

Abstract
Neurogenic detrusor overactivity (NDO) is a well known consequence of spinal cord injury (SCI), recognizable after spinal shock, during which the bladder is areflexic. NDO emergence and maintenance depend on profound plastic changes of the spinal neuronal pathways regulating bladder function. It is well known that neurotrophins (NTs) are major regulators of such changes. NGF is the best-studied NT in the bladder and its role in NDO has already been established. Another very abundant neurotrophin is BDNF. Despite being shown that, acting at the spinal cord level, BDNF is a key mediator of bladder dysfunction and pain during cystitis, it is presently unclear if it is also important for NDO. This study aimed to clarify this issue.
Results obtained pinpoint BDNF as an important regulator of NDO appearance and maintenance. Spinal BDNF expression increased in a time-dependent manner together with NDO emergence. In chronic SCI rats, BDNF sequestration improved bladder function, indicating that, at later stages, BDNF contributes NDO maintenance. During spinal shock, BDNF sequestration resulted in early development of bladder hyperactivity, accompanied by increased axonal growth of calcitonin gene-related peptide-labeled fibers in the dorsal horn. Chronic BDNF administration inhibited the emergence of NDO, together with reduction of axonal growth, suggesting that BDNF may have a crucial role in bladder function after SCI via inhibition of neuronal sprouting. These findings highlight the role of BDNF in NDO and may provide a significant contribution to create more efficient therapies to manage SCI patients.
Keywords: BDNF, incontinence, neurotrophins, spinal cord, urinary bladder
Introduction
One of the most important consequences of spinal cord injury (SCI) is severe urinary dysfunction (de Groat and Steers, 1990; Cruz and Cruz, 2011). Trauma is immediately followed by spinal shock, during which the bladder is areflexic and urinary retention occurs. Spinal shock is slowly replaced by an involuntary spinal micturition reflex controlled by a circuit located at lumbosacral spinal cord level. This new spinal micturition reflex does not guarantee the coordination between detrusor contraction and bladder outlet relaxation (Cruz and Cruz, 2011). Consequently, many SCI patients develop a functional bladder outlet obstruction called detrusor sphincter dyssynergia (DSD), which leads to long periods of high intravesical pressure. This increase in bladder pressure is punctuated by strong involuntary detrusor contractions, a condition termed neurogenic detrusor overactivity (NDO). Rises in intravesical pressure endanger urinary tract and renal function and can trigger episodes of autonomic dysreflexia (Rabchevsky, 2006). Therefore, initial management of SCI patients typically includes measures to protect kidney function by relieving DSD and reducing pressure. Once intravesical pressure is under control, incontinence emerges as a critical point to the quality of life of these patients. Results from a series of surveys are evident: after improving motor function, regaining bladder control is of the highest priority for SCI patients (Ku, 2006; French et al., 2010; Simpson et al., 2012). Antimuscarinic drugs and onabotulinum toxin A are used to reduce NDO and intravesical pressure (Cruz et al., 2011; Sahai et al., 2011) but these therapies are unable to reverse or to prevent the emergence of NDO.
It is currently assumed that NDO and DSD result from massive reorganization of neuronal pathways located in the lumbosacral spinal cord (Vizzard, 2006; Cruz and Cruz, 2011), although the molecular mechanisms are unclear. In the bladder significant cellular remodeling occurs, including smooth muscle hypertrophy, patchy denervation, and changes in interstitial cell (IC) populations (Johnston et al., 2012). Several studies suggest that neurotrophins (NTs) may be important mediators of such plastic changes (Ochodnický et al., 2011, 2012; Cruz, 2014), in particular, NGF, a neurotrophin that plays a key role in the regulation of the peripheral nervous system (Pezet and McMahon, 2006; Vizzard, 2006). Immunoneutralization of this NT has been shown to block bladder dysfunction in SCI rats (Seki et al., 2002, 2004). In contrast, the contribution of other NTs to NDO, most notably BDNF, remains poorly investigated. BDNF is extremely abundant in the CNS where it modulates neuroplasticity at spinal cord level. Its participation in central sensitization associated with chronic pain is well established (Merighi et al., 2004, 2008). Acute intrathecal administration of BDNF quickly produces cutaneous pain and bladder hyperactivity (Frias et al., 2013) whereas its intrathecal blockade was shown to reduce pain and bladder hyperactivity in rats with chronic cystitis (Frias et al., 2013). Therefore, the aim of the present study was to investigate the contribution of BDNF to the emergence of NDO using a rat model of SCI.
Materials and Methods
Animals.
Female Wistar rats from Charles River weighing 250–275 g were used. Animals were kept on a 12 h dark/light cycle, in a temperature-controlled environment with ad libitum access to food and water. All efforts were made to reduce animal stress and suffering as well as the number of animals used. The ethical guidelines for investigation of experimental pain in animals (Zimmermann, 1983) and the European Commission Directive of 22 September 2010 (2010/63/EU) were carefully followed in all procedures included in this study.
Chemicals and reagents.
All surgeries were performed under deep anesthesia induced by intraperitoneal injection of a mixture of medetomidine (0.25 mg/kg) and ketamine (60 mg/kg), diluted in sterile saline. For cystometries and terminal handling, rats received a subcutaneous bolus of urethane (1.2 g/kg) as anesthetic.
The recombinant protein TrkB-Ig2, which is an extracellular Ig-like domain of the receptor TrkB that binds to BDNF with picomolar affinity, was produced in-house and diluted in 20 mm Tris buffer, pH 8.2; 100 mm NaCl; and 10% glycerol (Banfield et al., 2001; Naylor et al., 2002). BDNF (Neuromics), saline, and TrkB-Ig2 were delivered into the subarachnoid space via osmotic mini-pumps (Alzet). Substances were delivered for 7 d (Model 2001) or 28 d (Model 2004).
For cell culture, DMEM-F12, FBS, penicillin/streptomycin (Pen/Strep), and l-glutamine were bought from Invitrogen. Collagenase (type IV-S), BSA, poly-l-lysine, and laminin were purchased from Sigma. B27, a serum-free supplement for neural cell culture, came from Gibco whereas NGF 2.5S came from Millipore.
Spinal cord transection and cystometry.
The model of SCI chosen for the present study was chronic spinal cord transection. One group of female Wistar rats were spinal intact and used as controls (Group A; Table 1). Two other groups were only submitted to spinal cord injury and left to recover for 1 week (Group B) or 4 weeks (Group C). The spinal cord was completely sectioned at T9 level and sterile Gelfoam was placed between the retracted ends of the cord. The remaining groups of rats underwent surgical implantation of a silicone catheter (SF Medical; internal diameter: 0.3 mm, outer diameter: 0.635 mm; Kerr et al., 1999; Thompson et al., 1999; Cruz et al., 2005, 2006; Frias et al., 2013) followed by complete spinal cord transection. Previously sterilized catheters were placed into the lumbar subarachnoid space at the L5/L6 spinal cord level. Briefly, a laminectomy was performed between T9 and 10 and the meninges were pierced. The catheter was inserted under the subarachnoid membrane and pushed until the tip reached the L5-L6 spinal cord segment. This was followed by spinal cord sectioning. The other end of the tip of the catheter was connected to an osmotic mini-pump (for continuous delivery of sterile saline, TrkB-Ig2, or BDNF; see below) placed between the shoulder blades and left for 1 week or 4 weeks.
Table 1.
Different experimental groups utilized in this study
| Experimental groups | Total number of animals |
|---|---|
| Spinal intact (A) | n = 6 (6) |
| SCI 1 week (B) | n = 6 (6) |
| SCI 4 weeks (C) | n = 6 (6) |
| SCI 4 weeks + (saline, 1, 10, and 20 μg) TrkB-Ig2 (D) | n = 6 (6) |
| SCI 1 week + chronic sterile saline (E) | n = 4 (4) |
| SCI 4 weeks + chronic sterile saline (F) | n = 4 (4) |
| SCI + TrkB-Ig2 20 μg/d for 1 week (G) | n = 7 (6) |
| SCI + BDNF 0.7 μg/d for 4 weeks (H) | n = 8 (6) |
| SCI + BDNF 2.1 μg/d for 4 weeks (I) | n = 8 (6) |
In Groups A–C, assessment of bladder function was performed in spinal-intact and in SCI animals at the end of first and fourth week post injury. In Group D, 4 week SCI animals were acutely treated, via an intrathecal catheter, with increasing amounts of a BDNF scavenger, TrkB-Ig2. Groups E and F received chronic intrathecal administration of saline for 1 week or 4 weeks post-SCI, respectively, via an osmotic mini-pump linked to a silicone intrathecal catheter. Group G was submitted to chronic delivery of TrkB-Ig2, using an intrathecal catheter connected to an osmotic mini-pump, for 1 week. Treatment was initiated immediately after SCI. SCI rats in Groups H and I were submitted to chronic intrathecal delivery of BDNF for four weeks post-SCI. Two different doses were tested, 0.7 and 2.1 μg/d. As in Groups E–G, treatment was initiated immediately after surgical cord lesion. Number of animals used for data analysis shown in parenthesis.
In one group of SCI animals, the subcutaneous end of the catheter was externalized for acute drug delivery 4 weeks after surgery (Group D). Animals received sequential intrathecal injections of sterile saline and TrkB-Ig2 (1 μg; 10 and 20 μg in a volume of 25 μl) every 30 min while bladder reflex activity was registered. Injection of TrkB-Ig2 was followed by a flush of the same volume of saline to assure that all recombinant protein was injected into the subarachnoid space.
