

Abstract
Viral proteins bind to numerous cellular and viral proteins throughout the infection cycle. However, the mechanisms by which viral proteins interact with such large numbers of factors remain unknown. Cellular proteins that interact with multiple, distinct partners often do so through short sequences known as molecular recognition features (MoRFs) embedded within intrinsically disordered regions (IDRs). In this study, we report the first evidence that MoRFs in viral proteins play a similar role in targeting the host cell. Using a combination of evolutionary modeling, protein–protein interaction analyses and forward genetic screening, we systematically investigated two computationally predicted MoRFs within the N-terminal IDR of the hepatitis C virus (HCV) Core protein. Sequence analysis of the MoRFs showed their conservation across all HCV genotypes and the canine and equine Hepaciviruses. Phylogenetic modeling indicated that the Core MoRFs are under stronger purifying selection than the surrounding sequence, suggesting that these modules have a biological function. Using the yeast two-hybrid assay, we identified three cellular binding partners for each HCV Core MoRF, including two previously characterized cellular targets of HCV Core (DDX3X and NPM1). Random and site-directed mutagenesis demonstrated that the predicted MoRF regions were required for binding to the cellular proteins, but that different residues within each MoRF were critical for binding to different partners. This study demonstrated that viruses may use intrinsic disorder to target multiple cellular proteins with the same amino acid sequence and provides a framework for characterizing the binding partners of other disordered regions in viral and cellular proteomes.
Keywords: virus, viral protein, hepatitis C virus, hepatitis virus, host–pathogen interactions, protein–protein interactions, intrinsically disordered proteins, systems biology
Introduction
Viruses interface with their host cell through physical interactions between viral and host proteins. The consequences of these interactions are diverse and contribute to changes in host cell behavior that produce an environment conducive to viral replication. In recent years, virus–host protein interaction networks have been described for many viruses. Although these studies demonstrated that even small viral proteins can interact with large numbers of cellular proteins, it remains unclear how viral proteins are able to participate in so many interactions.
Observations from cellular proteomes suggest that intrinsically disordered regions (IDRs) within proteins can facilitate interactions with multiple partners. Cellular proteins that interact with a large number of partners, known as “hubs”, are often partially or completely disordered.1–3 Many of these hub proteins function in cell signaling networks that require them to integrate upstream signals and regulate the activities of their downstream targets through numerous physical interactions. The binding diversity conferred by IDRs is critical to the topology and function of cellular signaling networks as it permits one-to-many binding.1,4–7 This is exemplified in the well-characterized tumor suppressor protein p53, a key signaling protein with 100 confirmed cellular binding partners, many of which bind its disordered N-terminal domain.3,7–10 A recent survey of existing protein structures identified over 25 other examples of cellular hubs that interact with multiple partners, suggesting disorder is a common strategy by which proteins maintain multiple partners.4
Much like hubs in cellular protein interaction networks, viral proteins establish themselves as global regulators of cellular function during infection, transforming the phenotype of the cell to create an environment conducive to viral replication. In RNA viruses, such as hepatitis C virus (HCV), where genetic capacity is limited, viral proteins must execute multiple functions in addition to their primary roles in virus replication or production. These functions often require physical interaction with other viral or host factors. One-to-many binding via viral IDRs would allow many virus–host interactions, and associated functions, to be compressed into small, overlapping regions of primary structure. This seems especially plausible as viral proteins exhibit higher levels of structural disorder and a higher density of linear motifs predicted to mediate protein–protein interactions compared to cellular proteins.11–24 However, the role of intrinsic disorder in mediating one-to-many binding between viral and host proteins has not been systematically investigated.
Binding via IDRs is frequently achieved through local segments that undergo disorder-to-order transitions upon forming complexes with their protein partners.25–29 IDR segments of 10–25 residues that become structured upon binding have been termed molecular recognition features (MoRFs).30 When unbound, MoRFs exist in a predominantly disordered state, exploring an ensemble of conformations. The bound conformation(s) of MoRFs depends on the contacts available in both partners.4 Although, binding can stabilize a single conformation,3,4,27–29,31 in some cases subregions of the IDR remain disordered, leading to formation of a “fuzzy” complex.27,32,33 The outsourcing of structural information to the binding partners allows the MoRF to be multispecific and to attain distinct, functional conformations specific to each binding partner.4,7,34
Although, bioinformatic studies and retrospective structural analyses have increased our appreciation of the ubiquity of intrinsic disorder in cellular4,13,35–39 and viral12–17,22,40 proteomes, the validation of predicted IDRs and MoRFs is critical to understanding their function. In this study, we report experimental characterization of the cellular binding partners of two computationally predicted MoRF regions within the N-terminal IDR of the HCV Core protein. Core binds to and packages the HCV RNA genome, and then interacts with the ER-associated viral glycoproteins, E1 and E2, to assist in formation of the mature viral particle.41 Two domains have been identified in HCV Core, each of which plays distinct roles in the HCV life cycle. The N-terminal domain, domain 1 (DI), is highly basic and hydrophilic.42 It mediates homotypic interactions, binds to the viral RNA genome during assembly of HCV particles, and is sufficient for nucleocapsid formation in vitro.43,44 In addition to its role in RNA encapsidation, HCV Core DI is essential for RNA chaperone activities that may guide the formation of specific structures of the HCV genome.45,46 Although a majority of Core is found in the cytoplasm, a subpopulation is targeted to the nucleolus, most likely via one or more nuclear localization signals present in DI.47,48 Core domain 2 (DII) constitutes the C-terminal, lipid droplet-associated domain of Core.49 It consists of two amphipathic helices that anchor Core to the surface of cytoplasmic lipid droplets before assembly.42,50,51
Using a combination of IDR and MoRF prediction, sequence analysis, and phylogenetic hypothesis testing, we identified HCV MoRFs that are under stronger purifying selection than the surrounding sequence, indicating important biological roles for these regions. Using the yeast two-hybrid (Y2H) assay along with forward genetics and site-directed mutagenesis, we demonstrated that the two predicted α-MoRFs in HCV Core were essential for binding to multiple unrelated host factors. The approach described in this study can be integrated into large-scale validation pipelines for computationally predicted MoRFs, leading to a better understanding of the diversity, function, and therapeutic potential of these important features of viral and cellular proteomes.
Results
Prediction of intrinsic disorder and MoRFs in HCV
To identify IDRs and MoRFs within the HCV polyprotein, we used the predictors of naturally disordered regions (PONDR®), PONDR®-VLXT52 and the “meta-predictor”, PONDR®-FIT.53 The PONDR®-FIT meta-predictor combines three predictors from the PONDR® family with three additional predictors of disorder, namely IUPred,54 FoldIndex,55 and TOP-IDP.56 This meta-predictor performs better than any of the individual predictors, providing one of the best predictions of disorder across a protein sequence.53 PONDR®-VLXT yields better sensitivity in predicting alpha-helical propensity and, in combination with the α-MoRF-Pred computational tool, can identify putative α-MoRFs.30 Although, several additional predictors have been developed to identify likely MoRFs within longer regions of disorder, we chose this particular predictor because of previous success with this approach. Additional discussion of the IDR and MoRF predictors can be found in Supporting Information.
These algorithms predicted increased disorder within the N-terminal 120 residues of the HCV Core protein and the C-terminal 280 residues of NS5A [Fig. 1(A)]. Two potential α-MoRFs were identified in the N-terminal IDR of Core (positions 21–38 and 82–99; Core MoRFs 1 and 2, respectively; residue positions based on HCV JFH-1 strain) and five putative α-MoRFs were found in the C-terminal IDR of NS5A (positions 200–217, 242–259, 306–323, 361–378, and 449–466; NS5A MoRFs 1–5, respectively) [Fig. 1(B–D)]. The PONDR® predictions of IDRs and MoRFs in Core and NS5A are consistent with previous bioinformatic and experimental analyses.12,16,42,57–61
Figure 1.

Sequence-based prediction of intrinsic disordered regions (IDRs) in the HCV polyprotein. (A) The PONDR®-FIT meta-predictor was used to estimate the level of disorder in the HCV JFH1 polyprotein. Higher PONDR scores indicate a higher propensity to be unstructured. Bar above the graph represents the HCV polyprotein. (B) PONDR®-VLXT scores were used along with the α-MoRF-pred tool to identify α-helical Molecular Recognition Features (α-MoRFs) in the HCV JFH1 polyprotein sequence. Grey blocks indicate the positions of predicted α-MoRFs. (C and D) PONDR®-FIT and –VLXT scores for HCV Core (C) and NS5A (D). Schematics of the Core and NS5A proteins are shown above the graphs. Brackets indicate domains.132
The features corresponding to Core MoRF 1 and 2 were conserved in all human HCV genotypes and in nonprimate Hepacivirus (NPHV) isolates from equine (AFJ20706)62 and canine hosts (AEC45560)63 but not in isolates from bat or rodent Hepaciviruses or GB virus B [Fig. 2(A)], suggesting that these elements emerged after the divergence of these more distantly related viral lineages. Features corresponding to the NS5A MoRFs were conserved among HCV genotypes but were not detected in more distantly related NS5A proteins [Fig. 2(B)]. Using Bayesian phylogenetic inference to estimate the site-specific ratios of nonsynonymous and synonymous substitution rates (dN/dS), we found that Core MoRFs 1 and 2 and NS5A MoRFs 1 and 3 had significantly lower dN/dS values than the surrounding IDRs [P < 0.01 for each, Tables1 and 2 and Fig. 2(C,D)]. This indicates that these MoRFs have undergone purifying selection and strongly suggests that they have a functional role in HCV replication. Further discussion of the phylogenetic analysis can be found in Supporting Information.
