

Abstract
Nucleotide-binding domain leucine-rich repeat-containing receptors (NLRs) are key proteins in the innate immune system. The 14 members of the NLRP family of NLRs contain an N-terminal pyrin domain which is central for complex formation and signal transduction. Recently, X-ray structures of NLRP14 revealed an unexpected rearrangement of the α5/6 stem-helix of the pyrin domain allowing a novel symmetric dimerization mode. We characterize the conformational transitions underlying NLRP oligomerization using molecular dynamics simulations. We describe conformational stability of native NLRP14 and mutants in their monomeric and dimeric states and compare them to NLRP4, a representative of a native pyrin domain fold. Thereby, we characterize the interplay of conformational dynamics, fold stability, and dimerization in NLRP pyrin domains. We show that intrinsic flexibility of NLRP pyrin domains is a key factor influencing their behavior in physiological conditions. Additionally, we provide further evidence for the crucial importance of a charge relay system within NLRPs that critically influences their conformational ensemble in solution.
Keywords: NLRP dimerization, NLRP14 signaling, pyrin domain, fold stability, entropy, conformational dynamics, charge relay system, molecular dynamics simulation
Introduction
Both, innate immune response and recognition of bacteria relies on cellular signaling mediated by members of the Nucleotide-inding domain Leucine-rich repeat-containing Receptor (NLR) family.1,2 NLRs contain multiple domains, each fulfilling specific roles in pattern recognition.3 Fourteen of the twenty-two human NLRs contain a pyrin domain and form the NLRP family (NLRP1-NLRP14).4,5
NLRP1 and NLRP3 are key components in the formation of inflammasomes, a multiprotein oligomer component of the innate immune system.6,7 Thus, NLRPs might be interesting drug targets to tackle inflammatory responses.8 Conditions ranging from Crohn's disease, rheumatoid arthritis to particular forms of malignancies have been linked to NLRP function and malfunction.9–11 Cancer implications are evident, as NLRPs are also involved in the cellular apoptotic machinery.12 Additionally, mutations in some NLRPs are associated with reproduction-related dysfunctions.13,14 In particular, mutations in NLRP14 were shown to impair spermatogenesis.15
NLRPs consist of three separate domains: an N-terminal pyrin domain, a NACHT (with reference to its initial identification in NAIP, CIITA, Het-E and TP1) domain, and a C-terminal leucine-rich repeat domain with the exception of NLRP1 that additionally presents a caspase activation and recruitment (CARD) domain at the C-terminus.16 The leucine-rich repeat domain is crucial for ligand binding and is considered to interact with the NACHT domain to keep the receptor in an auto-inhibited state.17 Upon ligand sensing, the leucine-rich repeat domain is thought to undergo conformational rearrangements allowing for NACHT-mediated NLRP oligomerization.18 This final complex then recruits down-stream signaling partners via the pyrin domain.19
High resolution X-ray structures of several pyrin domains of NLRPs revealed distinctly charged surface regions possibly reflecting the interaction sites. However, to date structural information on a pyrin–pyrin domain complex that would allow understanding of NLRP signalling at atomistic level is missing. Pyrin domains adopt the typical death domain fold formed by six α-helices wrapped around a conserved hydrophobic core.20 Recently, X-ray structures of the native and mutant NLRP14 pyrin domain showed an unexpected conformational transition.21 The compact fold of the six helix bundle is opened, thereby exposing parts of the hydrophobic core and forming an elongated α5/6 stem-helix. The exposed contact area allows for dimerization of NLRP pyrin domains, a novel type of homotypic interactions. A Glu-Arg-Asp charge relay system was proposed as a conformational regulation element in NLRPs. An intact charge bridge is thought to shift the conformational equilibrium of open, semi-open and closed state by favoring the closed conformation energetically. This hypothesis was strengthened by mutational experiments showing drastic changes in protein stability upon mutations involving residues in the charge relay system. The Glu-Arg-Asp charge relay system is broken in native NLRP14, but can be formed via L84R mutation. The resulting protein shows increased thermal stability and shifts the preferred conformation to the closed state. On the other hand, the physiologically relevant mutation D86V causing spermatogenic failure15 and affecting the third residue of the charge-relay system was shown to decrease protein stability without pronounced effects on the static three-dimensional structure.
Herein, we characterize conformational dynamics of the NLRP14 pyrin domain to investigate the underlying dynamic phenomena of NLRP signaling and oligomerization. We use molecular dynamics simulations22 to characterize the conformational ensemble of native and mutant NLRP14 (D86V and L84R) in monomeric and dimeric states and compare them to NLRP4, an NLRP with intact charge relay system (see Figure 1).
Figure 1.
