

Abstract
Antiretroviral therapy has increased the life span of HIV+ individuals; however, HIV-associated neurocognitive disorder (HAND) occurrence is increasing in aging HIV patients. Previous studies suggest HIV infection alters autophagy function in the aging CNS and HIV-1 proteins affect autophagy in monocyte-derived cells. Despite these findings, the mechanisms leading to dysregulated autophagy in the CNS remain unclear. Here we sought to determine how HIV Tat dysregulates autophagy in neurons. Tat caused a dose-dependent decrease in autophagosome markers, microtubule-associated protein-1 light chain β II (LC3II), and sequestosome 1(SQSTM1), in a membrane-enriched fraction, suggesting Tat increases autophagic degradation. Bafilomycin A1 increased autophagosome number, LC3II, and SQSTM1 accumulation; Tat cotreatment diminished this effect. Tat had no effect when 3-methyladenine or knockdown of beclin 1 blocked early stages of autophagy. Tat increased numbers of LC3 puncta and resulted in the formation of abnormal autophagosomes in vitro. Likewise, in vivo studies in GFAP-Tat tg mice showed increased autophagosome accumulation in neurons, altered LC3II levels, and neurodegeneration. These effects were reversed by rapamycin treatment. Tat colocalized with autophagosome and lysosomal markers and enhanced the colocalization of autophagosome with lysosome markers. Furthermore, co-IP studies showed that Tat interacts with lysosomal-associated membrane protein 2A (LAMP2A) in vitro and in vivo, and LAMP2A overexpression reduces Tat-induced neurotoxicity. Hence, Tat protein may induce autophagosome and lysosome fusion through interaction with LAMP2A leading to abnormal neuronal autophagy function and dysregulated degradation of critical intracellular components. Therapies targeting Tat-mediated autophagy alterations may decrease neurodegeneration in aging patients with HAND.
Keywords: autophagy, HIV, LAMP2A, Tat
Introduction
Currently >30 million people live with HIV worldwide. Modern treatment regimens result in HIV suppression and immune recovery; however, the prevalence of HIV-associated neurocognitive disorders (HAND) and neurodegeneration (Budka et al., 1987; Wiley and Achim, 1994; Gendelman et al., 1997; Cherner et al., 2007; Heaton et al., 2010) has remained the same or increased (Joska et al., 2010; Heaton et al., 2011), in particular among people over the age of 50. In the United States, the aging population represents one of the fastest growing groups with HIV (Scott et al., 2011).
Mechanisms of neurodegeneration causing HAND are not completely understood; however, recent studies have shown that HIV proteins interfere with clearance pathways such as macroautophagy (Alirezaei et al., 2008a, b; Zhou et al., 2011), a pathway necessary for recycling proteins or defective and older intracellular organelles (Cuervo, 2004). Macroautophagy (hereafter, autophagy) involves nucleation, initiation, elongation, and termination. Initially, autophagy-related proteins form a phagophore, which develops into the autophagosome, a double-membrane sac that delivers cytoplasmic material to the lysosomal compartment for degradation (Codogno et al., 2012). MAP1 light chain 3 (LC3I), one of the core autophagy proteins, is cleaved and conjugated into the membrane (LC3II) during autophagosomal formation. LC3II interaction with SQSTM1 mediates selective recruitment of ubiquitylated proteins to the autophagosome (Pankiv et al., 2007). Both commonly used at markers of autophagy, LC3II levels are affected by autophagy initiation as well as degradation, whereas SQSTM1 levels are inversely proportional to autophagy activity (Shvets et al., 2008).
Alterations in autophagy pathways have been described in AD (Nixon et al., 2005; Pickford et al., 2008), PD (Cuervo et al., 2004; Crews et al., 2010), and other CNS disorders (Cuervo, 2004). Similarly, neurodegeneration patterns during HIV infection have been linked to defects in autophagy (Alirezaei et al., 2008a, b; Zhou et al., 2011; Fields et al., 2013). Recently, HIV gp120 and Nef have been shown to affect autophagy, respectively (Kyei et al., 2009; Fields et al., 2013). HIV-1 Tat suppresses autophagy function in macrophages (Li et al., 2011) and bystander monocytes (Van Grol et al., 2010). Tat treatment caused altered endolysosome morphology and function leading to neurotoxic effects (Hui et al., 2012). Therefore, we aimed to investigate the molecular targets of the autophagy pathway in neurons that are affected by Tat.
In these studies we found that in in vitro or in GFAP-Tat tg mice Tat induces abnormal neuronal autophagosome formation, and associates with lysosome-associated membrane protein 2A (LAMP2A). Tat reversed Bafilomycin A1 (BafA1)-mediated block of degradation of autophagy markers and induced colocalization of autophagosome and lysosome markers. Last, Tat induced neurotoxicity in vitro and neurodegeneration in vivo, and Rapamycin (Rapam) reversed these effects. These findings suggest that Tat directly alters lysosome fusion to autophagosomes, possibly through interaction with LAMP2A. This mechanism could contribute, in concert with other Tat actions, HIV proteins, or inflammatory factors, to neurodegeneration in HAND and reduced neuronal autophagy in aged HIV patients.
Materials and Methods
Cell culture.
B103 cells (rat neuroblastoma) and primary mouse hippocampal neurons (E16) were cultured at 37 and 5% CO2. B103 rat neuroblastoma cells were used here for the cholinergic and GABAergic phenotypes (Schubert et al., 1974), both of which are implicated in frontal cortex and basal ganglia function (Stocco et al., 2010; Nguyen and Lin, 2014) and relevant to HAND (Masliah et al., 1994; Hesselgesser and Horuk, 1999). B103 and primary neurons were grown in DMEM with 5% or 2% FBS and special primary neuron media, respectively. Cells were treated with recombinant HIV Tat (10 ng/ml, 100 ng/ml, or 500 ng/ml; NIH AIDS Reagent Program, clade B, catalog #2222) alone or in combination with autophagy modulators for 24 h. B103 cells were cultured in media supplemented with 2% FBS when treated with recombinant Tat only, but with 5% FBS in experiments using autophagy modulators such as BafA1.
Autophagy modulators.
Cells were treated with Rapam (200 μm; Sigma-Aldrich; catalog #R0395), Torin 1 (250 nm; Tocris Biosciences; catalog #4247), tamoxifen (10 μm; Sigma-Aldrich; catalog #T5648), 3-methyl adenine (3MA; 1 mm; Sigma-Aldrich; catalog #M9281), BafA1 (30 nm; Sigma-Aldrich; catalog #11707), and chloroquine (Chloro; 30 μm; Millipore; catalog #193919).
Lentivirus construction and infection.
The shRNA BECN1 was designed against nucleotides 405–423 of human BECN1 and cloned into the pS1H1copGFP vector (System Biosciences). The cop GFP was replaced with the plum fluorescent protein to generate pLV-siBECN1-Plum. The mouse Lamp2 cDNA (Open Biosystems), HIV Tat (pTRE-Tat86 plasmid), and GFP-LC3 (a generous gift from Joshua Goldstein) were cloned into the third-generation self-inactivating lentivirus. Lentiviruses (LVs) were prepared by transient transfection in 293T cells (Naldini et al., 1996a, b; Tiscornia et al., 2006). B103 rat neuroblastoma cells or primary mouse neurons were grown on glass coverslips in 12-well plates, infected with the indicated LV at multiplicity of infection of 50 for 48 h, and then treated with Tat, BafA1, and Chloro alone or in combination. Cells were fixed in 4% paraformaldehyde for 30 min at 4°C before immunostaining, mounting on slides, and visualization.
Antibodies.
The following antibodies were used in immunoblot, immunohistochemistry, or both: light-chain 3 (LC3; MBL; catalog #PD014), anti-HIV-1 TAT (NIH AIDS Reagent Program; catalog #1974), cathepsin-D (CTSD; Cell Signaling Technology; catalog #2284), RAB7A (Abcam; catalog #ab50533), SQSTM1 (Sigma-Aldrich; catalog #P0067), GFAP (Cell Signaling Technology; catalog #3670), microtubule-associated protein 2 (MAP2; Millipore; catalog #MAB378), NeuN (Abcam; catalog #104225), and β-actin (BACT; Sigma-Aldrich; catalog #A2228).
Immunoblot.
Briefly, as previously described, cells were collected by trypsin digestion and centrifugation (Fields et al., 2013). Cell pellets were homogenized in RIPA lysis buffer by sonication and centrifuged at 5000 × g for 5 min. After determination of the protein content of all samples by BCA Protein assay (Thermo Fisher Scientific), homogenates were loaded (20 μg of total protein/lane), separated on 4–12% Bis-Tris gels, and electrophoresed in 5% HEPES running buffer and blotted onto Immobilon-P 0.45 μm membrane using NuPAGE transfer buffer. The membranes were blocked in 5% BSA in PBS-Tween 20 (PBST) for1 h. Membranes were incubated overnight at 4°C with primary antibodies. Following visualization, blots were stripped and probed with a mouse monoclonal antibody against BACT (1:2000; mab1501; Millipore) as a loading control. All blots were then washed in PBS and 0.05% Tween 20 and then incubated with secondary species-specific antibodies (American Qualex; 1:5000 in BSA-PBST) and visualized with enhanced chemiluminescence reagent (PerkinElmer). Images were obtained and semiquantitative analysis was performed with the VersaDoc gel imaging system and Quantity One software (Bio-Rad).
LAMP2A and Tat coimmunoprecipitation.
Briefly, homogenates from neuronal cells and mouse brains were prepared in RIPA buffer as for immunoblot analysis. Immunoprecipitation assays were performed essentially as previously described (Hashimoto et al., 2001). The lysates were then centrifuged for 5 min at 5000 g, and the protein concentrations were determined with a BCA protein assay kit. Three hundred micrograms of each of the supernatants was incubated with 2 μg of the antibody against LAMP2A or RAB7A overnight at 4°C. Then the immunocomplexes were adsorbed to protein G-Sepharose (GE Healthcare). After extensive washing with immunoprecipitation buffer, which contained 1% Trion X-100, samples were heated in NuPAGE SDS sample buffer (Invitrogen) for 5 min, resolved using 4–12% Bis-Tris gels, and blotted onto Immobilon-P 0.45 μm membrane using iBlot (Invitrogen) transfer buffer. Samples were then subjected to immunoblot analysis with mouse monoclonal antibodies against HIV-1 Tat and then secondary antibody linked to HRP for visualization as described above.
Immunohistochemistry and double immunolabeling.
B103 or primary mouse neurons were grown on acid-washed, poly-d-lysine-treated coverslips for 24 h. Untreated cells or those infected with lentivirus-expressing GFP-LC3 and/or LV-shBECN1-Plum (LV-GFP-LC3, MOI = 50) were treated with HIV recombinant proteins and/or autophagy-modulating compounds. Cells were fixed in 4% paraformaldehyde for 20 min at 4°C and then mounted with DAPI Immuno Mount. Alternatively, uninfected cells were incubated with appropriate primary antibody (Tat, LC3, CTSD, or LAMP2A) and secondary antibody and then mounted with DAPI Immuno Mount. The immunolabeled blind-coded sections were serially imaged with a laser-scanning confocal microscope (MRC-1024; Bio-Rad) and analyzed with ImageJ v1.43 software (NIH), as previously described (Crews et al., 2010). For each condition a total of 50 cells were analyzed. All slides were processed under the same standardized conditions. Immunostained sections were imaged with a digital Olympus microscope and the Image-Pro Plus program (version 4.5.1; Media Cybernetics).
