

Abstract
Rationale: Red blood cell (RBC) transfusions are associated with increased risk of acute respiratory distress syndrome (ARDS) in the critically ill, yet the mechanisms for enhanced susceptibility to ARDS conferred by RBC transfusions remain unknown.
Objectives: To determine the mechanisms of lung endothelial cell (EC) High Mobility Group Box 1 (HMGB1) release following exposure to RBCs and to determine whether RBC transfusion increases susceptibility to lung inflammation in vivo through release of the danger signal HMGB1.
Methods: In vitro studies examining human lung EC viability and HMGB1 release following exposure to allogenic RBCs were conducted under static conditions and using a microengineered model of RBC perfusion. The plasma from transfused and nontransfused patients with severe sepsis was examined for markers of cellular injury. A murine model of RBC transfusion followed by LPS administration was used to determine the effects of RBC transfusion and HMGB1 release on LPS-induced lung inflammation.
Measurements and Main Results: After incubation with RBCs, lung ECs underwent regulated necrotic cell death (necroptosis) and released the essential mediator of necroptosis, receptor-interacting serine/threonine-protein kinase 3 (RIP3), and HMGB1. RIP3 was detectable in the plasma of patients with severe sepsis, and was increased with blood transfusion and among nonsurvivors of sepsis. RBC transfusion sensitized mice to LPS-induced lung inflammation through release of the danger signal HMGB1.
Conclusions: RBC transfusion enhances susceptibility to lung inflammation through release of HMGB1 and induces necroptosis of lung EC. Necroptosis and subsequent danger signal release is a novel mechanism of injury following transfusion that may account for the increased risk of ARDS in critically ill transfused patients.
Keywords: RBC transfusion, necroptosis, endothelial, HMGB1, RIP3
At a Glance Commentary
Scientific Knowledge on the Subject
In critically ill subjects red blood cell (RBC) transfusions are associated with increased risk of the acute respiratory distress syndrome. However, whether RBC transfusions increase host susceptibility to injury and how this occurs remain unknown.
What This Study Adds to the Field
We demonstrate that banked RBCs induce regulated necrotic cell death (necroptosis) of human lung endothelial cells and subsequent release of the danger signal high-mobility group box 1 (HMGB1) and the mediator of necroptosis, receptor-interacting serine/threonine-protein kinase 3 (RIP3). RIP3 was elevated in nonsurvivors and transfused subjects with sepsis. Furthermore, we demonstrate that RBC transfusion enhances susceptibility to lung inflammation in vivo through HMGB1 release, confirming the notion that RBC transfusions confer increased risk of lung injury in susceptible populations. Collectively, our findings identify lung endothelial necroptosis as a novel mechanism of injury following transfusion.
Critically ill patients seem to be most vulnerable to the deleterious effects of red blood cell (RBC) transfusates, because studies have demonstrated increased morbidity and mortality associated with RBC transfusions in this population (1–4). Although epidemiologic studies have shown an association between the duration of red cell storage and adverse outcomes, it remains unclear whether the red cell storage lesion contributes to injury in susceptible hosts (5–7). Despite uncertainty whether the duration of storage contributes to the adverse effects of transfusates, it is well established that RBC transfusions are associated with increased risk of acute respiratory distress syndrome (ARDS). Multiple studies have demonstrated incremental risk with each RBC unit transfused, suggesting a possible causal relationship between RBC transfusion and ARDS in susceptible populations (2, 3, 8–10).
We previously investigated the interactions between allogenic RBCs and lung microvascular endothelial cells (ECs) and have found that banked human RBCs induce lung EC dysfunction marked by increased reactive oxygen species (ROS) generation (11). In vivo, syngeneic RBC transfusion led to increased lung endothelial activation and elevated levels of the proinflammatory danger signal high-mobility group box 1 (HMGB1) in the lung (12). HMGB1 is referred to as a classic damage-associated molecular pattern (DAMP) because intracellular HMGB1 is present in all nucleated cells and is critical in the homeostasis of most living cells, whereas extracellular HMGB1 possesses immunomodulatory capabilities. Extracellular HMGB1 can initiate and sustain the inflammatory response through ligation of pattern recognition receptors, including receptor for advanced glycation endproducts and toll-like receptor (TLR) 2 and TLR4 (13–17). Although HMGB1 can be secreted by immune cells, it is also released by necrotic cells (13, 15). Recently the release of DAMPs following regulated necrotic cell death (necroptosis) has garnered significant interest because it is increasingly appreciated that the form of cell death may alter the immunologic properties of DAMPs and modify the host response to injury (18, 19).
Based on our previous findings of vascular activation and increased lung HMGB1 expression following RBC transfusion we hypothesized that RBC transfusions enhance susceptibility to lung inflammation though HMGB1 release (12). We asked whether human lung EC released HMGB1 following interaction with RBCs and examined the role of regulated necrotic death as a mechanism of HMGB1 release from lung endothelium (11). Lastly, we determined the association of plasma levels of Receptor interacting serine/threonine-protein kinase 3 (RIP3), a critical regulator of necroptosis, with RBC transfusions and mortality in patients with severe sepsis. Some of the results of these studies have been previously reported in the form of an abstract (20).
Methods
A detailed description of the materials and methods can be found in the online supplement.
Preparation of Human RBCs
Studies involving human subjects were approved by the University of Pennsylvania Institutional Review Board. Leukoreduced RBC units were obtained from the blood bank at the Hospital of the University of Pennsylvania and used after 21 days of storage.
Detection of HMGB1 Release from ECs
Human lung microvascular ECs (HMVEC-L; Lonza) were grown to confluence and incubated with 1 × 108 RBCs in endothelial growth medium (Lonza, Basel, Switzerland) for 4 hours at 37°C. For select studies, cells were pretreated with 50 μM necrostatin-1 (Nec-1; Sigma Aldrich, St. Louis, MO) or 20 μM Z-VAD-FMK (Promega, Madison, WI) for 1 hour at 37°C. Supernatants and RBC lysates were collected and HMGB1 concentrations were assayed by ELISA (Chondrex, Redmond, WA) and reported as total HMGB1 per 106 EC.
