

Abstract
Folding coupled to binding is ubiquitous in biology. Nevertheless, the relationship of sequence to function for protein segments that undergo coupled binding and folding remains to be determined. Specifically, it is not known if the well-established rules that govern protein folding and stability are relevant to ligand-linked folding transitions. Upon small ligand biotinoyl-5′-AMP (bio-5′-AMP) binding the Escherichia coli protein BirA undergoes a disorder-to-order transition that results in formation of a network of packed hydrophobic side chains. Ligand binding is also allosterically coupled to protein association, with bio-5′-AMP binding enhancing the dimerization free energy by −4.0 kcal/mol. Previous studies indicated that single alanine replacements in a three residue hydrophobic cluster that contributes to the larger network disrupt cluster formation, ligand binding, and allosteric activation of protein association. In this work, combined equilibrium and kinetic measurements of BirA variants with alanine substitutions in the entire hydrophobic network reveal large functional perturbations resulting from any single substitution and highly non-additive effects of multiple substitutions. These substitutions also disrupt ligand-linked folding. The combined results suggest that, analogous to protein folding, functional disorder-to-order linked to binding requires optimal packing of the relevant hydrophobic side chains that contribute to the transition. The potential for many combinations of residues to satisfy this requirement implies that, although functionally important, segments of homologous proteins that undergo folding linked to binding can exhibit sequence divergence.
Keywords: folding upon binding, hydrophobic packing, isothermal titration calorimetry, sedimentation equilibrium, kinetics
Introduction
Coupled binding and folding contribute to the function of many biological macromolecules.1–3 This phenomenon has gained considerable attention with the discovery of intrinsically disordered proteins (IDPs) that fold upon association with a binding partner.4–6 Furthermore, disorder-to-order transitions can play critical roles in allosteric regulation.7,8 Despite their involvement in many cellular processes, little is known about the sequence–function relationship for protein segments that undergo these transitions.
The Escherichia coli (E. coli) biotin repressor, BirA, provides a model system to investigate the role of sequence in a coupled ligand-binding and folding process. BirA, a bifunctional protein that is the central component of the Biotin Regulatory System (Fig. 1), functions as both a metabolic enzyme, an activity that is required for viability, and a transcription repressor of the biotin biosynthetic operon.9–11 For both functions, apoBirA binds to biotin followed by ATP to synthesize the corepressor, bio-5′-AMP.12 The resulting holoBirA heterodimerizes with the biotin carboxyl carrier protein, BCCP, subunit of acetyl-CoA carboxylase, to catalyze post-translational biotin addition13,14 or homodimerizes and binds sequence-specifically to the biotin operator, bioO, to repress transcription of the biotin biosynthetic operon15,16 Although both dimerization reactions are energetically coupled to bio-5′-AMP binding, the coupling free energy of −4.0 kcal/mol is known only for the homodimerization reaction.17 High-resolution structures of apoBirA (1BIA) and complexes of BirA bound to biotin (1HXD) and a bio-5′-AMP analog, btnOH-AMP (2EWN), reveal disorder-to-order transitions in corepressor binding and allosteric activation.18–20 In apoBirA, two protein segments comprised of residues 116–124 and 206–234, referred to as the biotin binding loop (BBL) and adenylate binding loop (ABL), respectively, are disordered. Although biotin binding is coupled to BBL folding, this folding does not promote homodimerization.15,21 In the adenylate-bound structure, a network of hydrophobic residues from both the ABL and BBL is assembled around the ligand. The network includes a cluster formed by the side chains of residues V214, V219, and W223 of the ABL that assembles over the adenine ring and the side chains of residues F124 and P126 and M211 and V218 that bridge the BBL and ABL (Fig. 1). Charged and polar ABL side chains project away from the protein surface.
Figure 1.
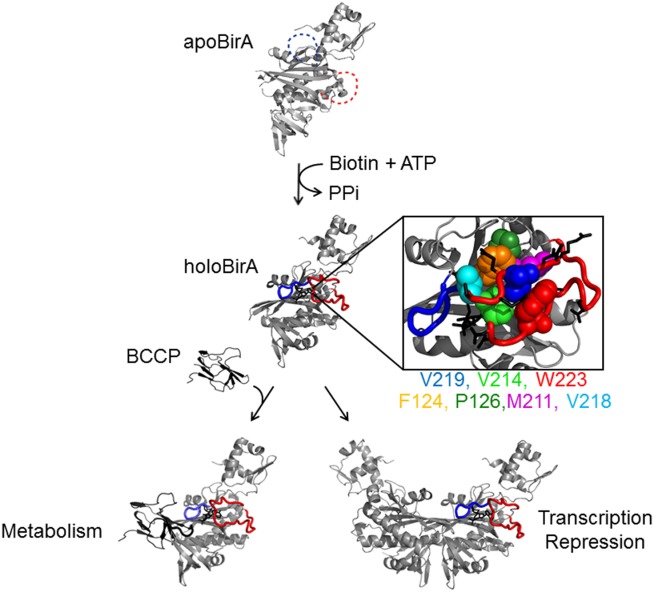
The E. coli biotin regulatory system illustrating the multiple functions of BirA (see text). Protein segments including the BBL (blue), residues 116–128, and the ABL (red), residues 210–234, are disordered (dashed line) in apoBirA and ordered in holoBirA.18,20 The boxed figure illustrates the hydrophobic network that assembles upon bio-5′-AMP binding. Color scheme: ABL cluster: V214 (light green), V219 (blue), W223 (red); ABL-BBL bridge: F124 (orange), P126 (dark green), M211 (magenta), V218 (cyan); the charged and polar side chains, which project away from the protein surface, are shown as black sticks. The models were generated using Pymol22 with pdb files 1BIA and 2EWN as input.
E. coli BirA is a member of a large enzyme family and alignment of BBL and ABL sequences of E. coli, Mycobacterium tuberculosis, and Staphylococcus aureus indicates little conservation at the hydrophobic network positions in E. coli BirA [Fig. 2(A)]. Moreover, this lack of sequence conservation extends to other bacterial ligases as well as eukaryotic orthologs (Supporting Information Fig. S1). The sequence divergence is difficult to reconcile with the demonstrated importance of the ABL cluster sequence for bio-5′-AMP binding and coupled dimerization observed for the E. coli enzyme. However, the high-resolution structures of the M.tb (4OP0) and S.au (3V8L) enzymes bound to bio-5′-AMP reveal, as observed for E. coli BirA, a network of hydrophobic side chains packed around the adenylate ligand [Fig. 2(B)].20,23,24 The sequence divergence and structural data suggest that, reminiscent of the hydrophobic cores that form upon protein folding, the function of the sequence in folding upon binding in biotin ligases may depend solely on the packing ability of the participating hydrophobic side chains. Protein folding studies have led to the general conclusion that a given fold can tolerate sequence variations provided that hydrophobic core side chain packing is retained.25–29 Moreover, packing between side chains in protein folding can be readily detected by non-additive effects of multiple amino acid substitutions on the folding free energy.30,31
Figure 2.