Figure 2.

The predicted MoRFs in Core and NS5A are conserved and are undergoing purifying selection. (A and B) Phylogenetic relationship (left) and PONDR scores (right) of Hepacivirus Core (A) and NS5A protein sequences (B). PONDR®-VLXT scores for each protein sequence are represented as heatmaps and aligned according to MUSCLE alignments of the same proteins. Predicted α-MoRF regions are shown as grey blocks. Thin black lines indicate gaps in the alignments. (C and D) Boxplots of the mean dN/dS ratios for MoRF and non-MoRF sequence partitions estimated from 86 Core-coding sequences (C) and 76 NS5A sequences (D) representing HCV genotypes 1–7. Dashed line indicates dN/dS = 1, or neutral selection.
Table 1.
Calculated dN/dS Values, Credible Intervals (CI) and Significance Levels for HCV Core Sequence Partitions
| Core partition | Median dN/dS | 95% CI | 99% CI | Significance | |
|---|---|---|---|---|---|
| MoRF1 | 0.37 | 0.28–0.51 | 0.25–0.61 | * | vs. Non-MoRF IDR |
| MoRF2 | 0.39 | 0.31–0.54 | 0.29–0.64 | * | vs. Non-MoRF IDR |
| Non-MoRF IDR | 1.24 | 1.00–1.53 | 0.93–1.67 | ||
| DI (IDR) | 0.97 | 0.80–1.19 | 0.75–1.30 | ||
| DII | 0.57 | 0.48–0.69 | 0.45–0.74 | * | vs. IDR |
| Core | 0.82 | 0.69–0.97 | 0.65–1.05 | ||
Indicates P < 0.01.
Table 2.
Calculated dN/dS Values, Credible Intervals (CI) and Significance Levels for HCV NS5A Sequence Partitions
| NS5A partition | Median dN/dS | 95% CI | 99% CI | Significance | |
|---|---|---|---|---|---|
| MoRF1 | 0.63 | 0.54–0.74 | 0.51–0.80 | * | vs. Non-MoRF IDR |
| MoRF2 | 0.86 | 0.78–0.96 | 0.75–1.00 | ns | vs. Non-MoRF IDR |
| MoRF3 | 0.56 | 0.49–0.63 | 0.47–0.67 | * | vs. Non-MoRF IDR |
| MoRF4 | 1.2 | 1.09–1.32 | 1.04–1.38 | ns | vs. Non-MoRF IDR |
| MoRF5 | 0.87 | 0.78–0.96 | 0.75–1.01 | ns | vs. Non-MoRF IDR |
| Non-MoRF IDR | 0.94 | 0.89–0.99 | 0.87–1.01 | ||
| DII/DIII (IDR) | 0.9 | 0.86–0.94 | 0.84–0.96 | ||
| DI | 0.67 | 0.64–0.71 | 0.63–0.73 | * | vs. DII/DIII (IDR) |
Indicates P < 0.01.
Identification of interacting partners of HCV MoRFs
Because regions of proteins that mediate protein–protein interactions have higher rates of purifying selection64–67 and no other functions have been described for the HCV MoRFs, we tested these sequences for interactions with cellular proteins. Seven Y2H constructs that included a single HCV MoRF and the flanking IDR sequence were used to screen a human liver cDNA library as a part of a previously reported large-scale Y2H screen for HCV–human interactions.68 The MoRF-containing fragments of the NS5A IDR were either self-activators or did not yield any reproducible interactions. However, screens using the two MoRF-containing fragments of HCV Core identified three cellular binding partners for each fragment [Fig. 3(A)]. Core fragment 1 (CF1) interacted with the cellular proteins DDX3X, NPM1, and TRIP11, whereas Core fragment 2 (CF2) interacted with GLRX5, EIF3L, and RILPL2 (see Supporting Information Table SI for the functions of each human protein binding partner of the Core MoRFs). All six interactions were confirmed by recloning the human gene fragment in freshly transformed yeast and retesting in pairwise Y2H assays. Four of the six interactions (DDX3X, NPM1, TRIP11, and RILPL2) were confirmed by in vitro split-luciferase assays [Fig. 3(B)]. It is not clear if the other two pairs represent false-positives in the Y2H assay or false-negatives in the split-luciferase assay; all protein–protein interaction assays appear to have high false-negative rates based on systematic comparisons with a gold standard set of interactions.69,70 Two of the cellular proteins identified in our screen, DDX3X71–76 and NPM1,77 were previously shown to interact with the N-terminus of Core and to co-localize with Core in human cells. RILPL2 and DDX3X are required for HCV replication.68,71,74,75,78
Figure 3.
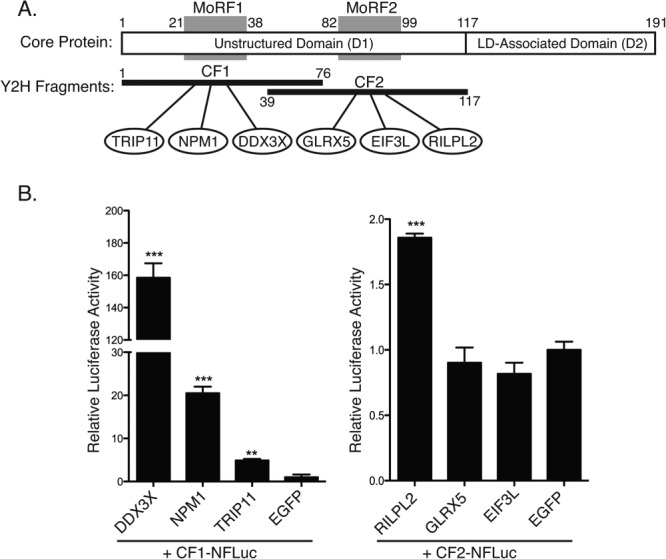
MoRF-containing fragments of HCV 2a Core protein mediate interactions with cellular proteins. (A) Yeast two-hybrid screening was used to identify cellular binding partners of HCV Core protein. The fragments used in Y2H screening (black bars, below) contain individual predicted α-MoRFs (grey blocks). Numbers indicate residue positions defining predicted α-MoRFs and Y2H fragments. Three cellular binding partners were found for each Core fragment (ovals). (B) Four of the six interactions were confirmed by split-luciferase assay. *, **, *** indicate P < 0.05, <0.005, and <0.0005, respectively, based on a one-tailed t-test (n = 3). The amounts of N-FLuc and C-FLuc fusion proteins were normalized to their respective GFP negative controls (Supporting Information Fig. S1).
Cellular proteins bind to core within the predicted MoRFs
To determine if the predicted MoRFs mediated binding to the cellular proteins, we used a forward genetic screen with a library of mutant Core fragments generated by error-prone PCR. A total of 17 variants of CF1 and 19 variants of CF2 were tested for interaction with each human partner in the Y2H assay (Fig. 4). Of the 36 variants, 33 had single amino acid substitutions and three had two substitutions. Five single amino acid substitutions fell within MoRF1 and seven were located in MoRF2.
Figure 4.
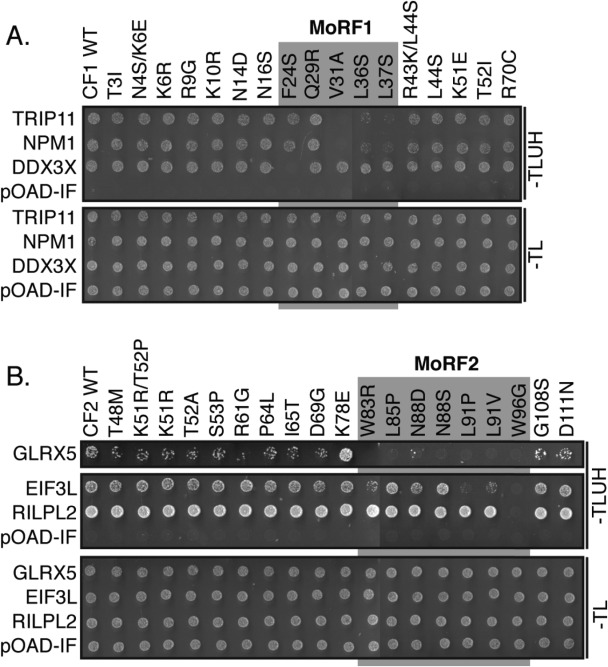
Cellular proteins bind to the predicted Core α-MoRF regions. Single or double residue substitutions were introduced into fragments containing Core MoRF1 (A) and 2 (B) by error-prone PCR. The effect of these substitutions on binding to cellular proteins was then assessed using the Y2H assay. Substitutions in the predicted α-MoRF regions (grey blocks) preferentially disrupted binding. pOAD-IF is a control plasmid, in which URA3 is in frame with the GAL4 activation domain and no human gene is present. –TLUH indicates Y2H selection medium lacking tryptophan, leucine, uracil, and histidine; –TL, growth control medium lacking tryptophan and leucine. All interactions were assessed in independent duplicate experiments; representative results are shown in this study.
All four variants in CF1 that displayed reduced binding to at least one partner had substitutions within MoRF1 (residues 21–38) [Fig. 4(A)]. Using a one-tailed Fisher's exact test to determine the likelihood of this result occurring by chance, we determined a P-value of 2.1 × 10−3. Similarly, 8 of the 19 variants in CF2 had reduced binding to at least one cellular binding partner [Fig. 4(B)]. All seven substitutions within MoRF2 (residues 82–99) disrupted binding to at least one cellular protein, whereas only one substitution outside of MoRF2 (position 61) reduced binding to GLRX5 specifically (P = 2.0 × 10−4). These results strongly suggest the Core MoRFs represent the binding sites for the cellular interacting proteins identified in this study.