In two groups of SCI animals, the intrathecal catheter was connected to an osmotic mini-pump for continuous delivery of either sterile saline (Group E) or TrkB-Ig2 (20 μg/d; Group G) for 1 week. On the seventh day, bladder reflex activity was evaluated by cystometry. In three other groups of SCI rats, as above, the silicone catheter was connected to an osmotic mini-pump for continuous delivery of either sterile saline (Group F) or BDNF (0.7 and 2.1 μg/d; Groups H and I, respectively) for a period of 4 weeks. The smallest dose was chosen as potentially producing effects on bladder function without cutaneous pain (Frias et al., 2013) whereas the highest dose was based on the effects observed with 0.7 μg/d. No side effects were registered in SCI animals submitted to either chronic BDNF sequestration or chronic administration of BDNF during the experimental period.
All animals were carefully monitored and received a daily intraperitoneal injection of antibiotic (ciprofloxacin, 1 mg/kg) for 2 weeks after surgery. To avoid urinary retention, bladders were manually emptied by abdominal compression twice every day. Because this procedure is more difficult to perform in male rats, female animals were preferred in the present study.
For cystometry, the bladder was exposed through a low abdominal midline incision and a 21 gauge needle was inserted into the bladder dome for saline infusion. Animals were left untouched for 15–30 min to allow bladder stabilization. Body temperature was maintained at 36–37°C with a heating pad. The urethra remained unobstructed throughout the experiment so that infused saline could easily be expelled by bladder contractions. After bladder stabilization, saline infusion was initiated and bladder reflex activity was recorded. Saline was infused through the dome needle at a constant rate of 6 ml/h while bladder contractions were registered by a pressure transducer (WPI) connected to a computer.
At the end of the experiments, animals were perfused and the position of the catheter verified. The cystometrograms obtained were analyzed. The frequency, peak and baseline pressures, and amplitude of bladder contractions were determined in the different phases of the experiment.
Perfusion and immunohistochemistry.
After cystometry, animals were perfused through the ascending aorta with cold, oxygenated calcium-free Tyrode's solution (0.12 m NaCl, 5.4 mm KCl, 1.6 mm MgCl2 · 6H2O, 0.4 mm MgSO4 · 7H2O, 1.2 mm NaH2PO4 · H2O, 5.5 mm glucose, and 26.2 mm NaHCO3) followed by 4% paraformaldehyde. The dissection of the perfused nervous tissue allowed the confirmation of the position of the intrathecal catheter. Only animals in which the catheter was correctly placed were considered for further analysis. Five animals, in which connection of the catheter to the osmotic mini-pump was broken (Groups G–I; Table 1), were excluded. The spinal cord segments L5-L6 were collected, postfixed for 4 h, and cryoprotected for 24 h in 30% sucrose with 0.1% sodium azide in 0.1 m phosphate buffer. Transverse 40 μm sections of the collected spinal cord segments were cut in a freezing microtome and stored in cryoprotective solution at −20°C until all tissue was collected.
The expressions of GAP-43 and phosphoJNK were tested using the ABC method. The specificity of the anti-phospho JNK was tested by incubating spinal sections from 4 week SCI rats in the absence of primary antibody. No staining was observed (Fig. 7A). Briefly, sections were thoroughly washed in PBS. After inhibition of endogenous peroxidase activity and further washes in PBS and PBST, sections were incubated in 10% normal swine serum in PBST for 2 h. Sections were then incubated for 48 h at 4°C with a specific antibody against GAP-43 made in rabbit (1:5000; Abcam) or against phosphoJNK (1:500; Cell Signaling Technology). All antibodies and the ABC complex were prepared in PBS 0.1 m containing 0.3% Triton X-100 (PBST). Subsequently, sections were washed in PBST and incubated for 1 h with polyclonal swine anti-rabbit biotin-conjugated antibody (1:200; Dakopatts). To visualize the immunoreactions, the ABC conjugated with peroxidase (1:200; Vector Laboratories) method was used with DAB tetrahydrochloride as chromogen (5 min in 0.05 m Tris buffer, pH 7.4, containing 0.05% DAB and 0.003% hydrogen peroxide). Sections were mounted on gelatin-coated slices and air dried for 12 h, cleared in xylene, mounted with Eukitt mounting medium, and coverslipped.
Figure 7.

A–I, Photomicrographs showing JNK expression in the superficial laminae I and II of the dorsal horn of the L5/L6 spinal cord segment of spinal-intact (B), 1 week (C), and 4 week (C) SCI animals; 1 week (F) and 4 week (G) SCI animals receiving chronic saline; 1 week SCI animals treated with chronic TrkB-Ig2 during the spinal shock (H); and 4 week SCI animals treated with chronic BDNF (I; 0.7 μg/d for 28 d). Scale bars: A–D, F–I, 100 μm; E, 50 μm. The specificity of the antibody was tested by incubating spinal sections in the absence of primary antibody. In these conditions, no immunostaining was observed (A). A time-dependent increase in JNK expression was observed in the superficial laminae I and II of the dorsal horn, when compared with spinal-intact animals (B–E, J, K; *p < 0.05). Immunoreaction was present in fibers and not observed in cells (E). Chronic intrathecal administration of saline did not induce any changes (F, G). Upon chronic treatment with TrkB-Ig2 for 1 week, JNK expression remained elevated, particularly in laminae II (*p < 0.05 vs spinal-intact animals), while chronic administration of BDNF was able to decrease JNK expression to similar levels of what was observed in spinal-intact animals. J, K, Bar graphs showing the mean intensity of JNK expression in laminae I and laminae II. Analysis of the intensity of phosphoJNK immunoreaction showed a time-dependent increase in the spinal cord of SCI rats and in cord sections from 1 week SCI animals treated with chronic TrkB-Ig2 (*p < 0.05 vs spinal intact). PhosphoJNK immunoreaction in cord sections from SCI rats treated with BDNF was no different from that observed in sections from spinal-intact animals.
Alternate spinal sections were used to determine the BDNF expression (1:1000; anti-rabbit; Millipore; AB1779) and colocalization between GAP-43 and calcitonin gene-related peptide (CGRP). The specificity of the anti-GAP-43 and anti-CGRP antibodies has been previously tested in other studies (Coelho et al., 2014). For this, sections were thoroughly washed in PBS and PBST, followed by a 2 h incubation in 10% normal horse serum in PBST. Sections were then incubated with anti-BDNF or a combination of anti-GAP-43 (1:5000) and anti-CGRP made in mouse (1:8000; Abcam) for 48 h. To control the specificity of immunodetection of BDNF, sections were incubated in the absence of primary antibody. No staining was observed (Fig. 2A). Afterward, sections were washed in PBST and incubated in Alexa Fluor 568 donkey anti-rabbit (1:1000; Invitrogen) and Alexa Fluor 488 goat anti-mouse (1:1000; Invitrogen) for 1 h. Sections were then washed, mounted in Prolong Gold mounting medium (Invitrogen), and observed in a Z4 Axio Imager Zeiss microscope.
Figure 2.

A–H, Photomicrographs showing BDNF expression in the L5–L6 spinal cord segment of spinal-intact and SCI animals. Scale bar, 20 μm. A, The specificity of the antibody was tested by incubating spinal cord sections in the absence of the primary antibody. No staining was observed in these conditions. B–D, In the spinal cord, BDNF expression was found in the dorsal horns of the spinal cord, particularly in the superficial layers of the cord. It significantly increased in a time-dependent manner after SCI (C, D, H; *p < 0.05 vs spinal intact, ***p < 0.01 vs spinal intact). E, Intrathecal sequestration of BDNF with TrkB-Ig2 for 1 week caused a marked decrease in BDNF expression in the dorsal horn compared with nontreated 1 week SCI rats (H; #p < 0.05 vs nontreated SCI rats at the same time point). F, G, BDNF intrathecal administration significantly increased BDNF expression in the superficial layers of the spinal cord in both experimental groups (H; &p < 0.05 vs nontreated SCI rats at the same time point). H, Bar graph depicting the mean intensity of BDNF expression in the L5/L6 spinal cord segment from all experimental groups. BDNF levels were upregulated at 1 and 4 weeks after SCI when compared with spinal-intact animals. Whereas BDNF sequestration with TrkB-Ig2 caused a decrease in its expression, chronic treatment with recombinant BDNF resulted in an increase of immunoreactivity.
In vitro studies: DRG cell culture and immunocytochemistry.
The experimental groups described above were replicated once for cell culture. In this case, four animals were used per experimental group. As before, all animals underwent cystometry under urethane anesthesia. After cystometry, animals were killed and the L5-S1 DRG collected and immediately placed in DMEM-F12, 10% FBS, and 1% Pen/Strep. DRGs were then incubated with 0.125% collagenase for 2 h at 37°C. After three washes in DMEM-F12 medium, DRGs neurons were resuspended and dissociated in DMEM-F12, 10% FBS, and 1% Pen/Strep by repeated pipetting. The resulting cell suspension was centrifuged at 1000 rpm for 10 min through a 2 ml cushion of 15% BSA for removal of cellular debris. The resulting pellet, containing the neurons, was resuspended in DMEM-F12 medium containing 1%Pen/Strep, l- glutamine (200 mm), B27 (20 μl/ml), and 0.05 μg/ml NGF. Cells obtained from each animal were plated in duplicate onto poly-l-lysine (20 μg/ml) and laminin-coated (5 μg/ml) coverslips and maintained at 37°C in a humidified 5% CO2 atmosphere.