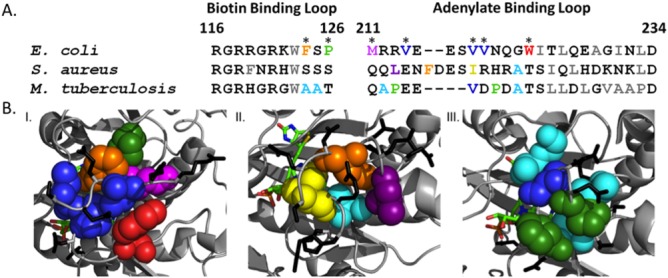
Sequence divergence for hydrophobic residues that assemble around the adenylate ligand in biotin ligase homologs. A. Alignment of the BBL and ABL segments of the Escherichia coli, Mycobacterium tuberculosis, and Staphylococcus aureus biotin protein ligases. The numbering system is from the E. coli protein. Color code: polar or charged residues (black), hydrophobic residues (gray), hydrophobic residues that assemble around the adenylate ligand (colored). B. Hydrophobic packing around the adenylate ligand of the I. E. coli (2EWN) II. S.a. (3V8L) and M.tb (4OP0) biotin protein ligases. The color code for the amino acids is identical to that used in A. Polar and charged side chains are shown as black sticks and the adenylate ligand as colored sticks. The alignments were extracted from alignments of the entire sequences of the three proteins using ClustalW32,33 and the protein structure figures were prepared using Pymol.22
In this work, the sequence–function relationships in BirA ligand-linked folding were investigated by measuring the consequences of introducing single and multiple alanine substitutions into the ABL cluster and bridging residues of the protein. Combined isothermal titration calorimetry (ITC) measurements of bio-5′-AMP binding and stopped-flow kinetic measurements of its synthesis reveal that any single alanine substitution results in large perturbations to adenylate binding and synthesis. Furthermore, effects of multiple substitutions are highly non-additive. Subtilisin-mediated proteolysis indicates that for the majority of the variant proteins the functional defects are accompanied by perturbation to ABL cluster folding. Consequences of the sequence changes for ligand-linked homodimerization, while relatively modest, are also non-additive for proteins with multiple alanine substitutions. These results indicate that, analogous to protein folding, full function of protein segments in binding-linked disorder-to-order transitions in BirA requires appropriate packing of the participating hydrophobic side chains. The results have implications for the evolution of sequences of protein segments that undergo folding coupled to binding.
Results
Alanine substitutions yield large non-additive effects on adenylate binding
Previous measurements indicate that any single alanine substitution in the ABL hydrophobic cluster significantly perturbs bio-5′-AMP binding while leaving biotin binding intact.34 These measurements were extended to proteins with alanine substitutions at residues that bridge the ABL and BBL and to variants with multiple alanine substitutions.
Alanine substitutions at bridging residues yield large perturbations to bio-5′-AMP binding. In contrast, the adenylate binding to wt BirA, which, due to its high affinity, requires use of the ITC displacement titration method,35 direct titrations sufficed for measuring bio-5′-AMP binding to the variants [Fig. 3(A)]. All titrations were performed at a total protein concentration of 2 μM, at which, even in the presence of saturating bio-5′-AMP, the major species is the monomer. For example, the variant that dimerizes most tightly, BirA F124A, is 80% monomer in its adenylate-bound form (Table3). The bio-5′-AMP binding isotherm for BirAV218A is well-described by a simple binding model and, consistent with 1:1 binding, data analysis yields an n-value of 0.98 ± 0.06 [Fig. 3(A), Table1]. Moreover, relative to wild-type BirA, the equilibrium dissociation constant and the Gibbs free energy of adenylate binding are perturbed by 450-fold and 3.5 kcal/mol, respectively. Alanine substitutions at the remaining bridge residues, P126, M211, and F124, resulted in changes to adenylate binding similar in magnitude to that measured for the V218A variant (Table1). By contrast, substitution of the charged ABL residues, R213 and E216, with alanine has minimal effects on bio-5′-AMP binding (Table1). Finally, consistent with previous studies of alanine substitutions in the ABL cluster residues, with the exception of the P126A variant, substitutions of ABL and bridge residues do not alter biotin binding (Supporting Information Table S1).
Figure 3.
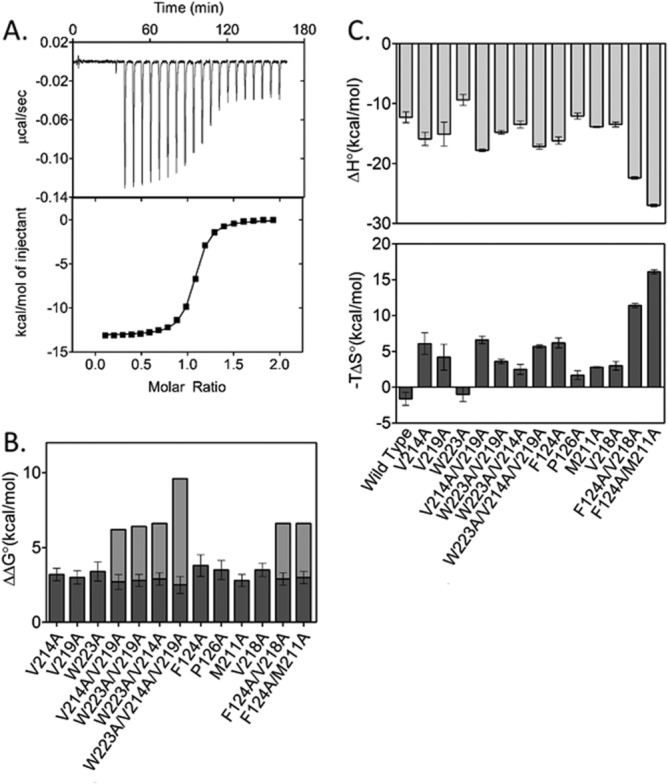
Alanine substitutions result in large, non-additive effects on bio-5′-AMP binding. A. Top panel: ITC trace for V218A BirA with bio-5′-AMP: 19 13 μL volumes of 20 μM bio-5′-AMP were injected into 2 μM V218A. Bottom panel: nonlinear regression of the binding isotherm obtained from A using a single site binding model. B. Perturbations to the Gibbs free energy of bio-5′-AMP binding to the alanine substituted BirA variants shown as dark gray bars with the expected additive effects of the multiple substitutions shown as the sum of the dark gray plus light gray bars. C. Enthalpic (ΔH°) and entropic (−TΔS°) contributions to bio-5′-AMP binding for the BirA variants. The error bars were obtained from standard propagation of errors associated with at least two independent measurements.
Table 3.
Dimerization Energetics of Bio-5′-AMP-Bound BirA Variants
| BirA variant | KDIM (M)a | ΔGºDIM (kcal/mol)b |
|---|---|---|
| WT | 6 (±2) × 10−6 | −7.0 ± (0.3) |
| V214Ac | 3 (±1) × 10−5 | −6.1 ± (0.2) |
| V219Ac | 8 (±1) × 10−5 | −5.5 ± (0.2) |
| W223Ac | 3 (±1) × 10−5 | −6.1 ± (0.2) |
| V214A/V219A | 3 (±2) × 10−5 | −6.1 ± (0.3) |
| W223A/V219A | 2 (±1) × 10−5 | −6.4 ± (0.3) |
| W223A/V214A | 2 (±1) × 10−5 | −6.2 ± (0.3) |
| W223A/V214A/V219A | 2 (±1) × 10−5 | −6.2 ± (0.2) |
| F124A | 1 (±0.4) × 10−5 | −6.6 ± (0.2) |
| P126A | 3 (±1) × 10−5 | −6.0 ± (0.2) |
| M211A | 7 (±3) × 10−5 | −5.6 ± (0.2) |
| V218A | 2 (±1) × 10−5 | −6.4 ± (0.3) |
| F124A/V218A | 3 (±2) × 10−5 | −6.1 ± (0.3) |
| F124A/M211A | 2 (±1) × 10−5 | −6.3 ± (0.3) |
Equilibrium dimerization constant are the average of at least two independent measurements performed as described in Materials and Methods. The errors were obtained by propagation the 65% confidence intervals associated with the equilibrium constant obtained from each individual measurement. In comparing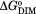 values for each variant with wild type the P values obtained from the unpaired t-test are <0.05 for all but the F124A/M211A variant.
values for each variant with wild type the P values obtained from the unpaired t-test are <0.05 for all but the F124A/M211A variant.