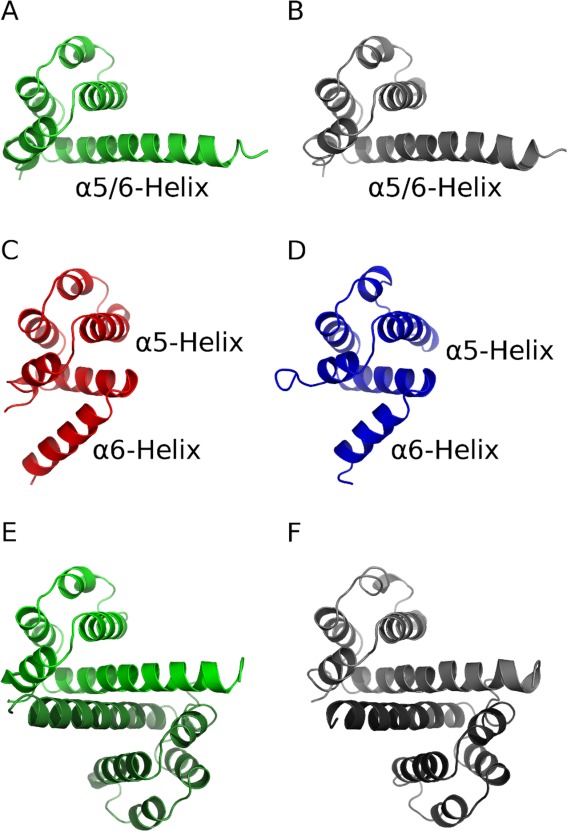
Overview of simulated systems: (A) monomeric native NLRP14 (light green cartoon) (B) monomeric physiological mutant NLRP14-D86V (light grey) (C) stabilized mutant NLRP14-L84R (red) (D) NLRP4 (blue) (E) dimeric native NLRP14 (light and dark green) (F) dimeric mutant NLRP14-D86V (light and dark grey).
Results
Molecular dynamics simulations yielded stable trajectories for all six systems over 100 ns. Still, we observed major differences in flexibility between the simulated systems. Simulations of native NLRP14 and NLRP14-D86V, both started in open state, show preservation of the hydrophobic core but pronounced flexibility in their solvent-exposed α6 helix. Thereby, both systems sample not only the complete open starting conformation but also a semi-open intermediate state (see Fig. 2). Transition from the open to the semi-open state is seen to be completely reversible for the D86V mutant on a 100 ns time scale. The kink point of the elongated α5/6 helix identified in molecular dynamics simulations is in agreement with experimental results from X-ray crystallography. We observe a highly dynamic structural ensemble resembling aspects of both closed and open conformation. The α5/6 helix appears kinked, but the hydrophobic core of the NLRP closed conformation is not yet fully formed.
Figure 2.
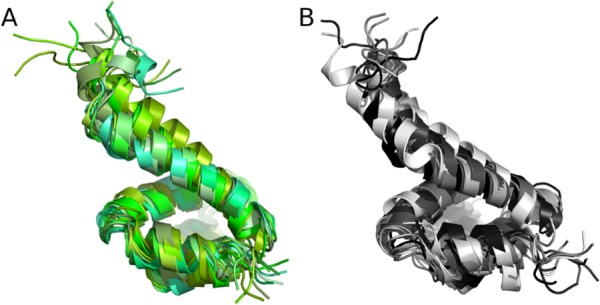
Conformational ensembles of monomeric NLRP14: Ten snapshots were extracted from simulations trajectories in 10 ns intervals and superposed. Both simulations of native NLRP14 (A, green shadings) and mutant NLRP14-D86V (B, grey shadings) sample semi-closed conformations within molecular dynamics trajectories.
We observe no major conformational transitions for all systems starting in a closed conformation or as dimeric systems. This behavior is underlined when analysing distances in the charge relay system of the pyrin domain (see Fig. 3). Systems in closed conformation (NLRP14-L84R, NLRP4) retain their stabilizing electrostatic interactions over the simulation time except for a conformational transition observed in the simulation of NLRP4 around 90 ns. Here, the side-chain of R81 reorients, thereby breaking parts of the salt bridges within the charge relay system. On the other hand, systems starting in open conformation do not form salt bridges and thus maintain higher distances in the charge relay system over the whole trajectory.
Figure 3.
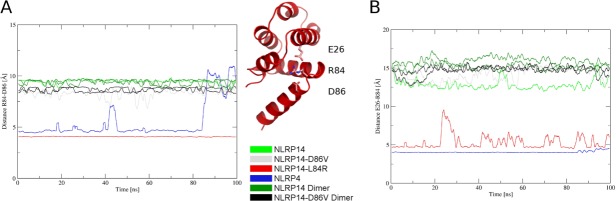
Geometry of the charge relay system: Distances of salt bridges within the simulations of NLRP pyrin domains are monitored over simulation time. (A) Distance of R84 and D86, (B) Distance of E26 and R84 for six simulations. NLRP14-L84R and NLRP4 both stay in the closed starting conformation with two fully formed salt bridges. Four other systems stay in their extended conformation and do not form contacts within these three residues.
