

Abstract
Insulin-regulated aminopeptidase (IRAP or oxytocinase) is a membrane-bound zinc-metallopeptidase that cleaves neuroactive peptides in the brain and produces memory enhancing effects when inhibited. We have determined the crystal structure of human IRAP revealing a closed, four domain arrangement with a large, mostly buried cavity abutting the active site. The structure reveals that the GAMEN exopeptidase loop adopts a very different conformation from other aminopeptidases, thus explaining IRAP's unique specificity for cyclic peptides such as oxytocin and vasopressin. Computational docking of a series of IRAP-specific cognitive enhancers into the crystal structure provides a molecular basis for their structure–activity relationships and demonstrates that the structure will be a powerful tool in the development of new classes of cognitive enhancers for treating a variety of memory disorders such as Alzheimer's disease.
Keywords: Alzheimer's disease, aminopeptidase, catalysis, computational modeling, crystallography, cyclic peptide, insulin signaling, memory enhancers
Introduction
Insulin-regulated aminopeptidase (IRAP, oxytocinase, EC 3.4.11.3) was originally identified in fat and muscle cells in association with the insulin-responsive glucose transporter GLUT41 as well as in the placenta as the enzyme that breaks down the peptide hormone oxytocin.2 It was subsequently found in many other organs, usually restricted to specific cell types.3 IRAP has been associated with tissue specific roles including: (i) maintenance of glucose homeostasis through its role in the regulation of the trafficking of GLUT4 in insulin-responsive cells; (ii) proteolytic control of oxytocin concentrations in pregnancy to prevent the onset of premature labor; (iii) regulation of brain oxytocin and vasopressin levels, modulating complex social behaviors such as trust, pair-bonding, anxiety, and (iv) participation in cognitive and memory processing in the brain.
IRAP's role in memory was identified through studies of central administration of the hexapeptide angiotensin IV (Ang-IV; VYIHPF), which enhances memory and learning in rodents.4–10 The binding site responsible for this activity was identified as IRAP.11 Subsequently, Ang-IV and a structurally distinct ligand, LVV-hemorphin-7 (LVVYPWTQRF, a degradation peptide of β-globin), were shown to be potent, competitive inhibitors of IRAP with Ki values in the high nanomolar range.12,13
IRAP has a distinct distribution pattern in the brain and is found in regions associated with cognitive, sensory, and motor function. In neurones of the hippocampus and cerebral cortex, IRAP is present in intracellular vesicles containing GLUT414 suggesting a comparable system exists for IRAP and GLUT4 in neurones as has been described for fat and muscle cells. In insulin-responsive tissues, IRAP and the glucose transporter are trafficked together to the cell surface after insulin stimulation. It is well established that glucose enhances cognitive performance.15 Thus, it has been proposed that IRAP inhibitors might exert their memory enhancing effects by either: (i) inhibiting IRAP's aminopeptidase activity, extending the half-life of endogenous neuropeptides that potentiate memory; (ii) regulating GLUT4 translocation and therefore enhancing glucose uptake; or (iii) modulating signal transduction as IRAP bears characteristics of a membrane-bound receptor.
The 1025 amino acid IRAP protein belongs to the M1 family of zinc metallopeptidases and is a Type II membrane-spanning protein with an N-terminal, 109 residue cytoplasmic domain and an 893 residue extracellular region including the catalytic domain [Fig. 1(a)]. Previously, we developed an homology model of the catalytic region of IRAP based on the structure of human leukotriene A4 hydrolase (LTA4H),16 though they shared limited overall sequence identity of ∼18%. Virtual screening of a ∼2 million compound library led to the identification of a lead series of drug-like compounds we called the HFI-142 series.17 A medicinal chemistry program yielded more than a dozen HFI-142 analogs with Ki's less than 10 µM, including three compounds with Ki's of less than 500 nM.17–19 One of those, HFI-419, was shown to enhance visual recognition and spatial working memory in rats.17
Figure 1.
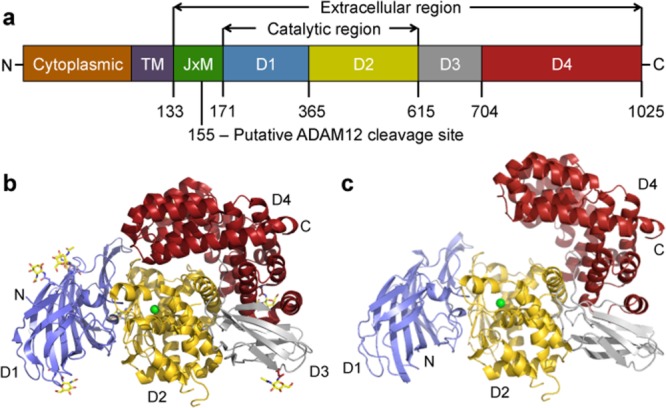
Three-dimensional structure of the IRAP monomer. (a) Domain organization of IRAP. From left to right are the cytoplasmic tail, transmembrane region, juxtamembrane region that includes the cleavage site for oxytocinase production, and the extracellular domains D1 to D4. (b) Crystal structure of the closed form. The domains are in different colors, the Zn ion is a green sphere, and glycosylation shown in stick fashion. (c) Homology model of the open form.An interactive view is available in the electronic version of the article
IRAP is the only documented M1 aminopeptidase that can cleave cyclic peptides such as vasopressin and oxytocin. Homology models based on related M1 aminopeptidases have been unable to explain how IRAP can specifically cleave cyclic peptides as the predicted active sites are too small to accommodate them. As the closest homolog to IRAP, endoplasmic reticulum aminopeptidase 1 (ERAP1), shares less than 50% sequence identity, these observations suggest that parts of the active site are poorly predicted in the available homology models.
Here, we report the crystal structure of IRAP and use it to understand the basis of recognition of physiological substrates and cognitive enhancers by the enzyme. We find that IRAP is dimeric. Each monomer, composed of four continuous domains, forms a closed hollow structure with the active site at its center. The crystal structure reveals the active site is completely remodeled compared to other M1 aminopeptidases with the GAMEN exopeptidase loop adopting a completely different conformation. Cyclic peptides oxytocin and vasopressin could be readily docked into the crystal structure. Furthermore, computational docking of members of the HFI-142 series into the IRAP structure provides a robust explanation of structure–activity relationships. Thus, the structure will prove a valuable resource for accelerating the discovery and development of IRAP-specific cognitive enhancers.
