

Abstract
Rationale: α1-Antitrypsin (A1AT) was identified as a plasma protease inhibitor; however, it is now recognized as a multifunctional protein that modulates immunity, inflammation, proteostasis, apoptosis, and cellular senescence. Like A1AT, protein phosphatase 2A (PP2A), a major serine-threonine phosphatase, regulates similar biologic processes and plays a key role in chronic obstructive pulmonary disease.
Objectives: Given their common effects, this study investigated whether A1AT acts via PP2A to alter tumor necrosis factor (TNF) signaling, inflammation, and proteolytic responses in this disease.
Methods: PP2A activity was measured in peripheral blood neutrophils from A1AT-deficient (PiZZ) and healthy (PiMM) individuals and in alveolar macrophages from normal (60 mg/kg) and high-dose (120 mg/kg) A1AT-treated PiZZ subjects. PP2A activation was assessed in human neutrophils, airway epithelial cells, and peripheral blood monocytes treated with plasma purified A1AT protein. Similarly, lung PP2A activity was measured in mice administered intranasal A1AT. PP2A was silenced in lung epithelial cells treated with A1AT and matrix metalloproteinase and cytokine production was then measured following TNF-α stimulation.
Measurements and Main Results: PP2A was significantly lower in neutrophils isolated from PiZZ compared with PiMM subjects. A1AT protein activated PP2A in human alveolar macrophages, monocytes, neutrophils, airway epithelial cells, and in mouse lungs. This activation required functionally active A1AT protein and protein tyrosine phosphatase 1B expression. A1AT treatment acted via PP2A to prevent p38 and IκBα phosphorylation and matrix metalloproteinase and cytokine induction in TNF-α–stimulated epithelial cells.
Conclusions: Together, these data indicate that A1AT modulates PP2A to counter inflammatory and proteolytic responses induced by TNF signaling in the lung.
Keywords: α1-antitrypsin, PP2A, phosphorylation, cell signaling, inflammation
At a Glance Commentary
Scientific Knowledge on the Subject
α1-Antitrypsin (A1AT) is a plasma antiprotease that counters the effects of neutrophil elastase to prevent lung elastin degradation and the development of emphysema. In addition to its role as a protease inhibitor, A1AT exerts biologic effects that can impact on several disease parameters in chronic obstructive pulmonary disease including inflammation, apoptosis, and protease expression. However, the mechanisms by which A1AT alters these factors in the lung are not completely understood.
What This Study Adds to the Field
Protein phosphatase 2A (PP2A) is a primary serine threonine phosphatase that regulates inflammation, protease expression, and apoptosis under smoking conditions in the lung. This study demonstrates that A1AT-deficient patients have reduced neutrophil PP2A activity and A1AT protein acts through protein tyrosine phosphatase 1B to dephosphorylate and thereby activate PP2A in the lung. PP2A is required for A1AT to counter tumor necrosis factor-α–induced lung inflammation. Thus, these findings establish a novel mechanism whereby A1AT activates PP2A to counter injurious tumor necrosis factor signaling responses that are linked to the development of inflammation and chronic obstructive pulmonary disease.
α1-Antitrypsin (A1AT) is the most abundant circulating serine protease inhibitor and misfolding or deficiency of this protein causes chronic liver and lung disease (1). The development of chronic obstructive pulmonary disease (COPD) in A1AT deficiency is classically associated with the destruction of the alveoli and small airway structures caused by protease expression, apoptosis, and an enhanced inflammatory response (2). This is believed to be caused by the unopposed actions of lung proteases, such as neutrophil elastase (3). Over the past decade, multiple extracellular roles of A1AT have emerged (4, 5). These studies indicate that A1AT prevents caspase activity (6), nitric oxide production (7), HIV type 1 infectivity and reproduction (8), tumor necrosis factor-α–converting enzyme activity (9), ER stress (10, 11), epithelial barrier disruption, and IL-8–mediated neutrophil chemotaxis (12). Thus, these findings show that A1AT counteracts key disease processes in COPD by a variety of mechanisms in addition to its role as an antiprotease.
Recently, we demonstrated that protein phosphatase 2A (PP2A), the primary eukaryotic serine-threonine phosphatase, is a key regulator of smoke-driven inflammation and protease expression in animal models of COPD (13, 14). PP2A activation prevents inflammation and proteolytic imbalances typically associated with COPD (13, 14). Furthermore, inhibiting PP2A exacerbated inflammatory and proteolytic responses in both primary human lung epithelial cells and mouse lung following cigarette smoke exposure (14). These studies suggest an important role for PP2A in modulating the development of COPD. The activity of PP2A is regulated by post-translational protein modifications (15). Specifically, PP2A becomes inactivated by phosphorylation of a tyrosine site at position 307 (Tyr307) in the catalytic subunit of the enzyme, which is offset by the activity of protein tyrosine phosphatase 1B (PTP1B) (13, 16). Given the common antiinflammatory effects of A1AT and PP2A, this study investigated whether A1AT acted via PTP1B and PP2A to counter lung inflammatory responses.
To address this hypothesis, we compared PP2A activity levels in neutrophils from healthy PiMM and A1AT-deficient PiZZ individuals and in alveolar macrophages and epithelial cells from normal (60 mg/kg) and high-dose (120 mg/kg) A1AT (Zemaira; CSL Behring, King of Prussia, PA)-treated PiZZ subjects. In addition, we determined how A1AT altered lung PP2A activity to inhibit the induction of proteases and cytokines following tumor necrosis factor (TNF)-α stimulation. A1AT down-regulates TNF-α expression (17, 18) and blocks TNF-α–mediated neutrophil activation in COPD (19). Thus, this study contributes to the growing knowledge of the diverse roles of A1AT in health and disease by identifying for the first time that PP2A acts via A1AT to counter key inflammatory responses in the lung. Some of the results of these studies have been previously reported in the form of an abstract (20).
Methods
Isolation of A1AT Protein from PiMM and PiZZ Subjects
A1AT protein was isolated from the blood of PiMM or PiZZ individuals as outlined in the online supplement. The institutional review board at Mount Sinai Roosevelt Medical Center approved all study protocols.
Neutrophil Isolation
Neutrophils were isolated from venous peripheral blood obtained from healthy (PiMM) and A1AT-deficient (PiZZ) volunteers as described previously (12) and outlined in the online supplement.