Core–cellular protein interactions are mediated by distinct residues
After establishing the requirement of the MoRF regions in host factor binding, we proceeded to map the binding sites of each human partner using site-directed mutagenesis. Our mutational analysis initially focused on conserved positions required for the interaction between Core and DDX3X (F24, G27, and Y35).71 Alanine substitutions were introduced at these positions and assessed for their impact on binding to each host factor by Y2H [Fig. 5(A)]. Each variant was expressed at levels similar to the wild type protein by western blotting [Fig. (5B)]. As previously reported,71 all three substitutions disrupted the interaction of CF1 and DDX3X. The substitution of the highly conserved tyrosine residue at position 35 in MoRF1 also disrupted the interaction with NPM1 and TRIP11. However, alanine substitutions at positions 24 and 27 were not broadly disruptive. The F24A substitution affected binding to DDX3X only, whereas the G27A substitution disrupted binding to TRIP11 and DDX3X but not NPM1.
Figure 5.
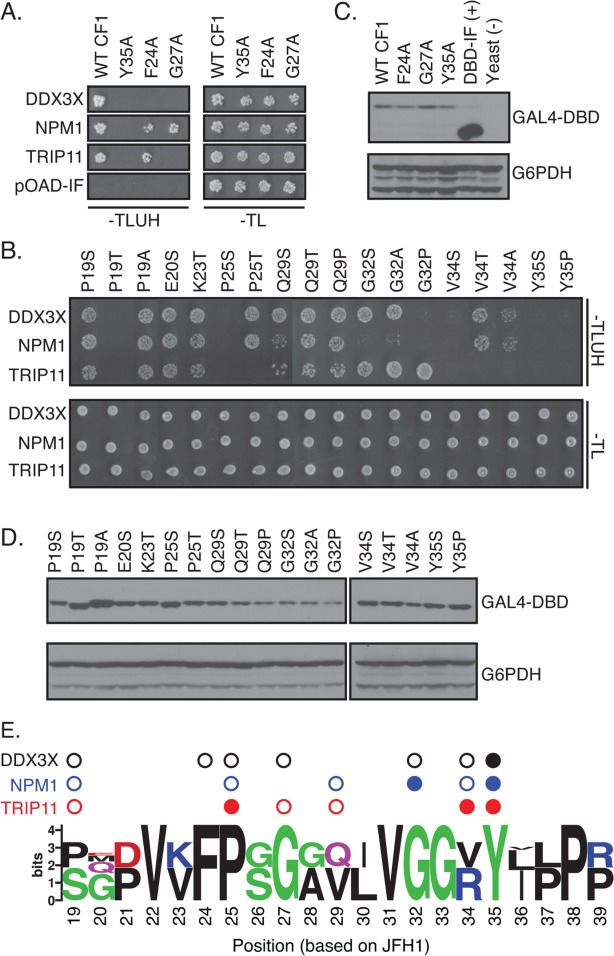
Distinct amino acids in MoRF1 are required for binding to cellular proteins. (A and B) Single residue substitutions in Core MoRF1 disrupt the binding to cellular proteins. (A) Substitutions previously shown to disrupt binding of Core to DDX3X71 were assayed for their impact on binding to cellular proteins using the Y2H assay. Top panel shows growth on Y2H selection medium whereas bottom panel shows growth on control medium. pOAD-IF is a control plasmid, in which URA3 is in frame with the GAL4 activation domain and no human gene is present. (B) Single residue substitutions were introduced into Core MoRF1 by site-directed mutagenesis and tested for their effect on binding to cellular proteins using the Y2H assay. Top panel, Y2H selection medium; bottom panel; growth control medium. (C and D) Western blots to confirm expression of mutant Core fragments. Upper blot was probed with anti-GAL4 DNA binding domain antibody (GAL4-DBD); lower blot was probed with anti-glucose-6-phosphate dehydrogenase antibody (G6PDH) as a loading control. (E) Summary of the effect of HCV Core MoRF1 substitutions on binding to human proteins. Web Logo of Core MoRF1 was derived from alignment of the six HCV genotypes and eight representative NPHV isolates. Circles indicate substitutions that disrupted binding to DDX3X (black), NPM1 (blue), or TRIP11 (red). Shaded circles indicate multiple substitutions at that position disrupted binding. All experiments were performed at least twice in independent replicates; representative results are shown.
Because the random mutagenesis and site-directed alanine mutagenesis of MoRF1 identified distinct patterns of disruption for the individual binding partners of MoRF1, we performed a more extensive mutagenesis of the MoRF1 region. To maximize the diversity in our library of mutants, we used degenerate primers to introduce multiple substitutions at individual positions across the MoRF1 and tested them for interactions with cellular partners in the Y2H assay [Fig. 5(B)]. All mutant proteins were expressed at similar levels [Fig. 5(D)]. As with the random mutagenesis experiment, distinct patterns of disruption were observed for each binding partner, suggesting that either distinct contacts or unique folds were required for interaction with each host factor. Although the substitutions P19T, P25S, V34S, Y35S, and Y35P disrupted the interactions with all of the human binding partners of MoRF1, four other substitutions disrupted the binding of only one or two proteins. Substitutions at positions 31, 36, and 37 disrupted interaction with TRIP11 and NPM1 but not DDX3X, whereas the F24A substitution only disrupted the interaction with DDX3X. Interestingly, substitution of proline residues at position 19 and 25 with either serine or threonine residues resulted in markedly different interaction patterns. There was no obvious bias of the broadly disruptive mutations toward more highly conserved amino acids [Fig. 5(E)]
In contrast to MoRF1, little has been performed to characterize the MoRF2 region. Therefore, we performed dual-alanine scanning mutagenesis to map the residue positions within MoRF2 required for binding. This screen identified six pairs of amino acids that, when substituted with alanine, disrupted binding to one or more of the three host factors that interacted with CF2 [Fig. 6(A)]. We then introduced single alanine substitutions at these positions to map the binding sites in higher resolution [Fig. 6(B)]. All MoRF2 variant were detected by western blotting, though some (e.g., P82A, N88A, and E89A) were expressed at slightly lower levels [Fig. 6(C)]. However, this level of expression was sufficient to enable the interaction with RILPL2 to be detected at levels indistinguishable from wild type. The conserved tryptophan residues at positions 93 and 96 were critical for binding to all three host factors [Fig. 6(D)]. These residues are part of a larger motif, GWAGWLxSP, present in all HCV genotypes. Intriguingly, these tryptophan residues are found within a similar but inverted sequence in the NPHVs, PxLQWxAWG (Supporting Information Fig. S2). Substitutions across MoRF2 disrupted binding with GLRX5. However, this interaction was weaker in the Y2H assay than the other pairs, which may have caused increased sensitivity to subtle variations in binding affinity or the level of protein expression of the variants.
Figure 6.
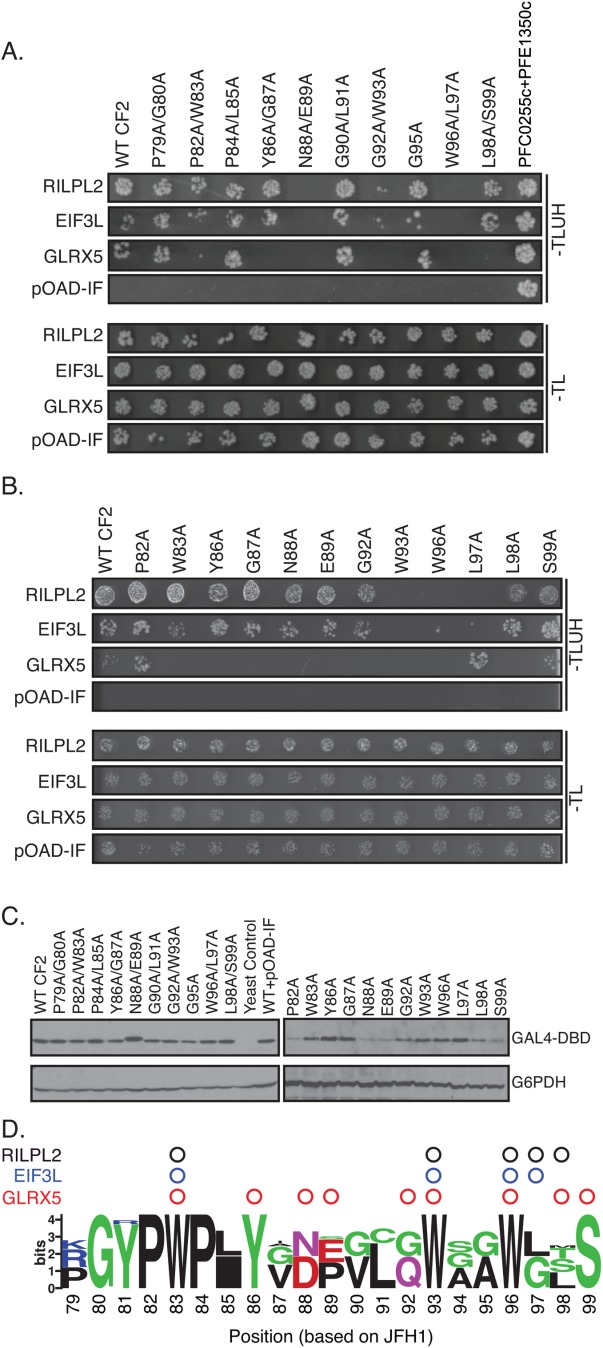
Distinct amino acids in MoRF2 are required for binding to cellular proteins. (A and B) Amino acid substitutions in Core MoRF2 disrupt the binding to cellular proteins. (A) Dual alanine substitutions were introduced into Core MoRF2 and assayed for their effect on binding to cellular proteins using the Y2H assay. Top and bottom panels show growth on Y2H selection medium and growth control media, respectively. pOAD-IF is a control plasmid in which URA3 is in frame with the GAL4 activation domain and no human gene is present. PFC0255c+PFE1350c is pair of interacting Plasmodium falciparum proteins that serve as a positive control. (B) Single alanine substitutions were generated for each dual alanine substitution that disrupted binding to at least one cellular protein and tested for their impact on binding to cellular proteins using the Y2H assay. (C) Western blots to confirm expression of mutant Core fragments. Upper blot was probed with anti-GAL4 DNA binding domain antibody (GAL4-DBD); lower blot was probed with anti-glucose-6-phosphate dehydrogenase antibody (G6PDH) as a loading control. (D) Summary of the effect of HCV Core MoRF2 substitutions on binding to human proteins. Web Logo of Core MoRF2 was derived from alignment of the six HCV genotypes and eight representative NPHV isolates. Circles indicate substitutions that disrupted binding to RILPL2 (black), EIF3L (blue), or GLRX5 (red). All experiments were performed at least twice in independent replicates; representative results are shown.