Results
Crystal structure of human IRAP
The crystal structure of IRAP (residues 155–1025) was determined to a resolution of 3.02 Å. Two molecules of IRAP were found in the asymmetric unit with the final model including: the 2 molecules (A and B), 1 Zn ion per monomer, 8 glycosylation sites per monomer, and 55 water molecules. Final data collection and refinement statistics are shown in Supporting Information Table SI. The two monomers superimpose with a root-mean-square deviation (rmsd) on Cα's of 0.44 Å, with the largest outliers located at either exposed loops or crystal contacts. As Molecule A is the more complete model, most of the following analyses focus on it.
The IRAP monomer has approximate dimensions of 93 Å × 62 Å × 55 Å [Fig. 1(b)]. Domain D1 (residues 171–365) forms an extensive β-sandwich with a seven-stranded β-saddle flanked on either side by three- and four-stranded β-sheets. Domain D2 (residues 366–615) adopts a thermolysin-like α/β-fold and contains the catalytic site with a Zn ion at its center and Zn binding HEXXH(X)18-E and GAMEN exopeptidase motifs. The active site is capped by domain D4 to form a large, mostly enclosed cavity (∼5300 Å3) adjacent to the Zn ion. Domain D3 (residues 616–704) adopts a β-sandwich fold consisting of three and four stranded β-sheets and forms a bridge between domains D2 and D4. Domain D4 (residues 705–1025) is entirely α-helical with 16 α-helices that assemble in a “bowl-like” shape and exhibit extensive interactions with domain D2. IRAP forms a dimer in the crystal that appears physiologically relevant (Supporting Information “Supplementary data.doc,” “Table SII,” and “Fig. S1”).
A DALI20 search of the Protein Data Bank reveals that the IRAP structure most closely resembles the closed forms of mammalian endoplasmic reticulum aminopeptidase 1 (ERAP1),21,22 ERAP2,23 aminopeptidase N (APN),24 and aminopeptidase A (APA)25 (Supporting Information Fig. S2). IRAP shares between 32% (APA) and 48% sequence identity (ERAP1) with them and superposition of the structures gives rmsd's on Cα's of 1.6–1.8 Å. A domain comparison reveals that D1 to D3 are very similar (rmsd's < 1.3 Å). In contrast D4 shows more variation with rmsd's ranging from 1.1 Å to 1.5 Å.
IRAP adopts an intermediate conformation between open and closed forms
A number of M1 aminopeptidases, including ERAP1, have been crystallized in two distinct conformational states: open and closed.21,22 The most significant difference between them is a large rotation of D3 relative to the catalytic D2, so that D4 is packed (closed state) or distant (open state) from D1 and the active site [cf. Fig. 1(b) vs. (c)]. For ERAP1, peptide substrates only bind in the open state and require a number of conformational changes for activity. These changes, which occur on transition from the open to the closed form, include reorientation of a key aromatic residue (Tyr549 in IRAP), movement of the GAMEN motif, and a loop-to-helix transition of a 17 residue segment (equivalent to IRAP residues 528–544 which form a helix). The IRAP structure appears to resemble an intermediate conformation between the fully open and closed forms seen for ERAP1 with the interdomain angles and position of the active site Tyr549 [Supporting Information Figs. S3(d) and 4] lying between the orientations seen in the two structures for ERAP1. In this state, the volume of the active site cavity in IRAP is ∼5300 Å3 compared to 2920 Å3 calculated for the closed form of ERAP1.21,22 We have previously shown that in the presence of Ang-IV IRAP becomes resistant to proteolysis, consistent with IRAP also adopting open and closed conformational states.26 We have modeled the open [Fig. 1(c)] and fully closed forms of IRAP based on the equivalent ERAP1 crystal structure. These models are consistent with published biochemical data on IRAP.
IRAP catalytic site
The initial IRAP crystals were grown in the presence of Ang-IV, a nanomolar inhibitor of IRAP with the first three residues (VYI) forming the minimal peptide fragment required for binding.27 In the related structure of human aminopeptidase N (APN) bound to Ang-IV24 only the first three residues were observed. Surprisingly, despite IRAP being crystallized in the presence of Ang-IV, there was no electron density consistent with either the intact peptide or the first three residues in the active site. Instead there is clear electron density associated with the Zn ion that we have interpreted as an alanine [Supporting Information Fig. S3(a)]. We have subsequently determined structures of IRAP from crystals grown in the absence of Ang-IV (3.90 Å resolution) and in the presence of a non-peptide inhibitor, 5,7-dichloro-2-[(dimethylamino)methyl]-8-quinolinol (2.96 Å resolution, see Supporting Information Table SI). In the latter, the electron density associated with the Zn ion is best modeled as a lysine [Supporting Information Fig. S3(b)], as observed in the structure of the related M1 aminopeptidase, ERAP2.23 Presumably the enzyme has picked up the amino acid during expression and purification.
In the crystal structure, the catalytic Zn ion is coordinated by His464, His468, and Glu487 of the HEXXH(X)18E zinc-binding motif [Supporting Information Fig. S3(c)] which superimpose almost identically between the related M1 aminopeptidases [Supporting Information Fig. S3(d)]. The fourth ligand is normally a bound water molecule, thought to be activated by Glu465 to perform a nucleophilic attack on the carbonyl carbon of the scissile bond [Supporting Information “Supplementary data.doc,” Fig. S3(e)]. Mutation of Glu465, or any of the Zn-binding residues, causes a complete loss of enzymatic activity.28 Glu465 is in an identical position to the equivalent residues of ERAP1 (Glu354) and ERAP2 (Glu371). However, the fourth ligand in IRAP appears to be an amino acid, modeled as an alanine [Supporting Information Fig. S3(a)] or lysine [Supporting Information Fig. S3(b)], and thus represents an enzyme–product complex. The amino acid has been modeled so that its carbonyl oxygen interacts directly with the metal and the amino group forms hydrogen bonds with Glu487 and Glu431, which provide the N-terminal anchor for peptide substrates [Supporting Information Fig. S3(c)]. This interpretation is consistent with how the fourth ligand binds in closely related aminopeptidases: bestatin in ERAP1 binds via a carbonyl oxygen,21,22 a lysine in ERAP2 binds via its carbonyl oxygen,23 and both oxygen atoms of an acetate ion bind to Zn in APN.24
A point of difference in the active site between IRAP and the other aminopeptidases is that the carbonyl oxygen of the ligands in the other active aminopeptidase structures also makes hydrogen bonding interactions with the hydroxyl group of a neighboring tyrosine residue [Supporting Information Fig. S3(d)]. This residue, Tyr549 in IRAP, is entirely conserved across M1 aminopeptidases and is responsible for polarizing the carboxyl group of the substrate peptide during the catalytic cycle [Supporting Information Fig. S3(e)]. In most M1 structures the equivalent residue is orientated toward the active site. However, in the IRAP structure, the hydroxyl group of Tyr549 is 6.1 Å distant from the carbonyl oxygen of the active site alanine and is therefore not in a position to form a hydrogen bond. Instead this residue occupies a position intermediate between that of Tyr438 of ERAP1 in the active closed and inactive open states [Supporting Information Figs. S3(d) and S4].21 The position of Tyr549 suggests the IRAP structure is a snapshot of an inactive state requiring a rotation of helix 7 that is located in domain D2 to bring the tyrosine back toward the IRAP active site. Based on the structure reported here a catalytic mechanism can be proposed (Supporting Information “Supplementary data.doc,” Fig. S3).