As a direct consequence of the stability of sampled conformational states, SASAs of the α6 helices differ significantly [Fig. 4(A)]. Whereas the elongated stem helix is readily accessible for the solvent in open conformation, SASA is reduced by 21–31% in closed conformation (NLRP14-L84R and NLRP4). Likewise, homo-dimerization in open conformation reduces accessibility of the α6 helix by 8–20%. Thereby, dimeric NLRP14-D86V retains higher solvent exposure and samples a broader conformational ensemble than the native system.
Figure 4.
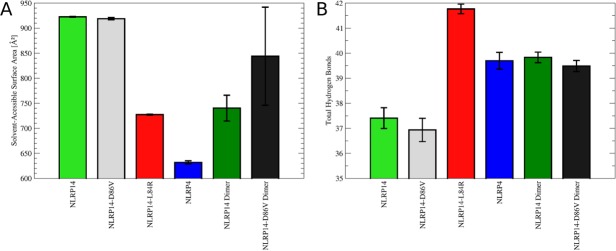
(A) Solvent exposure of the α6 helix: Ensemble-averaged solvent accessibilities were calculated for six simulated systems. NLRP14-L84R and NLRP4 show drastically reduced solvent exposure in closed conformation compared to monomers in open conformation (NLRP14, NLRP14-D86V). Dimers of these systems reduce solvent accessibilities to values close to these observed for monomers in the closed conformation. (B) Hydrogen bonding in NLRP pyrin domains: Total hydrogen bonds occurring in the conformational ensemble of NLRPs were counted. Open conformations of NLRP monomers (NLRP14, NLRP14-D86V) have a lower amount of hydrogen bonds compared to closed conformations (NLRP14-L84R, NLRP4). This loss is compensated by dimer formation recovering the hydrogen bond count to similar values as for closed state monomers.
The observed gain of contact area is also reflected in an increase of total hydrogen bonding in closed and dimeric systems [Fig. 4(B)]. Whereas NLRP14 and NLRP14-D86V form on average 37 hydrogen bonds per snapshot, this value is increased to between 39 and 42 for dimeric systems and systems in closed conformation. The stabilized mutant NLRP14-L84R shows the highest amount of hydrogen bonds per snapshot amongst all six systems simulated.
Residues involved in pyrin domain dimerization partially compensate for the loss of interaction partners in absence of the second domain by various mechanisms. Whilst exposed hydrophobic residues in NLRP14 mostly take part in the larger hydrophobic core of the closed conformation, hydrophilic amino acids fall in two distinct categories. E21 remains solvent-exposed and forms hydrogen bonds to the solvent. By contrast, R90 partially compensates for the loss of interactions in the dimer by intra-monomer interactions with D86. A similar trend is observed for E26 that is involved in the charge relay system. Here, hydrogen bonds with T29 and K85 are observed in monomeric state of NLRP14. Charge relay residue L86 of the NLRP wild type remains mostly solvent-exposed within the simulation time of 100 ns after removal of the second dimer sub-unit.
To assess global conformational stability we calculated average dihedral entropies for the backbone of all systems [Fig. 5(A)]. Lower entropies correspond to more ordered states. We observe the highest conformational entropy and thus most disorder for NLRP14-D86V followed by native NLRP14, both monomeric systems starting in open conformation. Dimerization rigidifies both systems to lower entropy values, although the NLRP14-D86V dimer still shows flexibility comparable to the NLRP14 monomer. Lowest entropies are observed for pyrin domains in closed conformation (NLRP14-L84R and NLRP4). Overall, entropies for systems in closed conformation are comparable to dimerized pyrin domains.
Figure 5.
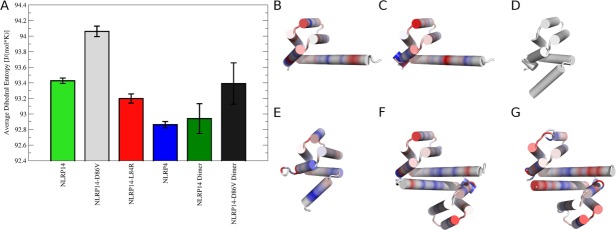
Conformational entropies of NLRPs: (A) Dihedral entropies were calculated over backbone torsions and averaged over the whole protein. Monomeric NLRPs in open form (NLRP14, NLRP14-D86V) show highest entropy, thus showing the broadest conformational ensemble. Conformational space is restricted for closed state monomers (NLRP14-L84R, NLRP4) and dimeric NLRPs. (B–G) Differences in conformational entropies in NLRPs: Residue-wise dihedral entropies were mapped to the starting structures in cartoon representation relative to the stabilized mutant NLRP14-L84R. Differences are shown on a color gradient from red (more flexible, +15 J (mol K−1)−1) over white (no change) to blue (more rigid, −15 J (mol K−1)−1). B) Monomeric native NLRP14 and (C) monomeric mutant NLRP14-D86V show mobilized kink regions in the elongated α5/6 stem helix. (D) Reference structure NLRP14-L84R (completely white). (E) NLRP4 is overall more rigid compared to NLRP14-L84R. Dimeric systems of native NLRP14 (F) and mutant NLRP14-D86V (G) show local rigidification in the dimerization interface. Dimeric NLRP14-D86V has still preserved residual flexibility in the α6 helix.