In an effort to understand the impact of individual substitutions within the MoRF regions, we used the PONDR-VLXT algorithm to quantify the predicted effect of each substitution on the propensity to form an α-helix. Marked differences were noted in the effects of substitutions on MoRF1 and MoRF2 (Fig. 7). In MoRF2, the five substitutions that caused loss of interaction with all three identified binding partners were among the eight substitutions with the greatest increase in the minimum VLXT scores, suggesting that these substitutions may disrupt binding by interfering with formation of an α-helical structure [Fig. 7(B)]. Substitutions in MoRF2 that disrupted specific interactions tended to have smaller effects on the minimum VLXT score, suggesting that the propensity of MoRF2 to form an α-helix was not affected. Rather, these amino acids may form partner-specific contacts with individual human proteins that are disrupted by the substitutions. The situation with MoRF1 is more complex. There is little correlation between the changes in minimum VLXT scores and the effect of substitutions on binding to human proteins. The presence of multiple helix-breaking residues (i.e., Gly and Pro) suggests that MoRF1 may have a more complex structure than the single α-helix suggested by the computational predictions. Consistent with this suggestion, a previous NMR analysis indicated that MoRF1 can form two separate α-helices.60
Figure 7.
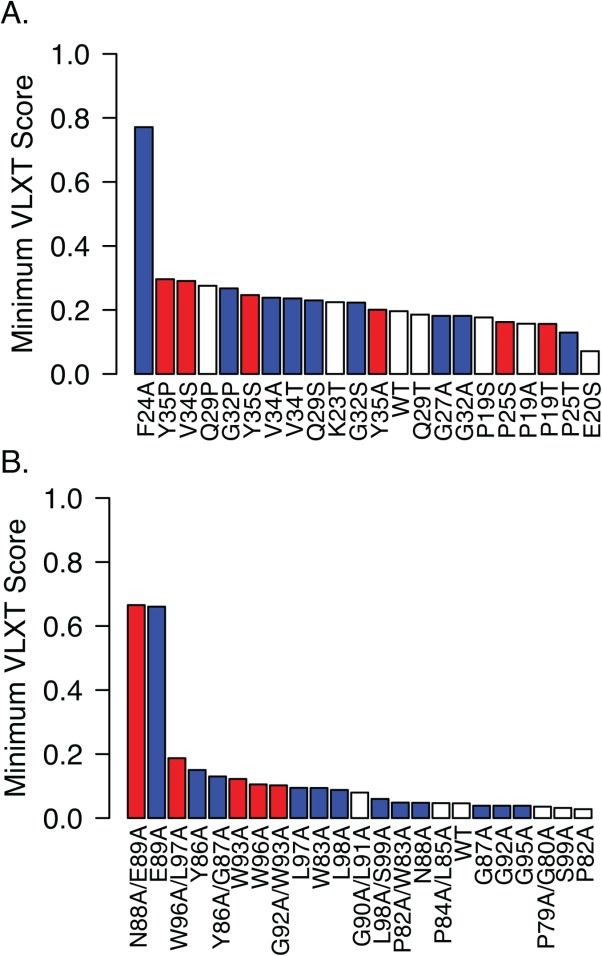
Effect of HCV Core substitutions on minimum VLXT scores. Mutant HCV core proteins from Figures 5 and 6 were analyzed for their propensity to form alpha helices using the VLXT algorithm. As an indication of the impact of the substitution on predicted helix formation, the minimum VLXT score within the predicted MoRF is plotted for each HCV Core MoRF1 (A) and Morf2 (B) variant. Substitutions that disrupted binding of all three cellular proteins (broadly disruptive) are indicated in red, whereas those that disrupted binding to one or two cellular proteins are shown in blue; substitutions that had no effect on binding are shown in white.
Discussion
IDRs often contain functional modules that mediate interactions with partner proteins.79 These features are common in cellular hub proteins and influence both the organization and function of cellular proteomes.1 Hubs can use the same region of disorder to bind to many different partners or, alternatively, can bind to many partners that contain disordered regions.7 Viral proteins, many of which act as hubs, are enriched in IDRs and predicted functional modules, suggesting that viral IDRs may mediate important protein–protein interactions with host proteins during infection.19,22,24
In this study, we tested the hypothesis that viral IDRs mediate one-to-many binding by mapping virus–host protein interactions of the computationally predicted MoRFs in HCV Core. These regions are dispensable for HCV RNA binding46 and formation of virus-like particles in vitro,80 but are conserved among distantly related Hepaciviruses and are undergoing higher rates of purifying selection than the surrounding IDRs [Fig. 2(C)]. Intriguingly, the flanking regions of the IDR that mediate interactions with the viral RNA are conserved among HCV and GB viruses, whereas portions of Core corresponding to the HCV MoRFs are absent from the GB virus Core protein,42 which emphasizes the modular nature of the Core MoRFs. Because the MoRF regions are dispensable for RNA binding and homotypic interactions, we hypothesized that the increased evolutionary constraint was due to their role in mediating protein–protein interactions, as demonstrated previously.64–67 Indeed, using a combination of bioinformatic, genetic, and computational tools, we identified three cellular proteins that bind to each of the Core MoRFs, two of which, DDX3X and RILPL2, are required for viral replication. These findings demonstrate that molecular recognition via distinct functional modules within IDRs is one strategy that enables viral proteins to interact with multiple viral and host proteins.
MoRFs are 10–25 amino acid segments within IDRs that have a higher likelihood of forming secondary structure than the surrounding IDR.30 However, in their unbound state they remain flexible and explore an ensemble of conformations. Only in their bound state do MoRFs achieve their “functional structure,” which is guided by contacts available in each partner. In the current study, we used computational predictors to identify MoRFs that tend to form α-helices.30,81 Consistent with these predictions, NMR studies demonstrated that HCV MoRF1 formed two α-helices in high concentrations of trifluoroethanol, which stabilizes local secondary structures.60 Although this result indicates that Core MoRF1 is capable of forming α-helices, it remains to be determined if the conformers identified in the presence of trifluoroethanol are also formed by MoRF1 in its bound state(s). In some cases, the same MoRF can form different secondary structures when bound to different partners.4,7
The complexity of the results from our mutational analysis [summarized in Figs. 5(E) and 6(D)] is consistent with the potential for MoRF1 to form diverse structures. Mutagenesis experiments showed that distinct residues in the MoRFs mediate binding to the individual cellular partners. In MoRF1, substitutions that disrupted the interaction with DDX3X and NPM1 were distinct from those that interfered with binding to TRIP11. Similarly, substitutions in MoRF2 that abrogated the interaction with GLRX5 were different from those that blocked binding to RILPL2 and EIF3L. These findings suggest that the predicted Core MoRFs bind to cellular partners through distinct amino acids and possibly distinct conformations, similar to cellular hub proteins that participate in one-to-many binding.4,7
Overall, our data is consistent with an analysis of amino acid substitutions that enhanced or disrupted the interaction between measles virus nucleoprotein (N) and the X domain of the viral phosphoprotein.82 Measles virus N interacts with the X domain via sequences within an IDR that become helical upon binding.83–86 All amino acid changes introduced into this region by random mutagenesis reduced binding to the X domain, suggesting that this sequence has been optimized for this interaction.82 Similarly, we show that the analogous regions in HCV Core—MoRFs 1 and 2—are undergoing purifying selection and that mutations within these regions preferentially disrupted interactions with cellular binding partners.
Intrinsic disorder is common in viral proteins, but the extent of disordered residues varies widely.14,19,87 Although viruses that have small genomes or that encode polyproteins tend to have more disorder, the best predictor of the extent of protein disorder in a given viral proteome is the family to which the virus belongs.19 However, even within virus families, individual species vary extensively in the amount of disorder.16,19,22,24 Our data and that of several other groups, suggest that interactions with viral and host proteins are primary roles of viral IDRs (reviewed by Ref.11). As discussed above, each HCV Core MoRF bound to three distinct cellular proteins. Given the large number of partners reported to bind to Core,88 it is likely that other proteins also bind to the Core MoRFs. Although our screens did not identify any reproducible interactions with NS5A fragments, the NS5A MoRFs have been previously shown to participate in protein–protein interactions. MoRF5 of HCV NS5A (residues 449–466) contains residues required for binding to HCV Core, suggesting that NS5A MoRF5 may mediate intraviral interactions during HCV assembly.89,90 The region of NS5A corresponding to the predicted MoRF3 (residues 306–323) binds to the cellular peptidyl-prolyl isomerase, cyclophilin A (CypA), and to HCV NS5B.59,91–93 Intriguingly, cyclosporine derivatives that block the interaction between CypA and HCV NS5A exhibit potent anti-HCV activity and have shown success in early clinical trials,94,95 suggesting that virus–host protein interactions mediated by MoRFs are potential targets for antiviral drugs.96–99 In measles viruses, a MoRF located in the C-terminal IDR of the N protein binds to both the virus P protein and to the cellular HSP70.83,100–104 It is not clear in the latter cases if binding of the viral and cellular proteins is regulated temporally or if discrete subpopulations bind to different partners. The potential for the same sequence within an IDR to engage multiple proteins provides an attractive mechanism by which the virus may compress essential functions into its limited proteome.