Detection of Necrosome Formation in Lung EC
Immunoprecipitation
HMVEC-L were treated with RBCs as described previously. Mouse monoclonal anti-RIP1 Ab (Abcam, Cambridge, MA) was used for immunoprecipitation and immunoblotting was performed using rabbit polyclonal anti-RIP3 and mouse monoclonal anti-RIP1 (Abcam).
Proximity ligation assay
RIP1 and RIP3 interactions were detected using the proximity ligation assay (Duolink; Sigma Aldrich). Quantification of proximity ligation assay puncta was performed using ImageJ (NIH) software.
Detection of RIP3 in Human Subjects
Subjects were selected from the Molecular Epidemiology of Severe Sepsis in the ICU cohort study at the University of Pennsylvania (21, 22) if they had plasma available from the date of admission to the medical intensive care unit and repeat plasma 2 days later. Details on the cohort can be found in the online supplement.
Plasma RIP3 levels were measured by ELISA (CUSABIO, Wuhan, China) and compared between categorical groups by Wilcoxon rank sum test.
Experimental Animals
C57Bl/6 animals were purchased from the Charles River Laboratories Inc. (Wilmington, MA). Experimental procedures were performed on 8- to 12-week-old male mice, 25–30 g in weight. Animal studies were conducted in accordance with the Institutional Animal Care and Use Committee at the University of Pennsylvania. Details of the in vivo transfusion model can be found in the online supplement.
Results
RBCs Induce HMGB1 Release from Human Lung ECs
Because we have previously observed increased lung HMGB1 expression following RBC transfusion and increased lung endothelial dysfunction following interaction with banked RBCs, we asked whether lung ECs were a potential source of HMGB1. We first tested the ability of multiple RBC units to induce HMGB1 release from HMVEC-L. Although there was significant heterogeneity in the ability of the RBCs to release HMGB1, we observed an increase in HMGB1 release in RBC-stimulated EC when compared with naive EC (P < 0.001) (Figure 1A). To examine the cellular localization of HMGB1, we performed imaging of naive and RBC-stimulated HMVEC-L. Although most naive ECs displayed nuclear HMGB1 staining, several RBC-stimulated ECs demonstrated no HMGB1 staining 4 hours following RBC stimulation (Figures 1B and 1C). Strikingly, RBC-treated ECs also displayed marked plasma membrane loss and nuclear swelling.
Figure 1.

Red blood cells (RBCs) induce high-mobility group box 1 (HMGB1) release from lung endothelial cells (ECs). (A) HMGB1 release following stimulation of human lung microvascular ECs with RBCs for 4 hours, *P < 0.001. Data represents two independent studies, eight different RBC units tested. (B) HMGB1 staining in lung EC 4 hours after RBC treatment. red = HMGB1; blue = DAPI; green = F-actin. (C) Quantification of cells displaying abnormal F-actin organization, RBC-treated ECs display a greater percentage of cells with cytoskeletal rearrangement, *P = 0.02. Images are representative of three independent experiments. Original magnification ×40. DAPI = 4′,6-diamidino-2-phenylindole.
Stored RBCs Decrease Lung EC Viability
The appearance of the RBC-treated ECs suggested that many of them had undergone regulated necrotic cell death, or necroptosis. Necroptosis is a recently described alternate cell death pathway that is critical in host defense, particularly in the setting of viral infections (23–25). In contrast to apoptosis, necroptosis is morphologically characterized by organelle swelling, lack of nuclear fragmentation, and ultimately plasma membrane rupture with consequent DAMP release. We first tested the effects of several different RBC units on lung EC viability as measured by counting the number of ECs following stimulation with RBC concentrates. The addition of red cell concentrates to lung ECs decreased recoverable lung ECs by a small (14%) but statistically significant amount (P = 0.009) (Figure 2A). We also measured lactate dehydrogenase release by lung EC as a marker of cell death. RBCs led to a significant increase in lactate dehydrogenase release (P < 0.01) (Figure 2B) that correlated with HMGB1 release (Spearman rank order correlation coefficient, r = 0.92; P < 0.001) (Figure 2C).
Figure 2.

Red blood cells (RBCs) induce lung endothelial cell (EC) death. (A) Following stimulation with RBCs, there is decreased recovery of lung EC, *P = 0.009. Eleven RBC units tested. Each open circle represents a different RBC unit, data representative of four independent studies. (B) RBC treatment increases lactate dehydrogenase (LDH) release, *P < 0.01. (C) Correlation between high-mobility group box 1 (HMGB1) release and LDH, Spearman rank sum correlation coefficient = 0.92, P < 0.001.
RBC-induced Necroptosis of Lung EC Mediates HMGB1 Release
Regulated necrotic cell death can be induced through the addition of noxious stimuli and through the engagement of pathogen-sensing receptors, such as the TLRs, particularly when caspases are inhibited (23, 26). The assembly of intracellular RIP kinase–containing complexes is necessary for programmed necrotic cell death. Although RIP1 is dispensable in some forms of necroptosis, an essential role of RIP3 and the downstream pseudokinase, mixed lineage kinase domain-like, in necroptosis is emerging (23–25, 27–29). To determine whether stored RBC units induced programmed cell death in lung EC, we measured the presence of RIP1-RIP3–containing death complexes in naive and RBC-treated ECs. One distinguishing feature of necroptosis is the formation of RIP1-RIP3–containing punctate structures (25). HMVEC-L stimulated with RBCs demonstrated colocalization of RIP1 and RIP3 and discrete puncta (Figure 3A) that were not observed in naive HMVEC-L (Figures 3A and 3B) (P = 0.015). We next tested the ability of multiple banked RBC units to increase RIP1 and RIP3 interactions. We performed immunoprecipitation of RIP1-containing complexes in naive and RBC-stimulated HMVEC-L under serum-free conditions and found increased RIP3 expression in RIP1-containing complexes isolated from RBC-treated HMVEC-L (Figures 3C and 3D) (P < 0.01). Enhanced RIP1-RIP3 interaction following RBC treatment was confirmed using the proximity ligation assay (Figures 3E and 3F).