Twelve hours later, the plated cells were fixed with ice-cold 4% paraformaldehyde for 15 min at room temperature. The cells were washed three times in PBS, followed by a 5 min incubation in PBS 0.2% Triton. Afterward, cells were again washed in PBS and incubated for 5 min in a solution of 0.1% of sodium borohydride. This was followed by three washes in PBS and 1 h incubation at room temperature in blocking solution (5% of FBS in 0.4% PBS-Tween 20). After blocking, cells were incubated in anti-β-III tubulin (1:2000 in blocking buffer; Promega) for 1 h at room temperature. The cells were then washed three times with PBS and incubated with the secondary antibody, Alexa Fluor 488 goat anti-mouse (1:1000 in blocking buffer; Invitrogen). This step was followed by washes in PBS and mounting in Vectashield medium (Vector Laboratories), after which slides were sealed. Representative images were collected in an Axioskop 40 microscope with the AxioVision 4.6 software (Carl Zeiss) for analysis of neurite branching, total neurite length, and area of the soma.
Immunodetection of bladder interstitial cells.
Bladder tissues (n = 5) from all experimental groups were fixed in 4% paraformaldehyde, washed in PBS, and blocked in 1% BSA/0.05% Triton X-100 in PBS before incubation with primary antibodies (anti-vimentin, 1:200, Sigma-Aldrich; anti-PDGFRα, 1:200, R&D Systems; or Phalloidin-TRITC) for 24 h. Tissues were washed in PBS for several hours and then incubated in secondary antibodies (Invitrogen, 1:200) with DAPI for 1 h. After overnight washing in PBS, tissues were mounted to glass slides with coverslips and Immuno-mount (Thermo Shandon). Slides were imaged with a Nikon C1 confocal microscope using an argon ion laser (488 nm), a green HeNe laser (543 nm), and a 408 nm laser diode. Fluorophores were imaged sequentially and images collected via appropriate filter sets to minimize bleedthrough. Similar imaging settings (laser intensity and gain) were used in all experiments. Series of optical sections were reconstructed in the acquisition and analysis software, EZ-C1 (Nikon). Vimentin immunofluorescence intensity was measured in ImageJ software. Guinea pig bladder was used as a positive control; negative controls included omission of all antibodies to test for autofluorescence. Significant fluorescence was not detected in negative controls.
Quantification and statistics.
Cystometrograms were analyzed using the LabScribe software (version 2.34900; iWorx Systems). The frequency and amplitude of bladder contractions, peak pressure, and baseline pressure were analyzed by using Kruskal–Wallis ANOVA or Kruskal–Wallis one-way repeated-measures ANOVA(in the 4 week SCI animals submitted to BDNF sequestration) followed by the Student–Newman–Keuls post hoc test in SigmaStat software (version 3.5.054). Data are presented as mean value ± SD and p < 0.05 was considered statistically significant.
The intensity of immunoreactivity was assessed using the Fiji Software (based on ImageJ; http://fiji.sc/Downloads#Fiji). In all cases, staining intensity was averaged from 8 to 10 sections per animal. A reference intensity of unstained tissue was measured to determine background intensity, which was deducted from the average intensity of each section to calculate the mean net staining intensity. Staining intensities determined in intact animals were used as standardizing controls. Data were analyzed by Kruskal–Wallis one-way ANOVA followed by the Student–Newman–Keuls post hoc test. Data are presented as mean value ± SD and p < 0.05 was considered statistically significant. Statistical analysis was SigmaStat software (version 3.5.054).
Neurite branching, total neurite length, and area of the soma were analyzed in MATLAB using SYnapse Detector (SynD; Schmitz et al., 2011), a synapse and neurite detection software program. SynD is able to quantify neurite branching by calculating the number of cellular processes crossing concentric circles placed around the cell body. Statistical analysis was performed in SigmaStat software (version 3.5.054) using one-way repeated-measures ANOVA followed by the Student–Newman–Keuls post hoc test for neurite branching and the Kruskal–Wallis ANOVA followed by the Dunn's test for the neurite length and for the area of the soma. In this case, values are presented as mean ± SEM as SynD automatically generates them. As above, p < 0.05 was considered statistically significant.
Results
Effect of BDNF on the emergence of NDO after spinal cord transection
The effects of SCI on bladder reflex activity were investigated 1 and 4 weeks after spinal cord lesion. The pattern of bladder reflex activity in spinal-intact animals, in what concerns the frequency, peak pressure, baseline pressure, and amplitude of bladder reflex contractions, is shown in Figures 1A and 4A–D. One week after SCI, the animals were in spinal shock and bladder reflex contractions were practically abolished (Figs. 1B, 4A–D). Four weeks after SCI, NDO was evident in all SCI animals (Fig. 1C), with significant increase in the values of frequency, peak pressure, baseline pressure, and amplitude of urinary function (Fig. 4A–D). This was accompanied by spinal upregulation of BDNF expression (see below; Fig. 2B–D), suggesting the involvement of BDNF in NDO emergence.
Figure 1.

A–H, Representative cystometrograms of spinal-intact and SCI animals. A, In spinal-intact animals, the pattern of bladder function was normal but significantly altered at 1 week after SCI (B), when bladder reflex activity was completely abolished. C, Four weeks after SCI, animals presented bladder hyperactivity. Placement of an indwelling intrathecal catheter linked to an osmotic mini-pump for continuous delivery of sterile saline to SCI animals for 1 week (D) or 4 (E) weeks did not affect bladder function. F, Chronic intrathecal BDNF sequestration with TrkB-Ig2, initiated immediately after SCI and lasting for 1 week, resulted in bladder hyperactivity. G, Intrathecal administration of BDNF (0.7 μg/d) for 4 weeks, initiated immediately after cord injury, resulted in alterations of bladder function compared with nontreated 4 week SCI rats. H, The highest dose of BDNF did not induce significant changes in bladder activity when compared with nontreated SCI-animals at the same time point.
Figure 4.
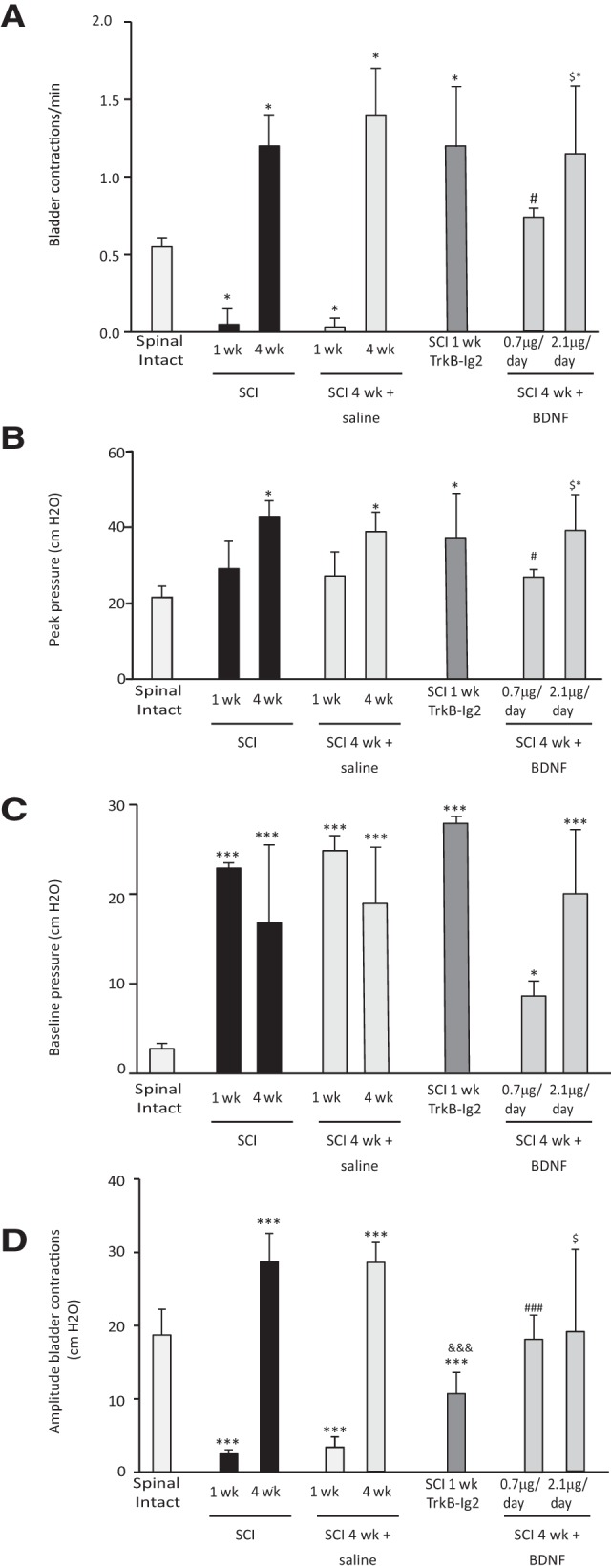
Bar graphs showing the mean frequency, peak pressure, baseline pressure, and amplitude of bladder contractions of spinal-intact and SCI injured animals. A, The frequency of bladder reflex contractions was significantly decreased 1 week after SCI but upregulated 4 weeks after SCI (*p < 0.05 vs spinal-intact animals). Chronic delivery of sterile saline to SCI animals for 1 and 4 weeks via an osmotic mini-pump connected to an indwelling intrathecal catheter did not alter bladder function compared with nonintervened SCI rats at the same time point. In these animals, there was a reduction of frequency of bladder contractions 1 week after SCI followed by an increase at 4 weeks post-SCI (*p < 0.05 vs spinal-intact animals). One week SCI animals intrathecally treated with chronic TrkB-Ig2 presented a significant increase in bladder reflex activity when compared with spinal-intact animals and with nontreated animals at the same time point (*p < 0.05). Chronic administration of BDNF (0.7 μg/d), lasting 4 weeks and initiated immediately after spinal lesion, reduced urinary frequency compared with nontreated SCI rats at the same time point (#p < 0.05). Urinary frequency was still elevated compared with spinal-intact animals (*p < 0.05). Administration of 2.1 μg/d for 4 weeks did not alter urinary frequency when compared with nontreated 4 week SCI rats (*p < 0.05 vs spinal-intact and $p < 0.05 vs 4 week SCI treated with 0.7 μg/d of BDNF). B, Four week SCI animals, with and without an