To determine the colocalization between Tat with different antibodies and with other cellular markers, double-labeling experiments were performed, as previously described (Spencer et al., 2009). For this purpose, paraformaldehyde-fixed B103 cells; primary mouse neurons immunolabeled with the mouse monoclonal antibody against human Tat (NIH AIDS Reagent Program, catalog #1974); and the rabbit polyclonal antibodies for LC3, CTSD, or LAMP2A were used. All slides were processed simultaneously under the same conditions and experiments were performed in triplicate to assess the reproducibility of results. Slides were imaged with a Zeiss high-magnification (×63) objective (NA 1.4) on an Axiovert 35 microscope (Zeiss) with an attached MRC1024 laser-scanning confocal microscope system (Bio-Rad).
Briefly, as previously described (Masliah et al., 2003), free-floating 40-μm-thick vibratome sections of mouse brains were washed with TBS, pH 7.4, pretreated in 3% H2O2, and blocked with 10% serum (Vector Laboratories), 3% BSA (Sigma), and 0.2% gelatin in TBS-T. Sections were incubated at 4°C overnight with the primary antibodies. Sections were then incubated in secondary antibody (1:75; Vector Laboratories), followed by Avidin D- HRP (ABC Elite; Vector Laboratories), and reacted with DAB (0.2 mg/ml) in 50 mm Tris, pH 7.4, with 0.001% H2O2. Control experiments consisted of incubation with pre-immune rabbit serum. Immunostained sections were imaged with a digital Olympus microscope and assessment of levels of Tat and GFAP immunoreactivity was performed using the Image-Pro Plus program (Media Cybernetics). For each case a total of three sections (10 images per section) was analyzed to estimate the average number of immunolabeled cells per unit area (mm2) and the average intensity of the immunostaining (corrected optical density). For analysis of neuronal cells, sections were immunostained with an antibody against NeuN (Millipore) and analyzed by stereology with the dissector method using the Stereo Investigator system as previously described. In addition, double-immunolabeling studies were performed as previously described (Spencer et al., 2009) to determine the cellular localization of Tat, LC3, CTSD, and LAMP2A. For this purpose, vibratome sections of mouse brains were immunostained with antibodies against HIV-1 Tat (red) and antibodies against GFAP, NeuN, LC3, LAMP2A, or CTSD (rabbit polyclonal, green). Sections were then reacted with secondary antibodies tagged with FITC to detect LC3, LAMP2A, or CTSD and with the Tyramide red amplification system (PerkinElmer) to detect HIV-1 Tat markers. Sections were mounted on SuperFrost slides (Fisher) and coverslipped with media containing DAPI. Sections were imaged with a Zeiss 63× (NA 1.4) objective on an Axiovert 35 microscope (Zeiss) with an attached MRC1024 laser-scanning confocal microscope system (Bio-Rad).
All experiments were conducted blind-coded, and code was broken after analysis was performed. Experiments were performed in duplicate to evaluate reproducibility of the effects of Tat and autophagy compounds in the in vitro and in vivo systems.
Neurotoxicity studies
LDH cytotoxicity assay was used (CytoTox96; Promega), as per the manufacturer's instruction, to determine Tat effects on neuron viability. Briefly, B103 neuronal cells were treated with Tat alone or in combination with BafA1, Torin 1, or Rapam for 24 h. Additionally, B103 neuronal cells were infected with LV or LVLamp2 for 72 h and then treated with Tat. Supernatants were collected; incubated with LDH reaction buffer in the dark, at room temperature for 30 min; and stop solution was added. Absorbance at 490 nm was taken on Molecular Devices FilterMax. Readings were normalized to lysis buffer-treated cells to obtain percentage cell death.
LysoTracker experiments
The LysoTracker assay (LysoTracker Yellow HCK-123; Life Technologies) was used to determine lysosomal acidification in B103 neuronal cells. Cells were treated alone or in combination with BafA1 for 20 min or Tat for 4 h, 8 h, or 24 h. Subsequently, cells were incubated with LysoTracker (65 nm) for 45 min at 37°C and imaged at an excitation/emission of 465/535 nm. Microscope slides were imaged with a Zeiss 63× (NA 1.4) objective on an Axiovert 35 microscope (Zeiss) with an attached MRC1024 laser-scanning confocal microscope system (Bio-Rad), and analyzed with ImageJ v1.43 software (NIH), as previously described (Crews et al., 2010).
Generation of inducible Tat transgenic mice, doxycycline, and Rapam infusion
Briefly, as previously described (Kim et al., 2003), inducible Tat transgenic mouse colonies were obtained by generation of two separate transgenic lines, Teton-GFAP mice (G-tg) and TRE-Tat86 mice (T-tg), and then cross-breeding these two lines of transgenic mice. Briefly, a DNA fragment (2238 bp) containing the Teton-GFAP gene, along with downstream simian virus 40 splicing and polyadenylation sequences, was released by XhoI and PvuII digestion of the pTeton-GFAP plasmid and purified by agarose gel electrophoresis and microinjected into fertilized eggs of F1 females obtained from mating between C3HeB and FeJ mice (The Jackson Laboratory). Founder transgenic animals were crossed with C57BL/6 mice to generate stable G-tg transgenic lines. Similarly, T-tg transgenic lines were obtained using a DNA fragment (1189 bp) released by XhoI and PvuII digestion of the pTRE-Tat86 plasmid. Founder animals and progeny carrying the transgenes were identified by PCR analysis of genomic DNA, which was extracted from mouse tail clippings (0.5–1 cm long) using the Wizard genomic DNA isolation kit (Promega). With this construct, mice express Tat upon doxycycline (DOX) treatment. For these experiments a total of n = 8 Non tg mice and n = 40 GFAP-Tat tg mice were used (7–8 months old). The GFAP-Tat tg mice were divided into five groups, and the first was not treated with DOX; however, the other four groups (n = 8 mice per group) were treated with DOX at 80 mg/kg (daily immunoprecipitation) for 2 weeks and then killed immediately after (week 2), and the other groups were killed at 2 week intervals after cessation of DOX (weeks 4, 6, and 8).
Additional experiments with the GFAP-Tat tg mice included treatments with the autophagy activator Rapam (Sigma-Aldrich). Because Rapam poorly crosses into the CNS, it was infused intracerebrally into the lateral ventricle of 9-month-old mice at a concentration of 20 mg/kg. Briefly, as previously described (Veinbergs, 2001), mice were anesthetized and under sterile conditions a 26 gauge stainless steel cannula was implanted stereotaxically into the lateral ventricle using the bregma as a reference (Franklin and Paxinos, 1997; bregma 0.5 mm, 1.1 mm lateral, depth 3 mm) and secured to the cranium using Superglue. The cannula was connected via a 5 mm coil of V3 Biolab vinyl to a model 1007D osmotic mini pump (Alzet) surgically placed subcutaneously beneath the shoulder. The solutions were delivered at a flow rate of 0.5 μl/h for 2 weeks. The pump was left for an additional 2 weeks and mice were killed 1 month after the initiation of the infusions. Brains were removed and divided sagittally. One hemibrain was postfixed in phosphate-buffered 4% PFA, pH 7.4, at 4°C for 48 h and sectioned at 40 μm with a Vibratome 2000 (Leica) and placed in cryosolution, while the other hemibrain was snap frozen and stored at −70°C for RNA and protein analysis.
RNA isolation and RT-PCR analysis of Tat expression
Total RNA was isolated using the TRIzol Reagents (Life Technologies) according to the manufacturer's instructions. Tat expression was analyzed using the Titan One Tube RT-PCR System kit (Boehringer Mannheim), with Tat-specific primers 5′-GGA ATT CAC CAT GGA GCC AGT AGA TCC T-3′ and 5′-CGG GAT CCC TAT TCC TTC GGG CCT GT-3′. RT-PCR was performed on a PE Thermal Cycler 9600 (Applied Biosystems) with a program of 50°C for 30 min and 94°C for 2 min, followed by 30 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s, and one cycle of 72°C for 7 min. Control RT-PCR s were performed in the absence of RT and genomic DNA templates to rule out the possibilities of genomic DNA contamination in RNA preparations and nonspecific amplification. In addition, the mouse GAPDH was included in the RT-PCR as an internal control, with GAPDH-specific primers 5′-CTC AGT GTA GCC CAG GAT GC-3′ and 5′-ACC ACC ATG GAG AAG GCT GG-3′.
Electron microscopy.
Briefly, B103 neuronal cells or primary mouse neurons were plated in 35 mm dishes with a coverslip in the bottom. After 48 h, cells were fixed in 2% paraformaldehyde and 1% glutaraldehyde, then fixed in osmium tetroxide and embedded in Epon Araldite. Blocks with the cells were detached from the coverslips and mounted for sectioning with an ultramicrotome (Leica). Similarly, sections from the GFAP-TAT tg mouse brains were fixed, embedded, and sectioned with the ultramicrotome. Grids were analyzed with a Zeiss OM 10 electron microscope as previously described (Rockenstein et al., 2001). To analyze the relative changes in average numbers of autophagic vesicles, a total of 25 cells were analyzed per condition. Cells were randomly acquired from three grids. Grids were analyzed with a Zeiss OM 10 electron microscope as previously described (Rockenstein et al., 2001). Electron micrographs were obtained at a magnification ×25,000.
Statistical analysis.
All the analyses were conducted on blind-coded samples. After the results were obtained, the code was broken and data were analyzed with the StatView program (SAS Institute). Comparisons among groups were performed with one-way ANOVA with post hoc Fisher's test, unpaired Student's t test, and χ2 analysis. All results were expressed as mean ± SEM. The differences were considered to be significant if p values were <0.05.
Results
HIV-1 Tat affects basal autophagy function in a neuronal cell line and primary neurons
Previous studies have shown that HIV-1 Tat can be secreted from infected macrophage/microglia cells and affect bystander cells, such as neurons (Toborek et al., 2003; Van Grol et al., 2010; Richard et al., 2013). To determine the acute effect of HIV-1 Tat on neuronal autophagy under basal conditions, we treated B103 neuronal cells (Fig. 1A–C) with increasing concentrations of recombinant HIV Tat for 24 h and assessed levels of markers of autophagy activity, SQSTM1, and LC3II. B103 rat neuroblastoma cells were treated with 10 ng/ml, 100 ng/ml, or 500 ng/ml of recombinant Tat. Treatment with Tat resulted in a dose-dependent decrease in LC3II and SQSTM1 levels (Fig. 1A). Densitometry analysis of the LC3II bands showed that Tat reduced LC3II levels to 80, 50, and 30% of control with 10, 100, and 500 ng/ml doses of Tat, respectively (Fig. 1B). Similarly, densitometry analysis of the SQSTM1 bands from the experiments using B103 cells showed that Tat reduced levels to 75, 45, and 35% of control with 10, 100, and 500 ng/ml doses of Tat, respectively (Fig. 1C). To further corroborate our results, primary mouse neurons (Fig. 1D–F) were treated with 100 ng/ml or 500 ng/ml recombinant Tat for 24 h and lysates were analyzed by immunoblot for LC3 and SQSTM1 (Fig. 1D). LC3II levels were decreased to 80 and 60% (p < 0.05 and p < 0.01, respectively), and SQSTM1 levels were decreased to 75 and 45% of control with 100 or 500 ng/ml recombinant Tat, respectively (Fig. 1E,F). These results indicate that Tat reduces autophagy in neuronal cells.