Figure 3.

Red blood cells (RBCs) induce formation of the necrosome complex in lung endothelial cells (ECs). (A) receptor-interacting serine/threonine-protein kinase 1 (RIP1) and RIP3 in naive and RBC-treated human lung microvascular ECs (HMVEC-L). (B) Quantification of colocalization, P = 0.015. (C) Detection of RIP1 and RIP3 containing complexes by immunoprecipitation. (D) Densitometery of RIP1-RIP3 complexes, P < 0.01. Data represent four independent studies, five RBC units tested, one representative RBC unit depicted in Figure 5C. (E) Proximity ligation assay for RIP1 and RIP3 in naive and RBC-treated HMVEC-L, cells treated with tumor necrosis factor (TNF)-α and ZVAD are depicted in the rightmost panel. (F) Quantification of PLA puncta, *P < 0.01 when compared with EC alone. PLA = proximity ligation assay.
To confirm that RBC-induced death of lung EC was secondary to necroptosis, we tested the ability of RBC units to induce EC death in the presence of the pancaspase inhibitor ZVAD-FMK, which would block apoptosis, prevent cleavage of RIP kinases, and allow for the initiation of necroptosis (26, 30). Although we observed significant heterogeneity in the ability of RBC units to induce necroptosis, 5 out of 10 RBC preparations tested induced caspase-independent cell death of HMVEC-L (Figure 4A). We next asked whether induction of lung EC necroptosis by RBC was a mechanism of HMGB1 release following RBC stimulation by using the chemical inhibitor Nec-1, which interferes with RIP1 activity and blocks necroptosis (31). Inhibition of necroptosis attenuated RBC-induced EC HMGB1 release (Figures 4B and 4C). Collectively, these data demonstrate that banked RBCs induce programmed necrotic death of lung EC and subsequent release of HMGB1.
Figure 4.

Red blood cells (RBCs) induce necroptosis of lung endothelial cells (ECs) and inhibition of necroptosis attenuates lung EC high-mobility group box 1 (HMGB1) release. (A) Viability of human lung microvascular ECs following RBC treatment, *P = 0.054. (B) HMGB1 release following RBC treatment, P = 0.03. necrostatin (Nec)-1–ZVAD significantly attenuates HMGB1 release with three of the six units tested, P ≤ 0.01. Data representative of three independent studies. (C) HMGB1 staining in lung EC 4 hours after RBC treatment or RBC treatment in the presence of Nec-1 (50 μM). red = HMGB1; blue = DAPI; green = F-actin. DAPI = 4′,6-diamidino-2-phenylindole.
Human ECs Lose HMGB1 after RBC Perfusion
Although previous studies have examined endothelial responses to RBC perfusion in murine isolated lung perfusion models, it remains unclear how allogeneic human RBCs affect the lung endothelium under physiologically relevant conditions that recapitulate dynamic blood flow (32). We therefore used a microengineered model that allowed for long-term microfluidic cell culture to produce intact human pulmonary microvascular endothelium (33, 34). To simulate physiologically relevant flow conditions during RBC transfusion, we integrated a programmable fluid pumping system in the microdevice. Although perfusion with media alone did not alter HMGB1 expression, introduction of RBCs led to loss of nuclear HMGB1 from multiple cells (see Figure E1 in the online supplement) (P < 0.001). These data confirm our previous observations of RBC-induced EC necroptosis and HMGB1 release.
RIP3 Is Released Extracellularly and Is Elevated in the Plasma of Transfused Patients
Although HMGB1 is released secondary to programmed necrosis, HMGB1 is not a specific marker of cellular necroptosis because HMGB1 can also be released from activated immune cells during inflammatory states including both sterile and pathogen-induced inflammation. Furthermore, apoptotic cells that were previously thought to be immunologically silent can release HMGB1 through various mechanisms (35, 36). Because it has previously been reported that RIP1-RIP3 proteins can form an ultrastable complex and others have detected extracellular release of RIP3 from cultured cells, we sought to determine if RIP3 is released extracellularly and could potentially serve as a marker of necrosis and cellular injury (23, 24, 37). We first examined supernatant RIP3 levels by immunoblot in naive ECs and ECs treated with RBCs. Under basal conditions, we detected minimal supernatant RIP3. However, following RBC stimulation, we were able to detect RIP3 in the supernatant (P = 0.021) (Figures 5A and 5B). Notably there was heterogeneity in the ability of the RBCs to induce RIP3 release. We next asked whether RIP3 release was dependent on necroptosis. ECs treated with RBCs demonstrated marked loss of RIP3, which was attenuated with the addition of Nec-1 (Figure 5C). These results were confirmed by ELISA because EC supernatant demonstrated increased RIP3 following incubation with RBCs that was attenuated with necroptosis inhibition (Figure 5D).
Figure 5.

Receptor interacting serine/threonine-protein kinase 3 (RIP3) is released extracellularly after treatment with red blood cells (RBCs). (A) Lung endothelial cells (ECs) supernatant (SN) RIP3 is increased following treatment with RBCs; note the heterogeneity in RIP3 release induced by RBCs. (B) SN densitometry, P = 0.021, total of nine RBC units tested. (C) Lung EC RIP3 decreases following treatment with RBC, addition of necrostatin (Nec)-1–ZVAD attenuates loss of EC RIP3. (D) Supernatant RIP3 is increased following RBC treatment and attenuated with Nec-1, *P = 0.002, +P = 0.045.
