems.
In summary, this is the first demonstration of the enzymatic formation of the nonnatural C2-C7 aromatic pentaketide derived from p-coumaroyl-CoA by a structure-based rationally designed single point mutant T197G of STS. We also demonstrate that the application of 13C labeled malonly-CoA and XCMS analysis can be used to identify small quantities of novel products being formed during the enzymatic reactions, with only the online HR-LCMS data.
Experimental Section
Site-directed mutagenesis
The plasmids expressing the VvSTS mutant was constructed with a QuikChange Site-Directed Mutagenesis Kit (Stratagene), according to the manufacturer's protocol, using the following pairs of primers: T197G Fw: 5′-GAGATCACAGTTGTTGGCTTTCGCGGCCCTTCC-3′ and Rv: 5′-GGAAGGGCCGCGAAAGCCAACAACTGTGATCTC-3′.
Protein expression and purification
After confirmation of the sequence, the plasmid was transformed into E. coli BL21* (De3). The cells harboring the plasmid were cultured to an OD600 of 0.6 in LB medium containing chloramphinecol (30 μg mL−1) at 37°C. Subsequently, isopropylthio-β-d-galactopyranoside (IPTG) (1.0 mM) was added to induce protein expression, and the cells were further cultured at 30°C for 4 h. All of the following procedures were performed at 4°C. E. coli cells were harvested by centrifugation at 4000g, and frozen at −80°C until further processing. The cells were disrupted by incubating with lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0) and lysozyme (10 mg ml−1) and culture volume of Benzonase® nuclease (3 U mL−1), at 4°C for 30 min. The lysate was then centrifuged at 12,000g for 30 min. The supernatant was loaded onto a Ni-NTA spin column (Qiagen) equilibrated with lysis buffer. The column was then washed with wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0). The His-tag was cleaved from the column using elution buffer (50 mM NaH2PO4, 300 mM NaCl, 300 mM imidazole, pH 8.0). The protein concentration was determined by the Bradford method (Protein Assay, Bio-Rad) with bovine serum albumin as the standard.
Enzymatic reaction
The reaction mixture contained 4-coumaroyl-CoA (54 μM), malonyl-CoA (108 μM) or 13C3-malonyl-CoA (108 μM) and the purified enzyme (20 μg) in potassium phosphate buffer (500 μL, 100 mM, pH 7.0). Incubations were performed at 30°C for 30 min and were stopped by the addition of ethyl acetate (500 μL) with 1% HCl. The products were then concentrated in a speed vacuum and resuspended in ethyl acetate (10 μL), and centrifuged at 14,000 rpm for 10 min before running on a column. The products were separated by reverse-phase HPLC (Agilent 1260) on a Zorbax C18 column (4.6 × 150 mm2, 5 μm, at a flow rate of 0.7 mL min−1). Gradient elution was performed with H2O and acetonitrile (ACN), both containing 0.2% trifluoroacetic acid: 0–7 min, 30% ACN; 5–17 min, linear gradient from 30 to 60% ACN; 17–25 min, 60% ACN; 25–27 min, linear gradient from 60% to 70% ACN. UV-detection was used for calculating fold-changes in production of resveratrol and naringenin by the Wt STS in comparison to T197G STS, using a freshly generated standard curve for resveratrol and naringenin. All calculations were based on triplicates. Three reactions were pooled into ethyl acetate (10 μL) for LC-MS analysis. Online HR–ESI-LCMS spectra were measured with an Agilent Technologies HPLC 1200 series HPLC coupled to a Thermo Scientific LTQ Orbitrap XLTM mass spectrometer fitted with an electrospray ionization (ESI) source. The ESI capillary temperature and the capillary voltage were 320°C and 4.0 V, respectively. The tube lens offset was set at 20.0 V. All spectra were obtained in the negative and positive mode, over a mass range of m/z 150–500, and at a range of one scan every 0.2 s. The collision gas was helium, and the relative collision energy scale was set at 30.0% (1.5 eV). Dependent MS/MS scans were acquired for the first four most abundant ions.