Figure 1.

Recombinant HIV-1 Tat reduces detectable LC3II and SQSTM1 levels in neuronal cells. A, Immunoblot specific for LC3, SQSTM1, and actin using lysates from B103 rat neuroblastoma cells treated with increasing doses (10 ng/ml, 100 ng/ml, or 500 ng/ml) of Tat for 24 h. B, Densitometry analysis of LC3II immunoblot in B103 rat neuroblastoma cells. C, Densitometry analysis of SQSTM1 immunoblot in B103 rat neuroblastoma cells. D, LC3 immunoblot of lysates from primary mouse neurons treated with 100 ng/ml or 500 ng/ml Tat for 24 h. E, Densitometry analysis of LC3II bands in primary mouse neurons. F, Densitometry analysis of SQSTM1 bands in primary mouse neurons. Statistical significance: *p < 0.05, **p < 0.01, one-way ANOVA, post hoc Fisher's test.
HIV-1 Tat mitigates BafA1-induced block of LC3II and SQSTM1 degradation in neuronal cells
Next we wanted to determine at which stage Tat interferes acutely with neuronal autophagy. To manipulate autophagy progression we treated B103 neuronal cells with autophagy inducers (Rapam, Torin 1, and tamoxifen) and inhibitors (BafA1, Chloro, and 3MA). As previously reported (Amadoro et al., 2014), BafA1 and Chloro treatment resulted in increased LC3II accumulation in neuronal cells (Fig. 2A). Densitometry analysis showed BafA1 (p < 0.001) and Chloro increased LC3II levels as high as ninefold and fourfold, respectively, compared with vehicle-treated cells, which is consistent with the concept that these molecules block autophagolysosome formation in neurons (Amadoro et al., 2014; Fig. 2B). In contrast, the highest concentrations of mTor inhibitors, Rapam and Torin 1, reduced LC3II levels by 40 and 50%, respectively (Fig. 2B). Next we sought to determine whether Tat is able to affect the activity of autophagy modulators by increasing or decreasing LC3II and SQSTM1 levels. Here, we treated neuronal cells with BafA1, 3MA, or Tat, alone or in conjunction, for 24 h. BafA1 increased LC3II band intensity, 3MA showed little effect, and Tat decreased LC3II levels after 24 h of treatment (Fig. 2C). However, cotreatment with Tat reduced BafA1 effects by 55% (p < 0.05; Fig. 2D). These results show that Tat counteracts BafA1 function and significantly induces autophagy degradation levels in BafA1-treated cells. To determine whether Tat can affect BafA1-mediated block on autophagosome substrate degradation, SQSTM1 levels were measured in BafA1-treated cells by immunoblot (Fig. 2E). Tat reduced SQSTM1 levels and BafA1 increased SQSTM1 levels, but cotreatment with BafA1 and Tat prevented SQSTM1 accumulation by 50% (Fig. 2F). Reduction in LC3II and SQSTM1 accumulation is indicative of increased autophagic degradative activity.
Figure 2.

Recombinant HIV-1 Tat reverses chemical blockade of neuronal autophagy in B103 cells. LC3-specific immunoblot of lysates from B103 cells treated with autophagy-modulating chemical compounds with or without Tat cotreatment. A, LC3 immunoblot of lysates from B103 cells treated with increasing concentrations of BafA1, Chloro, 3MA, Rapam, Torin 1, or tamoxifen (Tamox/Tam). B, Densitometry analysis of LC3II bands from lysates of chemical-treated B103 cells. C, Immunoblot for LC3 in lysates from B103 cells treated with BafA1 or 3MA with or without Tat cotreatment. D, Densitometry analysis of LC3II band in B103 lysates treated with BafA1 or 3MA with or without Tat cotreatment. E, Immunoblot for SQSTM1 in lysates from B103 cells treated with BafA1 with or without Tat cotreatment. F, Densitometry analysis of SQSTM1 band in B103 lysates treated with BafA1 with or without Tat cotreatment. Statistical significance: *p < 0.05, ***p < 0.001, one-way ANOVA, post hoc Fisher's test.
HIV-1 Tat reverses BafA1-mediated GFP-LC3 accumulation
The autophagy pathway is in constant flux, tagging cellular contents for autophagic degradation, forming autophagolysosomes, and subsequently degrading cargo. Infecting B103 neuronal cells (Fig. 3A,C,E) and primary neurons (Fig. 3B,D,F) with LV-GFP-LC3 facilitates visualization of autophagy in vitro. Under baseline conditions neuronal cells displayed distinct GFP-LC3 puncta; in contrast, Tat treatment resulted in an increase of GFP-LC3 puncta with several of them appearing merged or irregular (Fig. 3A). BafA1 and Chloro treatment also robustly increased GFP-LC3 puncta compared with baseline; however, Tat cotreatment reduced the number of GFP-LC3 puncta (Fig. 3A). Primary mouse neurons display abundant distinct GFP-LC3 puncta in a granular distribution throughout the cytoplasm; however, Tat-treated neurons displayed more abundant and dot-like GFP-LC3 puncta (Fig. 3B). BafA1 treatment resulted in a robust increase in distinct GFP-LC3 puncta throughout the cytoplasm, and similar to what was seen in neuronal cells, Tat cotreatment caused a large reduction in GFP-LC3 puncta (Fig. 3B). Chloro showed little effect on GFP-LC3 puncta number or appearance in primary neurons (Fig. 3B). Quantification of GFP-LC3 puncta showed that Tat, BafA1, and Chloro caused a 15-, 12-, and 6-fold increase in autophagosomes per cell, respectively (Fig. 3C). Tat cotreatment reduced BafA1- and Chloro-mediated increase in GFP-LC3 puncta by 75 and 40%, respectively (Fig. 3C). Primary neurons treated with Tat and BafA1 showed a 4.5- and 6-fold increase in GFP-LC3 puncta compared with untreated cells, but Chloro caused little change (Fig. 3D). Tat cotreatment reduced the number of GFP-LC3 puncta in primary neurons by 50 and 60% compared with BafA1 or Tat treatment alone, respectively (Fig. 3D). These studies reveal that Tat alters neuronal autophagy and may overcome BafA1 blockage of autophagolysosome formation. To determine whether induction of autophagy is necessary for Tat-mediated accumulation of GFP-LC3 puncta, we co-infected neuronal cells with LV-shBECN1-Plum and LV-GFP-LC3 followed by treatment with BafA1, Chloro, and Tat alone or in conjunction. Tat induced an increase in GFP-LC3 puncta in LV-shCtl-infected cells, but not in LV-shBECN1-Plum-infected cells (Fig. 3E). Tat treatment increased GFP-LC3 puncta fivefold in LV-shCtl-infected cells (p < 0.05), but no significant change was detected in LV-shBECN1-Plum-infected cells (Fig. 3F). These data indicate that initial stages of autophagosome formation are necessary for Tat-induced GFP-LC3 puncta accumulation.
Figure 3.

Recombinant HIV-1 Tat increases GFP-LC3 and reduces BafA1-induced autophagosome accumulation. Neuronal cell line (B103) and primary mouse neuron cells were infected with LV-GFP-LC3 to facilitate tracking and quantification of LC3 vesicles by laser confocal microscopy. A, GFP-LC3-positive vesicles (arrows) in B103 cells treated with vehicle, Tat, BafA1, Chloro, or Tat + BafA1 or Chloro. B, GFP-LC3-positive vesicles in primary hippocampal mouse neurons treated with vehicle, Tat, BafA1, Chloro, or Tat + BafA1 or Chloro. C, Computer-aided image analysis of GFP-LC3 autophagosomes per neuronal cell. D, Computer-aided image analysis of GFP-LC3 autophagosomes per primary mouse neuron. E, GFP-LC3-positive vesicles in B103 cells co-infected with LV-shCtl or LV-shBECN1-Plum and treated with Tat, BafA1, Chloro, or Tat + BafA1 or Chloro. F, Computer-aided image analysis of GFP-LC3 autophagosomes per neuronal cell (*p < 0.05 by one-way ANOVA, post hoc Dunnett's test when comparing vehicle to Tat and BafA1; #p < 0.05 by one-way ANOVA, post hoc Tukey–Kramer when comparing Tat or BafA1 vs Tat + BafA1). Scale bar, 5 μm.
HIV-Tat colocalizes with autophagosome and lysosome markers in neuronal cells and in primary neurons
Previous studies have shown that HIV-1 Tat can permeate the cell membrane of neurons and other cells where it may affect organelle function (Kaul and Lipton, 2006; Alirezaei et al., 2007). To determine whether HIV-1 Tat colocalizes with the autophagy pathway, we treated B103 cells (Fig. 4A–C,G,H) and primary mouse neurons (Fig. 4D,F) with recombinant Tat and then double immunolabeled for Tat and autophagy marker LC3 or CTSD, a lysosomal protease. Laser-scanning confocal microscopy for LC3 (green) and Tat (red) immunostaining in neuronal cells shows that in vehicle-treated cells no Tat signal was detected and LC3-immunoreactive structures appeared with a punctate pattern throughout the cytoplasm (Fig. 4A). Following Tat treatment we were able to detect Tat signal throughout the cytoplasm in a dot-like appearance similar to that of the LC3 immunostaining (Fig. 4A). Merged images showed Tat colocalizes (yellow) with LC3 puncta in the cytoplasm, and Tat signal was colocalized with LC3 signal in large punctate structures (Fig. 4A). In control neuronal cells, CTSD immunostaining (green) was detected in a granular pattern throughout the cytoplasm (Fig. 4B). In Tat-treated neuronal cells CTSD and Tat colocalized in punctate structures (Fig. 4B). Quantification of colocalization shows that Tat colocalizes with LC3 immunostaining in 40% of cells and with CTSD immunostaining in 60% of cells (Fig. 4C). In primary neurons LC3 immunostaining was robust and distributed throughout the cytoplasm (Fig. 4D); however, Tat treatment caused a consolidation of LC3 signal into distinct dot-like structures similar to autophagosomes (Fig. 4D). Tat-immunostained particles were colocalized (yellow) with these LC3+ well formed puncta (Fig. 4D). Likewise, Tat and CTSD signals were colocalized (yellow) in punctate structures (Fig. 4E) in the cytoplasm of primary neurons treated with Tat. Tat and LC3 immunostaining colocalized in 30% of these primary neurons, and Tat and CTSD immunostaining colocalized in 40% of the cells (Fig. 4F). To determine whether BafA1, Chloro, or Rapam affect Tat entry into the cytoplasm we treated neuronal cells with Tat alone or in conjunction with these molecules and then immunostained for MAP2 and Tat. Tat was detectable and colocalized with MAP2 by immunostaining in vehicle, BafA1, Chloro, or Rapam-treated cells (Fig. 4G). Quantification of Tat immunoreactivity was equal in vehicle, and BafA1, Chloro, or Rapam-treated cells (Fig. 4H). These results show that Tat closely associates with autophagy and lysosomal machinery in neuronal cells, in vitro.
Figure 4.