indwelling intrathecal catheter for chronic saline delivery, and 1 week SCI animals treated with TrkB-Ig2 presented an elevation of peak pressure when compared with spinal-intact animals (*p < 0.05). BDNF administration for 4 weeks' treatment with the lowest dose of BDNF produced a significant reduction compared with SCI at the same time point (#p < 0.05 vs 4 weeks SCI rats). Treatment with the highest dose of BDNF did not affect peak pressure, which was similar to that observed in nontreated 4 week SCI rats (*p < 0.05 spinal-intact animals and $p < 0.05 vs 4 week SCI animals treated with 0.7 μg/d of BDNF). C, One week and 4 week SCI animals, with and without an indwelling intrathecal catheter for chronic saline delivery, presented a significant increase in baseline pressure (***p < 0.001 vs spinal-intact animals). Chronic administration of TrkB-Ig2 did not change baseline pressure values when compared with 1 week SCI animals at the same time point, but it was still elevated compared with spinal-intact animals (***p < 0.001). Chronic intrathecal treatment with 0.7 and 2.1 μg/d for 4 weeks was also accompanied by high baseline pressure values (*p < 0.05, ***p < 0.001 vs spinal intact). D, Amplitude of bladder contractions was significantly reduced 1 week after spinal lesion and increased 4 weeks post injury (***p < 0.001 vs spinal-intact animals). Similar observations were made in SCI animals submitted to chronic delivery of saline via an indwelling silicone catheter linked to an osmotic mini-pump (***p < 0.001 vs spinal-intact animals). BDNF sequestration was accompanied by a reduction of amplitude of bladder contractions compared with spinal-intact animals (***p < 0.001). It was increased when compared with SCI animals at the same time point (&&&p < 0.001 vs 1 week SCI animals). Prolonged BDNF administration of 0.7 and 2.1 μg/d was accompanied by a slight reduction when compared with 4 week SCI rats (###p < 0.001 vs 4 week SCI animals and $p < 0.05 vs 4 week SCI animals treated with lowest dose of BDNF).
Chronic intrathecal BDNF sequestration was performed during the first week after spinal cord transection, by chronic delivery of TrkB-Ig2. Results indicate that BDNF scavenging promoted an earlier appearance of NDO (Fig. 1D), as shown by a significant increase of frequency and amplitude of bladder contractions, peak pressure in 1 week SCI animals treated with TrkB-Ig2 compared with nontreated 1 week SCI animals (which were in spinal shock; *p < 0.05; Fig. 4A–D). Baseline pressure remained elevated after treatment (Fig. 4C). Since BDNF sequestration during spinal shock resulted in early NDO, one could speculate that spinal BDNF may be counteracting the emergence of NDO. To confirm this hypothesis, SCI rats continuously received intrathecal BDNF. Treatment was initiated immediately after spinal injury. Preliminary experiments in which BDNF was administered for 1 week to SCI rats did not produce any effects on bladder function (data not shown). Thus, BDNF administration was prolonged for 4 weeks. Intrathecal administration of BDNF (0.7 μg/d) to SCI rats during 4 weeks had a marked effect on bladder function, with a signifi-cant decrease of frequency, amplitude, and peak pressure of bladder contractions compared with nontreated SCI animals at 4 weeks (Figs. 1G, 4A–D). Chronic BDNF administration at higher doses during 4 weeks (2.1 μg/d) did not affect bladder function and the pattern of bladder reflex contractions was similar to that observed in nontreated 4 week SCI animals ($p < 0.05; Figs. 1H, 4A–D). Hence, BDNF administration, initiated after spinal lesion, might exert a positive effect on bladder function. However, the dose of BDNF to be delivered is critical since bladder function was compromised and similar to 4 week SCI animals when higher doses of BDNF were used. Therefore, because of the poor effects of the high dose of BDNF on bladder function, the following experiments only focused on the 4 week SCI rats receiving the lower dose of BDNF (0.7 μg/d).
Chronic intrathecal administration of saline for 1 week or 4 weeks (Fig. 1D,E, respectively) did not change bladder function compared with SCI animals without a chronic indwelling catheter at the same time point (Fig. 4A–D). For that reason, we did not analyze BDNF levels in these animals and focused attention in groups with changes in bladder reflex contractions.
NDO emergence is accompanied by a time-dependent increase in spinal BDNF levels
After assessing the effects on bladder function, expression of BDNF in the L5-L6 segments of the spinal cord was performed to confirm BDNF sequestration or administration. BDNF was found throughout the gray matter of the spinal cord, the strongest immunoreaction was observed in the dorsal horns (Fig. 2B–G). The specificity of the antibody has been previously tested (Frias et al., 2013) and no immunoreaction was observed when sections were incubated in the absence of primary antibody (Fig. 2A). Analysis of the intensity of the immunostaining showed a time-dependent increase in spinal levels of BDNF after SCI, the strongest intensity observed 4 weeks after SCI in the superficial laminae of the cord (***p < 0.001 vs spinal intact; Fig. 2B–H). The efficacy of BDNF sequestration using TrkB-Ig2 was confirmed by a marked decrease in spinal BDNF staining in the dorsal horn (Fig. 2E,H). Chronic BDNF delivery resulted in stronger immunoreactivity in spinal sections from treated animals compared with nontreated SCI at the same time point (Fig. 2F–H), confirming delivery of exogenous BDNF.
BDNF sequestration improves bladder function in rats with established NDO
Given that BDNF expression was increased in animals with established NDO, the effect of BDNF sequestration on bladder function was assessed at this time point. For that, 4 week SCI rats received increasing doses of TrkB-Ig2 during cystometry. The recombinant protein was injected every 30 min in a cumulative manner. We found that a dose-dependent BDNF sequestration minimized the effects of NDO on bladder function (Fig. 3A–E). At doses of 10 μg TrkB-Ig2 bladder reflex contractions were practically abolished. These results suggest that BDNF is involved in the long-term maintenance of NDO arising after SCI.
Figure 3.

A, Representative cystometrograms of 4 week SCI animals following acute intrathecal injection of saline and TrkB-Ig2 and parameter analysis of bladder function. A, Intrathecal administration of saline did not alter bladder function when compared with baseline. In contrast, intrathecal injection of TrkB-Ig2 dose dependently abolished bladder hyperactivity. Bar graphs depict the mean frequency (B), peak pressure (C), baseline pressure (D), and amplitude (E) of bladder contractions of 4 week SCI animals treated with either saline or 1 μg, and 10 and 20 μg of TrkB-Ig2. The frequency and amplitude of bladder contractions significantly decreased after intrathecal injection of 10 and 20 μg of TrkB-Ig2 (*p < 0.05 vs baseline values). No changes were found in peak pressure, but baseline pressure was markedly augmented after administration of 1, 10, and 20 μg of TrkB-Ig2 (***p < 0.001 vs baseline values).
GAP-43 expression in the spinal cord is altered after SCI
The emergence of NDO after SCI has been previously linked to sprouting and synaptic reorganization of bladder sensory afferents (de Groat et al., 1990; de Groat and Yoshimura, 2006). These events can be monitored by analyzing GAP-43 expression, a marker of axonal sprouting (Benowitz and Routtenberg, 1997; Vizzard, 1999). GAP-43 immunoreaction was very modest in spinal sections from spinal-intact rats (Fig. 5A). On the contrary, GAP-43 expression significantly increased in a time-dependent manner after SCI in thin fibers coursing in the superficial laminae of the dorsal horn (***p < 0.001 vs spinal intact; Fig. 5B–D,I).
Figure 5.