To date, studies on MoRFs and linear motifs have been mostly retrospective, using previously collected data to identify and characterize binding sites3,4,27–29,31,105 and to develop binding site predictors.25,81,106–112 Indeed, these efforts have yielded over 50 bioinformatic predictors of disorder (reviewed in Ref.113) and several MoRF predictors.25,30,81,108 However, few studies have integrated binding site predictions to help identify and characterize protein–protein interaction binding interfaces.26,114,115 The analytical approaches and screening procedures implemented here provide a facile means to identify MoRF-mediated interactions that can be used to evaluate current predictors and further improve their performance.
Methods
A more complete description of the methods used in this study is provided in Supporting Information.
Informatic analyses
Predictions of disorder were performed using the PONDR® VL-XT52 and PONDR®-FIT53 algorithms, which can be accessed at http://pondr.com/index.html and through the DisProt database (http://www.disprot.org/pondr-fit.php), respectively.
Sequence analysis
Core and NS5A coding sequences from HCV genotypes 1–7 were chosen from the Los Alamos National Lab HCV sequence database116 and codon-aligned using MUSCLE117 implemented in SeaView.118 Protein sequences from HCV and NPHV isolates were aligned using MUSCLE117 with standard parameter settings. Block logos and sequence logos of MoRF regions were generated from these alignments using the BlockLogo program using default settings with the option to include gaps selected.119 Site-specific dN and dS substitution rates and dN/dS ratios were estimated using the renaissance counting method implemented in the BEAST Bayesian phylogenetics framework,120–122 using a random local clock,123 the HKY85 nucleotide substitution model124 and a constant population size coalescent as a tree prior. This Bayesian approach avoids conditioning the dN/dS estimates on a single phylogeny. Instead, the algorithm samples tree space to generate a distribution of dN/dS estimates. A Markov chain of 200 million states was sampled every 20,000 iterations with a 25% burn-in period to yield 7500 tree observations. The mean dN/dS estimates for each sequence partition were used to determine the 95 and 99% credible interval of the mean partition-specific dN/dS rates.
Y2H assays
MoRF-containing fragments of the HCV JFH1 polyprotein (UNIPROT: Q99IB8) were cloned into the yeast two-hybrid DNA-binding domain (DBD) plasmid pOBD2 by homologous recombination.125 Y2H screens were performed by mating as described.68,126–128
Split-luciferase assays
HCV Core fragments and human binding partners were cloned into p424-BYDV-NFLuc and p424-BYDV-C-Fluc-FLAG, respectively; EGFP was cloned into both vectors as negative controls. Split-luciferase assays were performed essentially as described in Ref.129, except that the binding reactions were incubated for 1 h. For an interaction to be considered significant, the luminescent signal must be higher than both control reactions as determined by a one-tailed t-test (P < 0.05).
Error-prone mutagenesis
Error-prone mutagenesis of the HCV Core MoRF-containing fragments was performed using nucleotide analogs 8-oxo-2' deoxyguanosine (8-oxo-dGTP; 1 µM) and 6-(2-deoxy-b-D-ribofuranosyl)−3,4-dihydro-8H-pyrimido-[4,5-c][1,2]oxazin-7-one (dPTP; 10 µM) as described.130,131
Acknowledgments
The authors thank David A. Sanders, C. Anders Olson, Whitney L. Dolan, Tzachi Hagai, and the Purdue Protein Biophysics Group for critically reading early versions of this manuscript. We thank Brian P. Dilkes, Michael Gribskov, and Philippe Lemey for thoughtful discussions that greatly improved this work.
Glossary
- α-MoRF
alpha-helical molecular recognition feature
- CF
core fragment
- CypA
cyclophlin A
- DBD
DNA-binding domain
- dN/dS
non-synonymous to synonymous substitution ratio
- HCV
hepatitis C virus
- IDR
intrinsically disordered region
- MoRF
molecular recognition feature
- NMR
nuclear magnetic resonance
- NPHV
non-primate hepacivirus
- PONDR
predictors of naturally disorder regions.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information
References
- Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005;272:5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323:573–584. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- Hsu WL, Oldfield CJ, Xue B, Meng J, Huang F, Romero P, Uversky VN, Dunker AK. Exploring the binding diversity of intrinsically disordered proteins involved in one-to-many binding. Protein Sci. 2013;22:258–273. doi: 10.1002/pro.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes C, Oldfield CJ, Ji F, Klitgord N, Cusick ME, Radivojac P, Uversky VN, Vidal M, Iakoucheva LM. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput Biol. 2006;2:e100. doi: 10.1371/journal.pcbi.0020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P. Intrinsically disordered proteins: a 10-year recap. Trends Biochem Sci. 2012;37:509–516. doi: 10.1016/j.tibs.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Oldfield CJ, Meng J, Yang JY, Yang MQ, Uversky VN, Dunker AK. Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics. 2008;9(Suppl 1):S1. doi: 10.1186/1471-2164-9-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells M, Tidow H, Rutherford TJ, Markwick P, Jensen MR, Mylonas E, Svergun DI, Blackledge M, Fersht AR. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc Natl Acad Sci USA. 2008;105:5762–5767. doi: 10.1073/pnas.0801353105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffill CR, Muller PA, Oh HK, Neo SP, Hogue KA, Cheok CF, Vousden KH, Lane DP, Blackstock WP, Gunaratne J. Mutant p53 interactome identifies nardilysin as a p53R273H-specific binding partner that promotes invasion. EMBO Rep. 2012;13:638–644. doi: 10.1038/embor.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunardi A, Di Minin G, Provero P, Dal Ferro M, Carotti M, Del Sal G, Collavin L. A genome-scale protein interaction profile of Drosophila p53 uncovers additional nodes of the human p53 network. Proc Natl Acad Sci USA. 2010;107:6322–6327. doi: 10.1073/pnas.1002447107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey NE, Trave G, Gibson TJ. How viruses hijack cell regulation. Trends Biochem Sci. 2011;36:159–169. doi: 10.1016/j.tibs.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Duvignaud JB, Savard C, Fromentin R, Majeau N, Leclerc D, Gagne SM. Structure and dynamics of the N-terminal half of hepatitis C virus core protein: an intrinsically unstructured protein. Biochem Biophys Res Commun. 2009;378:27–31. doi: 10.1016/j.bbrc.2008.10.141. [DOI] [PubMed] [Google Scholar]
- Garamszegi S, Franzosa EA, Xia Y. Signatures of pleiotropy, economy and convergent evolution in a domain-resolved map of human-virus protein-protein interaction networks. PLoS Pathog. 2013;9:e1003778. doi: 10.1371/journal.ppat.1003778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh GK, Dunker AK, Uversky VN. Protein intrinsic disorder toolbox for comparative analysis of viral proteins. BMC Genomics. 2008;9(Suppl 2):S4. doi: 10.1186/1471-2164-9-S2-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh GK, Dunker AK, Uversky VN. Protein intrinsic disorder and influenza virulence: the 1918 H1N1 and H5N1 viruses. Virol J. 2009;6:69. doi: 10.1186/1743-422X-6-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanyi-Nagy R, Lavergne JP, Gabus C, Ficheux D, Darlix JL. RNA chaperoning and intrinsic disorder in the core proteins of Flaviviridae. Nucleic Acids Res. 2008;36:712–725. doi: 10.1093/nar/gkm1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MR, Communie G, Ribeiro EA, Jr, Martinez N, Desfosses A, Salmon L, Mollica L, Gabel F, Jamin M, Longhi S, Ruigrok RW, Blackledge M. Intrinsic disorder in measles virus nucleocapsids. Proc Natl Acad Sci USA. 2011;108:9839–9844. doi: 10.1073/pnas.1103270108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CL, Marcotrigiano J, Rice CM. Bovine viral diarrhea virus core is an intrinsically disordered protein that binds RNA. J Virol. 2008;82:1294–1304. doi: 10.1128/JVI.01815-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushker R, Mooney C, Davey NE, Jacque JM, Shields DC. Marked variability in the extent of protein disorder within and between viral families. PLoS One. 2013;8:e60724. doi: 10.1371/journal.pone.0060724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuriki N, Oldfield CJ, Uversky VN, Berezovsky IN, Tawfik DS. Do viral proteins possess unique biophysical features? Trends Biochem Sci. 2009;34:53–59. doi: 10.1016/j.tibs.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Uversky V, Longhi S. Flexible Viruses: structural Disorder in Viral Proteins. New York: John Wiley; 2012. [Google Scholar]
- Uversky VN, Roman A, Oldfield CJ, Dunker AK. Protein intrinsic disorder and human papillomaviruses: increased amount of disorder in E6 and E7 oncoproteins from high risk HPVs. J Proteome Res. 2006;5:1829–1842. doi: 10.1021/pr0602388. [DOI] [PubMed] [Google Scholar]