Recombinant HIV-1 Tat colocalizes with LC3 and CTSD in neuronal cells and primary mouse neurons. Neuronal cell line (B103) and primary mouse neurons were treated with recombinant HIV-Tat for 24 h, fixed, double immunolabeled for Tat and LC3 or CTSD, and imaged by laser-scanning confocal microscopy. A, Neuronal cells immunostained for Tat (red channel) and LC3 (green channel). The merged images show colocalization of Tat and LC3 (yellow). B, B103 cells immunostained for Tat (red channel) and CTSD (green channel). The merged images show colocalization of Tat and CTSD (yellow). C, Computer-aided quantification of Tat colocalization with LC3 and CTSD. D, Primary mouse neurons immunostained for Tat (red channel) and LC3 (green channel). The merged images show colocalization of Tat and LC3 (yellow). E, Primary mouse neurons immunostained for Tat (red) and CTSD (green). The merged images show colocalization of Tat and CTSD (yellow). F, Computer-aided quantification of Tat colocalization with LC3 and CTSD in primary neurons. G, B103 cells immunostained for Tat (red) and MAP2 (green). The merged images show colocalization of Tat and MAP2 (yellow). H, Computer-aided quantification of Tat immunoreactivity in B103 cells treated with Tat alone or in conjunction with BafA1, Chloro, or Rapam. Scale bar, 10 μm. Dotted box represents detail presented at higher magnification to the right.
Ultrastructural analysis of Tat-treated neuronal cells confirms abnormal accumulation of autophagosomes
To further confirm the effect of HIV-1 Tat on the autophagy pathway, and to complement the immunocytochemical studies, next B103 neuronal cells (Fig. 5A,B) and primary mouse neurons (Fig. 5C,D) were analyzed by electron microscopy. Here we treated neuronal cells or primary neurons with Tat protein, BafA1, or both in combination. In vehicle-treated neuronal cells autophagic vesicles are scant and small compared with Tat or BafA1-treated cells (Fig. 5A). High-magnification micrographs of the neuronal cytoplasm show increased electron-dense structures in Tat-treated cells (Fig. 5A). Tat treatment increased the percentage of cells containing autophagosomes to 15% and altered morphology to more extensive autophagosomes in greater number. Interestingly, Tat + BafA1 treatment caused a reduction in well formed autophagosomes present in only 5% of the cells (Fig. 5B) accompanied by the presence of electron-dense structures and malformed autophagosomes. In primary neurons, distinct well formed autophagosomes were present in a lower percentage of the cells compared with the neuronal cell line (Fig. 5C). Tat treatment increased the number of autophagosome-positive cells from 1 to 8% of the population (Fig. 5D). BafA1 increased the number of cells with autophagosomes from 1 to 11% of the cell. Again, Tat + BafA1 increased the percentage of cells with autophagosomes to 5% (Fig. 5D), a reduction from either Tat or BafA1 treatment alone. These data suggest that HIV-1 Tat treatment increases the electron-dense autophagosome number in neurons.
Figure 5.

Ultrastructural analysis of Tat-treated neuronal cells confirms abnormal accumulation of autophagosomes. B103 neuronal cells and primary mouse neurons were treated with HIV-Tat and BafA1, alone or in conjunction, for 24 h. Cells were fixed, processed, and processed for transmitted electron microscopy analysis. A total of 50 cells were analyzed per condition. A, Electron micrographs of vehicle, Tat, and BafA1-treated B103 neuronal cells. B, Computer-aided image analysis of percentage of imaged neuronal cells displaying autophagosomes. C, Electron micrographs of vehicle, Tat, and BafA1-treated primary mouse neurons. D, Computer-aided image analysis of percentage of primary neurons displaying autophagosomes (*p < 0.05 by one-way ANOVA, post hoc Dunnett's test when comparing vehicle to Tat and BafA1; #p < 0.05 by one-way ANOVA, post hoc Tukey–Kramer when comparing Tat or BafA1 vs Tat+BafA1). Low power, 5000×. Scale bar, 5 μm. High power, 25,000×. Scale bar, 1 μm.
HIV-1 Tat increases autophagosome and lysosome fusion in neuronal cells
To determine whether Tat facilitates or blocks autophagolysosome formation, we treated B103 neuronal cells with Tat and then double immunolabeled against LC3 and CTSD and imaged using confocal microscopy. In vehicle-treated cells, CTSD and LC3 signals were similar with both distributed throughout the cell body in granular structures (Fig. 6A). CTSD and LC3 signal distribution observed in Tat-treated cells was similar to vehicle-treated cells; however, the merged image reveals more colocalization between the two signals (yellow) in the Tat-treated cells (Fig. 6A). In BafA1-treated cells the CTSD signal (red) was more distinct and dot like, while the LC3 immunostaining was robust but more dispersed versus associated with distinct puncta (Fig. 6A). In neuronal cells cotreated with Tat and BafA1, CTSD signal was observed in granular structures evenly distributed throughout the cell body. The LC3 signal observed in the cotreated cells was less and also well dispersed, but not so punctate in distribution as the CTSD signal (Fig. 6A). Double immunolabeling was determined by merging the red (CTSD) and green (LC3) images (Fig. 6B), and then quantified as percentage of the signal colocalized. In vehicle-treated cells ∼12% of the signal was colocalized; however, Tat treatment caused a significant (p < 0.05) increase in double immunolabeling to 30% (Fig. 6B). BafA1 treatment significantly (p < 0.01) reduced double immunolabeling to <5% of the signal (Fig. 6B). Cotreatment with Tat and BafA1 resulted in ∼25% double immunolabeling, a significant increase (p < 0.05) above BafA1 treatment alone. Next, we sought to investigate the role of Tat in lysosomal acidification over time by conducting a LysoTracker (yellow) assay using B103 cells treated alone or in combination with BafA1 and Tat. In Tat-treated cells, yellow signal appears to be present in higher quantity and in larger vesicles compared with vehicle-treated cells (Fig. 6D). As expected, in cells treated with BafA1 only, yellow signal was decreased compared with vehicle-treated cells, while this signal was emitted from multiple vesicles per cell in cells treated with a combination of BafA1 and Tat (Fig. 6C). Quantification of yellow particles showed a decrease in the number of acidified vesicles in BafA1-treated cells compared with the vehicle (Fig. 6D). The quantity of yellow particles in Tat-treated cells increased 3.7-fold during the first 4 h and remained increased until 24 h of treatment compared with vehicle or BafA1-treated cells (Fig. 6D). Interestingly, BafA1 and Tat cotreatment revealed similar results as Tat alone (Fig. 6D). These data suggest that Tat alters autophagy by enhancing autophagosome and lysosomal fusion.
Figure 6.
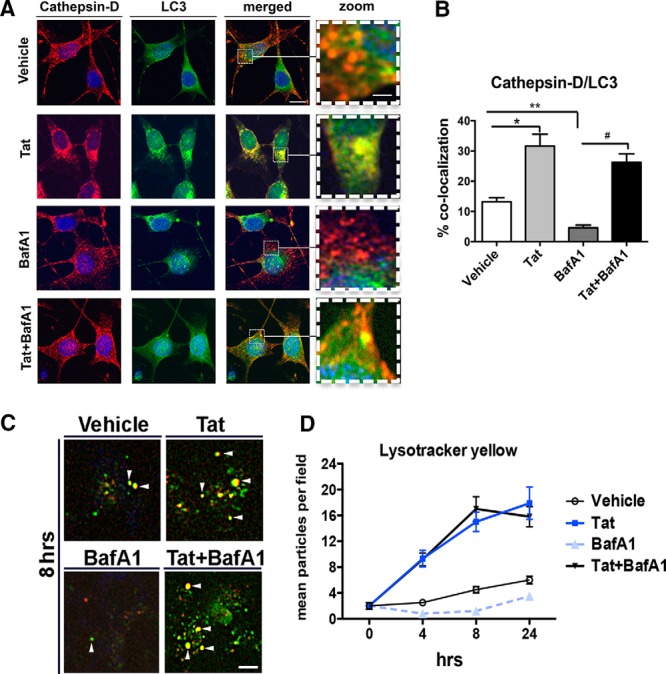
Recombinant HIV-1 Tat increases autophagosome and lysosomal fusion as measured by CTSD and LC3 colocalization as well as a LysoTracker assay. A neuronal cell line (B103) was treated with HIV-Tat and BafA1, alone or in combination, for 24 h and then fixed, double immunolabeled for CTSD and LC3, imaged by laser-scanning confocal microscopy, and quantified by computer-aided image analysis (average of 50 cells per condition). A, Immunostaining for CTSD (red channel), LC3 (green channel), and the merged signal (yellow), indicating colocalization. B, Computer-aided image analysis of the percentage of CTSD and LC3 colocalization in vehicle, Tat, BafA1, and cotreated cells. C, LysoTracker imaging of vehicle, BafA1, and HIV-Tat, alone or in combination, treated neuronal cell line (B103). Arrows point at acidified particles (yellow). D, Computer-aided image analysis of mean particles per field in vehicle, HIV-Tat, BafA1, and cotreated cells. Statistical significance: *p < 0.05, **p < 0.01, #p < 0.05 between indicated conditions, one-way ANOVA, post hoc Fisher's test. Scale bar, 10 μm. Dotted box represents detail presented at higher magnification to the right.
HIV-1 Tat associates with lysosomal protein with LAMP2A, but not with RAB7A in neuronal cells
The ultrastructural and confocal analysis suggests that Tat colocalizes with components of the autophagy and lysosomal pathways and alters autophagosome formation and acidification. To further investigate this possibility, we infected B103 neuronal cells with LV-Tat for 72 h. Coimmunoprecipitation of LAMP2A or RAB7A protein was performed with total lysates and then immunoblotted with Tat antibody. As expected, HIV-1 Tat was detectable in lysates from neuronal cells infected with LV-Tat, but not in lysates from LV-infected cells. Interestingly, Tat was also detectable at 14 kDa in the LAMP2A immunoprecipitation fraction from LV-Tat-infected cells, but not in LV-infected cells, suggesting Tat is directly associated with LAMP2A in these cells (Fig. 7A). LAMP2A was detectable in lysates from both LV- and LV-Tat-infected cells and in the LAMP2A immunoprecipitated fraction, but not in IgG immunoprecipitated fraction (Fig. 7B). Conversely, Tat was not detectable in the RAB7A immunoprecipitated fraction from neuronal cells infected with LV-Tat (Fig. 7C). RAB7A was detectable at 16 kDa in the total lysate and the RAB7A-immunoprecipitated fraction, but not in the IgG-immunoprecipitated fraction (Fig. 7D). To confirm that Tat was associated with LAMP2A, double-immunolabeling experiments and confocal microscopy were performed with B103 (Fig. 7E) neuronal cells and primary neurons (Fig. 7F). In neuronal cells LAMP2A and Tat colocalized in the punctate structures in the cytoplasm. Yellow signal in the overlay shows LAMP2A and Tat colocalization (Fig. 7E). LAMP2A (green) and Tat (red) signals were clearly localized to the soma of primary neurons (Fig. 7F). These data indicate that Tat interacts with lysosomal proteins such as LAMP2A and that this might play a role in the mechanisms through which Tat affects neuronal autophagy.
Figure 7.