To determine the clinical significance of our findings we asked whether RIP3 was detectable in the plasma of patients with severe sepsis. Plasma RIP3 was measured in 37 subjects on Day 0 and Day 2 following intensive care unit admission. Clinical characteristics of subjects are shown in Table 1. Compared with subjects who were never transfused, subjects who received blood transfusions had higher RIP3 on Day 2, median (interquartile range) was 358.7 (15–4,102.7) versus 2,692.0 (1,034.5–4,226.0) pg/ml, nontransfused versus transfused (P = 0.046) (Figure 6A). In addition, we tested for an association between plasma RIP3 and blood transfusion by quantile regression of Day 2 RIP3 levels by the volume of red cells transfused between Day 0 and Day 2. Plasma RIP3 levels increased with each additional milliliter of RBCs, with a coefficient of 1.27 (95% confidence interval, 0.11–2.42; P = 0.033) (see Figure E2). Furthermore, subjects who died had higher plasma RIP3 on Day 2 than survivors (2,962.0 [398.9–4,815.5] vs. 650.8 [50.8–1,129.1] pg/ml; nonsurvivors vs. survivors, P = 0.039), although their Day 0 levels were equivalent (Figures 6B and 6C). Although volume of blood transfused and incident ARDS were both associated with mortality in the parent cohort as described (22), RIP3 levels did not vary significantly by ARDS status in this small sample. Thus RIP3 is detectable in the plasma of patients and is associated with both RBC transfusion and mortality.
Table 1.
Clinical Characteristics of Subjects with Sepsis in Whom Plasma RIP3 Was Measured
| Transfused (n = 19) | Not Transfused (n = 18) | P Value | |
|---|---|---|---|
| Age | 56.5 ± 17.1 | 60.5 ± 17.3 | 0.48 |
| Race | |||
| African American | 7 (36.8%) | 3 (18.8%) | 0.18 |
| White | 12 (63.2%) | 12 (75.0%) | |
| Asian | 1 (6.3%) | ||
| APACHE 3 | 60 (43–93) | 60 (44–67) | 0.28 |
| Red blood cells, ml | 600 (600–900) | 0 (0–0) | <0.001 |
| ARDS by Day 5 | 14 (73.7%) | 10 (55.6%) | 0.25 |
| Hospital mortality | 12 (63.2%) | 13 (72.2%) | 0.56 |
Definition of abbreviations: APACHE = Acute Physiology and Chronic Health Evaluation; ARDS = acute respiratory distress syndrome; RIP3 = receptor-interacting serine/threonine-protein kinase 3.
The volume of packed red cells transfused over the first 3 days is shown in milliliters. ARDS was diagnosed in accordance with Berlin criteria. Variables are reported as mean ± SD, as median (interquartile range), or as number (proportion) and compared by Student t test, Wilcoxon rank sum test, or chi-square test as appropriate.
Figure 6.

Plasma receptor-interacting serine/threonine-protein kinase 3 (RIP3) levels in septic subjects are higher among patients who are transfused and nonsurvivors. (A) Box-and-whisker plots of RIP3 levels at Day 2 for patients with sepsis who were transfused (n = 19) versus never transfused (n = 18). The median RIP3 in nontransfused patients was 358.7 pg/ml versus 2,692.0 pg/ml in the transfused patients (P = 0.046.) RIP3 levels at (B) Day 0 and (C) Day 2 in patients with sepsis classified by survival (survivors, n = 12; nonsurvivors, n = 25). Levels were compared by Wilcoxon rank sum test. Boxes display medians with interquartile ranges; error bars indicate 10th to 90th percentiles.
RBC Transfusion Increases RIP1-RIP3 Interactions in the Lung of Transfused Mice and Plasma HMGB1 and RIP3 Is Increased after Transfusion
We next asked whether we could detect necrosome formation in murine lung following RBC transfusion. Naive mice were transfused with syngeneic RBCs as previously described (12). As depicted in Figure 7A, RBC transfusion led to enhanced RIP1-RIP3 interactions in murine lung. Because we detected both HMGB1 and RIP3 release from RBC-treated human ECs, we next measured plasma levels of HMGB1 and RIP3 following RBC transfusion. Plasma HMGB1 and RIP3 were significantly increased following RBC transfusion (35.2 ± 10.6 ng/ml vs. 82.3 ± 16.7 ng/ml, P = 0.027 for HMGB1; 40.8 ± 34.86 ng/ml vs. 469.4 ± 192.2 ng/ml, P = 0.034 for RIP3) (Figures 7C and 7D). We also observed a significant correlation between plasma HMGB1 and RIP3 (Pearson correlation coefficient, 0.70; P = 0.004, data not shown). Despite these observations and consistent with our previous findings, transfusion of stored RBCs to naive mice did not lead to increased lung or systemic chemokine release (Figure 8A; see Figure E3) (38).
Figure 7.

Receptor interacting serine/threonine-protein kinase 3 (RIP3) and high-mobility group box 1 (HMGB1) are increased following red blood cell (RBC) transfusion in mice. (A) Proximity ligation assay for RIP1 and RIP3 in the lungs of phosphate-buffered saline (PBS) or RBC transfused mice (n = 6). (B) Quantification of PLA puncta, *P < 0.001. Original magnification, ×40 (C) Plasma HMGB1 following transfusion, *P = 0.027. (D) Plasma RIP3 following transfusion (*P = 0.034). n = 9–12 mice per group. PLA = proximity ligation assay .
Figure 8.