- van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, Fuxreiter M, Gough J, Gsponer J, Jones DT, Kim PM, Kriwacki RW, Oldfield CJ, Pappu RV, Tompa P, Uversky VN, Wright PE, Babu MM. Classification of intrinsically disordered regions and proteins. Chem Rev. 2014;114:6589–6631. doi: 10.1021/cr400525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B, Williams RW, Oldfield CJ, Goh GK, Dunker AK, Uversky VN. Viral disorder or disordered viruses: do viral proteins possess unique features? Protein Pept Lett. 2010;17:932–951. doi: 10.2174/092986610791498984. [DOI] [PubMed] [Google Scholar]
- Garner E, Romero P, Dunker AK, Brown C, Obradovic Z. Predicting binding regions within disordered proteins. Genome Inform Ser Workshop Genome Inform. 1999;10:41–50. [PubMed] [Google Scholar]
- Callaghan AJ, Aurikko JP, Ilag LL, Gunter Grossmann J, Chandran V, Kuhnel K, Poljak L, Carpousis AJ, Robinson CV, Symmons MF, Luisi BF. Studies of the RNA degradosome-organizing domain of the Escherichia coli ribonuclease RNase E. J Mol Biol. 2004;340:965–979. doi: 10.1016/j.jmb.2004.05.046. [DOI] [PubMed] [Google Scholar]
- Demchenko AP. Recognition between flexible protein molecules: induced and assisted folding. J Mol Recogn. 2001;14:42–61. doi: 10.1002/1099-1352(200101/02)14:1<42::AID-JMR518>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Sugase K, Dyson HJ, Wright PE. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature. 2007;447:1021–1025. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol. 2002;12:54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- Oldfield CJ, Cheng Y, Cortese MS, Romero P, Uversky VN, Dunker AK. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry. 2005;44:12454–12470. doi: 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- Mohan A, Oldfield CJ, Radivojac P, Vacic V, Cortese MS, Dunker AK, Uversky VN. Analysis of molecular recognition features (MoRFs) J Mol Biol. 2006;362:1043–1059. doi: 10.1016/j.jmb.2006.07.087. [DOI] [PubMed] [Google Scholar]
- Fuxreiter M, Tompa P. Fuzzy complexes: a more stochastic view of protein function. Adv Exp Med Biol. 2012;725:1–14. doi: 10.1007/978-1-4614-0659-4_1. [DOI] [PubMed] [Google Scholar]
- Tompa P, Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem Sci. 2008;33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Erijman A, Aizner Y, Shifman JM. Multispecific recognition: mechanism, evolution, and design. Biochemistry. 2011;50:602–611. doi: 10.1021/bi101563v. [DOI] [PubMed] [Google Scholar]
- Davey NE, Edwards RJ, Shields DC. Computational identification and analysis of protein short linear motifs. Front Biosci. 2010;15:801–825. doi: 10.2741/3647. [DOI] [PubMed] [Google Scholar]
- Midic U, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Protein disorder in the human diseasome: unfoldomics of human genetic diseases. BMC Genomics 10(Suppl. 2009;1):S12. doi: 10.1186/1471-2164-10-S1-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–246. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- Le Gall T, Romero PR, Cortese MS, Uversky VN, Dunker AK. Intrinsic disorder in the Protein Data Bank. J Biomol Struct Dyn. 2007;24:325–342. doi: 10.1080/07391102.2007.10507123. [DOI] [PubMed] [Google Scholar]
- Sickmeier M, Hamilton JA, LeGall T, Vacic V, Cortese MS, Tantos A, Szabo B, Tompa P, Chen J, Uversky VN, Obradovic Z, Dunker AK. DisProt: the database of disordered proteins. Nucleic Acids Res. 2007;35:D786–D793. doi: 10.1093/nar/gkl893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh GK, Dunker AK, Uversky VN. A comparative analysis of viral matrix proteins using disorder predictors. Virol J. 2008;5:126. doi: 10.1186/1743-422X-5-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak SJ, Klein KC, Shoji I, Miyamura T, Lingappa JR. Assemble and interact: pleiotropic functions of the HCV core protein. In: Tan SL, editor. Hepatitis C viruses: genomes and molecular biology. Norfolk UK: Horizon Bioscience; 2006. [PubMed] [Google Scholar]
- Boulant S, Vanbelle C, Ebel C, Penin F, Lavergne JP. Hepatitis C virus core protein is a dimeric alpha-helical protein exhibiting membrane protein features. J Virol. 2005;79:11353–11365. doi: 10.1128/JVI.79.17.11353-11365.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel M, Lorinczi M, Rijnbrand R, Lemon SM, Watowich SJ. Self-assembly of nucleocapsid-like particles from recombinant hepatitis C virus core protein. J Virol. 2001;75:2119–2129. doi: 10.1128/JVI.75.5.2119-2129.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santolini E, Migliaccio G, La Monica N. Biosynthesis and biochemical properties of the hepatitis C virus core protein. J Virol. 1994;68:3631–3641. doi: 10.1128/jvi.68.6.3631-3641.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristofari G, Ivanyi-Nagy R, Gabus C, Boulant S, Lavergne JP, Penin F, Darlix JL. The hepatitis C virus Core protein is a potent nucleic acid chaperone that directs dimerization of the viral (+) strand RNA in vitro. Nucleic Acids Res. 2004;32:2623–2631. doi: 10.1093/nar/gkh579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanyi-Nagy R, Kanevsky I, Gabus C, Lavergne JP, Ficheux D, Penin F, Fosse P, Darlix JL. Analysis of hepatitis C virus RNA dimerization and core-RNA interactions. Nucleic Acids Res. 2006;34:2618–2633. doi: 10.1093/nar/gkl240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Realdon S, Gerotto M, Dal Pero F, Marin O, Granato A, Basso G, Muraca M, Alberti A. Proapoptotic effect of hepatitis C virus CORE protein in transiently transfected cells is enhanced by nuclear localization and is dependent on PKR activation. J Hepatol. 2004;40:77–85. doi: 10.1016/j.jhep.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Falcon V, Acosta-Rivero N, Chinea G, de la Rosa MC, Menendez I, Duenas-Carrera S, Gra B, Rodriguez A, Tsutsumi V, Shibayama M, Luna-Munoz J, Miranda-Sanchez MM, Morales-Grillo J, Kouri J. Nuclear localization of nucleocapsid-like particles and HCV core protein in hepatocytes of a chronically HCV-infected patient. Biochem Biophys Res Commun. 2003;310:54–58. doi: 10.1016/j.bbrc.2003.08.118. [DOI] [PubMed] [Google Scholar]
- Lyn RK, Hope G, Sherratt AR, McLauchlan J, Pezacki JP. Bidirectional lipid droplet velocities are controlled by differential binding strengths of HCV core DII protein. PLoS One. 2013;8:e78065. doi: 10.1371/journal.pone.0078065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulant S, Douglas MW, Moody L, Budkowska A, Targett-Adams P, McLauchlan J. Hepatitis C virus core protein induces lipid droplet redistribution in a microtubule- and dynein-dependent manner. Traffic. 2008;9:1268–1282. doi: 10.1111/j.1600-0854.2008.00767.x. [DOI] [PubMed] [Google Scholar]
- Boulant S, Targett-Adams P, McLauchlan J. Disrupting the association of hepatitis C virus core protein with lipid droplets correlates with a loss in production of infectious virus. J Gen Virol. 2007;88:2204–2213. doi: 10.1099/vir.0.82898-0. [DOI] [PubMed] [Google Scholar]
- Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim Biophys Acta. 2010;1804:996–1010. doi: 10.1016/j.bbapap.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- Prilusky J, Felder CE, Zeev-Ben-Mordehai T, Rydberg EH, Man O, Beckmann JS, Silman I, Sussman JL. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21:3435–3438. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- Campen A, Williams RM, Brown CJ, Meng J, Uversky VN, Dunker AK. TOP-IDP-scale: a new amino acid scale measuring propensity for intrinsic disorder. Protein Pept Lett. 2008;15:956–963. doi: 10.2174/092986608785849164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Kang CB, Yoon HS. Molecular and structural characterization of the domain 2 of hepatitis C virus non-structural protein 5A. Mol Cells. 2006;22:13–20. [PubMed] [Google Scholar]
- Hanoulle X, Badillo A, Verdegem D, Penin F, Lippens G. The domain 2 of the HCV NS5A protein is intrinsically unstructured. Protein Pept Lett. 2010;17:1012–1018. doi: 10.2174/092986610791498920. [DOI] [PubMed] [Google Scholar]
- Verdegem D, Badillo A, Wieruszeski JM, Landrieu I, Leroy A, Bartenschlager R, Penin F, Lippens G, Hanoulle X. Domain 3 of NS5A protein from the hepatitis C virus has intrinsic alpha-helical propensity and is a substrate of cyclophilin A. J Biol Chem. 2011;286:20441–20454. doi: 10.1074/jbc.M110.182436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angus AG, Loquet A, Stack SJ, Dalrymple D, Gatherer D, Penin F, Patel AH. Conserved glycine 33 residue in flexible domain I of hepatitis C virus core protein is critical for virus infectivity. J Virol. 2012;86:679–690. doi: 10.1128/JVI.05452-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerstein S, Solyom Z, Aladag A, Favier A, Schwarten M, Hoffmann S, Willbold D, Brutscher B. Transient structure and SH3 interaction sites in an intrinsically disordered fragment of the hepatitis C virus protein NS5A. J Mol Biol. 2012;420:310–323. doi: 10.1016/j.jmb.2012.04.023. [DOI] [PubMed] [Google Scholar]