A1AT Augmentation Therapy
Blood, bronchoalveolar lavage fluid (BALF), alveolar macrophages, and airway epithelial cells were taken from PiZZ A1AT subjects receiving intravenous A1AT augmentation (Zemaira) at standard dose (60 mg/kg/week) for 1 month, double dose (120 mg/kg/wk) the next month, and standard dose (60 mg/kg/wk) for the last month as outlined in the online supplement. The 120 mg/kg A1AT weekly dose was considered to be safe and well tolerated (21). Written consent was obtained from all study participants and the trial was approved by the institutional review board of the University of Miami School of Medicine.
Cell Culture
A549 cells that were stably transfected with lentivirus expressing shRNA for PP2AA or negative control shRNA (Santa Cruz Biotechnologies, Santa Cruz, CA) were treated with 2 μM A1AT (Abcam, Cambridge, MA) for 1 hour before 10 ng/ml TNF-α (Sigma, St. Louis, MO) exposure.
Inactivation of A1AT and Elastase Activity Determination
A1AT (1 mg) was oxidized and inactivated as outlined previously (22) (see online supplement). A1AT antiprotease activity was determined by titrating with neutrophil elastase and elastase activity was measured using the substrate N-methoxysuccinyl-Ala-Ala-Pro-Val-p-nitroanilide (Sigma) (13).
Animal Model
FVB/NJ and Ptp1b−/− mice (Mutant Mouse Regional Resource Center/Jackson Labs, Bar Harbor, ME) were intranasally administered either 2 μM of A1AT or albumin and euthanized 12 hours later as outlined in the online supplement. All animal experiments were performed with approval from Mount Sinai Roosevelt’s Medical Center’s Institutional Animal Care and Use Committee approval.
Phosphatase Activity Determination
PP2A, PTP1B, and mitogen-activated protein kinase phosphatase-1 (MKP-1) activity were determined from the tissue or cell as previously reported (13, 14).
Matrix Metalloproteinase and Cytokine Measurements
Matrix metalloproteinase (MMP) and cytokine levels were measured in cell media or BALF using a beads assay on the BioRad Bio-Plex 200 system (BioRad, Hercules, CA). Quantitative polymerase chain reaction was used to screen cytokine and protease expression profiles in cells.
Intracellular Signaling
Immunoblots were conducted on protein of lung and cells to determine levels of p-p38, p38, p-IκBα, IκBα, p-PP2AC(Tyr307), PP2AC, PP2AA, PTP1B, and actin (all antibodies from Cell Signaling, Danvers, MA). Nuclear factor (NF)-ĸB activation was measured on nuclear protein extracts using an assay from Active Motif (46096; Carlsbad, CA).
Statistical Analyses
Data are expressed as means ± SEM. For normally distributed data, Student t tests (two tailed) were used. For nonparametric data, Wilcoxon signed rank tests were used with GraphPad Prism (GraphPad Software, La Jolla, CA). All data sets are represented as mean ± SE.
Results
Neutrophils from A1AT-Deficient Subjects Have Deficient PP2A Activity Compared with Healthy Individuals
Neutrophils were isolated from healthy (MM) and A1AT-deficient (PiZZ) subjects not receiving A1AT augmentation therapy or who were at the end of their infusion cycle when AAT plasma levels were at the lowest (subject characteristics are presented in Table 1). Neutrophil PP2A relative activity levels were 48% less in PiZZ compared with PiMM individuals, without observing a change in cellular levels of PP2AC (Figure 1). As a control another serine-threonine phosphatase, MKP-1, had no altered activity in neutrophils from PiZZ individuals (Figure 1).
Table 1.
Demographics of Study Subjects
| PiMM Subjects | PiZZ (No Therapy) | PiZZ (before Therapy) | |
|---|---|---|---|
| Number | 9 | 9 | 8 |
| Age, yr | 41.1 ± 9.5 | 57.3 ± 5.0 | 60 ± 8.7 |
| Sex, male/female | 7/2 | 5/4 | 6/2 |
| A1AT genotype (MM/ZZ/ZS) | 9/0/0 | 0/9/0 | 0/7/1 |
| Baseline plasma A1AT, μM | 25.9 ± 2.9 | 3.62 ± 1.23 | 6.1 ± 2.3 |
| Former smokers, % | 0 | 66.6 | 62.5 |
| FEV1 % predicted | N/A | 59.7 ± 13.4 | 50.9 ± 10.1 |
| FVC % predicted | N/A | 93.8 ± 19.9 | 88.4 ± 14.1 |
| FEV1/FVC, % | N/A | 55.5 ± 14.6 | 44.8 ± 5.7 |
| DlCO % predicted | N/A | 68.78 ± 8.9 | 57.0 ± 14.4 |
Definition of abbreviations: A1AT = α1-antitrypsin; DlCO = diffusing capacity of carbon monoxide.
Figure 1.

Neutrophils from PiZZ subjects have reduced protein phosphatase 2A (PP2A) activity. Neutrophils were isolated from PiMM and PiZZ α1-antitrypsin (A1AT) homozygote individuals. Serine-threonine phosphatase activity for PP2A and mitogen-activated protein kinase phosphatase-1 (MKP-1) was determined from each individual and represented as picomoles of phosphate liberated per minute on the y axis. Graphs represent mean ± SEM of three measurements from each subject (n = 9 per group). P values shown, comparing both treatments connected by a line. Representative immunoblots are shown demonstrating equal cellular protein content.
A1AT Increased Cellular PP2A Activity
Because neutrophils from PiZZ subjects have reduced PP2A activity, we investigated whether A1AT could directly alter PP2A activity levels in these and other key cell types in the lung. In vitro, human plasma purified A1AT increased relative PP2A activity in neutrophils, monocytes, small airway epithelial cells, and A549 cells, without observing a change in cellular levels of PP2AC (Figure 2A). This induction of PP2A activity was dependent on the antielastase potential of A1AT and prior treatment and oxidation of A1AT with H2O2 negated its ability to activate PP2A (Figures 2B and 2C). Recombinant-inactive A1AT did not induce PP2A activity (Figure 2C). A1AT significantly increased PP2A activity in A549 cells at concentrations of 1 μM or greater (Figure 2D) and increased levels of PP2A activity were observed as early as 1 hour after stimuli (Figure 2E).
Figure 2.