Photomicrographs showing GAP-43 expression in the L5-L6 spinal cord segment of spinal-intact (A), 1 week (B), and 4 week (C) SCI animals; 1 week (E) and 4 week (F) SCI animals with an indwelling intrathecal catheter for saline delivery; 1 week SCI animals treated with chronic TrkB-Ig2 during the spinal shock (G); and 4 week SCI animals treated with chronic BDNF (0.7 μg/d for 28 d; H). Scale bars: A–C, E–H, 100 μm; D, 50 μm. GAP-43 was predominantly expressed in the superficial laminae of the dorsal horn, with a time-dependent increase after SCI (A–C), indicating the occurrence of fiber sprouting. GAP-43 immunoreaction was restricted to thin fibers running along the superficial dorsal horn (D). The expression of GAP-43 was not altered by chronic delivery of saline for 1 week (E) or 4 weeks (F). GAP-43 expression was significantly increased following administration of TrkB-Ig2 for 1 week (G) or BDNF (0.7 μg/d) for 4 weeks (F). I, Bar graph depicting the mean intensity of GAP-43 expression in the L5-L6 segment. The strongest intensity of GAP-43 was observed in 4 week SCI animals. The immunoreaction was also strong following BDNF sequestration TrkB-Ig2 compared with nontreated animals at the same time point. GAP-43 expression was unchanged in SCI animals that received chronic delivery of saline during 1 week or 4 weeks. BDNF administration induced a slight nonsignificant decrease of GAP-43 expression compared with nontreated 4 week SCI rats, but immunoreaction was still stronger than in spinal-intact animals (*p < 0.05, ***p < 0.001 vs spinal intact). J–O, GAP-43 expression colocalization with CGRP neuropeptide in the dorsal horn. Scale bar, 20 μm. In all experimental groups, there was a strict colocalization between GAP-43 and CGRP, which can be better observed in M.
Continuous BDNF sequestration during spinal shock resulted in intense GAP-43 expression, compared with spinal-intact rats and nontreated 1 week SCI animals (*p < 0.05 vs spinal intact; Fig. 5G,I).
Expression of GAP-43 was also analyzed after 4 weeks of intrathecal BDNF administration (0.7 μg/d) to SCI animals. Despite being more prominent than in spinal-intact rats and 1 week SCI animals (***p < 0.001), GAP-43 immunoreactivity was not as intense as that observed in nontreated 4 week SCI rats (Fig. 5H,I).
In all experimental animals, immunolabeling was found bilaterally in the superficial layers of the dorsal horn, in fibers coursing the superficial laminae suggesting that GAP-43-postive fibers were sensory afferents (Fig. 5D). To confirm this, we assessed if GAP-43 was colocalized with CGRP, a marker of sensory afferents. We found that GAP-43 immunoreaction was present in CGRP-positive structures, confirming our hypothesis (Fig. 5I–L).
To exclude the hypothesis that placement of the catheter, connected to an osmotic mini-pump, could by itself induce axonal sprouting, we analyzed GAP-43 expression in sections from SCI animals submitted to catheter placement and saline infusion for 1 and 4 weeks. Expression of GAP-43 was comparable to SCI animals without a chronic indwelling catheter at the same time point (Fig. 5E,F).
These findings indicate that changes in spinal BDNF levels, induced by SCI or exogenous manipulation of this NT, influence the sprouting of bladder afferents.
DRG cells presented elevated intrinsic growth ability
As increased levels of GAP-43 suggested increased axonal growth ability, DRG were collected from all experimental groups and cultured to confirm growth ability. Following dissociation, DRG neurons were cultured for 12 h to investigate their intrinsic growth ability. All neurons adhered to the cell substrate and emitted long neurites (Fig. 6A–E). Neurite branching was the parameter that showed more variation across the different experimental groups. Branching was significantly altered by SCI, as 1 week after injury the mean number of branches per cell was significantly lower than in cells obtained from spinal-intact rats (***p < 0.001 vs spinal intact; Fig. 6B,F). In contrast, branching was much more prominent in DRG neurons, particularly in the vicinity of the soma, obtained from 4 week SCI rats with NDO (Fig. 6C,G). In SCI animals submitted to BDNF sequestration during the first week after transection, neurite branching was significantly increased (*p < 0.05 vs spinal intact and SCI 1 week; Fig. 6D,F). In DRG neurons collected from 4 week SCI rats submitted to chronic BDNF administration, branching was also evident and slightly more prominent than in cells obtained from nontreated 4 week SCI rats, particularly in regions located >100 μm from the soma (Fig. 6E,G).
Figure 6.

Photomicrographs illustrating β3-tubulin expression of L5-S1 DRG neurons in culture. Scale bar, 50 μm. Neurons from 1 week SCI animals (B) presented shorter neurites when compared with those from spinal-intact animals (A). In cells obtained from 4 week SCI animals (C) neurites were long and ramified. Neurons from 1 week SCI animals treated with chronic TrkB-Ig2 had long neurites, which were extremely ramified (D). Similar observations were made in DRG neurons from 4 week SCI rats receiving chronic BDNF, although neurite branching was less exuberant (E). F, G, Graphs indicating the correlation between the mean number of branches with the distance from the soma (μm). F, One week SCI animals treated with TrkB-Ig2 presented a higher number of branches in short and long distances from the soma (up to 200 μm) when compared with spinal-intact and 1 week SCI animals (*p < 0.05). G, Four week SCI animals presented an increase in the number of branches until 60 μm from the soma, when compared with spinal-intact and 4 week SCI animals treated with BDNF (#p < 0.05). In contrast, 4 week SCI animals treated with chronic BDNF presented a significant higher number of neurites from 110 to 200 μm (*p < 0.05 vs spinal-intact and 4 week SCI animals).
Other parameters like neurite length and area of the soma showed less variation across experimental groups. Nevertheless, there was a significant reduction in the neurite length of DRG cells collected 1 week after SCI (*p < 0.05 vs spinal-intact animals; Table 2).
Table 2.
Mean total neurite length (μm) and mean soma area (μm2) from cultured DRG cells from spinal-intact animals, 1 and 4 weeks SCI animals, 1 week SCI animals treated with chronic TrkB-Ig2, and 4 week SCI animals treated with chronic BDNF (0.7 μg/d)
| Spinal intact | SCI 1 week | SCI 1 week + chronic TrkB-Ig2 | SCI 4 weeks | SCI 4 weeks + chronic BDNF (0.7 μg/d) | |
|---|---|---|---|---|---|
| Mean neurite length (μm) | 1723.3 ± 140.3 | 1545.7 ± 153.3* | 2307.3 ± 192.7# | 1892.9 ± 138.2 | 2020.4 ± 153.3 |
| Mean soma area (μm2) | 1383.0 ± 152.2 | 1188.2 ± 67.9 | 976.8 ± 70.1+ | 1237.4 ± 67.9++ | 1002.7 ± 39.2 |
Spinal lesion resulted in a decrease in growth ability as neurites were shorter 1 week after SCI compared with spinal intact rats (*p < 0.05 vs spinal intact). BDNF sequestration potentiated cell growth as cells from 1 week SCI animals treated with TrkB-Ig2 presented longer neurites, compared with nontreated animals at the same time point (#p < 0.05 vs SCI 1 week). In what concerns the soma area, DRG neurons from 1 week SCI animals treated with TrkB-Ig2 had slightly smaller cell bodies compared with nontreated SCI rats at the same time point (+p < 0.05 vs SCI 1 week). Four week SCI animals presented an increase of the soma area compared with other experimental groups (++p < 0.05 vs remaining groups).
Activation of JNK signaling pathways might be involved in the mechanisms regulating axonal sprouting
Several studies have provided evidence linking JNK activation at the tip of growing axons of sensory neurons to elongation of processes (Oliva et al., 2006; Barnat et al., 2010; Atkinson et al., 2011). To investigate if the same mechanism was operating in our SCI rats, we assessed the levels of JNK activation in the L6 spinal cord segment. In the absence of the primary antibody, no immunoreaction was observed (Fig. 7A). Little phosphoJNK immunoreaction was found in sections from spinal-intact animals (Fig. 7B,J,K). In contrast, positive immunoreaction occurred throughout the dorsal horn, particularly in laminae I–II; in SCI animals; and increased in a time-dependent manner after cord lesion (Fig. 7C–E,J,K). Treatment with BDNF scavenger, TrkB-Ig2, during the spinal shock caused a marked increase of phosphoJNK expression to levels comparable to those observed in 4 week nontreated SCI rats (*p < 0.05 vs spinal intact; Fig. 7H,J,K). Spinal phosphoJNK expression in 4 week SCI rats submitted to chronic treatment with BDNF was similar to that found in spinal-intact animals (Fig. 7I–K).
Expression of phosphoJNK was also analyzed in sections from SCI animals submitted to catheter placement and saline infusion for 1 and 4 weeks to exclude the occurrence of axonal growth due to placement of the intrathecal catheter. Immunoreaction was similar to that observed in sections from SCI animals without a chronic indwelling catheter at the same time point (Fig. 7F,G,J,K).
Bladder interstitial cells are differently expressed in the bladder after SCI
Previous work has shown that significant remodeling of ICs occurs in the chronic SCI rat bladder (Johnston et al., 2012). In the present study, it was of interest to determine whether there was an IC lesion at 1wk SCI and if IC populations could be restored with intrathecal BDNF sequestration or BDNF administration. Detrusor IC, labeled with anti-vimentin or anti-PDGFRα, which are normally closely associated with detrusor smooth muscle bundles (intramuscular IC, IC-IM), were notably reduced 1 and 4 weeks after SCI compared with spinal-intact rats (Fig. 8A–F). In contrast, interbundle ICs (IC-IBs) located between bundles were strikingly increased in acute SCI, but were diminished by 4 weeks post injury, assuming a more rounded morphology with loss of cell—cell contacts (Fig. 8B,C,E,F). While BDNF sequestration during spinal shock did not restore normal IC-IM or IC-IB populations (Fig. 8G,I), chronic BDNF treatment partially prevented loss of IC-IM within smooth muscle bundles with little effect on the IC-IB populations (Fig. 8H,J). Quantitative analysis of vimentin immunofluorescence is shown in the bar charts (Fig. 8). BDNF sequestration had no significant effect on vimentin fluorescence (n = 16 control images, n = 14 treatment images, p > 0.05). Chronic BDNF did not significantly affect vimentin fluorescence (n = 16 treatment images, n = 9 control images, p > 0.05).