Specificity and the S1 pocket
A large number of studies have explored the structural requirements of peptide binding to IRAP. The most crucial prerequisite is a free N-terminus; however, there are additional constraints on substrates provided by the S1 pocket. In the case of Ang-IV analogs it appears essential that the N-terminal residue has a straight, aliphatic side-chain29 and an aromatic ring at position 2 maximizes binding.30 In LVV-hemorphin-7 a valine residue at position 3 appears crucial and aromatic residues at positions 4 and 6 are major determinants of affinity.12 In contrast, C-terminal residues of either peptide do not appear important.12,29
The S1 pocket, adjacent to the active site, is responsible for determining the specificity of IRAP for the free N-termini of target peptides [Supporting Information Fig. S3(e)]. The GAMEN motif occupies one side of the S1 pocket. The motif not only assists in recognizing the N-termini of substrates but also helps align the scissile bond for catalysis. The importance of the GAMEN motif in IRAP has been demonstrated with mutations of any residues leading to decreased activity and mutation of Glu431 causing complete loss of activity.31 Glu431 interacts with the free amino group of the peptide substrate [Supporting Information Fig. S3(e)].
The similarities and differences in substrate specificity amongst IRAP and its closest homologs, ERAP1 and ERAP2, appear largely due to variations in the S1 pocket (Supporting Information Fig. S5).32 Although ERAP1 displays preference for hydrophobic and aromatic side-chains, ERAP2 displays preference for extended chains with positively charged ends. Surprisingly, IRAP displays aspects of specificity found in both enzymes. When compared to other M1 aminopeptidase structures, the loop containing the GAMEN motif in IRAP is significantly displaced with respect to the active site residues (Fig. 2). The first two residues of the GAMEN motif in ERAP1 (Gly317, Ala318), ERAP2 (Gly334, Ala335), APN (Gly352, Ala353), APA (Gly357, Ala558), and the equivalent residues of LTA4H (Gly268, Gly269) surround the N-terminal side-chain of bound substrate and, together with the adjacent amino acid (Ser316 of ERAP1, Pro333 of ERAP2, Ala351 of APN, Thr356 of APA, Tyr267 of LTA4H), make up one edge of the pocket (Supporting Information Fig. S5). In contrast, the rearrangement of the loop in IRAP results in the equivalent GAMEN residues, Gly428 and Ala429, sitting further back from the active site with the side-chain of adjacent Ala427 sitting completely out of the S1 pocket, enlarging the pocket sufficiently to fit all amino acid side-chains except tryptophan. Although homology models of IRAP have suggested Ala427 points into the active site, mutation of Ala427 to a large residue (tyrosine) had no effect on cleavage of physiological peptide substrates,33 consistent with the crystal structure.
Figure 2.
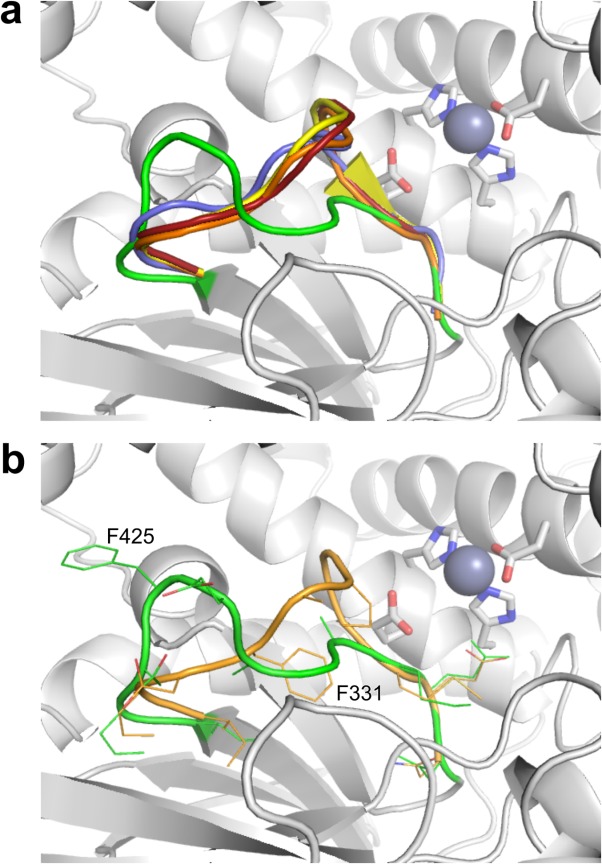
Comparison of the GAMEN motif loop conformation in several representative M1 aminopeptidase structures. The structures of ERAP1 open (PDB id: 3MDJ, blue), ERAP1 closed (PDB id: 2YD0, yellow), ERAP2 (PDB id: 4JBS, orange), and APN (PDB id: 4FYS, red) were superimposed on the IRAP crystal structure (green) using the catalytic site residues. (a) The different conformations of the GAMEN motif loops for the proteins colored as above. The rest of the IRAP structure is shown as a gray cartoon with the active site residues shown as rods colored by atom type. (b) Detailed representation of the IRAP and ERAP2 GAMEN motif loops. The two proteins are displayed as in (a) but with the residues of the loop shown as sticks colored by atom type. Displacement of the loop between the equivalent residues Ile422/328 and Met430/336 is substantial, with the difference in the side-chains of Phe425/331 exceeding 10 Å. An interactive view is available in the electronic version of the article.