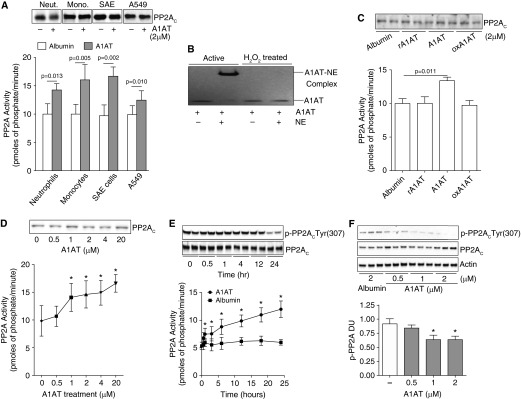
α1-Antitrypsin (A1AT) stimulation promotes cellular protein phosphatase 2A (PP2A) activity. (A) Treatment of human peripheral blood neutrophils and monocytes, human small airway epithelial (SAE) cells, and airway epithelial cell line A549 cells with 2 μM A1AT for 24 hours enhances PP2A activity. (B) Oxidized A1AT cannot form complexes with neutrophil elastase (NE) demonstrated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and could not (C) induce a PP2A cellular response in A549 cells. Recombinant/inactive A1AT (rA1AT) also had no effect on PP2A activity. (D) An A1AT dose response stimulation of A549 cells demonstrated that an A1AT concentration of greater than 1 μM significantly increases PP2A activity. (E) A1AT-induced PP2A activity was observed from 1 hour after stimuli with dephosphorylation of PP2AC greatest 24 hours after stimuli. (F) A1AT dephosphorylated PP2AC at Tyr site 307 in A549 cells. Densitometry analysis was performed for p-PP2AC(Tyr307) phosphorylation on immunoblots from 3 separate days and was represented as densitometry units (DU). Graphs represent mean ± SEM of three measurements from each subject (n = 6 per group). P values shown, comparing both treatments connected by a line. *P < 0.05 compared with baseline levels of activity. Representative PP2AC immunoblots are shown demonstrating equal cellular protein content.
A1AT Increased PP2A Activity in a PTP1B-Dependent Manner
PP2A activity is increased when the tyrosine site at position 307 (Tyr307) in the catalytic subunit PP2AC of the enzyme is dephosphorylated (16). This is the mechanism for A1AT stimulation as indicated in Figures 2E and 2F. This process is mediated by PTP1B (13, 16), which dephosphorylates PP2AC at this specific site. Because PTP1B regulates PP2A activity in the lung (13), we evaluated whether PTP1B activity was also sensitive to A1AT stimulation and could contribute to the PP2A activation observed in Figure 2. Interestingly, neutrophil PTP1B activity levels were 52% less in PiZZ compared with PiMM individuals (Figure 3A), which coincided with increased phosphorylation of PP2AC (Figure 3B). Silencing PTP1B expression in A549 cells prevented A1AT from activating PP2A (Figure 3C). Moreover, A1AT stimulation leads to the dephosphorylation of PP2A at tyrosine residue 307 only when PTP1B is expressed (Figure 3D).
Figure 3.

Protein tyrosine phosphatase 1B (PTP1B) is required for α1-antitrypsin (A1AT) to activate protein phosphatase 2A (PP2A). Neutrophils isolated from PiZZ A1AT homozygote individuals have (A) less PTP1B activity and (B) greater PP2AC phosphorylation than PiMM individuals (n = 10 per group). Loss of PTP1B expression in A549 cells prevents A1AT-induced (C) activation of PP2A and (D) dephosphorylation of p-PP2AC(Tyr307). Densitometry analysis was performed on immunoblots from 3 separate days. Graphs represent mean ± SEM of three measurements from each subject (n = 10 per group). P values shown, comparing both treatments connected by a line. Representative immunoblots are shown demonstrating equal cellular protein content. DU = densitometry units.
To further demonstrate the potential regulation of PTP1B-PP2A activity by A1AT, human A1AT protein was administered directly into the airways of wild-type and Ptp1b−/− mice and the impact on phosphatase activity was investigated. Intranasal delivery of A1AT significantly increased PP2A and PTP1B activity compared with mice administered albumin and PTP1B-deficient mice (Figure 4A). A1AT stimulation resulted in the dephosphorylation of PP2AC in airway tissue only in wild-type mice (Figure 4B). As observed with the human neutrophils, A1AT did not alter MKP-1 serine-threonine activity in mouse airways (see Figure E1 in the online supplement). Loss of PTP1B expression did impact on neutrophil death, with neutrophils from Ptp1b−/− mice undergoing apoptosis at a faster rate than wild-type neutrophils (Figure 4C). The B regulatory subunits are not altered with A1AT stimuli in A549 cells (Figures 5A and 5B). PP2A activity is primarily enhanced in the cytoplasm following A1AT stimuli but nonsignificant increases in activity also occur in other cellular locations (Figure 5C). Numerous signaling cascades can regulate phosphorylation of PP2A, thereby altering its activity. SET/I(2)PP2A binds to PP2A to prevent activity. A1AT stimulation did not alter SET/I(2)PP2A binding to PP2A (Figure 5D). However, inhibition of protein kinase A (PKA) prevented PTP1B and PP2A activity by A1AT stimulation (Figure 5E). Therefore, A1AT requires PKA to induce PTP1B and PP2A activation.
Figure 4.
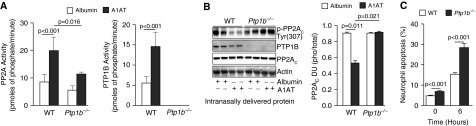
Airway administration of α1-antitrypsin (A1AT) protein activates protein tyrosine phosphatase 1B (PTP1B) and protein phosphatase 2A (PP2A) in mice. FVB/NJ and Ptp1b−/− mice were administered either 2 μM of albumin or A1AT intranasally. (A) PP2A and PTP1B activity levels and (B) PP2AC phosphorylation at Tyr site 307 was examined in mouse lungs. Densitometry analysis was performed for p-PP2AC(Tyr307) phosphorylation. (C) Neutrophils from Ptp1b−/− mice were undergoing cell death at a faster rate than wild-type (WT) neutrophils. Graphs represent mean ± SEM of three measurements from each animal (n = 10 per group). P values shown, comparing both treatments connected by a line. DU = densitometry units.
Figure 5.

Protein kinase A (PKA) inhibition prevents α1-antitrypsin (A1AT) activation of protein tyrosine phosphatase 1B (PTP1B)/protein phosphatase 2A (PP2A). A1AT did not alter PP2A B subunit (A) protein or (B) gene expression in A549 cells. (C) A1AT enhances PP2A activity primarily in the cytoplasm without altering translocation of the protein. (D) A1AT stimuli did not alter binding of the PP2A inhibitor, SET, to PP2A, demonstrated by IP of PP2AC and probing for SET and PP2AC. (E) Inhibition of PKA/cAMP activity alters A1AT’s ability to activate PP2A. Graphs represent mean ± SEM of three measurements. Representative immunoblots are shown demonstrating equal cellular protein content. *P < 0.05 for A1AT-treated versus albumin-treated A549 cells. **P < 0.05 for A1AT-treated versus A1AT+PKA inhibitor–treated A549 cells.