The Gibbs free energies were calculated using the equation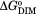 = RTlnKdim.
= RTlnKdim.
Previously published results.34
Table 1.
Thermodynamics of Bio-5′-AMP Binding Obtained from Isothermal Titration Calorimetry
| Protein | KDa | ΔG° (kcal/mol)b,c | ΔH° (kcal/mol)a | −TΔS° (kcal/mol)b | na |
|---|---|---|---|---|---|
| Wild type | 3.9(±1.9) × 10−11 | −13.9 ± 0.4 | −12.3 ± 0.9 | −1.6 ± 0.9 | 0.91 ± 0.01 |
| R213Ac | 1.4(±0.1) × 10−10 | −13.21 ± 0.04 | −10.16 ± 0.01 | −3.05 ± 0.04 | 0.96 ± 0.2 |
| E216Ac | 2.1(±0.2) × 10−10 | −12.96 ± 0.04 | −11.1 ± 0.5 | −1.8 ± 0.5 | 0.84 ± 0.01 |
| V214Ae | 9.7(±1.6) × 10−9 | −10.7 ± 0.1 | −15.9 ± 1.1 | 6.1 ± 1.5 | 0.88 ± 0.01 |
| V219Ae | 1.4(±0.9) × 10−8 | −10.9 ± 0.2 | −15.1 ± 2.0 | 4.2 ± 1.8 | 0.90 ± 0.01 |
| W223Ae | 6.9(±2.0) × 10−9 | −10.5 ± 0.5 | −9.4 ± 0.9 | −1.0 ± 1.0 | 0.93 ± 0.01 |
| V214A/V219A | 4.8(±0.1) × 10−9 | −11.2 ± 0.3 | −17.8 ± 0.2 | 6.6 ± 0.5 | 0.81 ± 0.01 |
| W223A/V219A | 5.1(±2.6) × 10−9 | −11.1 ± 0.1 | −14.8 ± 0.3 | 3.6 ± 0.3 | 0.88 ± 0.03 |
| W223A/V214A | 6.3(±0.8) × 10−9 | −11.0 ± 0.1 | −13.5 ± 0.6 | 2.5 ± 0.7 | 0.79 ± 0.01 |
| W223A/V214A/ V219A | 3.1(±0.9) × 10−9 | −11.4 ± 0.2 | −17.2 ± 0.4 | 5.7 ± 0.2 | 0.84 ± 0.06 |
| F124A | 3.2(±0.5) × 10−8 | −10.1 ± 0.1 | −16.2 ± 0.6 | 6.2 ± 0.7 | 1.00 ± 0.01 |
| P126A | 1.9(±0.4) × 10−8 | −10.4 ± 0.1 | −12.1 ± 0.5 | 1.7 ± 0.6 | 0.96 ± 0.04 |
| M211A | 5.1(±0.6) × 10−9 | −11.1 ± 0.1 | −13.9 ± 0.1 | 2.8 ± 0.1 | 1.00 ± 0.02 |
| V218A | 1.7(±0.6) × 10−8 | −10.4 ± 0.2 | −13.5 ± 0.4 | 3.0 ± 0.6 | 0.98 ± 0.06 |
| F124A/V218A | 5.8(±0.8) × 10−9 | −11.0 ± 0.1 | −22.4 ± 0.2 | 11.4 ± 0.3 | 0.86 ± 0.01 |
| F124A/M211A | 7.5(±1.1) × 10−9 | −10.9 ± 0.1 | −27.0 ± 0.2 | 16.1 ± 0.3 | 0.85 ± 0.01 |
All measurements were performed at 20°C in standard buffer (10 mM Tris HCl, 200 mM KCl, 2.5 mM MgCl2, pH 7.5 at 20°C).
Reported values, the average of at least two independent measurements, were obtained from non-linear least squares analysis using a single-site binding model in Origin 7.0 (MicroCal).
Gibbs free energy values, ΔG°, were obtained using the relationship ΔG° = −RTln(1/KD). Values of −TΔS° were calculated using the relationship ΔG° = ΔH° − TΔS°.
The reported uncertainties were calculated by propagating the confidence intervals of the independent measurements used to calculate the average reported value. The differences between the wild-type and variant Gibbs free energies of binding are significant (P value < 0.05) based on results of the unpaired t-test.
Measured using the displacement method as described in Materials and Methods.
Previously published results.34
Adenylate binding measurements performed on variants with multiple alanine substitutions reveal functional coupling between hydrophobic side chains that participate in the adenylate-linked disorder-to-order transition. Consistent with coupling, the free energy of adenylate binding to each multiply substituted variant is similar to that measured for each singly substituted parent [Fig. 3(B)]. The interaction energy between two residues in bio-5′-AMP binding can be calculated from the measured binding free energies for the wild type, each singly substituted protein and the doubly substituted protein. For example, interaction between V219 and W223 is calculated using the following expression:
in which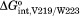 represents interaction energy between the V219 and W223 in adenylate binding and
represents interaction energy between the V219 and W223 in adenylate binding and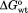 ,
,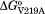 ,
,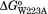 , and
, and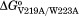 are the Gibbs free energies for binding of the wt, V219A, W223A, and V219A/W223A variants to bio-5′-AMP. For the doubly substituted variants, the coupling free energies range from −3 to −4 kcal/mol, while for the triply substituted variant, W223A/V219A/V214A, it is −7 kcal/mol. The absolute values of these coupling free energies are illustrated by the gray bars in Figure 3(B).
are the Gibbs free energies for binding of the wt, V219A, W223A, and V219A/W223A variants to bio-5′-AMP. For the doubly substituted variants, the coupling free energies range from −3 to −4 kcal/mol, while for the triply substituted variant, W223A/V219A/V214A, it is −7 kcal/mol. The absolute values of these coupling free energies are illustrated by the gray bars in Figure 3(B).
The partitioning of energetic perturbations to adenylate binding for the variants into enthalpic and entropic contributions is complex. However, for a majority of the variants, the enthalpic contribution to adenylate binding is more favorable than that measured for the wild-type protein and the entropic contribution less favorable [Fig. 3(C)]. Furthermore, with the exception of the binding enthalpy measured for the V214A/W223A doubly substituted variant, the enthalpic and entropic perturbations to adenylate binding are nonadditive for multiply substituted variants.
Alanine substitutions compromise ligand-linked ABL folding
Previous studies indicated that alanine substitutions in the BirA ABL hydrophobic cluster are accompanied by perturbations to ligand-linked ABL folding. Measurements of subtilisin-catalyzed BirA proteolysis, which initially occurs between residues 217 and 218 of the ABL,34,36 were used to determine if this is also the case for variants with substitutions in the bridging residues as well as for those with multiple alanine substitutions. BirA, variant or wild type, in the presence or absence of saturating bio-5′-AMP was combined with subtilisin and the resulting time-dependent decrease in the amount of intact protein was obtained from densitometric analysis of samples subjected to SDS-PAGE. For each time course analysis of the optical density versus time data using a single exponential model yields the rate of loss of intact protein [Fig. 4(A)]. The ratio of the rates measured in the absence and presence of bio-5′-AMP, kapo/kholo, for each protein indicates the magnitude of protection from digestion afforded by adenylate binding, which provides a measure of ABL folding upon binding.34 For the majority of the variants the kapo/kholo value is significantly decreased relative to that obtained for wild-type BirA [Fig. 4(B)]. Furthermore, alanine substitution at M211 and V218, which do not contribute directly to the ABL cluster, alters the ratio. Finally, consistent with the observed functional non-additivity, multiple alanine substitutions do not disrupt ABL folding any further than single substitutions. Indeed, the variant with the combined F124A (kapo/kholo = 7.0 ± 0.2) and M211A (kapo/kholo = 2.9 ± 0.1) substitutions has a kapo/kholo value similar to that measured for wild-type BirA.