HIV-1 Tat associates with lysosomal membrane protein, LAMP2A, in neuronal cell lines. The B103 neuronal cells were infected with LV-Tat for 72 h and then total lysate was isolated and LAMP2A was immunoprecipitated, resolved by SDS-PAGE, and then immunoblotted with Tat-specific antibody. A, LAMP2A immunoprecipitation (IP) of B103 lysate immunoblotted with Tat-specific antibody. B, LAMP2A immunoprecipitation of B103 lysate immunoblotted with LAMP2A-specific antibody. C, RAB7A immunoprecipitation of B103 lysate immunoblotted with Tat-specific antibody. D, RAB7A immunoprecipitation of B103 lysate immunoblotted with RAB7A-specific antibody. E, Neuronal cells infected with LV-Tat, double immunolabeled for Tat (red channel), LAMP2A (green channel), and the merged signals (yellow) and imaged by laser-scanning confocal microscopy. F, Primary mouse neurons infected with LV-Tat fixed and double immunolabeled for Tat (red), LAMP2A (green), and the merged signals (yellow) and imaged by laser-scanning confocal microscopy. Scale bar, 10 μm. Dotted box represents detail presented at higher magnification to the right. WB, Western blot.
HIV-1 Tat-induced acute neurotoxicity is enhanced by BafA1, but reversed with Rapam cotreatment or LAMP2A overexpression
HIV-1 Tat causes neurodegeneration and neuroinflammation in vitro (Nath et al., 1999; Nath and Steiner, 2014) and in tg mouse models (Kim et al., 2003). To determine acutely the extent to which Tat in combination with BafA1, Torin 1, or Rapam affect neuronal cell viability, we treated B103 cells with increasing doses of Tat and autophagy-modulating molecules alone or in conjunction, and measured LDH activity in the supernatant. Increasing doses of Tat protein caused an increase in cell death to 13% with 500 ng/ml of Tat (p < 0.01; Fig. 8A). BafA1 treatment resulted in 25% cell death, and Tat cotreatment increased cell death to 30% (Fig. 8B). Rapam or Torin 1 treatment alone showed no significant difference in cell death compared with untreated, but reduced, Tat-induced cell death equal to that of untreated cells (p < 0.05; Fig. 8B).
Figure 8.

HIV-1 Tat-induced neurotoxicity is enhanced with BafA1, but reversed with Rapam cotreatment or LAMP2A overexpression. B103 cells were treated with Tat protein with or without BafA1, Rapam, and Torin 1 treatment or LVLAMP2 infection, and then supernatant was assayed for LDH to determine percentage cell death. A, Graph of percentage cell death of untreated cells and cells treated with 10 ng/ml, 100 ng/ml, or 500 ng/ml Tat protein. B, Percentage cell death of untreated cells and cells treated with BafA1, Rapam, and Torin 1 with or without 500 ng/ml Tat protein. C, LAMP2A immunoblot of LV and LVLamp2-infected B103 cells. D, Percentage cell death of untreated cells and cells infected with LVLamp2 with or without 500 ng/ml Tat protein (*p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA, post hoc Dunnett's test when comparing to vehicle-treated cells).
Previous studies show increasing LAMP2A expression increases autophagy and cell survival (Zhang and Cuervo, 2008). To determine whether overexpressing LAMP2A in vitro affects Tat-induced neurotoxicity, we infected B103 neuronal cells with LV-LAMP2 for 48 h, and then treated cells with Tat. LAMP2A protein levels are increased in LV-LAMP2-infected cells (Fig. 8C). LAMP2A overexpression reduced Tat-induced neurotoxicity by 35% (Fig. 8D). These data demonstrate that increasing LAMP2A levels ameliorates Tat-induced neurotoxicity.
Increased neuronal autophagy in brains of GFAP-Tat tg mice by immunohistochemistry, immunoblot, and ultrastructural analysis
To investigate the effects of long-term expression of Tat on neuronal autophagy in vivo, DOX-dependent GFAP-Tat tg mice were fed DOX (tet-ON) for 2 weeks and then killed immediately after cessation of DOX treatment and at 4 weeks, 6 weeks, or 8 weeks after beginning of DOX treatment and analyzed for Tat mRNA, Tat protein (Fig. 9A), and LC3 expression. Tat mRNA was undetectable in Non-tg mice, but robustly increased immediately after DOX treatment and 4 weeks and to a lesser extent at 6 and 8 weeks post-DOX treatment (Fig. 9B,C). Immunohistochemical analysis for Tat was undetectable in Non tg mouse brains but detectable in all glial cells in the GFAP-Tat tg mice with the most robust signal detected immediately after DOX cessation and declining thereafter (Fig. 9D). To confirm Tat protein expression we used magnetic beads to immunoprecipitate Tat in Non tg and GFAP-Tat tg brain lysates. No Tat was detected in Non tg mice but a strong band was detected in GFAP-Tat tg mouse brains at 2 weeks of DOX treatment and 4 weeks after initial DOX treatment (Fig. 9E). To analyze the autophagy pathway in the presence of Tat, we immunostained Non tg and tg mouse brains for LC3. As expected in the neuronal cells of Non tg mouse brains and in GFAP-Tat tg mouse brains not treated with DOX, the LC3 signal was detected as granular structures in the cytoplasm; however, a robust LC3 signal, in well formed puncta, was detected in GFAP-Tat tg mouse brains after 2 weeks of DOX and 4 weeks after initial DOX treatment (Fig. 9F). LC3 immunostaining was similar to basal levels after 6 and 8 weeks of DOX treatment (Fig. 9F). Quantification of pixel intensity revealed LC3 staining increased fourfold in tg mouse brains immediately after 2 weeks of DOX and 4 weeks after initial DOX treatment (Fig. 9G). These data demonstrate increased autophagy in GFAP-Tat tg mice in both neurons and astrocytes.
Figure 9.

Analysis of autophagy alterations in GFAP-Tat mouse brains. A, DOX-inducible GFAP-Tat mice were treated with DOX for 2 weeks and then brain tissues were collected immediately or at 2 weeks, 4 weeks, or 6 weeks after cessation of DOX treatment. B, Tat and GAPDH mRNA amplified by RT-PCR and resolved on agarose gel. C, Quantification of Tat mRNA as normalized to GAPDH immediately or at 2 weeks, 4 weeks, or 6 weeks after cessation of DOX treatment. D, Tat immunostaining in brains from Non tg, untreated GFAP-Tat mice, and DOX-treated mice at 0 weeks, 2 weeks, 4 weeks, or 6 weeks post-DOX treatment. E, Tat immunoprecipitation and immunoblot from GFAP-Tat mouse brains. Brain sections from Non tg and GFAP-Tat tg animals immunostained for LC3 and image with laser-scanning confocal microscope. F, Granular cytoplasmic LC3 immunostaining (green) in Non tg and GFAP-Tat mouse brains at 0 weeks, 2 weeks, 4 weeks, or 6 weeks post-DOX treatment. Scale bar, 10 μm. G, Computer-aided image analysis of LC3 signal pixel intensity in Non tg (Ntg) and GFAP-Tat mouse brains at 0 weeks, 2 weeks, 4 weeks, or 6 weeks post-DOX treatment. H, Electron micrograph illustrating autophagosome ultrastructure in Non tg, untreated GFAP-Tat, and DOX-treated GFAP-Tat mouse brains. Magnification, 10,000×. Scale bar, 1 μm. I, Cytoplasmic LC3 and NeuN immunostaining in Non tg and GFAP-Tat mouse brains. J, Quantification of LC3+ grains (red) per NeuN-positive cell (green) in Non tg and GFAP-Tat mouse brains. K, Cytoplasmic LC3 and GFAP immunostaining in Non tg and GFAP-Tat mouse brains. L, Computer-aided image analysis of LC3+ grains (red) per GFAP-positive cell (green) in Non tg and GFAP-Tat mouse brains. Statistical significance: *p < 0.05 by one-way ANOVA, post hoc Dunnett's test when comparing Non tg to Tat tg treated with DOX; #p < 0.05 by one-way ANOVA, post hoc Tukey–Kramer after cessation of DOX, n = 4 mice per condition, 7–8 months old).
To further investigate the alterations in autophagy, mouse brains were analyzed by electron microscopy for autophagosome ultrastructure in neurons. Non tg and untreated GFAP-Tat tg mouse brains displayed the presence of discrete lysosomal and autophagosome structures (Fig. 9H). In contrast, neuronal cells in the GFAP-Tat tg mouse brains from mice treated with DOX for 2 weeks showed more electron-dense structures and numerous autophagosomes (Fig. 9H). Autophagosomes maintained this dense morphology at 6 weeks post initial DOX treatment (Fig. 9H). Furthermore we sought to characterize the cellular localization of autophagosome formation in GFAP-Tat tg mouse brains (Fig. 9I–L). LC3 signal (red) is robustly increased throughout the cytoplasm in NeuN+ (green) cells (Fig. 9I). LC3 signal (red) is increased in GFAP+ cells, but to a lesser extent compared with NeuN+ cells (Fig. 9K). Quantification of LC3 immunostaining was increased sevenfold in NeuN+ cells and 2.5-fold in GFAP+ cells in GFAP-Tat tg mouse brains compared with Non tg (Fig. 9 J,L) Interestingly, LC3+ puncta were threefold higher in NeuN+ cells compared with GFAP+ cells (Fig. 9J,L).
GFAP-Tat tg mice present neuroinflammation and neurodegeneration in the hippocampus
To determine whether the alterations in autophagy in GFAP-Tat tg mice are associated with neuroinflammation or neurodegeneration, brain sections were immunolabeled with antibodies against GFAP and NeuN. Non tg and untreated GFAP-Tat tg mouse brains showed moderate GFAP immunostaining, indicating low levels of neuroinflammation (Fig. 10A). In contrast, GFAP immunostaining was robust in mouse brains at 2 weeks of DOX and 2 weeks after DOX administration was discontinued (Fig. 10A). GFAP immunoreactivity increased by ∼50% after 2 weeks of DOX, and 40% 2 weeks after the last DOX administration (Fig. 10B). At 4 and 6 weeks after DOX initial treatment, GFAP immunostaining was equivalent to that of Non tg and untreated GFAP-Tat tg mouse brains (Fig. 10B). NeuN-immunoreactive neurons in the CA3/CA2 region of the hippocampus were decreased in GFAP-Tat tg mouse brains immediately after 2 weeks of DOX and the neurodegeneration continued to be apparent through 6 weeks after initial DOX treatment (Fig. 10C). NeuN+ cell numbers were decreased compared with Non tg and untreated GFAP-Tat tg mouse brains by ∼45 and 40% immediately after 2 weeks of DOX and 2 weeks after DOX treatment cessation, respectively (Fig. 10D). NeuN+ cell numbers were reduced through 6 weeks after initial DOX treatment ∼20% compared with Non tg or untreated GFAP-Tat tg mouse brains (Fig. 10D). These data indicate HIV-1 Tat expression in the brain promotes neuroinflammation and neurodegeneration.
Figure 10.