The structural basis for the different conformation of the GAMEN loop in IRAP compared to the other aminopeptidases is unclear. The open and closed forms of ERAP1 reveal the same restrictive GAMEN loop conformation, suggesting that the difference is not due to the transition between active and inactive conformers. Similarly, equivalent aminopeptidase structures with and without active site ligands display the same restrictive loop conformation, suggesting the nature of ligand in the active site is not the cause of the shift seen in IRAP. Crystal packing does not appear responsible as the GAMEN sequence itself is buried within the protein, while the adjacent loop residues (Ile422 to Ala427) are only partially exposed in the join between D2 and D4. The nearest symmetry related residue to the loop residues is more than 9 Å away. These observations suggest that the difference in GAMEN loop conformation between IRAP and the other aminopeptidases is a consequence of the local environment and hence a unique aspect of the IRAP active site structure. The most substantial rearrangement seen is for the side-chain of Phe425 with respect to its equivalents Phe314 (ERAP1) and Phe331 (ERAP2) (Fig. 2). In the ERAP structures, the aromatic side-chain of this phenylalanine residue sits beneath the adjacent GAMEN sequence, packing against the main-chain atoms of the GAMEN motif glycine residue. In IRAP, Phe425 has swung approximately 180° to point away from the GAMEN sequence and out toward D4, where it forms a π-stacking interaction with the guanidinium group of Arg920. In ERAP1 and ERAP2, the arginine residue at this position is replaced by a lysine residue that, while still capable of forming a π-stack with aromatic side-chains, is found to do so less frequently in protein interfaces.34 The removal of the bulky phenyl group from beneath the GAMEN sequence allows it to pack more closely to the fourth strand of the D2 β-sheet, resulting in the observed shift of the GAMEN loop and subsequent increase in size of the S1 pocket. Homology models of IRAP to date, including our own,17 do not include this altered GAMEN loop structure meaning that the size and shape of the S1 pocket has been incorrectly modeled. As such, accurate interpretation of putative substrate and inhibitor binding modes has been hampered.
Molecular basis for IRAP recognition of physiologically relevant peptides
IRAP has been shown to cleave a range of natural peptide hormones in vitro including oxytocin, vasopressin, Lys-bradykinin, Ang-III, Met-enkephalin, dynorphin A, cholecystokinin 8, and somatostatin.28,35–37 IRAP is unique however as it is the only documented M1 aminopeptidase that can cleave cyclic peptides such as vasopressin and oxytocin. Vasopressin and oxytocin were the first reported substrates of IRAP and their cleavage has been studied in the most detail.28,36,37 Initially, the N-terminal cysteine is cleaved at its peptide linkage and remains linked by its disulfide bond to an internal cysteine. Subsequently, sequential release of N-terminal amino acids is observed.
IRAP's unique ability to bind and cleave cyclic peptides is reflected in the structural differences of the IRAP active site compared to other aminopeptidases (Fig. 2). Docking of vasopressin and oxytocin into the IRAP structure [Fig. 3(a,b)], as well as into both open and fully closed models of IRAP, gives binding conformations consistent with substrate recognition and catalysis. The shifted GAMEN loop in IRAP allows the cyclic peptides to sit across the active site Zn ion in a productive orientation with the first peptide bond positioned ready for cleavage. Previous attempts by us to dock the cyclic peptides into homology models of IRAP were not successful because the GAMEN loop had been modeled in the conformation seen in related aminopeptidases. In the latter, the GAMEN loop impinges on the space required for the cyclic peptide to sit within the active site (Fig. 2). This provides a molecular basis for IRAP's specificity for cyclic peptides. IRAP is likely to recognize linear peptides in a similar way [Fig. 3(c), Supporting Information “Supplementary data.doc”].
Figure 3.
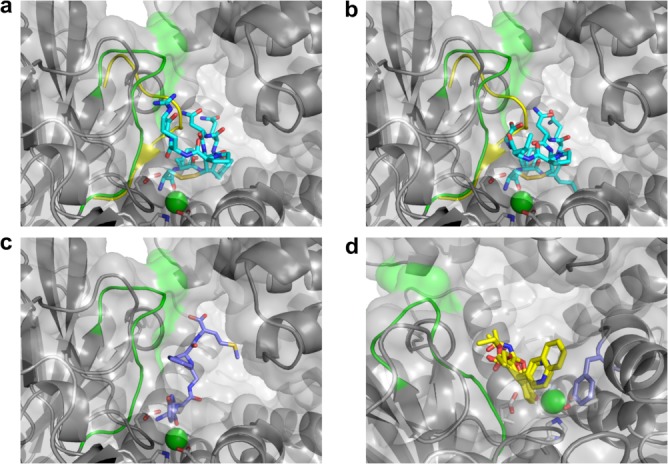
IRAP recognition of physiological ligands and cognitive enhancers. (a) Computational docking of the cyclic peptide oxytocin. IRAP is shown as a cartoon colored gray, except for the GAMEN loop residues which are colored green. Oxytocin and the active site residues are shown as rods colored by atom with carbon atoms either cyan (oxytocin) or white (IRAP) with the Zn ion shown as a green sphere. The molecular surface of IRAP is also shown. The GAMEN loop from ERAP1 (PDB id: 2YD0) has been superimposed (yellow). This position for the GAMEN loop, characteristic of other M1 aminopeptidase structures discussed in the text, impinges on the identified binding orientation of oxytocin, and prevents computational docking of cyclic peptides such as oxytocin. (b) Computational docking of the cyclic peptide vasopressin. View is as for oxytocin in panel a. (c) Docked conformation of Met-enkephalin in the IRAP crystal structure. Met-enkephalin displayed as rods with carbons purple for Met-enkephalin. (d) Molecular basis of recognition by IRAP of cognitive enhancers. The following inhibitors are shown in yellow bonds: HFI-142, HFI-419, HFI-435, HFI-437 with the side-chain of residue Phe544 shown in light purple.
Molecular basis for IRAP recognition of cognitive enhancers
Our previous studies have identified a series of IRAP-specific inhibitors that show activity in vivo as cognitive enhancers in normal rats and memory deficit rat models.17–19 These benzopyran-based compounds were originally identified through in silico screening of a homology model of IRAP based on the crystal structure of LTA4H.17 Further development has relied primarily on traditional medicinal chemistry in the absence of an experimental IRAP structure, resulting in compounds with Ki values as low as 40 nM.17,18
We have docked a selection of these compounds into the IRAP structure to find that compound ranking by docking score (a theoretical estimate of the KD for the formation of the docked complex) is consistent with the kinetic data (Supporting Information Table SIII). All compounds dock with almost identical poses, with the oxygen of the hydroxyl group on the chromene core interacting with the Zn ion and the chromene packed against the GAMEN loop [Fig. 3(d)]. Specificity of the compounds for IRAP over related aminopeptidases including ERAP1 and ERAP217 is therefore explained by IRAP's repositioning of this loop which impinges this site in the other enzymes preventing the compounds binding in this orientation. The common ethylcarboxylate group on the 3-position of the chromene core has a hydrogen bond to the backbone amide of the GAMEN loop alanine and the 4-position aromatic substituents form a ring stack against the side-chain of Phe544. The greater sensitivity of the quinolinyl compounds (HFI-435, HFI-437) than the pyridinyl compounds (HFI-142, HFI-419) to mutations at residue Phe544 (Phe544I/V)38 arises, in this model of binding, from the different degree of ring stacking to Phe544. The shorter pyridinyl group sits displaced from the Phe544 aromatic ring while the second ring of the quinolinyl group reaches partially over the ring to the β-carbon of the side-chain. This causes the greater degree of sensitivity seen for the quinolinyl compounds to substitution of the Phe by the β-branched Val and Ile residues. Overall, these results indicate that the IRAP structure provides a reliable basis for understanding the structure–activity relationships of IRAP-specific inhibitors.