Residue-wise dihedral entropies were mapped to the starting structures to identify regions of different mobility [Figure 5(B–G)]. The engineered mutant NLRP14-L84R was chosen as reference point with highest stability amongst NLRP14 monomers. Distinct behavior of the α5/6 region is observed for the individual systems. Monomeric native NLRP and mutant NLRP14-D86V show a highly mobile kink region at a slightly different region of the α5/6 helix. In contrast, NLRP4 in closed conformation shows an even more stabilized fold by reduced backbone dynamics. Dimerization of native NLRP14 and mutant NLRP14-D86V rigidifies the dimer interface region at the α5/6 helix, thus increasing stability. Mutant NLRP14-D86V still preserves some flexibility in the α6 helix in dimeric state indicating lower stability.
Discussion
Pyrin domains are central mediators of cellular signaling. In addition to static factors as shape or electrostatics, dynamic factors are increasingly recognized as important contributors to molecular recognition processes. Along with interpretation of thermal factors of NLRP X-ray structures,23 several studies used nuclear magnetic resonance (NMR) spectroscopy to investigate pyrin domain structure and dynamics. Conformational dynamics were analyzed for non-NLRP pyrin domains ASC24 and ASC2,25 revealing some disorder in the α6 helix of ASC2 through increased amide proton exchange. The major dynamic events in pyrin domain appear to be located in the α2-α3 loop. This loop region was described as highly flexible early on.20 Movements were found to take place on the micro second time scale and shown to be important for NLRP signaling.26 Dynamics of several NLRP pyrin domains were examined by NMR yielding different flexibility profiles in this region.27,28
In addition to experimental approaches, two molecular dynamics simulation studies on NLRPs were reported in literature. Using 10 ns sampling time Gattin and van Gunsteren aimed to explain effects of pH changes on the closed conformations of ASC and NLRP1 pyrin domains and identify alterations in salt bridges as key factors.29 Sahoo et al. rationalized co-factor binding to NACHT and LRR domains of NLRP3 using free energy calculations.30 To date, no study systematically comparing conformational dynamics on the NLRP pyrin domain in closed conformation with dynamics in the recently discovered open conformation and the related NLRP homo-dimers have been reported.
Herein, we describe how changes in molecular interactions within the charge relay system of NLRP pyrin domains propagate to major alterations in their conformational dynamics. We find a tight interplay of hydrogen bonding [Figs. 3 and 4(B)], solvent accessibility [Fig. 4(A)] and conformational flexibility (Fig. 5). Reduction of solvent accessibility of pyrin domains by an increase of hydrophobic interactions in closed state has already been described by Eibl et al. for single static structures.21 Herein, we show their propagation to conformational ensembles in solution and additionally capture effects related to dimerization of NLRP pyrin domains.
NLRPs show distinct behavior in solution by shifting between open, semi-open, and closed states in solution.21 Additionally, the open state of NLRPs facilitates homo-dimerization, again impacting conformational stability. As all these factors are crucial for NLRP signaling, we expect investigation of the pyrin domains' conformational dynamics highly valuable in rationalizing their functions in vivo.
Using molecular dynamics simulations we could show how the NLRP pyrin domain switches between open and semi-open state. The kink point in our simulation matches the region experimentally observed via X-ray crystallography. Nevertheless, we did not succeed in recording a complete transition between open and closed state of the pyrin domain. We expect sampling of this transition in NLRPs beyond sampling times currently reached with state-of-the-art molecular dynamics simulations. Still, we assume such major rearrangements soon in range given sampling times beyond the microsecond regime using dedicated simulation architectures.31 Additionally, enhanced sampling techniques32,33 are expected to further aid description of intermediate conformational states of the NLRP pyrin domain. Changes on the level of protein assembly state are increasingly recognized as key factors in biological systems and intrinsically require advanced sampling times and techniques.34
Gel filtration of the NLRP14 pyrin domain suggested that conformational transitions within the α5/6 helix are the rate limiting step for dimer formation.21 Thus, a full characterization of the free energy hypersurface of pyrin domain opening and closing would provide further insights into their dimerization and thus signalling properties. NMR experiments on pyrin domains reported in literature provide limited insights into the dynamic events underlying pyrin domain dimerization as the lower protein concentration is expected to shift the population of states towards the closed monomeric state.