GFAP-Tat mouse brains display advanced neuroinflammation and neurodegenerative pathology at highest Tat exposure. Non tg and GFAP-Tat mouse hippocampi were immunostained for astrogliosis marker GFAP and neural marker NeuN (green). A, GFAP immunolabeling in the hippocampus from Non tg, untreated GFAP-Tat, and mice at 0 weeks, 2 weeks, 4 weeks, or 6 weeks post-DOX treatment. B, Computer-aided image analysis of GFAP optical density in the hippocampus from Non tg (Ntg), untreated GFAP-Tat, and mice at 0 weeks, 2 weeks, 4 weeks, or 6 weeks post-DOX treatment. C, NeuN immunostaining in the hippocampus from Non tg, untreated GFAP-Tat, and mice at 0 weeks, 2 weeks, 4 weeks, or 6 weeks post-DOX treatment. D, Stereological image analysis using the dissector method to estimate total NeuN+ cells in the hippocampus from Non tg, untreated GFAP-Tat, and mice at 0 weeks, 2 weeks, 4 weeks, or 6 weeks post-DOX treatment. Statistical significance: *p < 0.05, **p < 0.01, #p < 0.05 between indicated conditions, one-way ANOVA, post hoc Fisher's test, n = 6 mice per condition, 7–8 months old. Scale bars: Top, 250 μm, bottom, 25 μm. Solid line box represents detail presented at higher magnification in the bottom.
Rapam treatment reduces neuroinflammation and neurodegeneration in GFAP-Tat tg mouse brains
In vitro studies in Figure 8 suggest activating pathways to enhance autophagy may prevent acute Tat-mediated neurotoxicity. To determine whether this may also occur in vivo in a long-term paradigm, Non tg and GFAP-Tat tg mice treated with DOX were infused with vehicle or Rapam; sections from these mice were immunostained for GFAP or NeuN. Non tg mice showed no difference in GFAP or NeuN immunostaining after Rapam treatment (Fig. 11A). In contrast, vehicle-treated GFAP-Tat tg mouse brains showed increased GFAP and decreased NeuN (Fig. 11A). Rapam treatment decreased GFAP and increased NeuN (Fig. 11A). Quantification of optical density revealed a twofold increase in GFAP immunostaining (p < 0.05) in GFAP-Tat tg mice, but Rapam treatment reduced this to levels comparable to those found in Non tg mice (Fig. 11B). NeuN+ cells were significantly reduced (p < 0.05) in GFAP-Tat tg mice compared with Non tg mice; however, Rapam infusion recovered NeuN levels in these GFAP-Tat tg mice to levels comparable to that of Non tg mice (Fig. 11C). To determine the cell-specific effects of Rapam treatment, Non tg and GFAP-Tat tg mouse brains were double immunostained for LC3 and GFAP or NeuN (Fig. 11D). Quantification of GFAP and LC3 colocalization showed that LC3 signal is increased by twofold (p < 0.05) in Non tg mice treated with Rapam, and vehicle-treated GFAP-Tat tg mice show a fourfold (p < 0.05) increase in LC3 signal (Fig. 11E). LC3 signal is reduced by 30% in Rapam-treated GFAP-Tat tg mice, similar to levels in Rapam-treated Non tg (Fig. 11E). LC3 colocalization with NeuN-positive cells increased by twofold (p < 0.05) in Rapam-treated Non tg mouse brains (Fig. 11F). LC3 signal in NeuN-positive cells in vehicle-treated GFAP-Tat tg mice was increased fourfold (p < 0.05) compared with vehicle-treated Non tg mice, while LC3 in NeuN-positive cells was increased by threefold in Rapam-treated GFAP-Tat tg mouse brains (Fig. 11F). These in vivo results show that Tat-mediated alterations in autophagy lead to cell death and that this effect is reversed with Rapam.
Figure 11.

Rapam treatment reduces neuroinflammation and neurodegeneration in GFAP-Tat tg mouse brains. Non tg and GFAP-Tat mouse brains were immunostained for GFAP, AIF1 (red), NeuN (green), and LC3. A, GFAP and NeuN immunostaining of brains from vehicle or Rapam-treated Non tg and GFAP-Tat tg mice. B, Computer-aided image analysis of GFAP optical density in brains from vehicle- and Rapam-treated Non tg and GFAP-Tat tg mice. C, Quantification of NeuN+ cells in vehicle- and Rapam-treated Non tg and GFAP-Tat tg mouse brains. D, GFAP, NeuN, and LC3 immunostaining of brains from vehicle or Rapam-treated Non tg and GFAP-Tat tg mice. E, Computer-aided quantification of LC3+ grains per GFAP+ cells in brains of vehicle- and Rapam-treated Non tg and GFAP-Tat tg mice. F, Computer-aided quantification of LC3+ grains per NeuN+ cells in brains of vehicle- and Rapam-treated Non tg and GFAP-Tat tg mice (*p < 0.05, one-way ANOVA, post hoc Fisher's test, n = 6 mice per condition, 7–8 months old). Scale bars: GFAP and NeuN, 25 μm; LC3, 10 μm.
HIV-1 Tat colocalizes with LAMP2A in GFAP-Tat tg mouse brains
To further confirm in vivo the in vitro studies suggesting Tat interacts with LAMP2A, homogenates from the brains of the GFAP-Tat tg mouse were analyzed by coimmunoprecipitation. Consistent with the in vitro results, we found that in the homogenates from the GFAP-Tat mice treated with DOX, Tat, and LAMP2A coimmunoprecipitated (Fig. 12A). Tat was not detected in Non tg brain lysates. LAMP2A immunostaining (green) was robust in Non tg and GFAP-Tat neurons; however, Tat signal (red) was only detectable in GFAP-Tat tg mouse brains and was strongly colocalized to LAMP2A (Fig. 12B). To confirm that Tat is associated with autophagosomes and lysosomes, as in neuronal cell lines and primary neurons, we double immunolabeled Tat with LC3 or CTSD. LC3 immunostaining was robust in Non tg and GFAP-Tat tg mouse brains, but Tat was detectable in GFAP-Tat tg mouse brains only (Fig. 12C). Tat colocalizes with LC3 in the cytoplasm, indicating Tat was closely associated with autophagosomes (Fig. 12C). To ensure that Tat is associated with lysosomes we double immunolabeled Tat with CTSD. Non tg and GFAP-Tat tg mice both strongly expressed CTSD (green) in neurons. Tat was detected in GFAP-Tat tg mouse brains only, and Tat (red) strongly colocalized with CTSD (yellow; Fig. 12D). To determine in which cell types Tat was expressed in the GFAP-Tat tg mice, brains from Non tg and GFAP tg mice were double immunolabeled for NeuN (green), GFAP (green), and Tat (red). Tat immunoreactivity was detected in GFAP-positive cells in GFAP-Tat tg, but not in Non tg mouse brains (Fig. 12E). Similarly, Tat signal was robust in NeuN-positive cells in GFAP-Tat tg, but not in Non tg mouse brains (Fig. 12F).These in vivo results show tat colocalizes with autophagosomal and lysosomal proteins in vivo.
Figure 12.

Tat associates with LC3, LAMP2A, and CTSD in GFAP-Tat mouse brains. GFAP-Tat mouse brains were isolated and used for LAMP2A and Tat coimmunoprecipitation of Tat double immunolabeling with LC3, LAMP2A, or CTSD. A, Non tg and GFAP-Tat mouse brains were homogenized and the lysates were immunoprecipitated (IP) for LAMP2A, resolved by SDS-PAGE and then immunoblotted for Tat. B, Non tg and GFAP-Tat mouse brains were double immunolabeled for Tat (red) and LAMP2A (green) and imaged by laser-scanning confocal microscopy. (Colocalization between Tat and LAMP2A is illustrated in yellow.) C, Non tg and GFAP-Tat mouse brains were double immunolabeled for Tat (red) and LC3 (green) and the images were merged (yellow) to illustrate colocalization. D, Non tg and GFAP-Tat mouse brains were double immunolabeled for Tat (red) and CTSD (green) and the images were merged (yellow) to illustrate colocalization. E, Non Tg and GFAP-Tat tg mouse brains were double immunolabeled for NeuN (green) and Tat (red). F, Non Tg and GFAP-Tat tg mouse brains were double immunolabeled for GFAP (green) and Tat (red). Scale bar, 10 μm. Dotted box represents detail presented at higher magnification to the right. WB, Western blot.
Discussion
The present study shows that HIV-1 Tat increases degradation of autophagic markers, and disrupts molecular inhibition of autophagy in neuronal cells. We found that Tat overcomes the actions of BafA1 on LC3II and SQSTM1 accumulation. Tat alters autophagosome morphology and quantity in neurons, though this effect was prevented when initial stages of autophagy were inhibited with shBECN1. Tat colocalizes with autophagy marker LC3 and lysosomal protease, CTSD, and induces LC3 and CTSD colocalization. Ultrastructure of Tat-treated neuronal cells and GFAP-Tat tg mice shows robust changes in autophagosome morphology and quantity. Moreover, Tat associates with neuronal LAMP2A in vitro and in brain homogenates from a GFAP-Tat tg mouse model of HAND. Importantly, Rapam or Torin 1 treatment and LAMP2A overexpression reduces Tat neurotoxic effects. Last, Rapam reversed the autophagy alterations and ameliorated the neurodegenerative and inflammatory phenotype in the GFAP-Tat tg mouse model. Our data suggest that Tat may facilitate abnormal autophagolysosome formation through interaction with lysosomal LAMP2A, leading to neurodegeneration.
The present findings are consistent with previous studies in which HIV proteins are secreted from infected cells and subsequently enter bystander cells (Benelli et al., 2000; Kandanearatchi et al., 2005; Kaul and Lipton, 2006; Alirezaei et al., 2007). HIV proteins affect cell processes through interaction with caspase machinery or NMDA receptors and thereby induce apoptosis, regulate calcium levels, alter glutamate excitotoxicity, and induce chemotaxis in bystander cells (Bonavia et al., 2001; Singh et al., 2005; Hargus and Thayer, 2013; Dalvi et al., 2014; Darbinian et al., 2014). Nef, a viral replication facilitator, prevents the destruction of HIV components by autophagolysosomes by inhibiting autophagic maturation in the host cells (Kyei et al., 2009). Gp120 was reported to increase autophagy in uninfected T-cells (Espert et al., 2007) as well as in LV-infected SK N SH cells (Zhou et al., 2011). Interestingly, in vitro experiments using HIV-infected microglial supernatant or gp120-transgenic mice showed a decrease in autophagy through possible decreases in Beclin levels and activity (Alirezaei et al., 2008a; Fields et al., 2013). Given that multiple HIV factors are expressed during HIVE, the combined impact of proteins, such as Tat, Nef, and gp120, on neuronal autophagy is likely to exacerbate neurodegeneration. Further investigations must be conducted to detect the singular as well as the aggregate effects of these proteins. The present studies follow up on a recent report that showed Tat affects the neuronal lysosome pathway and accumulation of autophagy proteins (Hui et al., 2012). Hui et al. (2012) found that Tat associates with lysosomes and reduces LC3II levels in neurons. LC3II levels are not sufficient to determine autophagy flux due to the fact that LC3II levels result from equilibrium of LC3II formation and degradation, and often must be analyzed in context with autophagosome presence and other markers. For example, BafA1 and Chloro disrupt autophagy by blocking acidification of lysosomes, and subsequent fusion of lysosomes with autophagosomes (Amadoro et al., 2014), resulting in accumulation of LC3II and SQSTM1. However, Tat reduced SQSTM1 and LC3II levels suggesting increased autophagic degradation. In contrast to the Hui et al. (2012) studies, and in support of the notion that Tat facilitates autophagosome and lysosome fusion, Tat is able to counter BafA1 and Chloro-mediated block in LC3II and SQSTM1 degradation. Tat may overcome this effect by promoting fusion of autophagosomes with lysosomes more rapidly than BafA1 or Chloro can block lysosome acidification (Fig. 13), leading to aberrant degradation of important neuronal organelles.