Discussion
Memory and cognitive impairments afflict more than 25% of people over 65 years due to a wide range of clinical conditions including Alzheimer's disease, head trauma, hypoxic damage, and stroke. Currently, most approved treatments are focused on cholinesterase inhibitors but these have proven limited efficacy. More innovative approaches are required and hence the development of memory-enhancing drugs is gaining momentum.19 Thus, the discovery that small molecule specific inhibitors of IRAP can enhance memory17 has generated much interest in academia and industry. As a part of this effort, we have determined the crystal structure of IRAP at atomic resolution.
The overall domain architecture seen in IRAP (Fig. 1) has previously been observed in a range of other M1 aminopeptidases including human ERAP1,21,22 ERAP2,23 APN,24 APA,25 and LTA4H16 (missing D3) (Supporting Information Fig. S2). Our comparison of the available crystal structures revealed that the first three extracellular domains in IRAP are closely similar to the other enzymes with more variability in the C-terminal domain that caps the active site (Supporting Information Fig. S2). Unlike its closest homologs, ERAP1 and ERAP2, IRAP is found to be a dimer in solution and in the crystal (Supporting Information “Supplementary data.doc,” Fig. S1). However, IRAP does use its C-terminal domain to dimerize [Supporting Information “Supplementary data.doc,” Fig. S1(b), Supporting Information Table SII] like human APA and APN. Modeling of the intact IRAP provides no compelling evidence that IRAP might act as a receptor to signal across membranes [Supporting Information “Supplementary data.doc,” Fig. S1(d)].
A comparison of the active site of IRAP with the related M1 aminopeptidases reveals many similarities [Supporting Information Fig. S3(d)] that, together with published mutagenesis studies, suggest a common catalytic mechanism [Supporting Information “Supplementary data.doc,” Fig. S3(e)]. However, key differences do exist including a major change in the conformation of the GAMEN loop that enlarges the S1 specificity pocket, thus explaining how IRAP can recognize cyclic peptides whereas the others cannot. Surprisingly, the IRAP structure revealed a conformation in which the C-terminal domain does not fully enclose the active site [Fig. 1(b)]. Modeling suggested a fully closed conformation of IRAP, like that seen in ERAP1,21,22 is possible. We also readily modeled an open form of IRAP based on ERAP1 [Fig. 1(c)]. The existence of fully open and closed IRAP states is consistent with published proteolysis data that revealed the apo enzyme is proteolytically sensitive but peptide-bound enzyme is insensitive.26 Interestingly, a key catalytic tyrosine, Tyr549, is orientated away from the active site and the crystallographic data suggest an amino acid is bound to the catalytic zinc ion. Thus, it appears that the crystal structure has captured IRAP as an enzyme–product complex with domain D4 starting to swing out from the active site to allow product release.
Despite numerous attempts, we have so far failed to soak in or co-crystallize IRAP with physiological ligands or synthetic inhibitors. However, computational docking into IRAP (in all three conformations) provides compelling functional insights. For example, docking of either the linear peptide Met-enkephalin or the cyclic peptides oxytocin and vasopressin results in tightly clustered solutions in which the first peptide bond is positioned over the Zn ion ready for catalysis (Fig. 3). These models provide the basis for understanding the extensive published structure-function studies of IRAP substrates. Vasopressin and oxytocin have been suggested to be physiological substrates of IRAP.39,40 The effect of vasopressin on facilitating memory consolidation and retrieval is well established and there is evidence that oxytocin is involved in long term memory.41 One hypothesis for the memory enhancing effects of IRAP inhibitors is that they prolong the half-life of these cyclic peptides by inhibiting their degradation by IRAP and hence promote memory through extended interaction of neuroactive peptides with their receptors. The modeling here provides a molecular basis for understanding IRAP's unique specificity toward cleavage of these cyclic neuropeptides.
Computational docking of selected benzopyran family members of cognitive enhancers into the IRAP structure demonstrated they dock in the same orientation relative to the active site in contrast to previous modeling studies that were hampered by incorrect modeling of the GAMEN loop.38 Strikingly, the docking scores correlate exceedingly well with experimentally derived Ki values (Supporting Information Table SIII). This result suggests the IRAP crystal structure could prove very useful for discovering and developing new families of cognitive enhancers.
Although very potent inhibitors of IRAP have been discovered, pharmacokinetic issues including good blood–brain barrier penetration remain to be solved.42 The crystal structure presented here will facilitate the development of new inhibitors with desirable properties that could be developed into cognitive enhancers to treat memory loss associated with a range of disorders like Alzheimer's disease, brain trauma, and stroke.
Materials and Methods
Protein production
Human IRAP (residues 155–1025) was cloned and expressed as previously described.26 Briefly, IRAP was expressed in Sf21 insect cells using a Sartorius Stedim Biotech Biostat-Cultibag RM Bioreactor system (Aubagne, France). The purified protein was dialyzed into 25 mM Tris–HCl (pH 7.2) and 150 mM NaCl, purified by size exclusion chromatography on a Superdex 200 column, and concentrated. One 10 L culture typically produced 1–2 mg of purified protein. The IRAP construct used here has been shown to be active using a fluorometric assay.26 IRAP was concentrated to 9 mg/mL for crystallization studies and stored at −80°C in 25 mM Tris pH 7.2, and 150 mM NaCl.