Methylation of reaction mixture
Two enzymatic reactions (500 μL-scale) (as previously described) were performed in tandem, combined, and completely dried before carrying out the methylation reaction. For complete methylation, trimethylsilyl diazomethane (50 μL) was added to the dried sample along with ethyl ether (1 mL) and methanol (100 μL). After a period of 1 h trimethylsilyl diazomethane (50 μL) was added.16,19 The reaction was rotated at room temperature over night and dried on a speed vacuum. The products resulting from the methylation reaction were re-suspended in methanol (10 μL) and analyzed by HR-LCMS in positive mode as previously described. Dependent MS/MS scans were acquired for the first three most abundant ions.
Substrates
The 4-Coumaroyl-CoA was chemically synthesized as previously described.20 13C3-malonyl-CoA, malonyl-CoA, resveratrol, and naringenin were purchased from Sigma.
XCMS and MZmine data processing
Raw data was converted to XML format using R processor (Supporting Information Script S1, obtained from http://www.metabolomics.strath.ac.uk/showPage.php?page=processingscripts), and analyzed using XCMS online.21 XCMS was utilized for calculating fold changes in CTAL and BNY. All calculations were based on triplicates.
Homology modeling and docking
The models of the WtVvSTS and T197GVvSTS were generated by the SWISS-MODEL package (http://expasy.ch/spdbv/) provided by the Swiss-PDB-Viewer program22 based on the crystal structure of wild-type STS from Acharis hypogaea (PDB code: 1Z1E). The model quality was assessed using PROCHECK.23 In the Ramachandran plot calculated for the model, most of the amino acid residues were present in the energetically allowed regions with only a few exceptions, primarily Gly residues that can adopt phi/psi angles in all four quadrants. The homology model created for VvSTS was uploaded onto SWISS-Dock. The mol2 file for malonyl-CoA was uploaded onto SWISS-Dock in the ligand selection tab. The mol2 file for malonyl-CoA was generated using UCSF Chimera, the. sdf file for malonyl-CoA obtained from PDB (PDB code: MLC) was opened using UCSF Chimera, and the hydrogen atoms were added using Tools/Structure Editing/AddH menu, and the file was then saved in the mol2 format. The best predicted malonyl-CoA docking model, based on energy minimization and the previously published structure of chalcone synthase complexed with malonyl-CoA structure (PDB code: 1CML) was then used to generate Figure 3. The cavity volume was calculated by the program CASTP (http://cast.engr.uic.edu/cast/).24 All protein structure figures were prepared with PYMOL (DeLano Scientific, http://www.pymol.org).
Acknowledgments
The authors would acknowledge the guidance of Dr. Dmitri Zagorevski for help with LC-MS analysis. The authors acknowledge no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information
References
- Funa N, Ohnishi Y, Fujii I, Shibuya M, Ebizuka Y, Horinouchi S. A new pathway for polyketide synthesis in microorganisms. Nature. 1999;400:897–899. doi: 10.1038/23748. [DOI] [PubMed] [Google Scholar]
- Katsuyama Y, Ohnishi Y. Type III polyketide synthases in microorganisms. In: Hopwood DA, et al., editors. Natural product biosynthesis by microorganisms and plant, Part A, Vol. 515: Methods in enzymology. San Diego: Elsevier Academic Press; 2012. pp. 359–377. [DOI] [PubMed] [Google Scholar]
- Yu DY, Xu FC, Zeng J, Zhan JX. Type III polyketide synthases in natural product biosynthesis. Iubmb Life. 2003;64:285–295. doi: 10.1002/iub.1005. [DOI] [PubMed] [Google Scholar]
- Austin MB, Noel AJP. The chalcone synthase superfamily of type III polyketide synthases. Nat Prod Rep. 2003;20:79–110. doi: 10.1039/b100917f. [DOI] [PubMed] [Google Scholar]
- Abe I. Novel applications of plant polyketide synthases. Curr Opin Chem Biol. 2012;16:179–185. doi: 10.1016/j.cbpa.2011.12.016. [DOI] [PubMed] [Google Scholar]