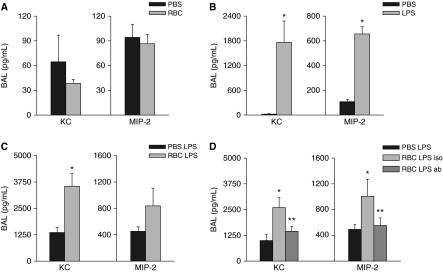
High-mobility group box 1 (HMGB1) mediates enhanced lung inflammatory responses following red blood cell (RBC) transfusion. (A) Bronchoalveolar lavage (BAL) KC and MIP-2 following transfusion alone, KC P = 0.71, MIP-2 P = 0.90. (B) BAL KC and MIP-2 following LPS injection, both *P < 0.01. Data represents two independent studies, n = 6 mice per group (for A and B). (C) BAL chemokines (KC *P < 0.01 and MIP-2 P = 0.35). Data represents five independent studies, n = 14–16 mice per group. (D) Transfusion with isotype antibody control increases BAL KC and MIP-2 (*P < 0.01 and *P = 0.03, respectively). Blockade of HMGB1 with blocking antibody attenuates BAL KC and MIP-2 (**P = 0.045 and **P = 0.037, respectively). Data represent three independent studies, n = 10–12 mice per group. KC = keratinocyte chemoattractant; MIP = macrophage inflammatory protein 2; PBS = phosphate-buffered saline.
RBC Transfusions Increase the Lung Inflammatory Response to LPS and Blockade of HMGB1 Attenuates Enhanced Lung Inflammatory Responses in RBC Transfused Mice
Previous studies have demonstrated augmentation of LPS-mediated inflammatory responses by HMGB1 (39, 40). We asked whether HMGB1 release following transfusion could augment LPS-induced lung inflammation. We first determined whether pretransfusion with RBCs augmented LPS-induced inflammatory responses in vivo, using a model of syngeneic RBC transfusion followed by low-dose endotoxin. Two hours following RBC transfusion, LPS was administered via tail vein. Plasma and lung chemokines were measured 4 hours later. Low-dose tail vein endotoxin administration to naive mice led to a predictable increase in bronchoalveolar lavage (BAL) chemokines 4 hours following LPS administration (Figure 8B). Transfusion of stored RBCs before LPS resulted in increased lung inflammation marked by BAL keratinocyte chemoattractant (KC) elevation compared with nontransfused LPS-treated mice (P = 0.01 and P = 0.35 for KC and macrophage inflammatory protein 2 [MIP2]) (Figure 8C). Transfusion of fresh blood did not significantly increase LPS-induced inflammatory responses (see Figure E4). Thus, transfusion with stored RBCs increases susceptibility to endotoxin-induced lung inflammation. HMGB1 was important functionally because administration of the HMGB1 neutralizing antibody significantly attenuated BAL chemokine release (P = 0.045 and P = 0.037 for KC and MIP-2 for RBC-LPS control antibody vs. RBC-LPS-HMGB1 blocking Ab) (Figure 8D). Thus, HMGB1 is released following transfusion and sensitizes the lung to subsequent injury.
Discussion
Multiple epidemiologic studies have demonstrated an association between RBC transfusion and the development of lung injury in susceptible hosts. In vitro, we demonstrate that lung ECs undergo regulated necrotic cell death and release HMGB1 and the critical regulator of necroptosis, RIP3, following incubation with banked RBCs. In transfused patients with sepsis and nonsurvivors of sepsis we demonstrate elevated plasma levels of RIP3. Furthermore, RBC transfusion led to increased RIP3 and HMGB1 release in naive mice and RBC transfusion sensitized the host to subsequent lung inflammation through a mechanism dependent on the danger signal, HMGB1. Thus, the induction of necroptosis and subsequent DAMP release is implicated as a novel mechanism of transfusion-mediated injury.
Extracellular HMGB1 is a potent mediator of inflammation whose release from activated inflammatory cells and necrotically injured cells has been well described (13, 14). In patients with septic shock persistent elevation of plasma HMGB1 differentiated survivors from nonsurvivors and elevated levels of HMGB1 were associated with mortality in a separate cohort of patients with ARDS (41, 42). Although we attempted to measure HMGB1 in the plasma of human subjects with severe sepsis, we were unable to detect plasma HMGB1 in more than half of our samples and HMGB1 was lower than anticipated in the few plasma samples we were able to measure (data not shown). We hypothesize that our difficulty may be related to the freeze-thaw cycle to which these samples were subjected, or it may be that among subjects with sepsis, other circulating mediators bind HMGB1 and make it difficult to detect by standard ELISA. HMGB1 has been described to activate microvascular endothelium and increase barrier dysfunction and permeability in lung endothelial monolayers, and lung endothelium itself has recently been described as a source of HMGB1 (43–46). We demonstrated that banked human RBCs can induce the release of HMGB1 from HMVECs.
To elucidate the mechanisms of HMGB1 release by RBC-treated EC, we noticed several striking features including increased EC death and morphologic characteristics of necroptosis. To confirm our morphologic observations of necroptosis, we identified RIP1-RIP3–containing death complexes displaying punctate-like structures following stimulation of lung EC with RBCs. Importantly, release of HMGB1 was attenuated when cells were treated with RIP1 kinase inhibitor Nec-1. Supporting a functional role of necroptosis in endothelium, recent reports have demonstrated palmitic acid induced necroptosis of human umbilical vein ECs that was independent of RIP1 but dependent on ubiquitin carboxyterminal hydrolyase (47). Because the endothelium is critical in innate immunity during sepsis, we speculate that activation of a necroptotic death program of lung EC in response to injury would enhance innate immune responses and propagation of signals systemically through the release of DAMPs. Indeed, the lung microvascular endothelium is unique because it provides a first defense against blood-borne pathogens and recent studies have suggested a critical role of lung endothelium in innate immune responses to intraalveolar stimuli (48).