We showed how the dominant conformations of NLRPs affect their thermodynamic stability. Monomers in open conformation tend to be unstable, thus having a higher tendency to oligomerization.21 The engineered, highly stable mutant NLRP14-L84R is experimentally solely observed in the monomeric state. This finding matches increased stability observed in our molecular dynamics simulations. Our data allows to rationalize the measured differences in thermal stability on atomistic level. Increased conformational restriction of NLRP-L84R leads to a higher melting temperature TM of 82.6° compared to the wild type protein (TM = 65.6°). The engineered intact charge relay system in NLRP14-L84R enforces a shift towards the closed conformation of the pyrin domain, thereby increasing compactness and stability. On the other hand, the mutant NLRP14-D86V has a significantly lower melting temperature (TM = 57.7°) correlating with increased local flexibility [Fig. 4(B)]. Dimerization of NLRP14 increases stability, although the dimer of D86V does not fully recover full compactness and conformational restriction by dimerization [Figs. 4(A) and 5(A)].
NLRP signaling critically depends on the conformational states of the pyrin domain, thereby highlighting the importance of protein flexibility in biomolecular recognition.35 The ensemble of thermally accessible conformational states is therefore crucial in understanding biological functions of proteins.36 Molecular dynamics simulations are an attractive alternative to experimental techniques as conformational transitions are accessible over several time scales ranging from femto-to microseconds.37 Thereby, simulations provide unique insights into structural biology due to extreme resolution in time and system size.38
We presented a molecular dynamics study of NLRPs in monomeric open, monomeric closed and dimeric state. We found that minor changes in the charge relay systems lead to major differences in conformational dynamics and stability of the systems. These differences particularly affect the α6 helix of open monomers that undergo major conformational changes. Thereby, our simulations show transitions between the experimentally observed open and a postulated semi-open state. These transitions do not occur in the closed state and in dimeric systems showing a more compact structure. The latter systems are characterized by increased conformational rigidity that compensates for the exposure of the α6 helix in case of the dimeric systems. By calculations of dihedral entropies our simulations show perfect agreement with experimental measures of protein thermal stability. Because of the central role of NLRPs in cellular signaling, a detailed characterization of conformational dynamics in the NLRP pyrin domain is crucial to understand their function. Herein, we demonstrate how computational methodologies can be exploited for characterization of the behavior of NLRPs in solution where these proteins are characterized by complex transitions between different conformational and oligomeric states.
Materials and Methods
Molecular dynamics simulations
We performed six independent all-atom molecular dynamics simulations using identical settings to ensure comparability of observables. Four simulations of pyrin domains were performed in monomeric state: native open NLRP14 (based on chain A of PDB: 4N1J21), open NLRP14-D86V (based on chain A of PDB 4N1K21) closed NLRP14-L84R (based on PDB 4N1L21), and closed NLRP4 (based on chain A in conformation A of PDB 4EWI28). NLRP4 was chosen as reference system with an all atom RMSD < 0.8 Å to NLRP14-L84R. Additionally, we simulated dimers of the open NLRP14 (based on chains A and B of PDB: 4N1J21) and open NLRP14-D86V (based on chains A and B of PDB 4N1K21). See Figure 1 for an overview of simulated systems. All molecular graphics were created using Pymol.39
We removed surface-bound glycerol and ions from all systems and replaced selenomethionine residues introduced for crystallography with standard methionine residues. Resolved water molecules were preserved for simulation. Systems were individually protonated for 300 K and pH = 7.0 using the tool protonate3d.40 Additionally, a periodic truncated octahedral water box with minimum wall distance of 10 Å was added to the systems using tleap from AmberTools.41
Simulations were performed using explicit TIP3P solvent42 and Particle Mesh Ewald43 for long-range electrostatics as implemented in pmemd in Amber10.41 The Amber force field 99SB-ILDN was used to describe the protein residues.44 Systems were energy minimized with harmonic restraints on heavy protein atoms and gradually heated from 100 to 300 K over 200 ps using an extensive equilibration protocol.45,46 After density equilibration over 1 ns in NpT ensemble at 1.0 bar, unrestrained simulations were performed at 300 K (maintained by Langevin thermostat47) for 100 ns for each system. Applying SHAKE algorithm48 on bonds involving hydrogen atoms, we used a time step of 2.0 fs for our simulations. About 5000 equal-spaced snapshots were saved to trajectory for each simulation.