PiZZ A1AT Protein’s Antielastase Activity Correlates with Its Ability to Activate PTP1B-PP2A
Individuals with the PiZZ A1AT homozygote genotype have lower circulating levels of A1AT with less antielastase activity (3). A549 cells treated with media supplemented with serum from PiMM or PiZZ individuals, with equivalent A1AT levels, have different PP2A activity profiles (Figure 6A). To examine whether the dysfunctional PiZZ A1AT protein changes airway PP2A and PTP1B activity responses, A1AT protein was isolated from PiMM and PiZZ individuals and antielastase responses were compared with potential phosphatase activity induction levels. Using an A1AT-specific resin, affinity chromatography was performed on serum from PiMM and PiZZ homozygote genotype individuals (n = 10 per group). Washes and elutions from A1AT affinity chromatography were examined by Coomassie blue gel electrophoresis and immunoblotting to confirm purity and specificity (see Figure E2).
Figure 6.

α1-Antitrypsin (A1AT) from the serum of PiZZ A1AT subjects has decreased antielastase activity and less ability to activate protein phosphatase 2A (PP2A) in cells. (A) A549 cells have increased PP2A activity following treatment with purified A1AT or serum from PiMM individuals but not with serum from PiZZ individuals. Equal A1AT levels were administered in each group. Affinity chromatography was performed to isolate A1AT from PiMM and PiZZ subjects. (B) A1AT levels were examined in both PiMM and PiZZ A1AT isolations by Coomassie blue staining and immunoblots. (C) Antineutrophil elastase activity assays demonstrated that PiZZ A1AT has less antielastase activity and binding potential to neutrophil elastase (NE) than PiMM A1AT. (D) Treatment of A549 cells for 24 hours with 2 μM albumin, commercially available A1AT (com), or A1AT isolated from PiMM subjects enhances protein tyrosine phosphatase 1B (PTP1B) and PP2A activities, unlike PiZZ A1AT protein. The ability of A1AT to increase PP2A activity in cells correlated with its antielastase activity (right). Graphs represent mean ± SEM of three measurements from each subject (n = 10 per group). P values shown, comparing both treatments connected by a line. Representative immunoblots are shown demonstrating equal cellular protein content.
As expected, A1AT is more abundant from PiMM compared with PiZZ individuals (Figure 6B) and, compared with equal amounts of PiZZ protein, PiMM A1AT has greater antielastase ability (Figure 6C). A1AT from PiZZ individuals binds less with neutrophil elastase to form complexes (Figure 6C). A549 cells have increased PTP1B and PP2A activity following stimulation with A1AT from PiMM homozygote genotype individuals and similar findings were observed with commercially available A1AT (Figure 6D). Interestingly, equal quantities of A1AT protein isolated from PiZZ homozygote genotype individuals did not activate PTP1B or PP2A in A549 cells (Figure 6D). A1AT-inducible PP2A activity correlated with the antielastase ability of A1AT (Figure 6D). This suggests that the functional activity and concentration of A1AT play are important in the activation of PP2A.
Targeting PP2A or PTP1B Expression Prevents A1AT Antiinflammatory Responses
Loss of PP2A enhances several inflammatory and proteolytic processes in the airways (13, 14) and A1AT inhibits lung inflammatory processes (12). TNF signaling triggers inflammatory responses in A1AT deficiency (19) and PP2A counters TNF signaling in the lung (14). Thus, an in vitro approach was used to examine if loss of PP2A or PTP1B expression reduced A1AT’s antiinflammatory antiprotease activity. Multiple kinases, proteases, and cytokines were examined by quantitative polymerase chain reaction, multiplex analysis, and immunoblotting of samples collected from A549 cells (control, PP2AA, and PTP1B shRNA stably transfected) (Figure 7A and see Figure E3 for PP2AA and PTP1B knockdown) treated with TNF-α and A1AT protein were performed.
Figure 7.

α1-Antitrypsin (A1AT) acts via protein phosphatase 2A (PP2A) to inhibit tumor necrosis factor (TNF)-α induced p38 and IκBα phosphorylation and subsequent nuclear factor (NF)-κB activation. A549 cells, which were stably transfected with lentivirus expressing negative control, PP2AA or protein tyrosine phosphatase 1B (PTP1B) shRNA, were exposed to 2 μM A1AT for 1 hour before stimulation with 10 ng/ml TNF-α (1 h). (A) Immunoblots and densitometry analysis was performed for p38 and IκBα phosphorylation. (B) Transcription factor, NF-κB, and relative activation were examined in each treatment group, compared with negative control shRNA-treated cells. Graphs represent mean ± SEM, where each measurement was performed three times (n = 6 per group). P values shown, comparing both treatments connected by a line. DU = densitometry units. *P < 0.05 compared with A549 cells treated only with TNF-α. **P < 0.05 compared with A1AT+TNF-α–treated control shRNA A549 cells.
A1AT significantly inhibited TNF-α–induced phosphorylation of p38 and IκBα (Figures 7A and 7B). Phosphorylation of IκBα by TNF-α resulted in the activation of NF-κB, which was partially prevented by A1AT only when PP2A or PTP1B were expressed (Figure 7B). As determined by lactate dehydrogenase release assay, cell viability was not affected by gene silencing or A1AT–TNF-α treatment (see Figure E3C). A1AT treatment prevented TNF-α–induced production of MMP-1 and -9 in a PTP1B-PP2A–dependent manner (Figures 8A and 8B). PTP1B and PP2A were also required for A1AT to inhibit the induction of monocyte chemotactic protein 1, IL-8, and IL-1β by TNF-α in A549 cells (Figure 8C). A1AT did prevent some inflammatory signaling responses independently of PP2A or PTP1B, with intercellular adhesion molecule 1 induction being prevented by A1AT in control, PP2AA, and PTP1B shRNA transfected cells (see Figure E4). Alternatively, the loss of PP2A enhanced cathepsin S, RANTES, and IL-6 expression but this was not sensitive to A1AT treatment (see Figure E4). Therefore, A1AT regulates PTP1B-PP2A activity to alter specific TNF-α–mediated signaling processes within the lung.
Figure 8.

Protein phosphatase 2A (PP2A) activity is required for α1-antitrypsin (A1AT) to inhibit tumor necrosis factor (TNF)-α induced proteases and cytokines. A549 cells, which were stably transfected with lentivirus expressing control PP2AA or protein tyrosine phosphatase 1B (PTP1B) shRNA, were exposed to 2 μM A1AT for 1 hour before 10 ng/ml TNF-α stimulation (24 h). Quantitative polymerase chain reaction and multiplex analysis were performed for (A) matrix metalloproteinase (MMP)-1 and (B) MMP-9. (C) Multiplex analysis was performed for IL-8 and monocyte chemotactic protein (MCP) 1 and quantitative polymerase chain reaction was performed for IL-1β. Graphs represent mean ± SEM, where each measurement was performed three times (n = 6 per group). *P < 0.05 compared with A549 cells treated only with TNF-α. **P < 0.05 compared with A1AT+TNF-α–treated control shRNA A549 cells.