Figure 8.

Photomicrographs of IC populations in the detrusor muscle. ICs were identified by anti-vimentin (green; A–C, G, H) or anti-PDGFRα (green; D–F, I, J) immunolabeling. Smooth muscle cells were labeled with phalloidin (red) and nuclei were counterstained with DAPI (blue). In the normal bladder (A, D), ICs were located within smooth muscle bundles (IC-IM) and between the bundles (IC-IB) in the normal rat bladder. One week post-SCI (B, E), there was marked upregulation of IC-IB and reduction in the IC-IM populations (*p < 0.05). Four weeks post-SCI (C, F) IC-IB presented disrupted morphology and IC-IM were apparently absent. BDNF sequestration, with intrathecal administration of TrkB-Ig2, during spinal shock (G, I) did not restore the IC populations. Prolonged administration of BDNF to SCI rats (0.7 μg/d for 4 weeks) partially restored IC-IM populations that were apparently absent in nontreated 4 week SCI rats, with little effect on IC-IB. Scale bars: 20 μm. K, Bar charts show summary quantitative analysis of detrusor IC vimentin immunolabeling after BDNF sequestration and chronic BDNF administration. Neither intervention significantly affected vimentin fluorescence (*p > 0.05).
Discussion
This work analyzed the contribution of BDNF to NDO emergence and maintenance caused by SCI. We propose a model in which SCI results in NDO via sprouting of bladder afferents (Fig. 9). In an early phase, spinal BDNF is upregulated in an attempt to downregulate abnormal axonal growth and prevent bladder dysfunction. Once the new micturition reflex is established, the protective role of BDNF is lost and this NT acts as a neurotransmitter, exacerbating bladder dysfunction.
Figure 9.

Proposed model of BDNF role in NDO emergence and maintenance. Our main experimental findings show that preventing the increased spinal BDNF, which occurs after cord injury, induces early NDO, as a consequence of premature sprouting of sensory afferents, This suggests that BDNF may exert a protective role on bladder function after SCI. Thus, chronic BDNF administration at the spinal cord level, also initiated immediately after spinal lesion, resulted in an amelioration of bladder function of SCI, compared with animals receiving vehicle. In this case, axonal growth was less prominent. We propose that SCI induces sprouting of sensory afferents, possibly as a consequence of upregulation of trophic factors, such as NGF. These afferents establish new synaptic connections at the spinal cord, which lead to an abnormal micturition reflex, totally located at the lumbosacral spinal cord. At the same time, spinal BDFN increases in an attempt to repress axonal sprouting at the spinal cord. Once the new connections are established, BDNF acts as a neurotransmitter, released upon stimulation of bladder afferents, and potentiates bladder dysfunction. Accordingly, BDNF sequestration in rats with established NDO blocked bladder reflex contractions.
BDNF is involved in SCI-induced bladder dysfunction
The observation that development of bladder dysfunction was accompanied by the upregulation of spinal BDNF expression suggested a link between BDNF and NDO. BDNF sequestration during cystometry in 4 week SCI rats leads to a dose-dependent reduction in the frequency and amplitude of bladder reflex contractions. Likewise, in rats with cystitis acute BDNF sequestration also produced a swift improvement of bladder function (Frias et al., 2013). The reason for this may relate to inactivation of second-order neurons. BDNF, released by sensory afferents, is a potent activator of the ERK pathway in spinal neurons (Pezet et al., 2002a; Lever et al., 2003b; Salio et al., 2007; Merighi et al., 2008). An early study showed that pharmacological dose-dependent ERK inhibition also improved bladder function, similar to what was found here (Cruz et al., 2006).
Our initial hypothesis, that time-dependent BDNF upregulation leads to NDO, implied that early BDNF sequestration would prevent bladder dysfunction. A similar hypothesis was proposed, which showed that NGF sequestration, initiated 10 d after SCI, prevented NDO and DSD (Seki et al., 2002, 2004). Surprisingly, BDNF sequestration during the spinal-shock period induced early NDO emergence. Using a model of dorsal root injury, Ramer et al. (2007) also verified that BDNF sequestration resulted in opposite effects depending on whether it was performed chronically and initiated immediately after lesion or only by a single intrathecal bolus at later stages. The early NDO phenotype was not explained by changes in the bladder IC population, already significantly altered in the spinal-shock period. It is therefore more likely to result from axonal reorganization and growth at the spinal cord level rather than local mechanisms within the bladder wall. Therefore, one can infer that spinal BDNF increase during early stages of SCI may modulate the reorganization of spinal neuronal circuits, in an attempt to block NDO emergence.
Chronic BDNF administration protects bladder function in SCI rats
To clarify the putative protective role of BDNF on bladder function, SCI animals were submitted to chronic BDNF administration initiated immediately after cord injury. We found that administration of 0.7 μg/d of BDNF for 4 weeks minimized bladder dysfunction, despite elevated urinary frequency and low compliance when compared with spinal-intact animals. Nevertheless, it is possible to infer that the BDNF role in mitigating NDO appearance and its maintenance is crucial. This moderating action of BDNF may occur via potentiation of the GABAergic neurotransmission. Because GABAergic neurons express TrkB receptor, the increased amount of BDNF at the spinal cord may have sufficed to increase GABA release (Pezet et al., 2002b; Lever et al., 2003a; Bardoni et al., 2007). One should recall that this neurotransmitter depresses bladder function and the potentiation of GABAergic neurotransmission has already been forwarded as a potential therapeutic measure for NDO (Miyazato et al., 2008, 2009). In addition, chronic BDNF may have been protective of the IC-IM. We have previously shown that loss of nerve/IC-IM contacts is associated with loss of IC-IM (Johnston et al., 2012) and it is possible that in the present study, BDNF preserved detrusor nerve varicosities, limiting the denervation that is typical in chronic SCI and minimizing IC-IM loss.
It is tempting to speculate a possible therapeutic application of BDNF in SCI patients. Accordingly, upregulation of BDNF following electrical stimulation has been linked to axonal motoneuron regeneration following peripheral nerve injury (Al-Majed et al., 2000). Given that spinal electrical stimulation via sacral neuromodulation (SNM) in SCI patients improved urinary incontinence (Sievert et al., 2010), one could propose that combining BDNF administration with SNM would be advantageous. However, it should be noted that a higher dose of this NT did not produce any improvement of bladder function. Presently, we cannot forward a conclusive explanation for these findings but it may be possible that the excess of BDNF administered could activate nonspecific NT receptors and/or other intracellular signaling pathways in spinal neurons.
Mechanisms modulating BDNF action on bladder function
Plastic reorganization of the sensory synaptic connections of bladder afferents in the spinal cord has been forwarded as one of the main causes for NDO (de Groat et al., 1990; Vizzard, 2006) and detrusor overactivity following bladder outlet obstruction (Steers et al., 1991; Gabella et al., 1992; Tuttle and Steers, 1992). Early studies have demonstrated that both conditions are associated with NGF upregulation, leading to hypertrophy of major pelvic ganglion and DRG neurons (Steers et al., 1991, 1996) and sprouting of central branches of bladder sensory afferents (Vizzard, 1999).
As before (Vizzard, 1999), we observed sprouting of peptidergic sensory afferents during the natural progression of the disease. Axonal growth was evaluated by analyzing the expression of GAP-43, a marker of axonal growth (Benowitz and Routtenberg, 1997), which gradually increased in the superficial laminae of the cord in 1 and 4 week SCI animals. This was confirmed by DRG cell culture assays, which demonstrated increase of neurite outgrowth and branching. The reason underlying sprouting of visceral afferents during the progression of the disease may be related to the reported elevation of spinal NGF levels (Seki et al., 2002; Brown et al., 2004), which stimulates DRG neuron growth (Gavazzi et al., 1999). Increased density of sensory afferents may lead to increased release of BDNF, leading to ERK activation in second-order neurons, previously linked to bladder dysfunction in SCI rats (Cruz et al., 2006). BDNF sequestration in 1 week SCI animals enhanced axonal growth as shown by the increase in GAP-43 expression. Likewise, in our cell culture assays, DRG cells showed a significant increase in neurite branching. In vitro studies had already documented the inhibitory effects of BDNF on NGF-driven neurite growth (Gavazzi et al., 1999; Kimpinski et al., 1999). Although not measured in the present study, it is very likely that in our SCI animals the amount of spinal NGF was very high and promoted axonal growth, which was more exuberant and occurred earlier in the absence of the inhibitory presence of BDNF.
As expected (Ondarza et al., 2003), we observed full colocalization of GAP-43 with CGRP, indicating that axonal sprouting of bladder peptidergic sensory afferents is the key event for bladder dysfunction after SCI (Zinck et al., 2007). Likewise, sprouting of peptidergic afferents after complete SCI is also associated with autonomic dysreflexia (Krenz and Weaver, 1998; Krenz et al., 1999; Weaver et al., 2001) as a positive correlation between hypertension and sprouting of CGRP-positive axons has already been identified (Cameron et al., 2006).