Figure 4.
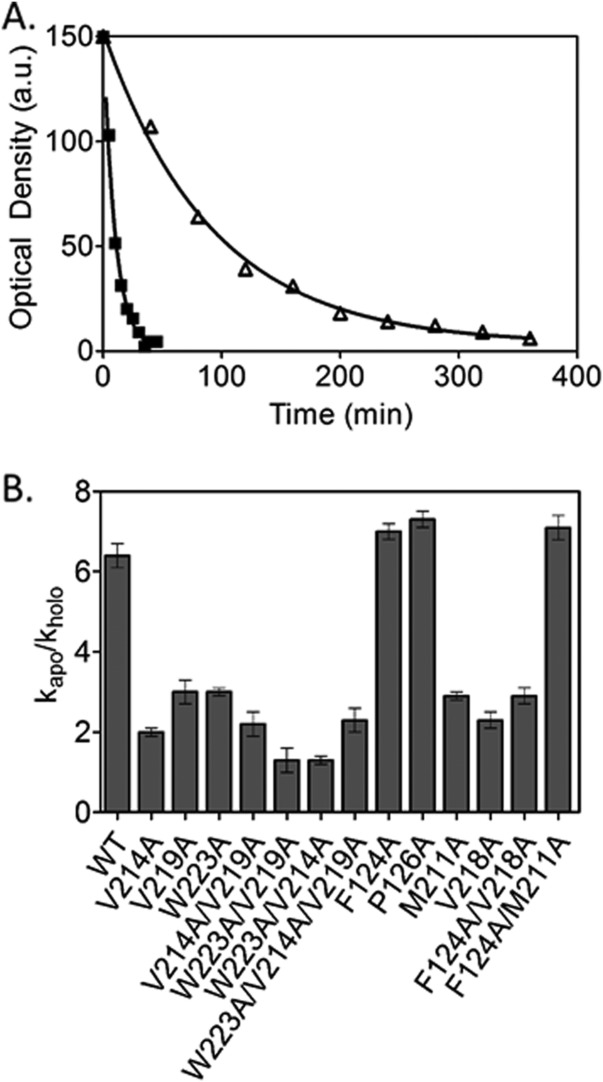
Ligand-linked folding is perturbed for the majority of the BirA variants. A. Time courses of subtilisin-catalyzed cleavage of P126A BirA measured in the absence (▪) and presence (Δ) of saturating bio-5′-AMP. The lines represent the best-fit of the data to a single exponential model. B. Ratio of the pseudo first-order rates of subtilisin-mediated cleavage in the absence, kapo, and presence, kholo, of saturating bio-5′-AMP.
Alanine substitutions perturb BirA-catalyzed bio-5′-AMP synthesis
As BirA catalyzes adenylate synthesis from biotin and ATP, the observed perturbations to bio-5′-AMP binding and loop folding for the alanine substituted proteins suggested that they may also be altered in catalyzing its synthesis. The time-dependence of adenylate synthesis is measured by monitoring the decrease in intrinsic BirA fluorescence that occurs upon rapid mixing of a solution containing the BirA.biotin complex with excess ATP. The resulting transient is analyzed using a single exponential model to obtain the apparent rate of adenylate synthesis (Fig. 5).12 For all variants, the measurements were performed at a single biotin and ATP concentration, with biotin present at sub-saturating but stoichiometric concentration and ATP at 1 mM, close to the Km value measured for wild-type BirA.12,37 The measurements reveal that bio-5′-AMP synthesis is compromised for all variants (Table2) with rate decreases ranging from 2-fold for V214A to 190-fold for the F124A/M211A double variant. For variants W223A and V219A/V214A, which catalyze adenylate synthesis too slowly for detection in stopped-flow measurements, synthesis was indirectly confirmed by mass spectrometric detection of biotinylated apoBCCP following overnight incubation with enzyme, biotin, and ATP (data not shown). The multiply substituted variants V214A/W223A and V219A/W223A, and V219A/V214A/W223A exhibited no detectable bio-5′-AMP synthesis. Although all BirA variants are compromised in bio-5′-AMP synthesis, the measured rates show no correlation with the bio-5′-AMP binding affinities.
Figure 5.
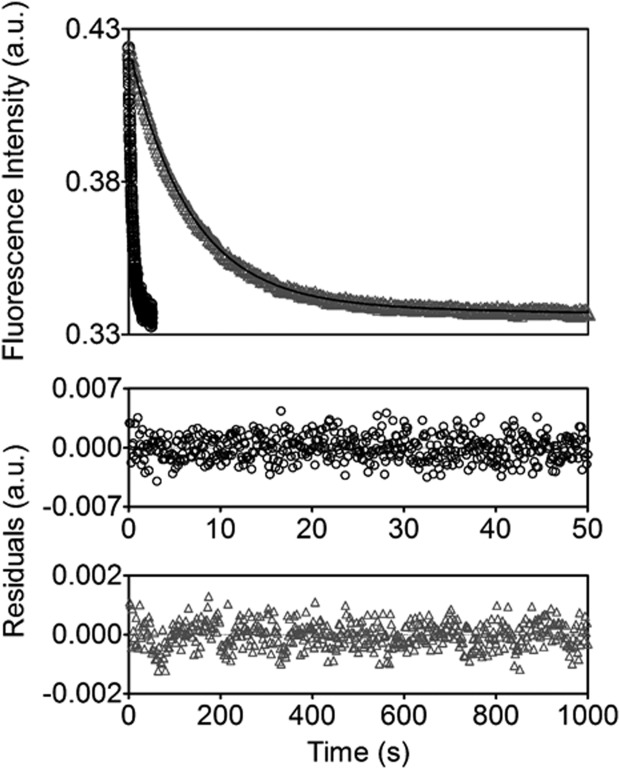
Alanine substituted variants are defective in catalyzing bio-5′-AMP synthesis. Kinetic transients for bio-5′-AMP synthesis by wild type ((○) and F124AV218A BirA ((Δ) with the lines representing the best-fits of the data to a single exponential equation and the residuals of the fits provided in the bottom panels.
Table 2.
Variants Are Defective in Catalyzing Bio-5′-AMP Synthesisa
| BirA variants | Apparent rate kapp (s−1)b | Fold decreasec |
|---|---|---|
| Wild type | 0.136 ± 0.004 | — |
| V214A | 0.0537 ± 0.0004 | 2.53 ± 0.03 |
| V219Ad | 0.0064 ± 0.0003 | 21.25 ± 0.06 |
| F124A | 0.0218 ± 0.0004 | 6.24 ± 0.03 |
| P126A | 0.052 ± 0.001 | 2.62 ± 0.04 |
| M211Ad | 0.0043 ± 0.0003 | 31.63 ± 0.08 |
| V218A | 0.043 ± 0.001 | 3.16 ± 0.04 |
| F124A/V218A | 0.0081 ± 0.0002 | 16.79 ± 0.04 |
| F124A/M211Ad | 0.00072 ± 0.00002 | 188.88 ± 0.04 |
Measurements were performed in Standard Buffer at 20°C as described in Materials and Methods.