A1AT Augmentation Therapy Enhances PTP1B-PP2A Activity and Reduces MMP-1 and -9 Levels in BALF of A1AT-Deficient Patients
PP2A counters the production of proteases in the lung (14). Because the activation of PP2A by A1AT impacted on several key proteolytic and inflammatory signaling processes in vitro, we examined MMP-1 and MMP-9 levels in BALF from PiZZ homozygote genotype individuals’ receiving A1AT augmentation therapy (subject characteristics are presented in Tables 1 and 2). BALF and plasma was collected from subjects receiving weekly 60 mg/kg A1AT, 30 days after subsequently receiving weekly 120 mg/kg A1AT and 30 days after returning to weekly 60 mg/kg A1AT treatment (Figure 9A). MMP-12 levels were also examined because PP2A can impact on cigarette smoke–induced MMP-12 expression (13).
Table 2.
Demographics of Study Subjects Receiving α1-Antitrypsin Augmentation Therapy
| 60 mg/kg | 120 mg/kg | |
|---|---|---|
| Number | 8 | 8 |
| FEV1 % predicted | 50.9 ± 10.1 | 53.9 ± 11.7 |
| FVC % predicted | 88.4 ± 14.1 | 87.9 ± 13.5 |
| FEV1/FVC, % | 44.8 ± 5.7 | 44.7 ± 6.7 |
Figure 9.

α1-Antitrypsin (A1AT) augmentation therapy enhances protein phosphatase 2A (PP2A) activity and subdues protease and cytokine production in humans. (A) A1AT-deficient (PiZZ) subjects were administered weekly A1AT intravenously (60 mg/kg for 1 mo, followed by 1 mo of 120 mg/kg weekly, and then returning to 60 mg/kg weekly for 1 mo). Plasma, bronchoalveolar lavage fluid (BALF), BALF cells, and airway epithelial cells were collected on the final day of each month of A1AT therapy (n = 8 subjects). (B) Plasma A1AT levels were observed to be enhanced following 120 mg/kg A1AT treatment, demonstrated by Coomassie blue staining and A1AT immunoblots. (C) BALF matrix metalloproteinase (MMP)-1, -9, and -12 levels were altered following A1AT augmentation therapy. Graphs represent individual subject MMP BALF concentrations. (D) A1AT did not alter protein tyrosine phosphatase 1B (PTP1B), PP2AA, or PP2AC expression in airway epithelial cells collected from the patients but subdued IL-8, IL-1β, monocyte chemotactic protein (MCP)-1, and tumor necrosis factor (TNF)-α expression. (E) The 120 mg/kg A1AT treatment induced dephosphorylation of p-PP2AC(Tyr307) in epithelial cells from patients. (F) Likewise, it enhanced PTP1B and PP2A activity in alveolar macrophages. Graphs represent mean ± SEM, where each measurement was performed three times (n = 8 per group). P values shown, comparing both treatments connected by a line. *P < 0.05.
Every subject had increased A1AT plasma levels and reduced MMP levels post double A1AT therapy (Figures 9B and 9C; see Figure E5). Weekly administration of 120 mg/kg A1AT significantly reduced MMP-1 and MMP-9 BALF levels. Reduced MMP-12 levels were observed also but only trend changes were detected. There was a tendency for MMP levels to increase 30 days after returning to the 60 mg/kg A1AT dose (Figure 9C), which coincided with reduced A1AT plasma levels (Figure 9B). Double dosing of A1AT treatment did not alter PP2A or PTP1B gene expression in airway epithelial cells collected from patients at the time of BALF collection but did alter expression of IL-8, IL-1β, monocyte chemotactic protein 1, and TNF-α (Figure 9D). PP2A was significantly dephosphorylated in airway epithelial cells following double dosing of A1AT (Figure 9E) thus indicating that high-dose treatment activated these cells. Enhanced PTP1B and PP2A activity was also observed in the alveolar macrophages from the same patients when administered weekly 120 mg/kg of A1AT compared with the 60 mg/kg dose (Figure 9F). These data provide further evidence that enhancing A1AT protein levels augments PTP1B-PP2A activation, which may down-regulate proteolytic responses in the lung. Future studies will address whether A1AT induction of PTP1B and PP2A is solely responsible for this decrease in lung protease production.
Discussion
COPD is associated with a protease-antiprotease imbalance (23) and this imbalance is accentuated in subjects with A1AT deficiency. Although A1AT protein is believed to counter the development of emphysema by inhibiting neutrophil elastase (24), it also deters other processes that facilitate disease progression, such as inflammation, oxidative stress (25), impaired endothelial cell integrity, and cell death (6). Deficiencies in PP2A activity play an important role in the development of asthma (26) and COPD (14) and this study identified for the first time that PP2A activity is diminished in neutrophils from A1AT-deficient subjects. In addition, it demonstrates that A1AT treatment acts via PTP1B to increase PP2A activity in key cell types in the lung in vitro and in vivo. These effects are significant, because A1AT requires PP2A expression to prevent TNF-α–inducible inflammation and protease production in lung epithelial cells. Our laboratory has recently published that PP2A is a key factor that protects against cigarette smoke–induced inflammation and tissue destruction (13, 14). Thus, these findings indicate that diminished PP2A responses contribute significantly to emphysema development in A1AT deficiency. Moreover, they suggest that targeting PP2A activity may be an effective therapeutic strategy in A1AT-deficient subjects.
A1AT inhibits neutrophil elastase and this function has been a primary area of investigation in the pathogenesis of A1AT-related emphysema (27, 28). A1AT augmentation therapy in A1AT deficiency generates blood and lung levels of A1AT that deter neutrophil chemotaxis (12) and protect against elastase-mediated degradation of the airways (21, 29). Here we identify that A1AT deficiency is associated with decreased PP2A activity in circulating neutrophils from PiZZ subjects. Furthermore, we report that administration of A1AT protein increases PP2A activity in neutrophils and other key cell types in the development of emphysema. PP2A is a critical regulator of inflammatory responses in neutrophils. Inactivating PP2A sustained NF-κB activation (30) and prolonged and intensified the oxidative burst produced by nicotinamide adenine dinucleotide phosphate reduced oxidase in human neutrophils (31). In addition, PP2A inhibition impeded the resolution of inflammation by blocking apoptosis (32) and exacerbating toll-like receptor signaling responses in myeloid cells (33). However, studies have shown that increasing PP2A activity can induce apoptosis in these cells by inactivating p38 (32).