Analysis of simulation data
We used ptraj and cpptraj from AmberTools for the analysis of trajectories.49 We performed standard stability checks for overall structures, secondary structures, and system energies. All further analyses were restricted to overlapping regions in all simulated systems corresponding to F9 to E93 in NLRP14 and F5 to R90 in NLRP4 (see Eibl et al.21 for the underlying sequence alignment).
We analyzed the total amount of hydrogen bonds within the proteins using cpptraj's default criteria for hydrogen bonds: a maximum distance of 3.0 Å between involved heavy atoms and a maximum angle of 45° between the three binding partners (acceptor-donor-hydrogen). Our calculations include hydrogen bonds of both backbone and side-chains. We report half the total amount of hydrogen bonds for dimeric systems, thus including half of inter-chain interactions. Hydrogen bond occupancies are reported as average and standard deviation over five trajectory portions.
To obtain an alignment-independent localized metric for flexibility, we calculated estimates for the total dihedral entropy by analyzing the distributions of backbone torsion angles φ, ψ, and ω. We extracted torsion angles from the simulation trajectory and derived a continuous probability density function using non-parametric kernel density estimation.50 Data was periodically duplicated to avoid bias at boundaries and integrated to yield a thermodynamic entropy arising from conformational flexibility.51 Ordered states correspond to low entropies with a single dihedral peak with a width of 1 degree corresponding to an entropy of 0 J (mol K−1)−1. Individual torsional entropies were summed up and divided by the number of residues to give an average dihedral entropy. For dimers averaged values over both chains are presented. In analogy to hydrogen bonds we analyzed dihedral entropies in five trajectory parts and report average and standard deviation.
Solvent-accessible surface area (SASA) was calculated for the α6 helix region of all simulations using the LCPO algorithm52 implemented in cpptraj. Following a sequence alignment by Eibl et al.21 we considered the last ten overlapping residues between simulations for this calculation. This corresponds to residues L84 to E93 in NLRP14 and R81 to R90 in NLRP4. Values for dimeric systems were again averaged over both subunits and are reported as average and standard deviation over five trajectory parts.
Additionally, we monitored distances and interactions within the charge relay system of the NLRP pyrin domain over simulation time. We measured the distance between atom Cγ of D86 and atom Cγ of R84 and the distance of latter point to atom Cδ of E26. All atoms were chosen to represent the atom farthest from the protein backbone conserved in all mutated proteins (D86V and L84R, respectively). All residues listed here are given as residue identifiers for NLRP14.
Acknowledgments
The authors acknowledge access to the Leo-II computer cluster provided by the platform High Performance Computing at University of Innsbruck. The authors thank Klaus Liedl (University of Innsbruck) and Florian Roessler (University of Cambridge) for fruitful discussions on the manuscript.
References
- Kufer TA, Fritz JH, Philpott DJ. NACHT-LRR proteins (NLRs) in bacterial infection and immunity. Trends Microbiol. 2005;13:381–388. doi: 10.1016/j.tim.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Barbe F, T Douglas, Saleh M. Advances in Nod-like receptor (NLR) biology. Cytokine Growth Factor Rev. doi: 10.1016/j.cytogfr.2014.07.001. (2014) 25: 681–697. [DOI] [PubMed] [Google Scholar]
- Kersse K, Bertrand MJ, Lamkanfi M, Vandenabeele P. NOD-like receptors and the innate immune system: coping with danger, damage, and death. Cytokine Growth Factor Rev. 2011;22:257–276. doi: 10.1016/j.cytogfr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Tschopp J, Martinon F, Burns K. NALPS: a novel protein family involved in inflammation. Nature Rev Mol Cell Biol. 2003;4:95–104. doi: 10.1038/nrm1019. [DOI] [PubMed] [Google Scholar]
- Zambetti LP, Mortellaro A. NLRPs, microbiota, and gut homeostasis: unravelling the connection. J Pathol. 2014;233:321–330. doi: 10.1002/path.4357. [DOI] [PubMed] [Google Scholar]
- Vyleta ML, Wong J, Magun BE. Suppression of ribosomal function triggers innate immune signaling through activation of the NLRP3 inflammasome. PLoS One. 2012;7:e36044. doi: 10.1371/journal.pone.0036044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambetti LP, Laudisi F, Licandro G, Ricciardo-Castagnoli P, Mortellaro A. The rhapsody of NLRPs: master players of inflammation…and a lot more. Immunol Res. 2012;53:78–90. doi: 10.1007/s12026-012-8272-z. [DOI] [PubMed] [Google Scholar]