Although the exact mechanisms of necroptosis execution remain unknown, mounting evidence suggests an essential role for RIP3 (23–25, 27, 29, 37, 49). Because RIP3 is an intracellular protein that undergoes nucleocytoplasmic shuttling and participates in programmed cell death pathways, extracellular release of RIP3 from HMVEC-L following necroptosis was an unexpected finding (50). We therefore sought to confirm these findings by examining the plasma of patients with sepsis for the presence of RIP3. RIP3 was not only detectable but was elevated in transfused patients with sepsis and associated with volume of RBC transfusion. RIP3 was also elevated in nonsurvivors of sepsis suggesting that continued production or abnormal clearance of RIP3 may portend a worse prognosis. However, larger studies are necessary to determine whether RIP3 can serve as a specific marker of necroptosis or predictor of mortality. Because we were powered only to detect a rather large effect size between groups, the lack of association between RIP3 and ARDS may be caused by low power, and this hypothesis should be tested in a larger population. Nonetheless, our findings of elevated RIP3 in nonsurvivors and transfused subjects also raise the possibility that RIP3 may function extracellularly to perpetuate inflammation similar to other alarmins, although the mechanisms by which this may occur remain unknown.
One unanswered question raised by our studies is how RBCs trigger necroptosis. Multiple stimuli including pathogens, death receptor engagement, oxidative damage, and aberrations in calcium homeostasis and ATP levels can trigger necroptosis. We observed increased oxidative stress in lung EC following stimulation with RBCs; however, in preliminary studies, ROS inhibition did not attenuate necroptosis or DAMP release in RBC-treated ECs (data not shown) (11). These findings are not unexpected because conflicting data regarding the induction of necroptosis by oxidants exist and suppression of necroptosis with ROS inhibition varies among different cell lines (24, 27, 51, 52). We also observed heterogeneity in the ability of RBC units to induce necroptosis and subsequent HMGB1 and RIP3 release. We speculate that this may be caused by donor factors or alterations of the RBC itself because we have previously observed heterogeneity in the expression of inflammatory ligands on banked RBCs and we did not observe a correlation between RBC storage duration and necroptosis induction or HMGB1 release (11).
Because we have previously observed increased EC release of RIP3 and HMGB1 following RBC-induced necroptosis, we asked whether RBC transfusion induced necroptosis and increased circulating HMGB1 and RIP3 in vivo. We found that transfusion alone led to enhanced RIP1-RIP3 interaction in the lung and we also observed increased plasma HMGB1, which correlated with RIP3 release following transfusion of naive mice. We speculate that enhanced release of HMGB1 and other DAMPs may reflect a subtle injury resulting from transfusion that is currently unrecognized. In this scenario, low levels of cell death may not result in clinical manifestations until the susceptible host sustains a subsequent insult resulting in injury. One mechanism by which this may occur is that DAMPs (e.g., HMGB1) released secondary to necroptosis may potentiate the innate immune response. Consistent with this theory is our observation that RBC transfusion increased host susceptibility to lung inflammation and inhibition of HMGB1 in LPS-treated animals that have been “primed” with RBC transfusion reduced lung inflammation. Thus, HMGB1 seems to be a mediator of lung inflammation following transfusion. Our findings of HMGB1-mediated lung inflammation following RBC transfusion are consistent with previous studies demonstrating HMGB1 as a potent mediator of lung injury (53).
In summary, we demonstrate that RBC transfusions can prime the host response to subsequent injury through release of the DAMP, HMGB1. Furthermore, we establish the induction of necroptosis by transfused RBCs as a novel mechanism of lung endothelial injury. Future studies examining the mechanisms of RBC-induced cell death and the pathways by which released mediators modify the host response may provide a greater understanding of the significance of RBC transfusions in contributing to disease in the critically ill and uncover novel approaches to prevent and treat transfusion-associated morbidity, including the development of ARDS, in this population.
Acknowledgments
Acknowledgment
Lung microvascular endothelial cells for studies in the microengineered model were provided by Drs. Horace Delisser, Kaori Ihida-Stansbury, and Steven Kawut under the Pulmonary Hypertension Breakthrough Initiative. Funding for the Pulmonary Hypertension Breakthrough Initiative is provided by the Cardiovascular Medical Research and Education Fund.
Footnotes
Supported by the National Heart, Lung and Blood Institute grants HL-102254 (N.J.M.) and HL-098362 (N.S.M.); University Research Foundation; University of Pennsylvania (N.J.M.); the National Institute of Diabetes and Digestive and Kidney Diseases grant DK-097307 (M.G.S.S.); the National Blood Foundation (N.S.M.); and American Lung Association (N.S.M.).
Author Contributions: D.Y.Q. performed the experiments. D.C., J.S., and D.H. designed and performed the microfluidics experiments and wrote parts of the manuscript. M.G.S.S., J.P.R., and N.J.M. conducted the clinical study with patients with severe sepsis and analyzed these data and wrote parts of the manuscript. G.S.W. analyzed and interpreted data. N.S.M. designed and performed the experiments, analyzed and interpreted data, and wrote the manuscript. All authors read and approved the manuscript.