In our studies, we found that the loss of PTP1B expression, a PP2A activator, significantly up-regulated neutrophil cell death under baseline, noninflamed conditions. We argue that this accelerated cell death under noninflamed conditions is dysfunctional and would expose the lung to harmful proteases and render it more susceptible to infection. However, the specific effects of A1AT and PTP1B-PP2A on neutrophil necrotic or apoptotic cell death need to be explored more fully in future studies.
The adequacy of current A1AT dosage guidelines (60 mg/kg/wk) represents an area of ongoing debate in this field of research (34). The previously mentioned dose slows the rate of decline and maintains serum levels of A1AT higher than 11 μM. However, we found that high-dose A1AT therapy increased PP2A activity in lung epithelial cells and alveolar macrophages from PiZZ subjects when compared with standard dose. Moreover, this increased PP2A activity was associated with down-regulation of proinflammatory cytokines and proteases in the lung lavage and within the lung epithelium. PP2A prevents the activation of the TNF pathway and A1AT was shown to counter TNF signaling in PiZZ neutrophils (19). Based on these findings, it is conceivable that targeting higher plasma A1AT levels in this disease may enhance PP2A activity levels to counter TNF signaling and preserve lung function and integrity in this disease. Indeed, our findings set the stage for future clinical studies to address the impact of high-dose A1AT on the progression of lung disease in A1AT deficiency.
Acute cigarette smoke exposure activates PP2A, which functions to deter smoke-mediated inflammation and protease expression in the lung (14). However, the activity of PTP1B and PP2A is diminished with chronic smoke exposure (13, 14) and this loss in activity coincides with the persistent inflammation that occurs in the advanced states of COPD (13). PP2A activation also prevents the inflammatory responses that result in tissue damage in acute pancreatitis (35), kidney injury (36), and gastric epithelial cells (37). Thus, it is conceivable that impaired PP2A responses may account for the systemic inflammatory responses that occur in A1AT deficiency (38). PP2A is a known regulator of JNK, AP-1 activation (39), and NF-κB signaling (40). The present study further establishes that A1AT acts via PP2A to counter p38 and NF-κB signaling in TNF-α–stimulated lung epithelial cells. TNF signaling plays an important role in the development of emphysema (41) and A1AT suppresses TNF-α protein expression. Recently, it was demonstrated that neutrophils from PiZZ subjects had increased TNF signaling responses, with higher neutrophil membrane levels of TNF-α, plasma levels of TNF receptor 1, and neutrophil-released secondary and tertiary granule proteins (19). Given our results and the known effects of PP2A on TNF signaling (42), this raises the question whether these stimulatory effects are related to deficient PP2A responses in neutrophils from PiZZ subjects.
It is important to note that PP2A activation was dependent on the presence of functionally active A1AT because recombinant or oxidized A1AT had no effect. We do not believe that this activation is dependent on the antielastase function of A1AT. Rather, we suspect that the natural conformation of A1AT is needed to engage cell surface receptors that activate PP2A. In this study, A1AT acted via PTP1B to dephosphorylate and activate PP2A. Furthermore, we found that inhibition of PKA prevented the activation of PP2A by A1AT. PKA can activate PP2A directly by phosphorylating the B56δ subunit (43) or indirectly by activating PTP1B (44). We suspect that PKA acts indirectly via PTP1B because silencing PTP1B completely prevented the activation of both PTP1B and PP2A by A1AT. However, the upstream mechanisms by which A1AT induces PKA or PTP1B to activate PP2A remain unidentified. PTP1B activity is regulated by receptor tyrosine kinases on the cell surface (45), whereas PKA activation is linked to G protein–coupled receptors (46). Thus, future studies will address whether A1AT acts on receptor tyrosine kinases or G protein–coupled receptors to increase PTP1B-PP2A activity and counter lung and systemic inflammatory diseases in A1AT deficiency.
In conclusion, we show that functionally active A1AT protein induces PP2A activation. A1AT activates PP2A by triggering PTP1B to dephosphorylate PP2A at tyrosine 307 in the catalytic subunit of the enzyme. Once activated by A1AT, PP2A can prevent the inflammatory and proteolytic responses triggered by TNF-α stimulation in the lung. Thus, these findings set the stage for future studies to explore whether increasing PP2A activity can effectively treat individuals with A1AT deficiency or other TNF-α–driven inflammatory diseases.
Acknowledgments
Acknowledgment
The authors gratefully acknowledge the generous support of the James P. Mara Center for Lung Disease and Dr. Gerard Turino. The authors also thank the α1-antitrypsin deficiency subjects and healthy blood donors who participated in this study.
Footnotes
Supported by Flight Attendant Medical Research Institute (YCSA 113380, P.G.; YCSA 24039, R.F.F.), CSL Behring (M.C.), CIA 074047 (R.F.F.), National Institutes of Health (5R01HL098528-05, R.F.F.), and the Alpha One Foundation Research Grant (R.F.F.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Contributions: P.G., study design and execution, data collection, and manuscript preparation. E.E., M.P., M.C., N.G.M., study design and execution. R.F.F., study design, execution, data collection, and overall manuscript preparation.