Structural studies
Crystallization was performed using the vapor-diffusion hanging-drop method at 22°C. Crystals appeared after several days in 25% (w/v) polyethylene glycol (PEG) 3000, and 100 mM Tris pH 8.5. Initially crystals of IRAP could only be grown in the presence of the inhibitor Ang-IV and the initial structure was determined from them. Crystals for data collection were transferred to artificial mother liquor containing 20% glycerol as cryo-protectant prior to freezing by rapid emersion into liquid nitrogen. Data were collected at the MX2 beamline at the Australian Synchrotron (Clayton, Victoria). Data collection was controlled using Blue-Ice software.43 The data were integrated with XDS,44 and scaled with SCALA from the CCP4 program suite.45 A dataset to a resolution of 3.02 Å was collected from a single crystal. The crystal belonged to the space group P21 with unit cell dimensions of a = 68.3 Å, b = 256.1 Å, c = 71.1 Å, β = 115.1°. Assuming two molecules in the asymmetric unit, the VM value for this crystal was 2.81 Å/Da with an estimated solvent content of 56%.46 Molecular replacement was performed using the program PHASER47 in the CCP4 program suite45 with the structures of the related M1 aminopeptidases ERAP1 (PDB id: 2YD0)21,22 and ERAP2 (PDB id: 3SES)23 as search models. A solution in which one molecule was located with ERAP1 and the second molecule in the asymmetric unit located with ERAP2 gave the best results. Alternate rounds of model building and refinement were performed with the programs COOT,48 REFMAC 5,49 and BUSTER.50 In total eight rounds of refinement were performed with the Rfactor and Rfree dropping after each round of refinement. Water molecules were built in during the last rounds of refinement. The final model yielded an Rcryst and Rfree of 20.0% and 26.0%, respectively. Molecule A consists of residues 159–596, 598–639, and 646–1025 and Molecule B consists of residues 160–222, 229–596, 598–638, 648–661, and 664–1025. It was apparent from the electron density maps that several asparagine residues were glycosylated: Asn 184, 215, 256, 266, 368, 682, 760, and 850. These residues were modified with either a mono- or di-saccharide of N-acetylglucosamine. The Ramachandran plot showed that 99.0% of the residues were in the allowed regions with 14 residues as outliers (Cys835, Asn638, Tyr972, Tyr784, Asp917, and Pro296 of Molecule A; Trp433, Asn693, Cys835, Pro162, Ser450, Ile718, Val373, Ile884 of Molecule B). All outlying residues were located in regions where the electron density is not well defined. Data and refinement statistics are listed in Supporting Information Table SI. The coordinates for the IRAP structures have been deposited in the Protein Data Bank (http://www.rcsb.org/pdb/) under the accession numbers 4P8Q and 4PJ6.
The PISA (Protein Interfaces, Surfaces and Assemblies) server (http://www.ebi.ac.uk/pdbe/pisa/) was used for all protein:protein surface interaction calculations. The DynDom program for protein domain motion analysis (http://fizz.cmp.uea.ac.uk/dyndom/) was used to calculate the conformational differences between dynamic proteins. The 3V: cavity, channel, and cleft volume calculator and extractor (http://3vee.molmovdb.org), was used for all protein cavity volume calculations. The program LSQMAN (Uppsala Software Factory) was used for alignment and comparison of proteins. Figures containing structural models were prepared in PyMOL.51
Computational modeling
Models of the “open” and fully “closed” conformations of IRAP were prepared by superimposing the individual domains of chain A of the IRAP crystal structure onto the equivalent domains of chain A from the ERAP1 open (PDB id: 3MDJ) and closed (PDB id: 2YD0) structures. In both cases, the domains were then reassembled and minor steric clashes resulting from the repacking of the domains removed through minimization using the MMF94s forcefield in Sybyl-X 2.1.1 (Certara L.P., St. Louis, MO).
Docking of ligands was performed using Surflex version 2.7 running in Sybyl-X 2.1.1. In cases where chiral centers were present, all possible enantiomers were docked. In all cases, the R-enantiomer docked better and thus the results are for this enantiomer. Ligand sites were defined on the basis of the crystallographic alanine associated with the catalytic zinc ion using a threshold of 0.5 and bloat value of 2.0. Molecules were docked in GeomX mode using default settings and inspected in Sybyl.
Acknowledgments
The authors thank Dr. Matthew Chung for his efforts in optimizing IRAP expression for structural studies. This research was partly undertaken on the MX2 beamline at the Australian Synchrotron, Victoria, Australia. The authors thank the beamline staff for their assistance. S.J.H., D.B.A., N.C.H., J.K.H., B.J.M., S.Y.C., and M.W.P. declare competing financial interests. These authors collaborated with the biopharmaceutical company Servier in the development of IRAP inhibitors. D.B.A was an Australian Postgraduate Award Scholar, a recipient of a St. Vincent's Institute Foundation Scholarship sponsored by Colin North and Major Engineering, a recipient of a Victoria Fellowship from the Victorian Government, and a recipient of a Leslie (Les) J. Fleming Churchill Fellowship from The Winston Churchill Memorial Trust. J.K.H. is a joint Cure Cancer/Leukaemia Foundation Postdoctoral Fellow. M.W.P. and S.Y.C. are NHMRC Research Fellows.