Originally Published in Press as DOI: 10.1164/rccm.201406-1095OC on October 20, 2014
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Hébert PC, Wells G, Blajchman MA, Marshall J, Martin C, Pagliarello G, Tweeddale M, Schweitzer I, Yetisir E Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med. 1999;340:409–417. doi: 10.1056/NEJM199902113400601. [DOI] [PubMed] [Google Scholar]
- 2.Gong MN, Thompson BT, Williams P, Pothier L, Boyce PD, Christiani DC. Clinical predictors of and mortality in acute respiratory distress syndrome: potential role of red cell transfusion. Crit Care Med. 2005;33:1191–1198. doi: 10.1097/01.ccm.0000165566.82925.14. [DOI] [PubMed] [Google Scholar]
- 3.Croce MA, Tolley EA, Claridge JA, Fabian TC.Transfusions result in pulmonary morbidity and death after a moderate degree of injury J Trauma 20055919–23.discussion 23–14 [DOI] [PubMed] [Google Scholar]
- 4.Corwin HL, Gettinger A, Pearl RG, Fink MP, Levy MM, Abraham E, MacIntyre NR, Shabot MM, Duh MS, Shapiro MJ. The CRIT Study: Anemia and blood transfusion in the critically ill—current clinical practice in the United States. Crit Care Med. 2004;32:39–52. doi: 10.1097/01.CCM.0000104112.34142.79. [DOI] [PubMed] [Google Scholar]
- 5.Kor DJ, Kashyap R, Weiskopf RB, Wilson GA, van Buskirk CM, Winters JL, Malinchoc M, Hubmayr RD, Gajic O. Fresh red blood cell transfusion and short-term pulmonary, immunologic, and coagulation status: a randomized clinical trial. Am J Respir Crit Care Med. 2012;185:842–850. doi: 10.1164/rccm.201107-1332OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koch CG, Li L, Sessler DI, Figueroa P, Hoeltge GA, Mihaljevic T, Blackstone EH. Duration of red-cell storage and complications after cardiac surgery. N Engl J Med. 2008;358:1229–1239. doi: 10.1056/NEJMoa070403. [DOI] [PubMed] [Google Scholar]
- 7.Janz DR, Zhao Z, Koyama T, May AK, Bernard GR, Bastarache JA, Young PP, Ware LB. Longer storage duration of red blood cells is associated with an increased risk of acute lung injury in patients with sepsis. Ann Intensive Care. 2013;3:33. doi: 10.1186/2110-5820-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silverboard H, Aisiku I, Martin GS, Adams M, Rozycki G, Moss M. The role of acute blood transfusion in the development of acute respiratory distress syndrome in patients with severe trauma. J Trauma. 2005;59:717–723. [PubMed] [Google Scholar]
- 9.Zilberberg MD, Carter C, Lefebvre P, Raut M, Vekeman F, Duh MS, Shorr AF. Red blood cell transfusions and the risk of acute respiratory distress syndrome among the critically ill: a cohort study. Crit Care. 2007;11:R63. doi: 10.1186/cc5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holena DN, Netzer G, Localio R, Gallop RJ, Bellamy SL, Meyer NJ, Shashaty MG, Lanken PN, Kaplan S, Reilly PM, et al. The association of early transfusion with acute lung injury in patients with severe injury. J Trauma Acute Care Surg. 2012;73:825–831. doi: 10.1097/TA.0b013e318256de38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mangalmurti NS, Chatterjee S, Cheng G, Andersen E, Mohammed A, Siegel DL, Schmidt AM, Albelda SM, Lee JS. Advanced glycation end products on stored red blood cells increase endothelial reactive oxygen species generation through interaction with receptor for advanced glycation end products. Transfusion. 2010;50:2353–2361. doi: 10.1111/j.1537-2995.2010.02689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mangalmurti NS, Friedman JL, Wang LC, Stolz D, Muthukumaran G, Siegel DL, Schmidt AM, Lee JS, Albelda SM. The receptor for advanced glycation end products mediates lung endothelial activation by RBCs. Am J Physiol Lung Cell Mol Physiol. 2013;304:L250–L263. doi: 10.1152/ajplung.00278.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 14.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 15.Wang H, Yang H, Czura CJ, Sama AE, Tracey KJ. HMGB1 as a late mediator of lethal systemic inflammation. Am J Respir Crit Care Med. 2001;164:1768–1773. doi: 10.1164/ajrccm.164.10.2106117. [DOI] [PubMed] [Google Scholar]
- 16.Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D, Sohn JW, Yamada S, Maruyama I, Banerjee A, et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290:C917–C924. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- 17.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 18.Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, Liu J, Antonelli A, Preti A, Raeli L, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. 2012;209:1519–1528. doi: 10.1084/jem.20120189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M, Dumitriu IE, Müller S, Iannacone M, Traversari C, et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. 2004;5:825–830. doi: 10.1038/sj.embor.7400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mangalmurti NS, Danielle Q, Worthen GS. Stored RBCs modulate the lung inflammatory response through HMGB1 release [abstract] Am J Respir Crit Care Med. 2014;189:A5759. [Google Scholar]
- 21.Wong HR, Lindsell CJ, Pettilä V, Meyer NJ, Thair SA, Karlsson S, Russell JA, Fjell CD, Boyd JH, Ruokonen E, et al. A multibiomarker-based outcome risk stratification model for adult septic shock. Crit Care Med. 2014;42:781–789. doi: 10.1097/CCM.0000000000000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reilly JP, Meyer NJ, Shashaty MG, Feng R, Lanken PN, Gallop R, Kaplan S, Herlim M, Oz NL, Hiciano I, et al. ABO blood type a is associated with increased risk of acute respiratory distress syndrome in caucasians following both major trauma and severe sepsis. Chest. 2014;145:753–761. doi: 10.1378/chest.13-1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288:31268–31279. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 26.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 27.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 28.Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–453. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 29.Xie T, Peng W, Yan C, Wu J, Gong X, Shi Y. Structural insights into RIP3-mediated necroptotic signaling. Cell Reports. 2013;5:70–78. doi: 10.1016/j.celrep.2013.08.044. [DOI] [PubMed] [Google Scholar]