Figure 13.

Diagrammatic representation of the potential mechanisms through which Tat might interfere with neuronal autophagy. Early during HIV infection the virus crosses the blood–brain barrier in monocytes. These monocytes disseminate progeny virus, cytokines, and toxic viral proteins. Viruses infect other permissive CNS cells, such as microglia and possible astrocytes, and a cascade of viral replication and neuroinflammation ensues. HIV-1 Tat is secreted by infected cells and gains access to neurons; a phenomenon that may lead to bystander neuron malfunction or death. Autophagy is an essential cellular process that allows neurons to maintain dynamic synapses and respond to stress and learning cues. Autophagy begins with (1) initiation of the phagophore (induced by Rapam, Torin 1, and Tamox; inhibited by 3MA), which is inhibited by mTor, and (2) then elongation and engulfment of targeted cytosolic components. (3) The autophagosome fuses with the lysosome (inhibited by BafA1 and Chloro), via interaction with LAMP2A, to form the autophagolysosome and promote degradation of the autophagic cargo. (4) Our data suggest Tat associates with LAMP2A and thereby alters autophagolysosome formation or CMA progression. This scenario may explain increased LC3II and SQSTM1 degradation while increasing cytoplasmic autophagosome numbers. Alternatively, Tat may inhibit LC3II formation by an unknown mechanism, while concomitantly increasing degradation of SQSTM1-conjugated proteins through CMA. (5) The final stage of autophagy includes degradation of the autophagolysosome, internal cargo, and associated proteins such as LC3II and SQSTM1-conjugated proteins. Tat may influence HAND by increasing autophagy progression and causing premature degradation of proteins essential for neural activity.
Autophagosome number is altered in the brains of HIV-infected persons (Alirezaei et al., 2008a); however, the nature of the autophagy pathway is such that a snapshot of protein levels is not sufficient to specify a pathologic mechanism. Our laboratory recently reported autophagy machinery levels are altered in postmortem brains of HIV patients (Zhou et al., 2011), and more recently that “young” (<50) HIV patients express high levels of autophagy machinery while levels are low in brains from “aged” (>50) patients (Fields et al., 2013). The aged and young distinctions are made in the context of HIV infection, consistent with other reports that suggest HIV patients over 50 are more susceptible to cognitive impairments (Wendelken and Valcour, 2012). This may be explained by our results showing that Tat increases GFP-LC3 autophagosomes present in neuronal cells and primary neurons; however, BafA1 had a similar effect on GFP-LC3 puncta. BafA1 increases GFP-LC3 puncta by inhibiting degradation of LC3II, but Tat may counteract this, and induce LC3II degradation by facilitating autophagolysosome formation. Accordingly, knockdown of BECN1 prevents Tat-induced GFP-LC3 puncta formation, suggesting macroautophagy is necessary for these effects. This may also explain how Tat increases the number of autophagosome-like structures per cell as imaged by electron microscopy.
Cargo-filled autophagosomes fuse with acidic lysosomes through interaction with LAMP2A (Bandyopadhyay and Cuervo, 2007; Zhang and Cuervo, 2008). Interestingly, Tat colocalizes with LC3 and CTSD, indicating association with both autophagosomes and lysosomes. Moreover, Tat enhances LC3 and CTSD double immunolabeling indicating increased autophagosome fusion with lysosomes. LAMP2A is a target of pathologic proteins such as α-syn, and this association may contribute to Parkinson's disease (Cuervo et al., 2004). Furthermore, LAMP2A is necessary for efficient macroautophagy and chaperone-mediated autophagy (CMA) progression (González-Polo et al., 2005; Zhang and Cuervo, 2008). Indeed, LAMP2A levels are reduced with age in some organs, and it has been suggested that restoring levels may reverse some aging effects (Saftig and Eskelinen, 2008; Saftig et al., 2008; Zhang and Cuervo, 2008). Importantly, Tat associates with LAMP2A in a way that may facilitate autophagolysosome formation and thereby increase LC3II degradation, which explains how Tat increases LC3+ puncta while decreasing LC3II protein levels in neurons. Accordingly, increased GFP-LC3 vesicles in Tat-treated cells may signify active autophagolysosomes, but GFP-LC3 puncta in BafA1-treated cells may represent accumulated LC3II+ autophagosomes. Conversely, LAMP2A is important for CMA (Cuervo et al., 2004; Bandyopadhyay and Cuervo, 2007; Zhang and Cuervo, 2008; Alvarez-Erviti et al., 2010). Therefore, Tat may interfere with CMA and thereby indirectly increase macroautophagy function as a feedback mechanism to maintain recycling of neuronal components. The fact that Rapam or Torin 1 treatment, and overexpression of LAMP2A, reduced Tat-mediated neurotoxicity suggests that Tat promotes deleterious and dysregulated autophagic degradation. This effect, along with other deleterious Tat mechanisms, may result in the observed neurodegeneration.
The inducible GFAP-Tat tg mouse model provides a unique method for studying the role of Tat in neuropathogenesis. The model is associated with neurodegeneration and neurotoxicity similar to that seen in brains of HIV patients (Kim et al., 2003). In our studies, increased LC3 puncta coincided with 2 weeks of Tat expression. This corroborates data in our cell models by showing that in vivo Tat expression affects neurons over an extended period. In vitro, upon Tat treatment we found decreased LC3II and SQSTM1 levels while LC3+ puncta were increased, suggesting an increase in autophagic flux. Similarly, in GFAP-Tat tg mice after 2 weeks of Tat expression we found increased LC3+ puncta in neurons and increased neurodegeneration. This mechanism may explain differences in the brain; autophagy protein levels increased in brains of young and decreased in brains of aged HIV patients (Fields et al., 2013). Tat appears to be closely associated with the autophagy machinery in neurons from the GFAP-Tat tg model, suggesting a similar mechanism as seen in our cell models. Our studies confirm Tat causes neurodegeneration in vivo and in vitro, both of which are reversed by Rapam treatment. Paradoxically, Rapam and Tat both increase autophagic flux, but Rapam reverses Tat-induced neurodegeneration. This effect may be due to robust induction of neuronal macroautophagy by Rapam that overcomes the multiple insults Tat causes in neurons. Another possibility is that mTor inhibition ameliorates Tat-induced neurotoxicity through a pathway other than autophagy induction; future studies will focus on determining this mechanism. Tat-induced autophagy alterations may occur at the autophagolysosome formation stage in a way that leads to neurodegeneration through aberrant degradation. These data may suggest Tat plays a specific role, along with other pathogenic mechanisms, promoting deleterious neuronal autophagy alterations that are rectified through normal macroautophagy induction or increased LAMP2A, which may sequester Tat. Nonetheless, it is clear that our results do not provide conclusive evidence that LAMP2A is involved in Tat-mediated effects on autophagy. Future investigations will focus on how Tat and LAMP2A interactions, and which portions, affect autophagolysosome formation in neurons. Moreover, astrocyte LC3+ puncta are also increased, suggesting Tat affects glial autophagy. Increased astrocyte activation and LC3 signal are both reversed following Rapam treatment, and this may contribute to reduced neurotoxicity in vivo by rectifying astrocyte autophagy dysfunction. Further studies are necessary to fully understand the role of Tat on astrocyte autophagy as well as the mechanism at play in neurons.
Our current data may partially explain mechanisms that lead to HAND; Tat, in concert with other factors, may initially alter neuronal autophagy in a deleterious manner, while other processes or chronic exposure to Tat and overuse of the autophagy system may result in lower autophagy levels and concomitant neurodegeneration in late stages of HIV infection. Therapies that increase LAMP2A function through blocking interactions with Tat or increasing LAMP2A expression and increasing autophagy efficiency may be useful in combatting HAND.
Footnotes
We thank National Institute of Aging (AG043384 to E.M.), the National Institute of Mental Health (MH062962, MH5974, and MH83506 to E.M.), and the National Institute for Neurological Disorders and Stroke (1F32NS083426-01 to J.F.). We would also like to thank the National Institutes of Health AIDS Reagents Program for providing recombinant Tat (catalog #2222).
The authors declare no competing financial interests.
References
- Alirezaei M, Watry DD, Flynn CF, Kiosses WB, Masliah E, Williams BR, Kaul M, Lipton SA, Fox HS. Human immunodeficiency virus-1/surface glycoprotein 120 induces apoptosis through RNA-activated protein kinase signaling in neurons. J Neurosci. 2007;27:11047–11055. doi: 10.1523/JNEUROSCI.2733-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alirezaei M, Kiosses WB, Fox HS. Decreased neuronal autophagy in HIV dementia: a mechanism of indirect neurotoxicity. Autophagy. 2008a;4:963–966. doi: 10.4161/auto.6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alirezaei M, Kiosses WB, Flynn CT, Brady NR, Fox HS. Disruption of neuronal autophagy by infected microglia results in neurodegeneration. PLoS One. 2008b;3:e2906. doi: 10.1371/journal.pone.0002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, Schapira AH. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol. 2010;67:1464–1472. doi: 10.1001/archneurol.2010.198. [DOI] [PubMed] [Google Scholar]
- Amadoro G, Corsetti V, Florenzano F, Atlante A, Ciotti MT, Mongiardi MP, Bussani R, Nicolin V, Nori SL, Campanella M, Calissano P. AD-linked, toxic NH2 human tau affects the quality control of mitochondria in neurons. Neurobiol Dis. 2014;62:489–507. doi: 10.1016/j.nbd.2013.10.018. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay U, Cuervo AM. Chaperone-mediated autophagy in aging and neurodegeneration: lessons from alpha-synuclein. Exp Gerontol. 2007;42:120–128. doi: 10.1016/j.exger.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Benelli R, Barbero A, Ferrini S, Scapini P, Cassatella M, Bussolino F, Tacchetti C, Noonan DM, Albini A. Human immunodeficiency virus transactivator protein (Tat) stimulates chemotaxis, calcium mobilization, and activation of human polymorphonuclear leukocytes: implications for Tat-mediated pathogenesis. J Infect Dis. 2000;182:1643–1651. doi: 10.1086/317597. [DOI] [PubMed] [Google Scholar]
- Bonavia R, Bajetto A, Barbero S, Albini A, Noonan DM, Schettini G. HIV-1 Tat causes apoptotic death and calcium homeostasis alterations in rat neurons. Biochem Biophys Res Commun. 2001;288:301–308. doi: 10.1006/bbrc.2001.5743. [DOI] [PubMed] [Google Scholar]
- Budka H, Costanzi G, Cristina S, Lechi A, Parravicini C, Trabattoni R, Vago L. Brain pathology induced by infection with the human immunodeficiency virus (HIV). A histological, immunocytochemical, and electron microscopical study of 100 autopsy cases. Acta Neuropathol. 1987;75:185–198. doi: 10.1007/BF00687080. [DOI] [PubMed] [Google Scholar]
- Cherner M, Cysique L, Heaton RK, Marcotte TD, Ellis RJ, Masliah E, Grant I. Neuropathologic confirmation of definitional criteria for human immunodeficiency virus-associated neurocognitive disorders. J Neurovirol. 2007;13:23–28. doi: 10.1080/13550280601089175. [DOI] [PubMed] [Google Scholar]
- Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol. 2012;13:7–12. doi: 10.1038/nrm3249. [DOI] [PubMed] [Google Scholar]
- Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, Hansen L, Adame A, Galasko D, Masliah E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One. 2010;5:e9313. doi: 10.1371/journal.pone.0009313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70–77. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Dalvi P, Wang K, Mermis J, Zeng R, Sanderson M, Johnson S, Dai Y, Sharma G, Ladner AO, Dhillon NK. HIV-1/cocaine induced oxidative stress disrupts tight junction protein-1 in human pulmonary microvascular endothelial cells: role of Ras/ERK1/2 pathway. PLoS One. 2014;9:e85246. doi: 10.1371/journal.pone.0085246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbinian N, Khalili K, Amini S. Neuroprotective activity of pDING in response to HIV-1 Tat. J Cell Physiol. 2014;229:153–161. doi: 10.1002/jcp.24392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Department of Health and Human Services, Public Health Service: Centers for Disease Control and Prevention. 2005. HIV/AIDS surveillance report, pages 1–63.