The apparent rates are the average of the best fit rates obtained from nonlinear least squares analysis of at least five (stopped-flow) or three (fluorimeter) traces with the standard error of the mean. Comparison of wild type with variant rates using the unpaired t-test yielded P values < 0.05. With the exception of those obtained for the wild-type protein, all transients were analyzed using the model that included a linear term for photobleaching along with the single exponential.
Fold decrease is the rate measured for wild-type BirA divided by that measured for the variant.
Measured using the fluorimeter.
Alanine substitutions yield modest but non-additive effects on homodimerization
Dimerization of BirA bound to bio-5′-AMP, holoBirA, is energetically more favorable than apoBirA dimerization by −4.0 kcal/mol and measurements performed on adenylate-bound variants with single alanine substitutions in the ABL cluster indicate penalties to this coupling ranging from 1 to 1.5 kcal/mol (Table3).17,34 In this work, the dimerization measurements were extended to proteins with single alanine substitutions in the BBL-ABL bridging residues and those with multiple alanine substitutions in the hydrophobic network.
Dimerization measurements were performed on the bio-5′-AMP bound variants using sedimentation equilibrium. Centrifugation of M211A variant was performed at three speeds on protein prepared at three concentrations. Analysis of the data using a single species model indicated average molecular weights higher than expected for the monomeric protein. Global analysis using a monomer–dimer model, which exhibited excellent agreement between the best-fit curves and the data [Fig. 6(A)], indicated that the alanine replacement results in a 1.5 kcal/mol penalty to the dimerization free energy (Table3). Alanine substitutions of the remaining bridging residues resulted in perturbations to dimerization energetics ranging from 0.5 to 1.0 kcal/mol.
Figure 6.
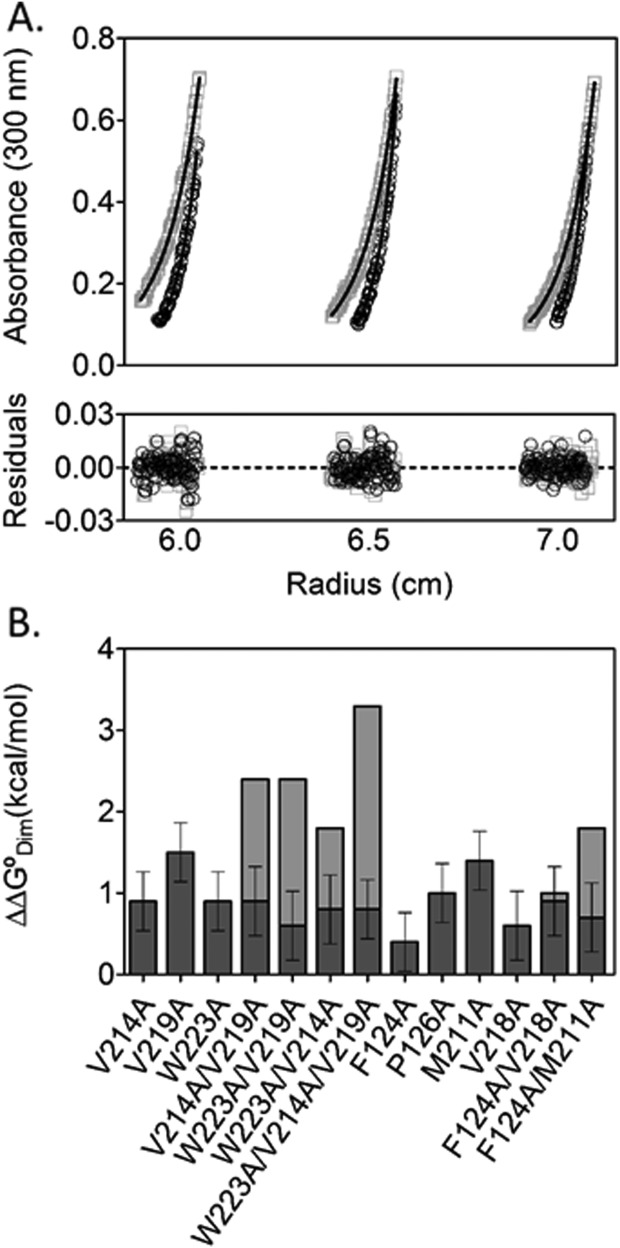
Effects of alanine substitutions on ligand-linked dimerization are modest but non-additive. A. Absorbance versus radius profiles for bio-5′-AMP bound M211A variant prepared at 40, 50, and 60 μM and centrifuged at 21,000 (□) and 24,000 (○) rpm along with the best-fit curves obtained from the global analysis of nine datasets to a monomer–dimer model using WinNonLin.3 The lower panel shows the residuals of the fits for each of the six datasets that are shown. B. Energetic penalties to holoBirA dimerization indicated as dark gray bars with anticipated additive effects of multiple alanine substitutions provided as the sum of the dark and light gray bars.
The effects on dimerization resulting from multiple alanine substitutions in the hydrophobic network were also measured. Data obtained on all proteins with two or three alanine substitutions indicated good agreement with a monomer–dimer model. The dimerization free energies for the variants indicate non-additive effects of the multiple substitutions [Fig. 6(B)]. For multiple substitutions in the hydrophobic cluster the non-additivity is large, as indicated by the 1–3 kcal difference between the anticipated additive effects and the measured values of the dimerization free energies. For the bridging residues addition of the substitution at position 124 to that at 211 reverts the dimerization free energy of the latter protein to a value closer to that measured for wild-type BirA. Only the combination of the V218A and F124A substitutions shows additive effects on dimerization.
Discussion
The hydrophobic network in BirA that assembles concomitant with bio-5′-AMP binding provides an opportunity to examine the relationship between the sequence and function in a ligand-linked disorder-to-order transition. The measurements performed on the alanine-substituted BirA variants indicate that, reminiscent of the hydrophobic cores that assemble in protein folding, full function requires cooperative packing of all contributing side chains. These results imply that although the sequence of the network for a single biotin ligase from a particular species is, due to packing constraints, conserved, the network residues for ligases from distant homologs are free to diverge so long as packing can be achieved.
The large perturbations to adenylate binding resulting from single alanine substitutions in the hydrophobic network coupled with the non-additivity of multiple substitutions indicate that the disorder-to-order transition in BirA adheres to the rules that govern hydrophobic packing in protein folding. All variants with both single and multiple alanine substitutions exhibit similar large perturbations to adenylate binding free energy. Moreover, the majority of the variants, as judged by the loss of adenylate-mediated protection from subtilisin cleavage, are defective in adenylate binding loop folding. Even the variant with an alanine substitution at position M211, which does not form part of the ABL cluster, shows decreased ligand-mediated protection. These results are consistent with cooperative assembly of the hydrophobic network in which any single substitution affects the integrity of the entire network.