Glossary
- Ang-IV
angiotensin-IV
- APA
aminopeptidase A
- APN
aminopeptidase N
- ERAP
endoplasmic reticulum aminopeptidase
- IRAP
insulin-regulated aminopeptidase
- LTA4H
leukotriene A4 hydrolase
- SEC-MALS
size exclusion chromatography-multi-angle light scattering.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
References
- Keller SR, Scott HM, Mastick CC, Aebersold R, Lienhard GE. Cloning and characterization of a novel insulin-regulated membrane aminopeptidase from Glut4 vesicles. J Biol Chem. 1995;270:23612–23618. doi: 10.1074/jbc.270.40.23612. [DOI] [PubMed] [Google Scholar]
- Rogi T, Tsujimoto M, Nakazato H, Mizutani S, Tomoda Y. Human placental leucine aminopeptidase/oxytocinase. A new member of type II membrane-spanning zinc metallopeptidase family. J Biol Chem. 1996;271:56–61. doi: 10.1074/jbc.271.1.56. [DOI] [PubMed] [Google Scholar]
- Keller SR. Role of the insulin-regulated aminopeptidase IRAP in insulin action and diabetes. Biol Pharm Bull. 2004;27:761–764. doi: 10.1248/bpb.27.761. [DOI] [PubMed] [Google Scholar]
- Braszko JJ, Kupryszewski G, Witczuk B, Wisniewski K. Angiotensin II-(3–8)-hexapeptide affects motor activity, performance of passive avoidance and a conditioned avoidance response in rats. Neuroscience. 1988;27:777–783. doi: 10.1016/0306-4522(88)90182-0. [DOI] [PubMed] [Google Scholar]
- Wright JW, Miller-Wing AV, Shaffer MJ, Higginson C, Wright DE, Hanesworth JM, Harding JW. Angiotensin II(3–8) (ANG IV) hippocampal binding: potential role in the facilitation of memory. Brain Res Bull. 1993;32:497–502. doi: 10.1016/0361-9230(93)90297-o. [DOI] [PubMed] [Google Scholar]
- Pederson ES, Harding JW, Wright JW. Attenuation of scopolamine-induced spatial learning impairments by an angiotensin IV analog. Regul Pept. 1998;74:97–103. doi: 10.1016/s0167-0115(98)00028-7. [DOI] [PubMed] [Google Scholar]
- Wright JW, Stubley L, Pederson ES, Kramár EA, Hanesworth JM, Harding JW. Contributions of the brain angiotensin IV-AT4 receptor subtype system to spatial learning. J Neurosci. 1999;19:3952–3961. doi: 10.1523/JNEUROSCI.19-10-03952.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson ES, Krishnan R, Harding JW, Wright JW. A role for the angiotensin AT4 receptor subtype in overcoming scopolamine-induced spatial memory deficits. Regul Pep. 2001;102:147–156. doi: 10.1016/s0167-0115(01)00312-3. [DOI] [PubMed] [Google Scholar]
- Albiston AL, Pederson ES, Burns P, Purcell B, Wright JW, Harding JW, Mendelsohn FA, Weisinger RS, Chai SY. Attentuation of scopolamine-induced learning deficits by LVV-hemorphin-7 in rats in the passive avoidance and Morris water maze paradigms. Behav Brain Res. 2004;154:239–243. doi: 10.1016/j.bbr.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Lee J, Albiston AL, Allen AM, Mendelsohn FA, Ping SE, Barrett GL, Murphy M, Morris MJ, McDowall SG, Chai SY. Effect of intracerebroventricular injection of AT4 receptor ligands, Nle1-angiotensin IV and LVV-hemorphin 7, on spatial learning in rats. Neuroscience. 2004;124:341–349. doi: 10.1016/j.neuroscience.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Albiston AL, McDowall SG, Matsacos D, Sim P, Clune E, Mustafa T, Lee J, Mendelsohn FA, Simpson RJ, Connolly LM, Chai SY. Evidence that the angiotensin IV (AT4) receptor is the enzyme insulin regulated aminopeptidase. J Biol Chem. 2001;276:48263–48266. doi: 10.1074/jbc.C100512200. [DOI] [PubMed] [Google Scholar]
- Lee J, Mustafa T, McDowall SG, Mendelsohn FA, Brennan M,LL, Chai SY. Structure-activity study of LVV-hemorphin-7: angiotensin AT4 receptor ligand and inhibitor of insulin-regulated aminopeptidase. J Pharmacol Exper Ther. 2003;305:205–211. doi: 10.1124/jpet.102.045492. [DOI] [PubMed] [Google Scholar]
- Lew RA, Mustafa T, Ye S, McDowall SG, Chai SY, Albiston AL. Angiotensin AT4 ligands are potent, competitive inhibitors of insulin regulated aminopeptidase (IRAP) J Neurochem. 2003;86:344–350. doi: 10.1046/j.1471-4159.2003.01852.x. [DOI] [PubMed] [Google Scholar]
- Fernando RN, Albiston AL, Chai SY. The insulin-regulated aminopeptidase IRAP is colocalised with GLUT4 in the mouse hippocampus—potential role in modulation of glucose uptake in neurons? Eur J Neurosci. 2008;28:588–598. doi: 10.1111/j.1460-9568.2008.06347.x. [DOI] [PubMed] [Google Scholar]
- Ragozzino ME, Unick KE, Gold PE. Hippocampal acetylcholine release during memory testing in rats: augmentation by glucose. Proc Natl Acad Sci USA. 1996;93:4693–4698. doi: 10.1073/pnas.93.10.4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thunnissen MM, Nordlund P, Haeggstrom JZ. Crystal structure of human leukotriene A(4) hydrolase, a bifuntional enzyme in inflammation. Nat Struct Biol. 2001;8:131–135. doi: 10.1038/84117. [DOI] [PubMed] [Google Scholar]
- Albiston AL, Morton CJ, Ng HL, Pham V, Yeatman HR, Ye S, Fernando RN, De Bundel D, Ascher DB, Mendelsohn FA, Parker MW, Chai SY. Identification and characterization of a new cognitive enhancer based on inhibition of insulin-regulated aminopeptidase. FASEB J. 2008;22:4209–4217. doi: 10.1096/fj.08-112227. [DOI] [PubMed] [Google Scholar]
- Mountford S, Albiston AL, Charman WN, Ng HL, Holien JK, Parker MW, Nicolazzo JA, Thompson PE, Chai SY. Synthesis, structure-activity relationships and brain uptake of a novel series of benzopyran inhibitors of insulin-regulated aminopeptidase. J Med Chem. 2014;57:1368–1377. doi: 10.1021/jm401540f. [DOI] [PubMed] [Google Scholar]
- Albiston AL, Diwakarla S, Fernando RN, Mountford SJ, Yeatman HR, Morgan B, Pham V, Holien JK, Parker MW, Thompson PE, Chai SY. Identification and development of specific inhibitors for insulin-regulated aminopeptidase as a new class of cognitive enhancers. Br J Pharmacol. 2011;164:37–47. doi: 10.1111/j.1476-5381.2011.01402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- Kochan G, Krojer T, Harvey D, Fischer R, Chen L, Vollmar M, von Delft F, Kavanagh KL, Brown MA, Bowness P, Wordsworth P, Kessler BM, Oppermann U. Crystal structures of the endoplasmic reticulum aminopeptidase-1 (ERAP1) reveal the molecular basis for N-terminal peptide trimming. Proc Natl Acad Sci USA. 2011;108:7745–7750. doi: 10.1073/pnas.1101262108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TT, Chang SC, Evnouchidou I, York IA, Rock KL, Goldberg AL, Stratikos E, Stern LJ. Structural basis for antigenic peptide precursor processing by the endoplasmic reticulum aminopeptidase ERAP1. Nat Struct Mol Biol. 2011;18:604–613. doi: 10.1038/nsmb.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birtley JR, Saridakis E, Stratikos E, Mavridis IM. The crystal structure of human endoplasmic reticulum aminopeptidase 2 reveals the atomic basis for distinct roles in antigen processing. Biochemistry. 2012;51:286–295. doi: 10.1021/bi201230p. [DOI] [PubMed] [Google Scholar]