- MacDonald JA, Wijekoon CP, Liao KC, Muruve DA. Biochemical and structural aspects of the ATP-binding domain in inflammasome-forming human NLRP proteins. IUBMB Life. 2013;65:851–862. doi: 10.1002/iub.1210. [DOI] [PubMed] [Google Scholar]
- Kufer TA, Sansonetti PJ. NLR functions beyond pathogen recognition. Nat Immunol. 2011;12:121–128. doi: 10.1038/ni.1985. [DOI] [PubMed] [Google Scholar]
- Carlstrom M, Ekman AK, Petersson S, Soderkvist P, Enerback C. Genetic support for the role of the NLRP3 inflammasome in psoriasis susceptibility. Exper Dermatol. 2012;21:932–937. doi: 10.1111/exd.12049. [DOI] [PubMed] [Google Scholar]
- Lei Y, Lui VWY, Grandis JR, Egloff AM. Identification of mutations in the PYRIN-containing NLR genes (NLRP) in head and neck squamous cell carcinoma. PLoS One. 2014;9:e85619. doi: 10.1371/journal.pone.0085619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inohara N, Nunez G. NODS: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol. 2003;3:374–382. doi: 10.1038/nri1086. [DOI] [PubMed] [Google Scholar]
- Tian X, Pascal G, Monget P. Evolution and functional divergence of NLRP genes in mammalian reproductive systems. BMC Evol Biol. 2009;9:202. doi: 10.1186/1471-2148-9-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duenez-Guzman EA, Haig D. The evolution of reproduction-related NLRP genes. J Mol Evol. 2014;78:194–201. doi: 10.1007/s00239-014-9614-3. [DOI] [PubMed] [Google Scholar]
- Westerveld GH, Korver CM, van Pelt AMM, Leschot NJ, van der Veen F, Repping S, Lombardi MP. Mutations in the testis-specific NALP14 gene in men suffering from spermatogenic failure. Hum Reprod. 2006;21:3178–3184. doi: 10.1093/humrep/del293. [DOI] [PubMed] [Google Scholar]
- Proell M, Riedl SJ, Fritz JH, Rojas AM, Schwarzenbacher R. The Nod-like receptor (NLR) family: a tale of similarities and differences. PLoS One. 2008;3:e2119. doi: 10.1371/journal.pone.0002119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Yan C, Liu P, Huang Z, Ma R, Zhang C, Wang R, Zhang Y, Martinon F, Miao D, Deng H, Wang J, Chang J, Chai J. Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science. 2013;341:172–175. doi: 10.1126/science.1236381. [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- Martinon F, Hofmann K, Tschopp J. The pyrin domain: a member of the death domain-fold family implicated in apoptosis and inflammation. Curr Biol. 2001;10:R118–R120. doi: 10.1016/s0960-9822(01)00056-2. [DOI] [PubMed] [Google Scholar]
- Hiller S, Kohl A, Fiorito F, Herrmann T, Wider G, Tschopp J, Grutter MG, Wuthrich K. NMR structure of the apoptosis-and inflammation-related NALP1 pyrin domain. Structure. 2003;11:1199–1205. doi: 10.1016/j.str.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Eibl C, Hessenberger M, Wenger J, Brandstetter H. Structures of the NLRP14 pyrin domain reveal a conformational switch mechanism regulating its molecular interactions. Acta Cryst. 2014;D70:2007–2018. doi: 10.1107/S1399004714010311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus M, Petsko GA. Molecular dynamics simulations in biology. Nature. 1990;347:631–639. doi: 10.1038/347631a0. [DOI] [PubMed] [Google Scholar]
- Bae JY, Park HH. Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammosome assembly. J Biol Chem. 2011;286:39528–39536. doi: 10.1074/jbc.M111.278812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liepinsh E, Barbals R, Dahl E, Sharipo A, Staub E, Otting G. The death-domain fold of the ASC PYRIN domain, presenting a basis for PYRIN/PYRIN recognition. J Mol Biol. 2004;332:1155–1163. doi: 10.1016/j.jmb.2003.07.007. [DOI] [PubMed] [Google Scholar]
- Natarajan A, Ghose R, Hill JM. Structure and dynamics of ASC2, a pyrin domain-only protein that regulates inflammatory signaling. J Biol Chem. 2006;281:31863–31875. doi: 10.1074/jbc.M605458200. [DOI] [PubMed] [Google Scholar]
- Pinheiro AS, Proell M, Eibl C, Page R, Schwarzenbacher R, Peti W. Three-dimensional structure of the NLRP7 pyrin domain: insight into pyrin-pyrin-mediated effector domain signaling in innate immunity. J Biol Chem. 2010;285:27402–27410. doi: 10.1074/jbc.M110.113191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro AS, Eibl C, Ekman-Vural Z, Schwarzenbacher R, Peti W. The NLRP12 pyrin domain—structure, dynamics and functional insights. J Mol Biol. 2011;413:790–803. doi: 10.1016/j.jmb.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eibl C, Grigoriu S, Hessenberger M, Wenger J, Puehringer S, Pinheiro AS, Wagner RN, Proell M, Reed JC, Diederichs K, Peti W. Structural and functional analysis of the NLRP4 pyrin domain. Biochemistry. 2012;51:7330–7341. doi: 10.1021/bi3007059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattin Z, van Gunsteren WF. A molecular dynamics study of the ASC and NALP1 pyrin domains at neutral and low pH. Chem Bio Chem. 2008;8:923–933. doi: 10.1002/cbic.200700434. [DOI] [PubMed] [Google Scholar]