The perturbations to the detailed thermodynamics of adenylate binding to the alanine substituted variants are consistent with disruption of folding. All alanine substitutions in the network result in Gibbs free energies of bio-5′-AMP binding similar in magnitude to the measured free energy of biotin binding by wild-type BirA, a process that is accompanied by BBL folding only. Moreover, the patterns of enthalpic and entropic contributions to adenylate binding by the variants are more similar to those measured for wtBirA binding to biotin, not bio-5′-AMP. Wild-type BirA binds to biotin with a relatively modest Gibbs free energy of −10 kcal/mol, a highly favorable enthalpy of −20 kcal/mol and an unfavorable entropic contribution to the binding free energy at 20°C of +10 kcal/mol. By contrast adenylate binding occurs with a more favorable Gibbs free energy of −14 kcal/mol but a less favorable −12 kcal/mol enthalpy and more favorable −1.6 kcal/mol entropy at 20°C. The differences in the energetic signatures for binding of the two ligands are consistent with the additional folding that accompanies adenylate binding with the less favorable enthalpy reflecting dehydration of the hydrophobic side chains that form the network and the more favorable entropy release of the water to the bulk. With the exception of W223ABirA, the thermodynamic signatures of adenylate binding to the variants, with their more favorable enthalpies and unfavorable entropies, are more consistent with wtBirA binding to biotin than to bio-5′-AMP.
In contrast to adenylate binding, the impacts of single alanine substitutions on homodimerization are relatively small, ranging from 0.5 to 1.5 kcal/mol penalties to the Gibbs free energy for the process. Moreover, unlike the effects on bio-5′-AMP binding which are consistently in the +4 kcal/mol range, the perturbations to dimerization vary for the proteins studied. In general, multiple alanine substitutions show non-additive effects on dimerization, with the V218A/F124A double variant the only exception. Given the distance of the hydrophobic cluster from the dimerization surface it is reasonable to assume that all the measured energetic penalties reflect perturbations to the coupling free energy between corepressor binding and dimerization. The modest effects of alanine substitutions on dimerization reflect the fact that (Fig. 1), although optimal packing of the hydrophobic network is required for achieving the full −4.0 kcal/mol coupling between ligand binding and dimerization, the coupling free energy provides only a fraction of the total free energy of holoBirA dimerization.
The results of these studies are consistent the lack of sequence conservation for residues that assemble around the adenylate ligand in BirA homologs (Fig. 2). Like the amino acid residues that form the hydrophobic cores in protein folding, the requirement for optimal packing provides the major selective pressure for these segments.29 In principle, a number of combinations of hydrophobic side chains can pack sufficiently well to support function. Nonetheless, the lack of sequence conservation in the cluster residues does not imply for any single ligase that the sequence of this region is unimportant. Indeed, kinetic measurements reveal that even single alanine substitutions in the hydrophobic network of E. coli BirA can have large consequences for bio-5′-AMP synthesis (Table2). The requirement of biotin transfer to BCCP for viability predicts that for a ligase from a single species the identity of the hydrophobic network sequence is highly constrained. This prediction was born out in the results of genetic screens for BirA mutants, which yielded no mutations in the coding sequences for the hydrophobic network residues.9,38 The requirement of biotin transfer for viability precluded survival mutants belonging to this class.
The results of this work indicate that the sequences of regions that undergo ligand-linked disorder-to-order transitions in BirA are, like the hydrophobic cores in protein folding, constrained by the requirement for optimal packing. This result predicts that although a protein segment that undergoes folding upon binding may be of great functional significance, its sequence need not be evolutionarily conserved.
Materials and Methods
Chemicals and biochemicals
All chemicals were at least reagent grade. The d-biotin, ATP, isopropyl β-d-thiogalactoside (IPTG), phenylmethanesulfonyl fluoride (PMSF), and polyethyleneimine (PEI) were purchased from Sigma–Aldrich and 1,4-dithio-dl-threitol (DTT) was obtained from Research Organics. The bio-5′-AMP was synthesized and purified as previously described39,40 and d-biotin stock solutions were prepared by dissolving the dry powder in Standard Buffer (SB: 10 mM Tris HCl, 200 mM KCl, 2.5 mM MgCl2, followed by adjusting the pH to 7.5 at 20°C). The ATP was dissolved in water, the solution pH was adjusted to 7.5, and after spectrophotometric determination of the concentration 250 μL aliquots were stored at −70°C.
Site-directed mutagenesis, expression, and purification of BirA variants
Site-directed mutagenesis of the C-terminally (His)6-tagged BirA coding sequence in the pBtac2 vector was performed using the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer's instructions. The C-terminal (His)6 tag has previously been shown to have no effect on BirA function.37 Sequences were verified by dideoxy sequencing at the University of Maryland College Park DNA Sequencing Facility and ACGT Inc. Each protein was expressed in E. coli JM109 cells transformed with the appropriate plasmid. Cultures were grown at 30°C in LB media supplemented with 100 μg/mL ampicillin to an O.D.600 of 0.6–0.9, at which point protein expression was induced by addition of IPTG to a final concentration of 1 mM. After 4 h the cells were harvested by centrifugation, lysed by sonication, and the resulting cell debris was removed by centrifugation at 5000 rpm at 4°C for 30 min. Polyethyleneimine (PEI) was added to the cell extract at a 0.2% (v/v) ratio and the precipitate was pelleted by centrifugation. After chromatography on Ni-NTA resin (Qiagen) and followed by SP Sepharose resin34 (GE Healthcare), the pure protein was dialyzed against storage buffer (10 mM Tris HCl, 200 mM KCl, 2.5 mM MgCl2, 5% [v/v] glycerol, pH 7.5 at 20°C) and stored in 1 mL aliquots at −70°C. Protein purity was estimated to be >95% based on Coomassie brilliant blue staining of samples subjected to electrophoresis on SDS-polyacrylamide gels. The fractional activity in bio-5′-AMP binding was determined by stoichiometric titrations monitored by steady-state fluorescence spectroscopy.40
Isothermal titration calorimetry
BirA binding to biotin and bio-5′-AMP binding was measured by ITC using a VP-ITC microcalorimeter (Microcal, Northampton, MA). Each protein was first exchanged into Standard Buffer (10 mM Tris HCl, pH 7.50 ± 0.01 at 20°C, 200 mM KCl, 2.5 mM MgCl2) either by dialysis or chromatography using a Micro Bio-Spin 6 column (Bio-Rad), filtered through a 0.45 μm PTFE membrane syringe filter (PALL), and subjected to spectrophotometric concentration determination. Final ligand and protein solutions were prepared by dilution of concentrated stocks into SB that had been filtered through a 0.22 μm PVDF membrane syringe filter (Millipore). All samples were degassed for at least 10 min at 20°C using a Thermovac (MicroCal) prior to sample cell and syringe loading. Titrations were performed at 20°C by injecting 19–25, 10–13 μL volumes of a 20 μM biotin or bio-5′-AMP solution into 1.4 mL of a 2 μM protein solution at a stirring rate of 310 rpm. Displacement titrations were performed by injecting 25–10 μL volumes of 20 μM bio-5′-AMP into a solution containing 2 μM BirA saturated with biotin.
Sedimentation equilibrium
Protein dimerization was measured by equilibrium analytical ultracentrifugation using an Optima XL-I Analytical Ultracentrifuge (Beckman Coulter). Each protein was exchanged into SB by dialysis or chromatography on a Micro Bio-Spin 6 column (Bio-Rad). Samples were then filtered through 0.45 μm PTFE membrane syringe filters (PALL) before determining the concentration spectrophotometrically. Protein samples at three concentrations, each combined with bio-5′-AMP in a 1:1:15 molar ratio under stoichiometric conditions, were centrifuged in cells equipped with 12 mm six-hole or 3 mm two-hole charcoal-filled Epon centerpieces in a four-hole An-60 rotor (Beckman Coulter) at 3 speeds ranging from 18,000 to 24,000 rpm.