- Austin MB, Bowman ME, Ferrer JL, Schroder J, Noel JP. An aldol switch discovered in stilbene synthases mediates cyclization specificity of type III polyketide synthases. Chem Biol. 2004;11:1179–1194. doi: 10.1016/j.chembiol.2004.05.024. [DOI] [PubMed] [Google Scholar]
- Hrazdina G, Kreuzaler F, Hahlbrock K, Grisebach H. Substrate specificity of flavanone synthase from cell suspension cultures of parsley and structure of release products in vitro. Arch Biochem Biophys. 1976;175:392–399. doi: 10.1016/0003-9861(76)90526-9. [DOI] [PubMed] [Google Scholar]
- Abe I. Engineering of plant polyketide biosynthesis. Chem Pharm Bull. 2008;56:1505–1514. doi: 10.1248/cpb.56.1505. [DOI] [PubMed] [Google Scholar]
- Abe I. Engineered biosynthesis of plant polyketides: structure-based and precursor-directed approach. Top Curr Chem. 2009;297:45–66. doi: 10.1007/128_2009_22. [DOI] [PubMed] [Google Scholar]
- Morita H, Yamashita M, Shi S-P, Wakimoto T, Kondo S, Kato R, Sugio S, Kohno T, Abe I. Synthesis of unnatural alkaloid scaffolds by exploiting plant polyketide synthase. 2011;108:13504–13509. doi: 10.1073/pnas.1107782108. Proc Natl Acad Sci USA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemler JA, Lim CG, Daiss JL, Koffas MA. A versatile microbial system for biosynthesis of novel polyphenols with altered estrogen receptor binding activity. Chem Biol. 2010;17:392–401. doi: 10.1016/j.chembiol.2010.03.010. [DOI] [PubMed] [Google Scholar]
- Chemler JA, Koffas MAG. Metabolic engineering for plant natural product biosynthesis in microbes. Curr Opin Biotech. 2008;19:597–605. doi: 10.1016/j.copbio.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Bhan N, Xu P, Koffas MAG. Pathway and protein engineering approaches to produce novel and commodity small molecules. Curr Opin Biotech. 2013;24:1137–1143. doi: 10.1016/j.copbio.2013.02.019. [DOI] [PubMed] [Google Scholar]
- Tautenhahn R, Bottcher C, Neumann S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinform. 2008;9:504. doi: 10.1186/1471-2105-9-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CA, Want EJ, O'Maille G, Abagyan R, Siuzdak G. XCMS: processing mass spectrometry data for metabolite profiling using Nonlinear peak alignment, matching, and identification. Anal Chem. 2006;78:779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- Eckermann C, Schroder G, Eckermann S, Strack D, Schmidt E, Schneider B, Schroder J. Stilbenecarboxylate biosynthesis: a new function in the family of chalcone synthase-related proteins. Phytochem. 2003;62:271–286. doi: 10.1016/s0031-9422(02)00554-x. [DOI] [PubMed] [Google Scholar]
- Funa N, Awakawa T, Horinouchi S. Pentaketide resorcylic acid synthesis by type III polyketide synthase from Neurospora crassa. J Biol Chem. 2007;282:14476–14481. doi: 10.1074/jbc.M701239200. [DOI] [PubMed] [Google Scholar]
- Gorham J. In: The stilbenoids. Harbome JBST, Reinhold L, et al., editors. Oxford: Pergamon Press; 1980. pp. 203–252. [Google Scholar]
- Hirano K, Sakai S, Ishikawa T, Avci FY, Linhardt RJ, Toida T. Preparation of the methyl ester of hyaluronan and its enzymatic degradation. Carb Res. 2005;340:2297–2304. doi: 10.1016/j.carres.2005.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockigt J, Zenk MH. Chemical syntheses and properties of hydroxycinnamoyl coenzyme A derivative. Z Naturforschung. 1975;30:352–358. doi: 10.1515/znc-1975-5-609. [DOI] [PubMed] [Google Scholar]
- Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G. XCMS online: a web-based platform to process untargeted metabolomic data. Anal Chem. 2012;84:5035–5039. doi: 10.1021/ac300698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Liang J, Edelsbrunner H, Woodward C. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Sci. 1998;7:1884–1897. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Edelsbrunner H, Woodward C. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Sci. 1998;7:1884–1897. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information