Originally Published in Press as DOI: 10.1164/rccm.201405-0872OC on October 23, 2014
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Hepper NG, Black LF, Gleich GJ, Kueppers F. The prevalence of alpha 1-antitrypsin deficiency in selected groups of patients with chronic obstructive lung disease. Mayo Clin Proc. 1969;44:697–710. [PubMed] [Google Scholar]
- 2.Kalsheker NA. Molecular pathology of alpha 1-antitrypsin deficiency and its significance to clinical medicine. QJM. 1994;87:653–658. [PubMed] [Google Scholar]
- 3.Ogushi F, Fells GA, Hubbard RC, Straus SD, Crystal RG. Z-type alpha 1-antitrypsin is less competent than M1-type alpha 1-antitrypsin as an inhibitor of neutrophil elastase. J Clin Invest. 1987;80:1366–1374. doi: 10.1172/JCI113214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hunt JM, Tuder R. Alpha 1 anti-trypsin: one protein, many functions. Curr Mol Med. 2012;12:827–835. doi: 10.2174/156652412801318755. [DOI] [PubMed] [Google Scholar]
- 5.Bergin DA, Hurley K, McElvaney NG, Reeves EP. Alpha-1 antitrypsin: a potent anti-inflammatory and potential novel therapeutic agent. Arch Immunol Ther Exp (Warsz) 2012;60:81–97. doi: 10.1007/s00005-012-0162-5. [DOI] [PubMed] [Google Scholar]
- 6.Petrache I, Fijalkowska I, Medler TR, Skirball J, Cruz P, Zhen L, Petrache HI, Flotte TR, Tuder RM. alpha-1 antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosis. Am J Pathol. 2006;169:1155–1166. doi: 10.2353/ajpath.2006.060058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan ED, Pott GB, Silkoff PE, Ralston AH, Bryan CL, Shapiro L. Alpha-1-antitrypsin inhibits nitric oxide production. J Leukoc Biol. 2012;92:1251–1260. doi: 10.1189/jlb.0212071. [DOI] [PubMed] [Google Scholar]
- 8.Shapiro L, Pott GB, Ralston AH. Alpha-1-antitrypsin inhibits human immunodeficiency virus type 1. FASEB J. 2001;15:115–122. doi: 10.1096/fj.00-0311com. [DOI] [PubMed] [Google Scholar]
- 9.Lockett AD, Kimani S, Ddungu G, Wrenger S, Tuder RM, Janciauskiene SM, Petrache I. α₁-Antitrypsin modulates lung endothelial cell inflammatory responses to TNF-α. Am J Respir Cell Mol Biol. 2013;49:143–150. doi: 10.1165/rcmb.2012-0515OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carlson JA, Rogers BB, Sifers RN, Finegold MJ, Clift SM, DeMayo FJ, Bullock DW, Woo SL. Accumulation of PiZ alpha 1-antitrypsin causes liver damage in transgenic mice. J Clin Invest. 1989;83:1183–1190. doi: 10.1172/JCI113999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carroll TP, Greene CM, O’Connor CA, Nolan AM, O’Neill SJ, McElvaney NG. Evidence for unfolded protein response activation in monocytes from individuals with alpha-1 antitrypsin deficiency. J Immunol. 2010;184:4538–4546. doi: 10.4049/jimmunol.0802864. [DOI] [PubMed] [Google Scholar]
- 12.Bergin DA, Reeves EP, Meleady P, Henry M, McElvaney OJ, Carroll TP, Condron C, Chotirmall SH, Clynes M, O’Neill SJ, et al. α-1 Antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8. J Clin Invest. 2010;120:4236–4250. doi: 10.1172/JCI41196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geraghty P, Hardigan AA, Wallace AM, Mirochnitchenko O, Thankachen J, Arellanos L, Thompson V, D'Armiento JM, Foronjy RF. The glutathione peroxidase 1-protein tyrosine phosphatase 1B-protein phosphatase 2A axis: a key determinant of airway inflammation and alveolar destruction. Am J Respir Cell Mol Biol. 2013;49:721–730. doi: 10.1165/rcmb.2013-0026OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wallace AM, Hardigan A, Geraghty P, Salim S, Gaffney A, Thankachen J, Arellanos L, D’Armiento JM, Foronjy RF. Protein phosphatase 2A regulates innate immune and proteolytic responses to cigarette smoke exposure in the lung. Toxicol Sci. 2012;126:589–599. doi: 10.1093/toxsci/kfr351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science. 1992;257:1261–1264. doi: 10.1126/science.1325671. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Parsons S, Brautigan DL. Tyrosine phosphorylation of protein phosphatase 2A in response to growth stimulation and v-src transformation of fibroblasts. J Biol Chem. 1994;269:7957–7962. [PubMed] [Google Scholar]
- 17.Churg A, Wang RD, Xie C, Wright JL. alpha-1-Antitrypsin ameliorates cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med. 2003;168:199–207. doi: 10.1164/rccm.200302-203OC. [DOI] [PubMed] [Google Scholar]
- 18.Chidwick K, Whichelow CE, Zhang Z, Fairburn K, Sachs JA, Blake DR, Winyard PG. Relationship between alpha 1-antitrypsin inactivation and tumor necrosis factor alpha concentration in the synovial fluid of patients with rheumatoid arthritis. Arthritis Rheum. 1994;37:1723–1726. doi: 10.1002/art.1780371203. [DOI] [PubMed] [Google Scholar]
- 19.Bergin DA, Reeves EP, Hurley K, Wolfe R, Jameel R, Fitzgerald S, McElvaney NG.The circulating proteinase inhibitor alpha-1 antitrypsin regulates neutrophil degranulation and autoimmunity Science Translational Medicine 20146217ra211 [DOI] [PubMed] [Google Scholar]
- 20.Geraghty P, Eden E, Pillai M, Campos M, McElvaney N, Foronjy R.Alpha-1 antitrypsin counters inflammation by modulating the activity of protein phosphatase 2a [abstract]. Am J Respir Crit Care Med 2014;189:A4012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campos MA, Kueppers F, Stocks JM, Strange C, Chen J, Griffin R, Wang-Smith L, Brantly ML. Safety and pharmacokinetics of 120 mg/kg versus 60 mg/kg weekly intravenous infusions of alpha-1 proteinase inhibitor in alpha-1 antitrypsin deficiency: a multicenter, randomized, double-blind, crossover study (SPARK) COPD. 2013;10:687–695. doi: 10.3109/15412555.2013.800852. [DOI] [PubMed] [Google Scholar]
- 22.Taggart CC, Greene CM, McElvaney NG, O’Neill S. Secretory leucoprotease inhibitor prevents lipopolysaccharide-induced IkappaBalpha degradation without affecting phosphorylation or ubiquitination. J Biol Chem. 2002;277:33648–33653. doi: 10.1074/jbc.M203710200. [DOI] [PubMed] [Google Scholar]
- 23.Stockley RA. Neutrophils and protease/antiprotease imbalance. Am J Respir Crit Care Med. 1999;160:S49–S52. doi: 10.1164/ajrccm.160.supplement_1.13. [DOI] [PubMed] [Google Scholar]