Kinetic measurements of bio-5′-AMP synthesis
Single turnover measurements of BirA-catalyzed bio-5′-AMP synthesis were performed using either a Kintek SF-2001 stopped-flow instrument equipped with fluorescence detection or an ISS-PC1 fluorimeter. For all measurements, the excitation wavelength was set at 295 nm and emission was monitored above 340 nm using a cutoff filter (Corion) for the stopped-flow measurements and at 333 nm for measurements performed using the fluorimeter. The time-dependent decrease in the intrinsic protein fluorescence was monitored upon mixing BirA (wt or variant) combined with biotin in SB at a 1:0.9 molar ratio with ATP to achieve final BirA:biotin and nucleotide concentrations of 0.9 μM and 1 mM.
Subtilisin-catalyzed proteolysis
The rates of subtilisin-catalyzed cleavage of BirA (wt or variant) were determined at 20°C in SB in the presence and absence of saturating bio-5′-AMP. Solutions of protein or protein and ligand were first equilibrated for 30 min at 20°C. Subtilisin prepared in SB was then added to the protein to achieve a final BirA:subtilisin (w:w) ratio of 41.5:1. Aliquots of 20 μL were removed from each reaction at regular time intervals and combined with 1 μL freshly prepared 100 mM PMSF in absolute ethanol and 12 μL of Laemmli sample loading buffer to stop proteolysis. Digestion products were then separated by electrophoresis in a 15% SDS-polyacrylamide gel. After staining with Coomassie brilliant blue the optical density of bands corresponding the intact BirA were quantified using a Molecular Dynamics Laser Scanning Personal Densitometer (GE Healthcare). The rates of subtilisin-catalyzed cleavage of BirA were obtained by nonlinear least squares analysis using the following equation:
| 1 |
in which BirAOD,t is the integrated optical density for the band corresponding to intact BirA at time t in minutes, and k is the pseudo-first order rate of cleavage.
Data analysis
ITC
Analysis of the bio-5′-AMP binding data was performed using the following single-site binding model in Origin 7.0 (MicroCal):
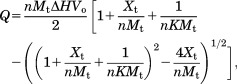 |
2 |
in which Q is the total heat content of the solution contained in Vo, the active cell volume, ΔH is the molar binding enthalpy, K is the equilibrium association constant, n is the binding stoichiometry, and Mt and Xt are the bulk concentration of macromolecule and ligand, respectively. Binding free energies and entropies were calculated using the relationships ΔG° = −RTlnK, ΔG° = ΔH° − TΔS° and the parameters obtained from the analysis. For the displacement titrations the equilibrium constants and enthalpies obtained from the data analysis are apparent values, which can be related to the true values for bio-5′-AMP binding using the following equations:
| 3 |
| 4 |
in which the A and B subscripts are relevant to the parameters associated with bio-5′-AMP and biotin binding, respectively. Thus, the parameters associated with adenylate binding are obtained from the apparent values obtained from analysis of the displacement titrations and the values obtained for biotin binding in independent titrations (Supporting Information Table S1).
Sedimentation equilibrium
The absorbance versus radius profiles obtained for each scan were first analyzed in WinNONLIN41 using a single species model to obtain σ, the reduced molecular mass, from which the weight average molecular weight was calculated using the following equation:
| 5 |
where M is the molecular weight, is the partial specific volume of the protein, ρ is the density of the buffer, ω is the angular velocity of the rotor, R is the gas constant, and T is the temperature in Kelvin. Values of the BirA monomer partial specific volume and the buffer density are 0.755 mL/g and 1.007 g/mL, respectively.15
is the partial specific volume of the protein, ρ is the density of the buffer, ω is the angular velocity of the rotor, R is the gas constant, and T is the temperature in Kelvin. Values of the BirA monomer partial specific volume and the buffer density are 0.755 mL/g and 1.007 g/mL, respectively.15
Absorbance versus radius profiles were also globally analyzed to obtain the equilibrium association constant for dimerization, Ka, using the following monomer–dimer model:
| 6 |
in which Ct is the total concentration at each radial position r, δ is the baseline offset, cm(ro) is the monomer concentration at reference radial position, ro. The reduced molecular weight of the monomer, σmon, of 1.22 was previously determined by sedimentation equilibrium measurements performed on apoBirA. The quality of the fits was assessed from the magnitude of the square root of the variance and the distribution of the residuals of the fit about zero.
Kinetic measurements of bio-5′-AMP synthesis
Analysis of kinetic transients was performed either using the software provided by KinTek Corporation or in Prism. Apparent rates of bio-5′-AMP synthesis were obtained by nonlinear regression of the fluorescence versus time data using a single exponential model. For the variants that catalyzed bio-5′-AMP with rates significantly slower than wild-type BirA the time-dependent decrease in fluorescence included a contribution from photobleaching. In these cases, the data were analyzed using a model that included a linear term for the photobleaching along with the single exponential decrease associated with adenylate synthesis. Inclusion of this additional phase yielded more random distribution of the residuals of the fits but did not significantly change the best-fit values of the apparent rates.
Glossary
- ABL
adenylate binding loop
- BBL
biotin binding loop
- BCCP
biotin carboxyl carrier protein subunit
- Bio-5′-AMP
biotinoyl-5′-adenylate
- BirA
biotin protein ligase/biotin repressor
- btnOH-AMP
biotinol-5′-adenylate
- E. coli
Escherichia coli
- IDP
intrinsically disordered protein
- ITC
isothermal titration calorimetry
- Tris
tris-hydroxymethyl amino methane.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
References
- Malabanan MM, Amyes TL, Richard JP. A role for flexible loops in enzyme catalysis. Curr Opin Struct Biol. 2010;20:702–710. doi: 10.1016/j.sbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risueno RM, Gil D, Fernandez E, Sanchez-Madrid F, Alarcon B. Ligand-induced conformational change in the T-cell receptor associated with productive immune synapses. Blood. 2005;106:601–608. doi: 10.1182/blood-2004-12-4763. [DOI] [PubMed] [Google Scholar]
- Reichheld SE, Yu Z, Davidson AR. The induction of folding cooperativity by ligand binding drives the allosteric response of tetracycline repressor. Proc Natl Acad Sci USA. 2009;106:22263–22268. doi: 10.1073/pnas.0911566106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turoverov KK, Kuznetsova IM, Uversky VN. The protein kingdom extended: ordered and intrinsically disordered proteins, their folding, supramolecular complex formation, and aggregation. Prog Biophys Mol Biol. 2010;102:73–84. doi: 10.1016/j.pbiomolbio.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P. Unstructural biology coming of age. Curr Opin Struct Biol. 2011;21:419–425. doi: 10.1016/j.sbi.2011.03.012. [DOI] [PubMed] [Google Scholar]
- Larion M, Salinas RK, Bruschweiler-Li L, Miller BG, Bruschweiler R. Order-disorder transitions govern kinetic cooperativity and allostery of monomeric human glucokinase. PLoS Biol. 2012;10:e1001452. doi: 10.1371/journal.pbio.1001452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilser VJ, Thompson EB. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc Natl Acad Sci USA. 2007;104:8311–8315. doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DF, Campbell AM. The birA gene of Escherichia coli encodes a biotin holoenzyme synthetase. J Mol Biol. 1981;146:451–467. doi: 10.1016/0022-2836(81)90042-5. [DOI] [PubMed] [Google Scholar]