- Burbelo PD, Dubovi EJ, Simmonds P, Medina JL, Henriquez JA, Mishra N, Wagner J, Tokarz R, Cullen JM, Iadarola MJ, Rice CM, Lipkin WI, Kapoor A. Serology-enabled discovery of genetically diverse hepaciviruses in a new host. J Virol. 2012;86:6171–6178. doi: 10.1128/JVI.00250-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A, Simmonds P, Gerold G, Qaisar N, Jain K, Henriquez JA, Firth C, Hirschberg DL, Rice CM, Shields S, Lipkin WI. Characterization of a canine homolog of hepatitis C virus. Proc Natl Acad Sci USA. 2011;108:11608–11613. doi: 10.1073/pnas.1101794108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana E, Fu Y, Chen J, Gerstein M. Interpretation of genomic variants using a unified biological network approach. PLoS Comput Biol. 2013;9:e1002886. doi: 10.1371/journal.pcbi.1002886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HB, Hirsh AE, Steinmetz LM, Scharfe C, Feldman MW. Evolutionary rate in the protein interaction network. Science. 2002;296:750–752. doi: 10.1126/science.1068696. [DOI] [PubMed] [Google Scholar]
- Khurana E, Fu Y, Colonna V, Mu XJ, Kang HM, Lappalainen T, Sboner A, Lochovsky L, Chen J, Harmanci A, Das J, Abyzov A, Balasubramanian S, Beal K, Chakravarty D, Challis D, Chen Y, Clarke D, Clarke L, Cunningham F, Evani US, Flicek P, Fragoza R, Garrison E, Gibbs R, Gumus ZH, Herrero J, Kitabayashi N, Kong Y, Lage K, Liluashvili V, Lipkin SM, MacArthur DG, Marth G, Muzny D, Pers TH, Ritchie GR, Rosenfeld JA, Sisu C, Wei X, Wilson M, Xue Y, Yu F, Genomes Project Consortium, Dermitzakis ET, Yu H, Rubin MA, Tyler-Smith C, Gerstein M. Integrative annotation of variants from 1092 humans: application to cancer genomics. Science. 2013;342:1235587. doi: 10.1126/science.1235587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MW, Kern AD. Comparative genomics of centrality and essentiality in three eukaryotic protein-interaction networks. Mol Biol Evol. 2005;22:803–806. doi: 10.1093/molbev/msi072. [DOI] [PubMed] [Google Scholar]
- Dolan PT, Zhang C, Khadka S, Arumugaswami V, Vangeloff AD, Heaton NS, Sahasrabudhe S, Randall G, Sun R, LaCount DJ. Identification and comparative analysis of hepatitis C virus-host cell protein interactions. Mol Biosyst. 2013;9:3199–3209. doi: 10.1039/c3mb70343f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun P, Tasan M, Dreze M, Barrios-Rodiles M, Lemmens I, Yu H, Sahalie JM, Murray RR, Roncari L, de Smet AS, Venkatesan K, Rual JF, Vandenhaute J, Cusick ME, Pawson T, Hill DE, Tavernier J, Wrana JL, Roth FP, Vidal M. An experimentally derived confidence score for binary protein-protein interactions. Nat Methods. 2009;6:91–97. doi: 10.1038/nmeth.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan K, Rual JF, Vazquez A, Stelzl U, Lemmens I, Hirozane-Kishikawa T, Hao T, Zenkner M, Xin X, Goh KI, Yildirim MA, Simonis N, Heinzmann K, Gebreab F, Sahalie JM, Cevik S, Simon C, de Smet AS, Dann E, Smolyar A, Vinayagam A, Yu H, Szeto D, Borick H, Dricot A, Klitgord N, Murray RR, Lin C, Lalowski M, Timm J, Rau K, Boone C, Braun P, Cusick ME, Roth FP, Hill DE, Tavernier J, Wanker EE, Barabasi AL, Vidal M. An empirical framework for binary interactome mapping. Nat Methods. 2009;6:83–90. doi: 10.1038/nmeth.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angus AG, Dalrymple D, Boulant S, McGivern DR, Clayton RF, Scott MJ, Adair R, Graham S, Owsianka AM, Targett-Adams P, Li K, Wakita T, McLauchlan J, Lemon SM, Patel AH. Requirement of cellular DDX3 for hepatitis C virus replication is unrelated to its interaction with the viral core protein. J Gen Virol. 2010;91:122–132. doi: 10.1099/vir.0.015909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamiya N, Worman HJ. Hepatitis C virus core protein binds to a DEAD box RNA helicase. J Biol Chem. 1999;274:15751–15756. doi: 10.1074/jbc.274.22.15751. [DOI] [PubMed] [Google Scholar]
- Owsianka AM, Patel AH. Hepatitis C virus core protein interacts with a human DEAD box protein DDX3. Virology. 1999;257:330–340. doi: 10.1006/viro.1999.9659. [DOI] [PubMed] [Google Scholar]
- Ariumi Y, Kuroki M, Abe K, Dansako H, Ikeda M, Wakita T, Kato N. DDX3 DEAD-box RNA helicase is required for hepatitis C virus RNA replication. J Virol. 2007;81:13922–13926. doi: 10.1128/JVI.01517-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall G, Panis M, Cooper JD, Tellinghuisen TL, Sukhodolets KE, Pfeffer S, Landthaler M, Landgraf P, Kan S, Lindenbach BD, Chien M, Weir DB, Russo JJ, Ju J, Brownstein MJ, Sheridan R, Sander C, Zavolan M, Tuschl T, Rice CM. Cellular cofactors affecting hepatitis C virus infection and replication. Proc Natl Acad Sci USA. 2007;104:12884–12889. doi: 10.1073/pnas.0704894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You LR, Chen CM, Yeh TS, Tsai TY, Mai RT, Lin CH, Lee YH. Hepatitis C virus core protein interacts with cellular putative RNA helicase. J Virol. 1999;73:2841–2853. doi: 10.1128/jvi.73.4.2841-2853.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai RT, Yeh TS, Kao CF, Sun SK, Huang HH, Wu Lee YH. Hepatitis C virus core protein recruits nucleolar phosphoprotein B23 and coactivator p300 to relieve the repression effect of transcriptional factor YY1 on B23 gene expression. Oncogene. 2006;25:448–462. doi: 10.1038/sj.onc.1209052. [DOI] [PubMed] [Google Scholar]
- Li Q, Brass AL, Ng A, Hu Z, Xavier RJ, Liang TJ, Elledge SJ. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc Natl Acad Sci USA. 2009;106:16410–16415. doi: 10.1073/pnas.0907439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein KC, Dellos SR, Lingappa JR. Identification of residues in the hepatitis C virus core protein that are critical for capsid assembly in a cell-free system. J Virol. 2005;79:6814–6826. doi: 10.1128/JVI.79.11.6814-6826.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Oldfield CJ, Meng J, Romero P, Uversky VN, Dunker AK. Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry. 2007;46:13468–13477. doi: 10.1021/bi7012273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruet A, Dosnon M, Vassena A, Lombard V, Gerlier D, Bignon C, Longhi S. Dissecting partner recognition by an intrinsically disordered protein using descriptive random mutagenesis. J Mol Biol. 2013;425:3495–3509. doi: 10.1016/j.jmb.2013.06.025. [DOI] [PubMed] [Google Scholar]
- Bourhis JM, Johansson K, Receveur-Brechot V, Oldfield CJ, Dunker KA, Canard B, Longhi S. The C-terminal domain of measles virus nucleoprotein belongs to the class of intrinsically disordered proteins that fold upon binding to their physiological partner. Virus Res. 2004;99:157–167. doi: 10.1016/j.virusres.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Bourhis JM, Receveur-Brechot V, Oglesbee M, Zhang X, Buccellato M, Darbon H, Canard B, Finet S, Longhi S. The intrinsically disordered C-terminal domain of the measles virus nucleoprotein interacts with the C-terminal domain of the phosphoprotein via two distinct sites and remains predominantly unfolded. Protein Sci. 2005;14:1975–1992. doi: 10.1110/ps.051411805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson K, Bourhis JM, Campanacci V, Cambillau C, Canard B, Longhi S. Crystal structure of the measles virus phosphoprotein domain responsible for the induced folding of the C-terminal domain of the nucleoprotein. J Biol Chem. 2003;278:44567–44573. doi: 10.1074/jbc.M308745200. [DOI] [PubMed] [Google Scholar]
- Longhi S, Receveur-Brechot V, Karlin D, Johansson K, Darbon H, Bhella D, Yeo R, Finet S, Canard B. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J Biol Chem. 2003;278:18638–18648. doi: 10.1074/jbc.M300518200. [DOI] [PubMed] [Google Scholar]
- Xue B, Dunker AK, Uversky VN. Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. J Biomol Struct Dyn. 2012;30:137–149. doi: 10.1080/07391102.2012.675145. [DOI] [PubMed] [Google Scholar]
- Kwofie SK, Schaefer U, Sundararajan VS, Bajic VB, Christoffels A. HCVpro: hepatitis C virus protein interaction database. Infect Genet Evol. 2011;11:1971–1977. doi: 10.1016/j.meegid.2011.09.001. [DOI] [PubMed] [Google Scholar]