- Wong AHM, Zhou D, Rini JM. The X-ray crystal structure of human aminopeptidase N reveals a novel dimer and the basis for peptide processing. J Biol Chem. 2012;287:36804–36814. doi: 10.1074/jbc.M112.398842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu C, Lin YL, Li F. Structural insights into central hypertension regulation by human aminopeptidase A. J Biol Chem. 2013;288:25638–25645. doi: 10.1074/jbc.M113.494955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascher DB, Cromer BA, Morton CJ, Volitakis I, Cherny RA, Albiston AL, Chai SY, Parker MW. Regulation of insulin-regulated membrane aminopeptidase activity by its C-terminal domain. Biochemistry. 2011;50:2611–2622. doi: 10.1021/bi101893w. [DOI] [PubMed] [Google Scholar]
- Sardinia MF, Hanesworth JM, Krebs LT, Harding JW. AT4 receptor binding characteristics: D-amino acid- and glycine-substituted peptides. Peptides. 1993;14:949–954. doi: 10.1016/0196-9781(93)90071-n. [DOI] [PubMed] [Google Scholar]
- Matsumoto H, Rogi T, Yamashiro K, Kodama S, Tsuruoka N, Hattori A, Takio K, Mizutani S, Tsujimoto M. Characterization of a recombinant soluble form of human placental leucine aminopeptidase/oxytocinase expressed in Chinese hamster ovary cells. Eur J Biochem. 2000;267:46–52. doi: 10.1046/j.1432-1327.2000.00949.x. [DOI] [PubMed] [Google Scholar]
- Sardinia MF, Hanesworth JM, Krishnan F, Harding JW. AT4 receptor structure-binding relationship: N-terminal-modified angiotensin IV analogues. Peptides. 1994;15:1399–1406. doi: 10.1016/0196-9781(94)90115-5. [DOI] [PubMed] [Google Scholar]
- Krishnan R, Hanesworth JM, Wright JW, Harding JW. Structure-binding studies of the adrenal AT4 receptor: analysis of position two- and three-modified angiotensin IV analogs. Peptides. 1999;20:915–920. doi: 10.1016/s0196-9781(99)00081-9. [DOI] [PubMed] [Google Scholar]
- Laustsen PG, Vang S, Kristensen T. Mutational analysis of the active site of human insulin-regulated aminopeptidase. Eur J Biochem. 2001;268:98–104. doi: 10.1046/j.1432-1327.2001.01848.x. [DOI] [PubMed] [Google Scholar]
- Zervoudi E, Papakyriakou A, Georgiadou D, Evnouchidou I, Gajda A, Poreba M, Salvesen GS, Drag M, Hattori A, Swevers L, Vourloumis D, Stratikos E. Probing the S1 specificity pocket of the aminopeptidases that generate antigenic peptides. Biochem J. 2011;435:411–420. doi: 10.1042/BJ20102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye S, Chai SY, Lew RA, Ascher DB, Morton CJ, Parker MW, Albiston AL. Identification of modulating residues defining the catalytic cleft of insulin-regulated aminopeptidase. Biochem. Cell Biol. 2008;86:251–261. doi: 10.1139/o08-037. [DOI] [PubMed] [Google Scholar]
- Crowley PB, Golovin A. Cation-π interactions in protein-protein interfaces. Proteins. 2005;59:231–239. doi: 10.1002/prot.20417. [DOI] [PubMed] [Google Scholar]
- Herbst JJ, Ross SA, Scott HM, Bobin SA, Morris NJ, Lienhard GE, Keller SR. Insulin stimulates cell surface aminopeptidase activity toward vasopressin in adipocytes. Am J Physiol. 1997;272:E600–606. doi: 10.1152/ajpendo.1997.272.4.E600. [DOI] [PubMed] [Google Scholar]
- Matsumoto H, Hattori A, Mizutani S, Tsujimoto M. Cleavage of peptide hormones by placental leucine aminopeptidase/oxytocinase. In: Mizutani S, Turner AJ, Nomura S, Ino K, et al., editors. Cell-surface aminopeptidases: basic and clinical aspects. Amsterdam: Elsevier Science BV; 2001. pp. 295–299. [Google Scholar]
- Matsumoto H, Nagasaka T, Hattori A, Rogi T, Tsuruoka N, Mizutani S, Tsujimoto M. Expression of placental leucine aminopeptidase/oxytocinase in neuronal cells and its action on neuronal peptides. Eur J Biochem. 2001;268:3259–3266. doi: 10.1046/j.1432-1327.2001.02221.x. [DOI] [PubMed] [Google Scholar]
- Albiston AL, Pham V, Ye S, Ng HL, Lew RA, Thompson PE, Holien JK, Morton CJ, Parker MW, Chai SY. Phenylalanine-544 plays a key role in substrate and inhibitor binding by providing a hydrophobic packing point at the active site of insulin-regulated aminopeptidase. Mol Pharmacol. 2010;78:600–607. doi: 10.1124/mol.110.065458. [DOI] [PubMed] [Google Scholar]
- Wallis MG, Lankford MF, Keller SR. Vasopressin is a physiological substrate for the insulin-regulated aminopeptidase IRAP. Am J Physiol Endocrinol Metab. 2007;293:E1092–E1102. doi: 10.1152/ajpendo.00440.2007. [DOI] [PubMed] [Google Scholar]
- Albiston AL, Fernando RN, Yeatman HR, Burns P, Ng HL, Daswani D, Diwakarla S, Pham V, Chai SY. Gene knockout of insulin-regulated aminopeptidase: loss of the specific binding site for angiotensin IV and age-related deficit in spatial memory. Neurobiol Learn Mem. 2010;93:19–30. doi: 10.1016/j.nlm.2009.07.011. [DOI] [PubMed] [Google Scholar]
- Engelmann M, Wotjak CT, Neumann I, Ludwig M, Landgraf R. Behavioral consequences of intracerebral vasopressin and oxytocin: focus on learning and memory. Neurosci Biobehav Rev. 1996;20:341–358. doi: 10.1016/0149-7634(95)00059-3. [DOI] [PubMed] [Google Scholar]
- Andersson H, Hallberg M. Discovery of inhibitors of insulin-regulated aminopeptidase as cognitive enhancers. Int J Hypertens. 2012;2012:789671. doi: 10.1155/2012/789671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. Blu-Ice and the distributed control system: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart O, Vonrhein C, Womack T. BUSTER, Version 1.10.0. Cambridge, United Kingdom: Global Phasing, Ltd; 2011. [Google Scholar]
- DeLano WL. The PYMOL user's manual. San Carlos, CA: Delano Scientific; 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information