- 30.Wu YT, Tan HL, Huang Q, Sun XJ, Zhu X, Shen HM. zVAD-induced necroptosis in L929 cells depends on autocrine production of TNFα mediated by the PKC-MAPKs-AP-1 pathway. Cell Death Differ. 2011;18:26–37. doi: 10.1038/cdd.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 32.Kiefmann R, Rifkind JM, Nagababu E, Bhattacharya J. RBC induce hypoxic lung inflammation. Blood. 2008;111:5205–5214. doi: 10.1182/blood-2007-09-113902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huh D, Leslie DC, Matthews BD, Fraser JP, Jurek S, Hamilton GA, Thorneloe KS, McAlexander MA, Ingber DE. A human disease model of drug toxicity-induced pulmonary edema in a lung-on-a-chip microdevice. Sci Transl Med. 2012;4:159ra147. doi: 10.1126/scitranslmed.3004249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Reconstituting organ-level lung functions on a chip. Science. 2010;328:1662–1668. doi: 10.1126/science.1188302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bell CW, Jiang W, Reich CF, III, Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol. 2006;291:C1318–C1325. doi: 10.1152/ajpcell.00616.2005. [DOI] [PubMed] [Google Scholar]
- 36.Jiang W, Bell CW, Pisetsky DS. The relationship between apoptosis and high-mobility group protein 1 release from murine macrophages stimulated with lipopolysaccharide or polyinosinic-polycytidylic acid. J Immunol. 2007;178:6495–6503. doi: 10.4049/jimmunol.178.10.6495. [DOI] [PubMed] [Google Scholar]
- 37.Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–350. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mangalmurti NS, Xiong Z, Hulver M, Ranganathan M, Liu XH, Oriss T, Fitzpatrick M, Rubin M, Triulzi D, Choi A, et al. Loss of red cell chemokine scavenging promotes transfusion-related lung inflammation. Blood. 2009;113:1158–1166. doi: 10.1182/blood-2008-07-166264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Wang H, Mason JM, Levine J, Yu M, Ulloa L, Czura CJ, Tracey KJ, Yang H. Recombinant HMGB1 with cytokine-stimulating activity. J Immunol Methods. 2004;289:211–223. doi: 10.1016/j.jim.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 40.Hreggvidsdottir HS, Ostberg T, Wähämaa H, Schierbeck H, Aveberger AC, Klevenvall L, Palmblad K, Ottosson L, Andersson U, Harris HE. The alarmin HMGB1 acts in synergy with endogenous and exogenous danger signals to promote inflammation. J Leukoc Biol. 2009;86:655–662. doi: 10.1189/jlb.0908548. [DOI] [PubMed] [Google Scholar]
- 41.Gibot S, Massin F, Cravoisy A, Barraud D, Nace L, Levy B, Bollaert PE. High-mobility group box 1 protein plasma concentrations during septic shock. Intensive Care Med. 2007;33:1347–1353. doi: 10.1007/s00134-007-0691-2. [DOI] [PubMed] [Google Scholar]
- 42.Nakamura T, Sato E, Fujiwara N, Kawagoe Y, Maeda S, Yamagishi S. Increased levels of soluble receptor for advanced glycation end products (sRAGE) and high mobility group box 1 (HMGB1) are associated with death in patients with acute respiratory distress syndrome. Clin Biochem. 2011;44:601–604. doi: 10.1016/j.clinbiochem.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 43.Wolfson RK, Chiang ET, Garcia JG. HMGB1 induces human lung endothelial cell cytoskeletal rearrangement and barrier disruption. Microvasc Res. 2011;81:189–197. doi: 10.1016/j.mvr.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, Suffredini AF. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–2660. doi: 10.1182/blood-2002-05-1300. [DOI] [PubMed] [Google Scholar]
- 45.Treutiger CJ, Mullins GE, Johansson AS, Rouhiainen A, Rauvala HM, Erlandsson-Harris H, Andersson U, Yang H, Tracey KJ, Andersson J, et al. High mobility group 1 B-box mediates activation of human endothelium. J Intern Med. 2003;254:375–385. doi: 10.1046/j.1365-2796.2003.01204.x. [DOI] [PubMed] [Google Scholar]
- 46.Wolfson RK, Mapes B, Garcia JG. Excessive mechanical stress increases HMGB1 expression in human lung microvascular endothelial cells via STAT3. Microvasc Res. 2014;92:50–55. doi: 10.1016/j.mvr.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khan MJ, Rizwan Alam M, Waldeck-Weiermair M, Karsten F, Groschner L, Riederer M, Hallström S, Rockenfeller P, Konya V, Heinemann A, et al. Inhibition of autophagy rescues palmitic acid-induced necroptosis of endothelial cells. J Biol Chem. 2012;287:21110–21120. doi: 10.1074/jbc.M111.319129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Andonegui G, Zhou H, Bullard D, Kelly MM, Mullaly SC, McDonald B, Long EM, Robbins SM, Kubes P. Mice that exclusively express TLR4 on endothelial cells can efficiently clear a lethal systemic Gram-negative bacterial infection. J Clin Invest. 2009;119:1921–1930. doi: 10.1172/JCI36411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang Y, Ma J, Chen Y, Wu M. Nucleocytoplasmic shuttling of receptor-interacting protein 3 (RIP3): identification of novel nuclear export and import signals in RIP3. J Biol Chem. 2004;279:38820–38829. doi: 10.1074/jbc.M401663200. [DOI] [PubMed] [Google Scholar]
- 51.Festjens N, Vanden Berghe T, Cornelis S, Vandenabeele P. RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell Death Differ. 2007;14:400–410. doi: 10.1038/sj.cdd.4402085. [DOI] [PubMed] [Google Scholar]
- 52.Vanlangenakker N, Vanden Berghe T, Vandenabeele P. Many stimuli pull the necrotic trigger, an overview. Cell Death Differ. 2012;19:75–86. doi: 10.1038/cdd.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. HMG-1 as a mediator of acute lung inflammation. J Immunol. 2000;165:2950–2954. doi: 10.4049/jimmunol.165.6.2950. [DOI] [PubMed] [Google Scholar]