- Department of Health and Human Services, Public Health Service: Centers for Disease Control and Prevention. 2007. HIV/AIDS surveillance report, pages 1–54.
- Espert L, Denizot M, Grimaldi M, Robert-Hebmann V, Gay B, Varbanov M, Codogno P, Biard-Piechaczyk M. Autophagy and CDR+ T lymphocyte destruction by HIV-1. 2007 doi: 10.4161/auto.3275. [DOI] [PubMed] [Google Scholar]
- Fields J, Dumaop W, Rockenstein E, Mante M, Spencer B, Grant I, Ellis R, Letendre S, Patrick C, Adame A, Masliah E. Age-dependent molecular alterations in the autophagy pathway in HIVE patients and in a gp120 tg mouse model: reversal with beclin-1 gene transfer. J Neurovirol. 2013;19:89–101. doi: 10.1007/s13365-012-0145-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. San Diego: Academic; 1997. [Google Scholar]
- Gendelman HE, Persidsky Y, Ghorpade A, Limoges J, Stins M, Fiala M, Morrisett R. The neuropathogenesis of the AIDS dementia complex. AIDS. 1997;11(Suppl A):S35–S45. [PubMed] [Google Scholar]
- González-Polo RA, Boya P, Pauleau AL, Jalil A, Larochette N, Souquère S, Eskelinen EL, Pierron G, Saftig P, Kroemer G. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci. 2005;118:3091–3102. doi: 10.1242/jcs.02447. [DOI] [PubMed] [Google Scholar]
- Hargus NJ, Thayer SA. Human immunodeficiency virus-1 Tat protein increases the number of inhibitory synapses between hippocampal neurons in culture. J Neurosci. 2013;33:17908–17920. doi: 10.1523/JNEUROSCI.1312-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, Rockenstein E, Mante M, Mallory M, Masliah E. beta-Synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron. 2001;32:213–223. doi: 10.1016/S0896-6273(01)00462-7. [DOI] [PubMed] [Google Scholar]
- Heaton RK, Clifford DB, Franklin DR, Jr, Woods SP, Ake C, Vaida F, Ellis RJ, Letendre SL, Marcotte TD, Atkinson JH, Rivera-Mindt M, Vigil OR, Taylor MJ, Collier AC, Marra CM, Gelman BB, McArthur JC, Morgello S, Simpson DM, McCutchan JA, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology. 2010;75:2087–2096. doi: 10.1212/WNL.0b013e318200d727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol. 2011;17:3–16. doi: 10.1007/s13365-010-0006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselgesser J, Horuk R. Chemokine and chemokine receptor expression in the central nervous system. J Neurovirol. 1999;5:13–26. doi: 10.3109/13550289909029741. [DOI] [PubMed] [Google Scholar]
- Hui L, Chen X, Haughey NJ, Geiger JD. Role of endolysosomes in HIV-1 Tat-induced neurotoxicity. ASN Neuro. 2012;4:243–252. doi: 10.1042/AN20120017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joska JA, Gouse H, Paul RH, Stein DJ, Flisher AJ. Does highly active antiretroviral therapy improve neurocognitive function? A systematic review. J Neurovirol. 2010;16:101–114. doi: 10.3109/13550281003682513. [DOI] [PubMed] [Google Scholar]
- Kandanearatchi A, Nath A, Lipton SA, Masliah E, Everall IP. Protection against HIV-1 gp120 and HIV-1 tat neurotoxicity. In: Gendelman HE, Grant AD, Everall IP, Lipton SA, Swindells S, editors. The neurology of AIDS. Ed 2. Oxford, UK: Oxford UP; 2005. pp. 200–210. [Google Scholar]
- Kaul M, Lipton SA. Mechanisms of neuronal injury and death in HIV-1 associated dementia. Curr HIV Res. 2006;4:307–318. doi: 10.2174/157016206777709384. [DOI] [PubMed] [Google Scholar]
- Kim BO, Liu Y, Ruan Y, Xu ZC, Schantz L, He JJ. Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 Tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am J Pathol. 2003;162:1693–1707. doi: 10.1016/S0002-9440(10)64304-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, Wu L, Kominami E, Ueno T, Yamamoto A, Federico M, Panganiban A, Vergne I, Deretic V. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol. 2009;186:255–268. doi: 10.1083/jcb.200903070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JC, Au KY, Fang JW, Yim HC, Chow KH, Ho PL, Lau AS. HIV-1 trans-activator protein dysregulates IFN-gamma signaling and contributes to the suppression of autophagy induction. AIDS. 2011;25:15–25. doi: 10.1097/QAD.0b013e328340fd61. [DOI] [PubMed] [Google Scholar]
- Masliah E, Achim C, DeTeresa R, Ge N, Wiley C. Cellular neuropathology in HIV encephalitis. In: Price R, editor. HIV, AIDS and the brain. New York: Raven; 1994. pp. 119–131. [PubMed] [Google Scholar]
- Masliah E, Alford M, Adame A, Rockenstein E, Galasko D, Salmon D, Hansen LA, Thal LJ. Abeta1-42 promotes cholinergic sprouting in patients with AD and Lewy body variant of AD. Neurology. 2003;61:206–211. doi: 10.1212/01.WNL.0000073987.79060.4B. [DOI] [PubMed] [Google Scholar]
- Naldini L, Blömer U, Gage FH, Trono D, Verma IM. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci U S A. 1996a;93:11382–11388. doi: 10.1073/pnas.93.21.11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996b;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- Nath A, Steiner J. Synaptodendritic injury with HIV-Tat protein: what is the therapeutic target? Exp Neurol. 2014;251:112–114. doi: 10.1016/j.expneurol.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A, Conant K, Chen P, Scott C, Major EO. Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes. A hit and run phenomenon. J Biol Chem. 1999;274:17098–17102. doi: 10.1074/jbc.274.24.17098. [DOI] [PubMed] [Google Scholar]
- Nguyen DP, Lin SC. A frontal cortex event-related potential driven by the basal forebrain. eLife. 2014;3:e02148. doi: 10.7554/eLife.02148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–2199. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J, Pham TN, Ishizaka Y, Cohen EA. Viral protein R upregulates expression of ULBP2 on uninfected bystander cells during HIV-1 infection of primary CD4+ T lymphocytes. Virology. 2013;443:248–256. doi: 10.1016/j.virol.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Mante M, Sisk A, Masliaha E. Early formation of mature amyloid-b proteins deposits in a mutant APP transgenic model depends on levels of Ab1–42. J Neurosci Res. 2001;66:573–582. doi: 10.1002/jnr.1247. [DOI] [PubMed] [Google Scholar]
- Saftig P, Eskelinen EL. Live longer with LAMP-2. Nat Med. 2008;14:909–910. doi: 10.1038/nm0908-909. [DOI] [PubMed] [Google Scholar]
- Saftig P, Beertsen W, Eskelinen EL. LAMP-2: a control step for phagosome and autophagosome maturation. Autophagy. 2008;4:510–512. doi: 10.4161/auto.5724. [DOI] [PubMed] [Google Scholar]
- Schubert D, Heinemann S, Carlisle W, Tarikas H, Kimes B, Patrick J, Steinbach JH, Culp W, Brandt BL. Clonal cell lines from the rat central nervous system. Nature. 1974;249:224–227. doi: 10.1038/249224a0. [DOI] [PubMed] [Google Scholar]
- Scott JC, Woods SP, Carey CL, Weber E, Bondi MW, Grant I. Neurocognitive consequences of HIV infection in older adults: an evaluation of the “cortical” hypothesis. AIDS Behav. 2011;15:1187–1196. doi: 10.1007/s10461-010-9815-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevts E, Fass E, Scherz-Shouval R, Elazar Z. The N-terminus and Phe52 residue of LC3 recruit p62/SQSTM1 into autophagosomes. J Cell Sci. 2008;121:2685–2695. doi: 10.1242/jcs.026005. [DOI] [PubMed] [Google Scholar]
- Singh IN, El-Hage N, Campbell ME, Lutz SE, Knapp PE, Nath A, Hauser KF. Differential involvement of p38 and JNK MAP kinases in HIV-1 Tat and gp120-induced apoptosis and neurite degeneration in striatal neurons. Neuroscience. 2005;135:781–790. doi: 10.1016/j.neuroscience.2005.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson's and Lewy body diseases. J Neurosci. 2009;29:13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocco A, Lebiere C, Anderson JR. Conditional routing of information to the cortex: a model of the basal ganglia's role in cognitive coordination. Psychol Rev. 2010;117:541–574. doi: 10.1037/a0019077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiscornia G, Singer O, Verma IM. Design and cloning of lentiviral vectors expressing small interfering RNAs. Nat Protoc. 2006;1:234–240. doi: 10.1038/nprot.2006.36. [DOI] [PubMed] [Google Scholar]
- Toborek M, Lee YW, Pu H, Malecki A, Flora G, Garrido R, Hennig B, Bauer HC, Nath A. HIV-Tat protein induces oxidative and inflammatory pathways in brain endothelium. J Neurochem. 2003;84:169–179. doi: 10.1046/j.1471-4159.2003.01543.x. [DOI] [PubMed] [Google Scholar]
- Van Grol J, Subauste C, Andrade RM, Fujinaga K, Nelson J, Subauste CS. HIV-1 inhibits autophagy in bystander macrophage/monocytic cells through Src-Akt and STAT3. PLoS One. 2010;5:e11733. doi: 10.1371/journal.pone.0011733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veinbergs I, Van Uden E, Mallory M, Alford M, McGiffert C, DeTeresa R, Orlando R, Masliah H. Role of apolipoprotein E receptors in regulating the differential in vivo neurotrophic effects of apolipoprotein E. Exp Neurol. 2001;170:15–26. doi: 10.1006/exnr.2001.7684. [DOI] [PubMed] [Google Scholar]
- Wendelken LA, Valcour V. Impact of HIV and aging on neuropsychological function. J Neurovirol. 2012;18:256–263. doi: 10.1007/s13365-012-0094-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley CA, Achim C. HIV encephalitis is the pathologic correlate of dementia in AIDS. Ann Neurol. 1994;36:673–676. doi: 10.1002/ana.410360422. [DOI] [PubMed] [Google Scholar]
- Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med. 2008;14:959–965. doi: 10.1038/nm.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Masliah E, Spector SA. Autophagy is increased in postmortem brains of persons with HIV-1-associated encephalitis. J Infect Dis. 2011;203:1647–1657. doi: 10.1093/infdis/jir163. [DOI] [PMC free article] [PubMed] [Google Scholar]