- Masaki T, Suzuki R, Murakami K, Aizaki H, Ishii K, Murayama A, Date T, Matsuura Y, Miyamura T, Wakita T, Suzuki T. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J Virol. 2008;82:7964–7976. doi: 10.1128/JVI.00826-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawlik K, Baugh J, Chatterji U, Lim PJ, Bobardt MD, Gallay PA. HCV core residues critical for infectivity are also involved in core-NS5A complex formation. PLoS One. 2014;9:e88866. doi: 10.1371/journal.pone.0088866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grise H, Frausto S, Logan T, Tang H. A conserved tandem cyclophilin-binding site in hepatitis C virus nonstructural protein 5A regulates Alisporivir susceptibility. J Virol. 2012;86:4811–4822. doi: 10.1128/JVI.06641-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanoulle X, Badillo A, Wieruszeski JM, Verdegem D, Landrieu I, Bartenschlager R, Penin F, Lippens G. Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyl cis/trans isomerase activity of cyclophilins A and B. J Biol Chem. 2009;284:13589–13601. doi: 10.1074/jbc.M809244200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosnoblet C, Fritzinger B, Legrand D, Launay H, Wieruszeski JM, Lippens G, Hanoulle X. Hepatitis C virus NS5B and host cyclophilin A share a common binding site on NS5A. J Biol Chem. 2012;287:44249–44260. doi: 10.1074/jbc.M112.392209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartenschlager R, Lohmann V, Penin F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat Rev Microbiol. 2013;11:482–496. doi: 10.1038/nrmicro3046. [DOI] [PubMed] [Google Scholar]
- Gallay PA, Lin K. Profile of alisporivir and its potential in the treatment of hepatitis C. Drug Des Dev Ther. 2013;7:105–115. doi: 10.2147/DDDT.S30946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Cheng Y, LeGall T, Oldfield CJ, Mueller JP, Van YY, Romero P, Cortese MS, Uversky VN, Dunker AK. Rational drug design via intrinsically disordered protein. Trends Biotechnol. 2006;24:435–442. doi: 10.1016/j.tibtech.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Neduva V, Russell RB. Peptides mediating interaction networks: new leads at last. Curr Opin Biotechnol. 2006;17:465–471. doi: 10.1016/j.copbio.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Perkins JR, Diboun I, Dessailly BH, Lees JG, Orengo C. Transient protein–protein interactions: structural, functional, and network properties. Structure. 2010;18:1233–1243. doi: 10.1016/j.str.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Kingston RL, Hamel DJ, Gay LS, Dahlquist FW, Matthews BW. Structural basis for the attachment of a paramyxoviral polymerase to its template. Proc Natl Acad Sci USA. 2004;101:8301–8306. doi: 10.1073/pnas.0402690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Communie G, Habchi J, Yabukarski F, Blocquel D, Schneider R, Tarbouriech N, Papageorgiou N, Ruigrok RW, Jamin M, Jensen MR, Longhi S, Blackledge M. Atomic resolution description of the interaction between the nucleoprotein and phosphoprotein of Hendra virus. PLoS Pathog. 2013;9:e1003631. doi: 10.1371/journal.ppat.1003631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blocquel D, Habchi J, Gruet A, Blangy S, Longhi S. Compaction and binding properties of the intrinsically disordered C-terminal domain of Henipavirus nucleoprotein as unveiled by deletion studies. Mol Biosyst. 2012;8:392–410. doi: 10.1039/c1mb05401e. [DOI] [PubMed] [Google Scholar]
- Couturier M, Buccellato M, Costanzo S, Bourhis JM, Shu Y, Nicaise M, Desmadril M, Flaudrops C, Longhi S, Oglesbee M. High affinity binding between Hsp70 and the C-terminal domain of the measles virus nucleoprotein requires an Hsp40 co-chaperone. J Mol Recogn. 2010;23:301–315. doi: 10.1002/jmr.982. [DOI] [PubMed] [Google Scholar]
- Zhang X, Bourhis JM, Longhi S, Carsillo T, Buccellato M, Morin B, Canard B, Oglesbee M. Hsp72 recognizes a P binding motif in the measles virus N protein C-terminus. Virology. 2005;337:162–174. doi: 10.1016/j.virol.2005.03.035. [DOI] [PubMed] [Google Scholar]
- Vacic V, Oldfield CJ, Mohan A, Radivojac P, Cortese MS, Uversky VN, Dunker AK. Characterization of molecular recognition features, MoRFs, and their binding partners. J Proteome Res. 2007;6:2351–2366. doi: 10.1021/pr0701411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey NE, Cowan JL, Shields DC, Gibson TJ, Coldwell MJ, Edwards RJ. SLiMPrints: conservation-based discovery of functional motif fingerprints in intrinsically disordered protein regions. Nucleic Acids Res. 2012;40:10628–10641. doi: 10.1093/nar/gks854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkel H, Van Roey K, Michael S, Davey NE, Weatheritt RJ, Born D, Speck T, Kruger D, Grebnev G, Kuban M, Strumillo M, Uyar B, Budd A, Altenberg B, Seiler M, Chemes LB, Glavina J, Sanchez IE, Diella F, Gibson TJ. The eukaryotic linear motif resource ELM: 10 years and counting. Nucleic Acids Res. 2014;42:D259–266. doi: 10.1093/nar/gkt1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disfani FM, Hsu WL, Mizianty MJ, Oldfield CJ, Xue B, Dunker AK, Uversky VN, Kurgan L. MoRFpred, a computational tool for sequence-based prediction and characterization of short disorder-to-order transitioning binding regions in proteins. Bioinformatics. 2012;28:i75–83. doi: 10.1093/bioinformatics/bts209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meszaros B, Simon I, Dosztanyi Z. Prediction of protein binding regions in disordered proteins. PLoS Comput Biol. 2009;5:e1000376. doi: 10.1371/journal.pcbi.1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauer JC, Yaffe MB. Computational prediction of protein–protein interactions. Method Mol Biol. 2004;261:445–468. doi: 10.1385/1-59259-762-9:445. [DOI] [PubMed] [Google Scholar]
- Oldfield CJ, Cheng Y, Cortese MS, Romero P, Uversky VN, Dunker AK. Coupled folding and binding with α-helix-forming molecular recognition elements. Biochemistry. 2005;44:12454–12470. doi: 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- He B, Wang K, Liu Y, Xue B, Uversky VN, Dunker AK. Predicting intrinsic disorder in proteins: an overview. Cell Res. 2009;19:929–949. doi: 10.1038/cr.2009.87. [DOI] [PubMed] [Google Scholar]
- Neduva V, Linding R, Su-Angrand I, Stark A, de Masi F, Gibson TJ, Lewis J, Serrano L, Russell RB. Systematic discovery of new recognition peptides mediating protein interaction networks. PLoS Biol. 2005;3:e405. doi: 10.1371/journal.pbio.0030405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran V, Luisi BF. Recognition of enolase in the Escherichia coli RNA degradosome. J Mol Biol. 2006;358:8–15. doi: 10.1016/j.jmb.2006.02.012. [DOI] [PubMed] [Google Scholar]
- Kuiken C, Yusim K, Boykin L, Richardson R. The Los Alamos hepatitis C sequence database. Bioinformatics. 2005;21:379–384. doi: 10.1093/bioinformatics/bth485. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- Olsen LR, Kudahl UJ, Simon C, Sun J, Schonbach C, Reinherz EL, Zhang GL, Brusic V. BlockLogo: visualization of peptide and sequence motif conservation. J Immunol Methods. 2013;400–401:37–44. doi: 10.1016/j.jim.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P, Minin VN, Bielejec F, Kosakovsky Pond SL, Suchard MA. A counting renaissance: combining stochastic mapping and empirical Bayes to quickly detect amino acid sites under positive selection. Bioinformatics. 2012;28:3248–3256. doi: 10.1093/bioinformatics/bts580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. BEAST: bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Suchard MA. Bayesian random local clocks, or one rate to rule them all. BMC Biol. 2010;8:114. doi: 10.1186/1741-7007-8-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, Qureshi-Emili A, Li Y, Godwin B, Conover D, Kalbfleisch T, Vijayadamodar G, Yang M, Johnston M, Fields S, Rothberg JM. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- Khadka S, Vangeloff AD, Zhang C, Siddavatam P, Heaton NS, Wang L, Sengupta R, Sahasrabudhe S, Randall G, Gribskov M, Kuhn RJ, Perera R, LaCount DJ. A physical interaction network of dengue virus and human proteins. Mol Cell Proteomics. 2011;10:M111 012187. doi: 10.1074/mcp.M111.012187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaCount DJ. Interactome mapping in malaria parasites: challenges and opportunities. Method Mol Biol. 2012;812:121–145. doi: 10.1007/978-1-61779-455-1_7. [DOI] [PubMed] [Google Scholar]
- LaCount DJ, Vignali M, Chettier R, Phansalkar A, Bell R, Hesselberth JR, Schoenfeld LW, Ota I, Sahasrabudhe S, Kurschner C, Fields S, Hughes RE. A protein interaction network of the malaria parasite Plasmodium falciparum. Nature. 2005;438:103–107. doi: 10.1038/nature04104. [DOI] [PubMed] [Google Scholar]
- Brown HF, Wang L, Khadka S, Fields S, LaCount DJ. A densely overlapping gene fragmentation approach improves yeast two-hybrid screens for Plasmodium falciparum proteins. Mol Biochem Parasitol. 2011;178:56–59. doi: 10.1016/j.molbiopara.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasila TS, Pajunen MI, Savilahti H. Critical evaluation of random mutagenesis by error-prone polymerase chain reaction protocols, Escherichia coli mutator strain, and hydroxylamine treatment. Anal Biochem. 2009;388:71–80. doi: 10.1016/j.ab.2009.02.008. [DOI] [PubMed] [Google Scholar]
- Zaccolo M, Williams DM, Brown DM, Gherardi E. An approach to random mutagenesis of DNA using mixtures of triphosphate derivatives of nucleoside analogues. J Mol Biol. 1996;255:589–603. doi: 10.1006/jmbi.1996.0049. [DOI] [PubMed] [Google Scholar]
- Moradpour D, Penin F. Hepatitis C virus proteins: from structure to function. Curr Top Microbiol Immunol. 2013;369:113–142. doi: 10.1007/978-3-642-27340-7_5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information