Molecular mechanisms governing afferent sprouting at dorsal horns
An important intracellular signaling pathway involved in neurite outgrowth and elongation is the JNK pathway. This signaling cascade is a member of the MAPKs, a large family of signaling kinases (Widmann et al., 1999; Johnson and Lapadat, 2002; Krishna and Narang, 2008). It was recently demonstrated that JNK activation was required for axon elongation (Oliva et al., 2006; Barnat et al., 2010; Atkinson et al., 2011), to stabilize and modulate the microtubule dynamics at the growth cone and neurite tips (Barnat et al., 2010). Accordingly, we found stronger expression of phosphoJNK in laminae I–II of the experimental groups in which axonal sprouting was more pronounced. This was the spinal location where GAP-43 immunoreactivity was also stronger, corresponding to the areas of sprouting of bladder sensory afferents. Interestingly, administration of BDNF was also associated to neuronal sprouting, albeit branching was not so exuberant. In this case, JNK activation was not different from controls, indicating that BDNF-mediated axonal sprouting was JNK independent.
Conclusions
Here, we used the complete transection model and identified BDNF as an important modulator of sensory afferent sprouting, the key mechanism underlying NDO emergence. This SCI model reflects symptoms of complete SCI patients, not necessarily the most common injury, but it is highly reproducible, frequently used when studying NDO and well established in our research group (de Groat et al., 1990; Kruse et al., 1995; Yoshiyama et al., 1999; Cruz et al., 2006, 2008; Cruz and Cruz, 2011; Santos-Silva et al., 2012). We propose that upregulation of BDNF during disease progression may act to control exaggerated axonal sprouting. Once sprouting has occurred, acute BDNF sequestration may serve to depress bladder hyperactivity, although restoration of bladder cellular architecture is unlikely achieved. Thus, BDNF is an attractive therapeutic target to be differentially manipulated at different stages of NDO establishment. Future studies will analyze the most effective BDNF treatment, in terms of dosages, time of delivery, and combination with other therapeutic measures, including the use of biomaterials and undifferentiated cells for spinal regeneration.
Footnotes
Initial financial support came from InComb (FP7 HEALTH project 223234). Funds also came from the International PhD Program on Neurosciences, University of Porto, Portugal. B.F. was supported by FCT–Fundação para a Cięncia e Tecnologia (SFRH/BD/63225/2009). S.M.Y.G. was supported by a BBSRC-Pfizer CASE Partnership Grant (BB/G530176/1) to K.D.M., S.A. acknowledges support from the Medical Research Council. M.M.S. was supported by FEDER through COMPETE and by national funds through FCT (FCOMP-01-0124-FEDER-017455, HMS ICT/0020/2010). We would like to acknowledge the technical support of Sérgio Carvalho-Barros.
The authors declare no competing financial interests.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Al-Majed AA, Brushart TM, Gordon T. Electrical stimulation accelerates and increases expression of BDNF and trkB mRNA in regenerating rat femoral motoneurons. Eur J Neurosci. 2000;12:4381–4390. doi: 10.1111/j.1460-9568.2000.01341.x. [DOI] [PubMed] [Google Scholar]
- Atkinson PJ, Cho CH, Hansen MR, Green SH. Activity of all JNK isoforms contributes to neurite growth in spiral ganglion neurons. Hear Res. 2011;278:77–85. doi: 10.1016/j.heares.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfield MJ, Naylor RL, Robertson AG, Allen SJ, Dawbarn D, Brady RL. Specificity in Trk receptor:neurotrophin interactions: the crystal structure of TrkB-d5 in complex with neurotrophin-4/5. Structure. 2001;9:1191–1199. doi: 10.1016/S0969-2126(01)00681-5. [DOI] [PubMed] [Google Scholar]
- Bardoni R, Ghirri A, Salio C, Prandini M, Merighi A. BDNF-mediated modulation of GABA and glycine release in dorsal horn lamina II from postnatal rats. Dev Neurobiol. 2007;67:960–975. doi: 10.1002/dneu.20401. [DOI] [PubMed] [Google Scholar]
- Barnat M, Enslen H, Propst F, Davis RJ, Soares S, Nothias F. Distinct roles of c-Jun N-terminal kinase isoforms in neurite initiation and elongation during axonal regeneration. J Neurosci. 2010;30:7804–7816. doi: 10.1523/JNEUROSCI.0372-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz LI, Routtenberg A. GAP-43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci. 1997;20:84–91. doi: 10.1016/S0166-2236(96)10072-2. [DOI] [PubMed] [Google Scholar]
- Brown A, Ricci MJ, Weaver LC. NGF message and protein distribution in the injured rat spinal cord. Exp Neurol. 2004;188:115–127. doi: 10.1016/j.expneurol.2004.03.017. [DOI] [PubMed] [Google Scholar]
- Cameron AA, Smith GM, Randall DC, Brown DR, Rabchevsky AG. Genetic manipulation of intraspinal plasticity after spinal cord injury alters the severity of autonomic dysreflexia. J Neurosci. 2006;26:2923–2932. doi: 10.1523/JNEUROSCI.4390-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho A, Oliveira R, Rossetto O, Cruz CD, Cruz F, Avelino A. Intrathecal administration of botulinum toxin type A improves urinary bladder function and reduces pain in rats with cystitis. Eur J Pain. 2014;18:1480–1489. doi: 10.1002/ejp.513. [DOI] [PubMed] [Google Scholar]
- Cruz CD, Cruz F. Spinal cord injury and bladder dysfunction: new ideas about an old problem. ScientificWorldJournal. 2011;11:214–234. doi: 10.1100/tsw.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz CD. Neurotrophins in bladder function: what do we know and where do we go from here? Neurourol Urodyn. 2014;33:39–45. doi: 10.1002/nau.22438. [DOI] [PubMed] [Google Scholar]
- Cruz CD, Avelino A, McMahon SB, Cruz F. Increased spinal cord phosphorylation of extracellular signal-regulated kinases mediates micturition overactivity in rats with chronic bladder inflammation. Eur J Neurosci. 2005;21:773–781. doi: 10.1111/j.1460-9568.2005.03893.x. [DOI] [PubMed] [Google Scholar]
- Cruz CD, McMahon SB, Cruz F. Spinal ERK activation contributes to the regulation of bladder function in spinal cord injured rats. Exp Neurol. 2006;200:66–73. doi: 10.1016/j.expneurol.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Cruz CD, Charrua A, Vieira E, Valente J, Avelino A, Cruz F. Intrathecal delivery of resiniferatoxin (RTX) reduces detrusor overactivity and spinal expression of TRPV1 in spinal cord injured animals. Exp Neurol. 2008;214:301–308. doi: 10.1016/j.expneurol.2008.08.016. [DOI] [PubMed] [Google Scholar]
- Cruz F, Herschorn S, Aliotta P, Brin M, Thompson C, Lam W, Daniell G, Heesakkers J, Haag-Molkenteller C. Efficacy and safety of onabotulinumtoxinA in patients with urinary incontinence due to neurogenic detrusor overactivity: a randomised, double-blind, placebo-controlled trial. Eur Urol. 2011;60:742–750. doi: 10.1016/j.eururo.2011.07.002. [DOI] [PubMed] [Google Scholar]
- de Groat WC, Steers WD. Autonomic regulation of the urinary bladder and sex organs. In: Loewy AD SK, editor. Central regulation of autonomic functions. London: Oxford UP; 1990. pp. 310–333. [Google Scholar]
- de Groat WC, Yoshimura N. Mechanisms underlying the recovery of lower urinary tract function following spinal cord injury. Prog Brain Res. 2006;152:59–84. doi: 10.1016/S0079-6123(05)52005-3. [DOI] [PubMed] [Google Scholar]
- de Groat WC, Kawatani M, Hisamitsu T, Cheng CL, Ma CP, Thor K, Steers W, Roppolo JR. Mechanisms underlying the recovery of urinary bladder function following spinal cord injury. J Auton Nerv Syst. 1990;(30 Suppl):S71–S77. doi: 10.1016/0165-1838(90)90105-R. [DOI] [PubMed] [Google Scholar]
- French JS, Anderson-Erisman KD, Sutter M. What do spinal cord injury consumers want? A review of spinal cord injury consumer priorities and neuroprosthesis from the 2008 neural interfaces conference. Neuromodulation. 2010;13:229–231. doi: 10.1111/j.1525-1403.2009.00252.x. [DOI] [PubMed] [Google Scholar]
- Frias B, Allen S, Dawbarn D, Charrua A, Cruz F, Cruz CD. Brain-derived neurotrophic factor, acting at the spinal cord level, participates in bladder hyperactivity and referred pain during chronic bladder inflammation. Neuroscience. 2013;234:88–102. doi: 10.1016/j.neuroscience.2012.12.044. [DOI] [PubMed] [Google Scholar]
- Gabella G, Berggren T, Uvelius B. Hypertrophy and reversal of hypertrophy in rat pelvic ganglion neurons. J Neurocytol. 1992;21:649–662. doi: 10.1007/BF01191726. [DOI] [PubMed] [Google Scholar]