- 24.Janoff A. Elastases and emphysema. Current assessment of the protease-antiprotease hypothesis. Am Rev Respir Dis. 1985;132:417–433. doi: 10.1164/arrd.1985.132.2.417. [DOI] [PubMed] [Google Scholar]
- 25.Brantly M. Alpha1-antitrypsin: not just an antiprotease: extending the half-life of a natural anti-inflammatory molecule by conjugation with polyethylene glycol. Am J Respir Cell Mol Biol. 2002;27:652–654. doi: 10.1165/rcmb.F250. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi Y, Mercado N, Barnes PJ, Ito K. Defects of protein phosphatase 2A causes corticosteroid insensitivity in severe asthma. PLoS ONE. 2011;6:e27627. doi: 10.1371/journal.pone.0027627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hubbard RC, Fells G, Gadek J, Pacholok S, Humes J, Crystal RG. Neutrophil accumulation in the lung in alpha 1-antitrypsin deficiency. Spontaneous release of leukotriene B4 by alveolar macrophages. J Clin Invest. 1991;88:891–897. doi: 10.1172/JCI115391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma S, Lin YY, He J, Rouhani FN, Brantly M, Turino GM. Alpha-1 antitrypsin augmentation therapy and biomarkers of elastin degradation. COPD. 2013;10:473–481. doi: 10.3109/15412555.2013.771163. [DOI] [PubMed] [Google Scholar]
- 29.Wewers MD, Crystal RG. Alpha-1 antitrypsin augmentation therapy. COPD. 2013;10:64–67. doi: 10.3109/15412555.2013.764402. [DOI] [PubMed] [Google Scholar]
- 30.Miskolci V, Castro-Alcaraz S, Nguyen P, Vancura A, Davidson D, Vancurova I. Okadaic acid induces sustained activation of NFkappaB and degradation of the nuclear IkappaBalpha in human neutrophils. Arch Biochem Biophys. 2003;417:44–52. doi: 10.1016/s0003-9861(03)00336-9. [DOI] [PubMed] [Google Scholar]
- 31.Lu DJ, Takai A, Leto TL, Grinstein S. Modulation of neutrophil activation by okadaic acid, a protein phosphatase inhibitor. Am J Physiol. 1992;262:C39–C49. doi: 10.1152/ajpcell.1992.262.1.C39. [DOI] [PubMed] [Google Scholar]
- 32.Alvarado-Kristensson M, Andersson T. Protein phosphatase 2A regulates apoptosis in neutrophils by dephosphorylating both p38 MAPK and its substrate caspase 3. J Biol Chem. 2005;280:6238–6244. doi: 10.1074/jbc.M409718200. [DOI] [PubMed] [Google Scholar]
- 33.Qiu Q, Zheng Z, Chang L, Zhao YS, Tan C, Dandekar A, Zhang Z, Lin Z, Gui M, Li X, et al. Toll-like receptor-mediated IRE1α activation as a therapeutic target for inflammatory arthritis. EMBO J. 2013;32:2477–2490. doi: 10.1038/emboj.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stockley RA, Miravitlles M, Vogelmeier C Alpha One International Registry (A.I.R.) Augmentation therapy for alpha-1 antitrypsin deficiency: towards a personalised approach. Orphanet J Rare Dis. 2013;8:149. doi: 10.1186/1750-1172-8-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandoval J, Escobar J, Pereda J, Sacilotto N, Rodriguez JL, Sabater L, Aparisi L, Franco L, López-Rodas G, Sastre J. Pentoxifylline prevents loss of PP2A phosphatase activity and recruitment of histone acetyltransferases to proinflammatory genes in acute pancreatitis. J Pharmacol Exp Ther. 2009;331:609–617. doi: 10.1124/jpet.109.157537. [DOI] [PubMed] [Google Scholar]
- 36.Jin Jung K, Hyun Kim D, Kyeong Lee E, Woo Song C, Pal Yu B, Young Chung H. Oxidative stress induces inactivation of protein phosphatase 2A, promoting proinflammatory NF-κB in aged rat kidney. Free Radic Biol Med. 2013;61C:206–217. doi: 10.1016/j.freeradbiomed.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 37.Chung HY, Cha B, Kim H. Inhibition of serine-threonine protein phosphatases in monocyte chemoattractant protein-1 expression in Helicobacter pylori-stimulated gastric epithelial cells. Ann N Y Acad Sci. 2007;1095:220–227. doi: 10.1196/annals.1397.026. [DOI] [PubMed] [Google Scholar]
- 38.Escobar J, Pereda J, Arduini A, Sandoval J, Sabater L, Aparisi L, Lopez-Rodas G, Sastre J. Protein phosphatases and chromatin modifying complexes in the inflammatory cascade in acute pancreatitis. World J Gastrointest Pharmacol Ther. 2010;1:75–80. doi: 10.4292/wjgpt.v1.i3.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Avdi NJ, Malcolm KC, Nick JA, Worthen GS. A role for protein phosphatase-2A in p38 mitogen-activated protein kinase-mediated regulation of the c-Jun NH(2)-terminal kinase pathway in human neutrophils. J Biol Chem. 2002;277:40687–40696. doi: 10.1074/jbc.M204455200. [DOI] [PubMed] [Google Scholar]
- 40.Miskolci V, Castro-Alcaraz S, Nguyen P, Vancura A, Davidson D, Vancurova I. Okadaic acid induces sustained activation of NFkappaB and degradation of the nuclear IkappaBalpha in human neutrophils. Arch Biochem Biophys. 2003;417:44–52. doi: 10.1016/s0003-9861(03)00336-9. [DOI] [PubMed] [Google Scholar]
- 41.Churg A, Dai J, Tai H, Xie C, Wright JL. Tumor necrosis factor-alpha is central to acute cigarette smoke-induced inflammation and connective tissue breakdown. Am J Respir Crit Care Med. 2002;166:849–854. doi: 10.1164/rccm.200202-097OC. [DOI] [PubMed] [Google Scholar]
- 42.Härmälä-Braskén AS, Mikhailov A, Söderström TS, Meinander A, Holmström TH, Damuni Z, Eriksson JE. Type-2A protein phosphatase activity is required to maintain death receptor responsiveness. Oncogene. 2003;22:7677–7686. doi: 10.1038/sj.onc.1207077. [DOI] [PubMed] [Google Scholar]
- 43.Ahn JH, McAvoy T, Rakhilin SV, Nishi A, Greengard P, Nairn AC. Protein kinase A activates protein phosphatase 2A by phosphorylation of the B56delta subunit. Proc Natl Acad Sci USA. 2007;104:2979–2984. doi: 10.1073/pnas.0611532104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tao J, Malbon CC, Wang HY. Insulin stimulates tyrosine phosphorylation and inactivation of protein-tyrosine phosphatase 1B in vivo. J Biol Chem. 2001;276:29520–29525. doi: 10.1074/jbc.M103721200. [DOI] [PubMed] [Google Scholar]
- 45.Dadke S, Kusari A, Kusari J. Phosphorylation and activation of protein tyrosine phosphatase (PTP) 1B by insulin receptor. Mol Cell Biochem. 2001;221:147–154. doi: 10.1023/a:1010909031310. [DOI] [PubMed] [Google Scholar]
- 46.Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev. 2005;85:1303–1342. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]