- Sahoo BR, Maharana J, Bhoi GK, Lenka SK, Patra MC, Dikhit MR, Dubey PK, Pradhan SK, Behera BK. A conformational analysis of mouse Nalp3 domain structures by molecular dynamics simulations, and binding site analysis. Mol Biosyst. 2014;10:1104–1116. doi: 10.1039/c3mb70600a. [DOI] [PubMed] [Google Scholar]
- Shaw DE, Maragakis P, Lindorff-Larsen K, Piana S, Dror RO, Eastwood MP, Bank JA, Jumper JM, Salmon JK, Shan Y, Wriggers W. Atomic-level characterization of the structural dynamics of proteins. Science. 2010;330:341–346. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- Pierce LC, Salomon-Ferrer R, Augusto F. de Oliveira C, McCammon JA, Walker RC. Routine access to millisecond time scale events with accelerated molecular dynamics. J Chem Theory Comput. 2012;8:2997–3002. doi: 10.1021/ct300284c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs JE, Fuchs D, Liedl KR. Dynamic regulation of phenylalanine hydroxylase. Pteridines. 2014;2:33–39. [Google Scholar]
- von Grafenstein S, Wallnoefer HG, Kirchmair J, Fuchs JE, Huber RG, Schmidtke M, Sauerbrei A, Rollinger JM, Liedl KR. Interface dynamics explain assembly dependency of influenza neuraminidase catalytic activity. J Biomol Struct Dynam. 2015;1:104–120. doi: 10.1080/07391102.2013.855142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr DD, Nussinov R, Wright PE. The role of dynamics conformational ensembles in biomolecular recognition. Nat Chem Biol. 2009;5:789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- Klepeis JL, Lindorff-Larsen K, Dror RO, Shaw DE. Long-timescale molecular dynamics simulations of protein structure and function. Curr Opin Struct Biol. 2009;19:120–127. doi: 10.1016/j.sbi.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Dror RO, Green HF, Valant C, Borhani DW, Valcourt JR, Pan AC, Arlow DH, Canals M, Lane JR, Rahmani R, Baell JB, Sexton PM, Christopoulos A, Shaw DE. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature. 2013;503:295–299. doi: 10.1038/nature12595. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The Pymol molecular graphics system, version 1.5.0.2. San Carlos, CA: De Lano Scientific; 2008. [Google Scholar]
- Labute P. Protonate3D: assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins. 2009;75:187–205. doi: 10.1002/prot.22234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, et al. AMBER 10. San Francisco: University of California; 2008. [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura J, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- Darden T, York D, Pedersen L. Particle mesh Ewald: an N log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- Lindorff-Larsen K, Paian S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallnoefer HG, Handschuh S, Liedl KR, Fox T. Stabilizing of a globular protein by a highly complex water network: a molecular dynamics study on factor Xa. J Phys Chem B. 2010;114:7405–7412. doi: 10.1021/jp101654g. [DOI] [PubMed] [Google Scholar]
- Fuchs JE, Huber RG, von Grafenstein S, Wallnoefer HG, Fuchs D, Liedl KR. Dynamic regulation of phenylalanine hydroxylase by simulated redox manipulation. PLoS One. 2012;7:e53005. doi: 10.1371/journal.pone.0053005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grest GS, Kremer K. Molecular-dynamics simulation for polymers in the presence of a heat bath. Phys Rev A. 1986;33:3628–3631. doi: 10.1103/physreva.33.3628. [DOI] [PubMed] [Google Scholar]
- Ciccotti G, Ryckaert JP. Molecular dynamics simulations of rigid molecules. Comput Phys Rep. 1986;4:346–362. [Google Scholar]
- Roe DR, Cheatham TE., III PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Huber RG, Fuchs JE, von Grafenstein S, Laner M, Wallnoefer HG, Abdelkader N, Kroemer RT, Liedl KR. Entropy from state probabilities: hydration entropy of cations. J Phys Chem B. 2013;117:6466–6472. doi: 10.1021/jp311418q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs JE, Huber RG, Waldner BJ, Kahler U, von Grafenstein S, Kramer C, Liedl KR. (Submitted) Dynamics govern specificity of a protein–protein interface: substrate recognition by thrombin. [DOI] [PMC free article] [PubMed]
- Weiser J, Shenkin PS, Still WC. Approximate atomic surfaces from linear combination of pairwise overlaps (LCPO) J Comput Chem. 1999;20:217–230. [Google Scholar]