- Barker DF, Campbell AM. Genetic and biochemical characterization of the birA gene and its product: evidence for a direct role of biotin holoenzyme synthetase in repression of the biotin operon in Escherichia coli. J Mol Biol. 1981;146:469–492. doi: 10.1016/0022-2836(81)90043-7. [DOI] [PubMed] [Google Scholar]
- Cronan JE., Jr The E. coli bio operon: transcriptional repression by an essential protein modification enzyme. Cell. 1989;58:427–429. doi: 10.1016/0092-8674(89)90421-2. [DOI] [PubMed] [Google Scholar]
- Xu Y, Beckett D. Kinetics of biotinyl-5′-adenylate synthesis catalyzed by the Escherichia coli repressor of biotin biosynthesis and the stability of the enzyme-product complex. Biochemistry. 1994;33:7354–7360. doi: 10.1021/bi00189a041. [DOI] [PubMed] [Google Scholar]
- Weaver LH, Kwon K, Beckett D, Matthews BW. Competing protein:protein interactions are proposed to control the biological switch of the E. coli biotin repressor. Protein Sci. 2001;10:2618–2622. doi: 10.1110/ps.32701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagautdinov B, Matsuura Y, Bagautdinova S, Kunishima N. Protein biotinylation visualized by a complex structure of biotin protein ligase with a substrate. J Biol Chem. 2008;283:14739–14750. doi: 10.1074/jbc.M709116200. [DOI] [PubMed] [Google Scholar]
- Eisenstein E, Beckett D. Dimerization of the Escherichia coli biotin repressor:corepressor function in protein assembly. Biochemistry. 1999;38:13077–13084. doi: 10.1021/bi991241q. [DOI] [PubMed] [Google Scholar]
- Streaker ED, Beckett D. Coupling of protein assembly and DNA binding:biotin repressor dimerization preceeds biotin operator binding. J Mol Biol. 2003;325:937–948. doi: 10.1016/s0022-2836(02)01308-6. [DOI] [PubMed] [Google Scholar]
- Streaker ED, Gupta A, Beckett D. The biotin repressor: thermodynamic coupling of corepressor binding, protein assembly, and sequence-specific DNA binding. Biochemistry. 2002;41:14263–14271. doi: 10.1021/bi0203839. [DOI] [PubMed] [Google Scholar]
- Wilson KP, Shewchuk LM, Brennan RG, Otsuka AJ, Matthews BW. Escherichia coli biotin holoenzyme ynthetase/bio repressor crystal structure delineates the biotin- and DNA- binding domains. Proc Natl Acad Sci USA. 1992;89:9257–9261. doi: 10.1073/pnas.89.19.9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver LH, Kwon K, Beckett D, Matthews BW. Corepressor-induced organization and assembly of the biotin repressor: a model for allosteric activation of a transcriptional regulator. Proc Natl Acad Sci USA. 2001;98:6045–6050. doi: 10.1073/pnas.111128198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood Z, Weaver LH, Brown PH, Beckett D, Matthews BW. Co-repressor induced order and biotin repressor dimerization: a case for divergent followed by convergent evolution. J Mol Biol. 2006;357:509–523. doi: 10.1016/j.jmb.2005.12.066. [DOI] [PubMed] [Google Scholar]
- Brown PH, Cronan JE, Grotli M, Beckett D. The biotin repressor: modulation of allostery by corepressor analogs. J Mol Biol. 2004;337:857–869. doi: 10.1016/j.jmb.2004.01.041. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL molecular graphics system. Schrödinger, LLC: 2002. , Version 1.2r3pre. [Google Scholar]
- Pendini NR, Yap MY, Traore DA, Polyak SW, Cowieson NP, Abell A, Booker GW, Wallace JC, Wilce JA, Wilce MC. Structural characterization of Staphylococcus aureus biotin protein ligase and interaction partners: an antibiotic target. Protein Sci. 2013;22:762–773. doi: 10.1002/pro.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Akhter Y, Wilmanns M, Ehebauer MT. Active site conformational changes upon reaction intermediate biotinyl-5′-AMP binding in biotin protein ligase from Mycobacterium tuberculosis. Protein Sci. 2014;23:932–939. doi: 10.1002/pro.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim WA, Sauer RT. Alternative packing arrangements in the hydrophobic core of lambda repressor. Nature. 1989;339:31–36. doi: 10.1038/339031a0. [DOI] [PubMed] [Google Scholar]
- Lim WA, Sauer RT. The role of internal packing interactions in determining the structure and stability of a protein. J Mol Biol. 1991;219:359–376. doi: 10.1016/0022-2836(91)90570-v. [DOI] [PubMed] [Google Scholar]
- Katz B, Kossiakoff AA. Crystal structures of subtilisin BPN' variants containing disulfide bonds and cavities: concerted structural rearrangements induced by mutagenesis. Proteins. 1990;7:343–357. doi: 10.1002/prot.340070406. [DOI] [PubMed] [Google Scholar]
- Sondek J, Shortle D. Accommodation of single amino acid insertions by the native state of staphylococcal nuclease. Proteins. 1990;7:299–305. doi: 10.1002/prot.340070402. [DOI] [PubMed] [Google Scholar]
- Lesk AM, Chothia C. How different amino acid sequences determine similar protein structures: the structure and evolutionary dynamics of the globins. J Mol Biol. 1980;136:225–270. doi: 10.1016/0022-2836(80)90373-3. [DOI] [PubMed] [Google Scholar]
- Chen J, Stites WE. Energetics of side chain packing in staphylococcal nuclease assessed by systematic double mutant cycles. Biochemistry. 2001;40:14004–14011. doi: 10.1021/bi011268l. [DOI] [PubMed] [Google Scholar]
- Hurley JH, Baase WA, Matthews BW. Design and structural analysis of alternative hydrophobic core packing arrangements in bacteriophage T4 lysozyme. J Mol Biol. 1992;224:1143–1159. doi: 10.1016/0022-2836(92)90475-y. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010;38:W695–699. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganathan S, Beckett D. Nucleation of an allosteric response via ligand-induced loop folding. J Mol Biol. 2007;373:96–111. doi: 10.1016/j.jmb.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PH, Beckett D. Use of binding enthalpy to drive an allosteric transition. Biochemistry. 2005;44:3112–3121. doi: 10.1021/bi047792k. [DOI] [PubMed] [Google Scholar]
- Xu Y, Nenortas E, Beckett D. Evidence for distinct ligand-bound conformational states of the multifunctional Escherichia coli repressor of biotin biosynthesis. Biochemsitry. 1995;34:16624–16631. doi: 10.1021/bi00051a010. [DOI] [PubMed] [Google Scholar]
- Kwon K, Beckett D. Function of a conserved sequence motif in biotin holoenzyme synthetases. Protein Sci. 2000;9:1530–1539. doi: 10.1110/ps.9.8.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buoncristiani MR, Howard PK, Otsuka AJ. DNA-binding and enzymatic domains of the bifunctional biotin operon repressor (BirA) of Escherichia coli. Gene. 1986;44:255–261. doi: 10.1016/0378-1119(86)90189-7. [DOI] [PubMed] [Google Scholar]
- Lane MD, Rominger KL, Young DL, Lynen F. The enzymatic synthesis of holotranscarboxylase from apotranscarboxylase and (+)-biotin. II. Investigation of the reaction mechanism. J Biol Chem. 1964;239:2865–2871. [PubMed] [Google Scholar]
- Abbott J, Beckett D. Cooperative binding of the Escherichia coli repressor of biotin biosynthesis to the biotin operator sequence. Biochemistry. 1993;32:9649–9456. doi: 10.1021/bi00088a017. [DOI] [PubMed] [Google Scholar]
- Johnson ML, Correia JJ, Yphantis DA, Halvorson HR. Analysis of data from the analytical ultracentrifuge by nonlinear least-squares techniques. Biophys J. 1981;36:575–588. doi: 10.1016/S0006-3495(81)84753-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information