- Gavazzi I, Kumar RD, McMahon SB, Cohen J. Growth responses of different subpopulations of adult sensory neurons to neurotrophic factors in vitro. Eur J Neurosci. 1999;11:3405–3414. doi: 10.1046/j.1460-9568.1999.00756.x. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Johnston L, Cunningham RM, Young JS, Fry CH, McMurray G, Eccles R, McCloskey KD. Altered distribution of interstitial cells and innervation in the rat urinary bladder following spinal cord injury. J Cell Mol Med. 2012;16:1533–1543. doi: 10.1111/j.1582-4934.2011.01410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr BJ, Bradbury EJ, Bennett DL, Trivedi PM, Dassan P, French J, Shelton DB, McMahon SB, Thompson SW. Brain-derived neurotrophic factor modulates nociceptive sensory inputs and NMDA-evoked responses in the rat spinal cord. J Neurosci. 1999;19:5138–5148. doi: 10.1523/JNEUROSCI.19-12-05138.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimpinski K, Jelinski S, Mearow K. The anti-p75 antibody, MC192, and brain-derived neurotrophic factor inhibit nerve growth factor-dependent neurite growth from adult sensory neurons. Neuroscience. 1999;93:253–263. doi: 10.1016/S0306-4522(99)00156-6. [DOI] [PubMed] [Google Scholar]
- Krenz NR, Weaver LC. Sprouting of primary afferent fibers after spinal cord transection in the rat. Neuroscience. 1998;85:443–458. doi: 10.1016/S0306-4522(97)00622-2. [DOI] [PubMed] [Google Scholar]
- Krenz NR, Meakin SO, Krassioukov AV, Weaver LC. Neutralizing intraspinal nerve growth factor blocks autonomic dysreflexia caused by spinal cord injury. J Neurosci. 1999;19:7405–7414. doi: 10.1523/JNEUROSCI.19-17-07405.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna M, Narang H. The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cellular and molecular life sciences: CMLS. 2008;65:3525–3544. doi: 10.1007/s00018-008-8170-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse MN, Bray LA, de Groat WC. Influence of spinal cord injury on the morphology of bladder afferent and efferent neurons. J Auton Nerv Syst. 1995;54:215–224. doi: 10.1016/0165-1838(95)00011-L. [DOI] [PubMed] [Google Scholar]
- Ku JH. The management of neurogenic bladder and quality of life in spinal cord injury. BJU Int. 2006;98:739–745. doi: 10.1111/j.1464-410X.2006.06395.x. [DOI] [PubMed] [Google Scholar]
- Lever IJ, Pezet S, McMahon SB, Malcangio M. The signaling components of sensory fiber transmission involved in the activation of ERK MAP kinase in the mouse dorsal horn. Mol Cell Neurosci. 2003b;24:259–270. doi: 10.1016/S1044-7431(03)00200-8. [DOI] [PubMed] [Google Scholar]
- Lever I, Cunningham J, Grist J, Yip PK, Malcangio M. Release of BDNF and GABA in the dorsal horn of neuropathic rats. Eur J Neurosci. 2003a;18:1169–1174. doi: 10.1046/j.1460-9568.2003.02848.x. [DOI] [PubMed] [Google Scholar]
- Merighi A, Carmignoto G, Gobbo S, Lossi L, Salio C, Vergnano AM, Zonta M. Neurotrophins in spinal cord nociceptive pathways. Prog Brain Res. 2004;146:291–321. doi: 10.1016/S0079-6123(03)46019-6. [DOI] [PubMed] [Google Scholar]
- Merighi A, Salio C, Ghirri A, Lossi L, Ferrini F, Betelli C, Bardoni R. BDNF as a pain modulator. Prog Neurobiol. 2008;85:297–317. doi: 10.1016/j.pneurobio.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Miyazato M, Sasatomi K, Hiragata S, Sugaya K, Chancellor MB, de Groat WC, Yoshimura N. Suppression of detrusor-sphincter dysynergia by GABA-receptor activation in the lumbosacral spinal cord in spinal cord-injured rats. Am J Physiol Regul Integr Comp Physiol. 2008;295:R336–R342. doi: 10.1152/ajpregu.90315.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazato M, Sugaya K, Goins WF, Wolfe D, Goss JR, Chancellor MB, de Groat WC, Glorioso JC, Yoshimura N. Herpes simplex virus vector-mediated gene delivery of glutamic acid decarboxylase reduces detrusor overactivity in spinal cord-injured rats. Gene Ther. 2009;16:660–668. doi: 10.1038/gt.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor RL, Robertson AG, Allen SJ, Sessions RB, Clarke AR, Mason GG, Burston JJ, Tyler SJ, Wilcock GK, Dawbarn D. A discrete domain of the human TrkB receptor defines the binding sites for BDNF and NT-4. Biochem Biophys Res Commun. 2002;291:501–507. doi: 10.1006/bbrc.2002.6468. [DOI] [PubMed] [Google Scholar]
- Ochodnický P, Cruz CD, Yoshimura N, Michel MC. Nerve growth factor in bladder dysfunction: contributing factor, biomarker, and therapeutic target. Neurourol Urodyn. 2011;30:1227–1241. doi: 10.1002/nau.21022. [DOI] [PubMed] [Google Scholar]
- Ochodnicky P, Cruz CD, Yoshimura N, Cruz F. Neurotrophins as regulators of urinary bladder function. Nat Rev Urol. 2012;9:628–637. doi: 10.1038/nrurol.2012.178. [DOI] [PubMed] [Google Scholar]
- Oliva AA, Jr, Atkins CM, Copenagle L, Banker GA. Activated c-Jun N-terminal kinase is required for axon formation. J Neurosci. 2006;26:9462–9470. doi: 10.1523/JNEUROSCI.2625-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondarza AB, Ye Z, Hulsebosch CE. Direct evidence of primary afferent sprouting in distant segments following spinal cord injury in the rat: colocalization of GAP-43 and CGRP. Exp Neurol. 2003;184:373–380. doi: 10.1016/j.expneurol.2003.07.002. [DOI] [PubMed] [Google Scholar]
- Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci. 2006;29:507–538. doi: 10.1146/annurev.neuro.29.051605.112929. [DOI] [PubMed] [Google Scholar]
- Pezet S, Malcangio M, Lever IJ, Perkinton MS, Thompson SW, Williams RJ, McMahon SB. Noxious stimulation induces Trk receptor and downstream ERK phosphorylation in spinal dorsal horn. Mol Cell Neurosci. 2002a;21:684–695. doi: 10.1006/mcne.2002.1205. [DOI] [PubMed] [Google Scholar]
- Pezet S, Cunningham J, Patel J, Grist J, Gavazzi I, Lever IJ, Malcangio M. BDNF modulates sensory neuron synaptic activity by a facilitation of GABA transmission in the dorsal horn. Mol Cell Neurosci. 2002b;21:51–62. doi: 10.1006/mcne.2002.1166. [DOI] [PubMed] [Google Scholar]
- Rabchevsky AG. Segmental organization of spinal reflexes mediating autonomic dysreflexia after spinal cord injury. Prog Brain Res. 2006;152:265–274. doi: 10.1016/S0079-6123(05)52017-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramer LM, McPhail LT, Borisoff JF, Soril LJ, Kaan TK, Lee JH, Saunders JW, Hwi LP, Ramer MS. Endogenous TrkB ligands suppress functional mechanosensory plasticity in the deafferented spinal cord. J Neurosci. 2007;27:5812–5822. doi: 10.1523/JNEUROSCI.0491-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai A, Cortes E, Seth J, Khan MS, Panicker J, Kelleher C, Kessler TM, Fowler CJ, Dasgupta P. Neurogenic detrusor overactivity in patients with spinal cord injury: evaluation and management. Curr Urol Rep. 2011;12:404–412. doi: 10.1007/s11934-011-0221-1. [DOI] [PubMed] [Google Scholar]
- Salio C, Averill S, Priestley JV, Merighi A. Costorage of BDNF and neuropeptides within individual dense-core vesicles in central and peripheral neurons. Dev Neurobiol. 2007;67:326–338. doi: 10.1002/dneu.20358. [DOI] [PubMed] [Google Scholar]
- Santos-Silva A, Charrua A, Cruz CD, Gharat L, Avelino A, Cruz F. Rat detrusor overactivity induced by chronic spinalization can be abolished by a transient receptor potential vanilloid 1 (TRPV1) antagonist. Auton Neurosci. 2012;166:35–38. doi: 10.1016/j.autneu.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Schmitz SK, Hjorth JJ, Joemai RM, Wijntjes R, Eijgenraam S, de Bruijn P, Georgiou C, de Jong AP, van Ooyen A, Verhage M, Cornelisse LN, Toonen RF, Veldkamp W. Automated analysis of neuronal morphology, synapse number and synaptic recruitment. J Neurosci Methods. 2011;195:185–193. doi: 10.1016/j.jneumeth.2010.12.011. [DOI] [PubMed] [Google Scholar]
- Seki S, Sasaki K, Fraser MO, Igawa Y, Nishizawa O, Chancellor MB, de Groat WC, Yoshimura N. Immunoneutralization of nerve growth factor in lumbosacral spinal cord reduces bladder hyperreflexia in spinal cord injured rats. J Urol. 2002;168:2269–2274. doi: 10.1016/S0022-5347(05)64369-8. [DOI] [PubMed] [Google Scholar]
- Seki S, Sasaki K, Igawa Y, Nishizawa O, Chancellor MB, De Groat WC, Yoshimura N. Suppression of detrusor-sphincter dyssynergia by immunoneutralization of nerve growth factor in lumbosacral spinal cord in spinal cord injured rats. J Urol. 2004;171:478–482. doi: 10.1097/01.ju.0000088340.26588.74. [DOI] [PubMed] [Google Scholar]
- Sievert KD, Amend B, Gakis G, Toomey P, Badke A, Kaps HP, Stenzl A. Early sacral neuromodulation prevents urinary incontinence after complete spinal cord injury. Ann Neurol. 2010;67:74–84. doi: 10.1002/ana.21814. [DOI] [PubMed] [Google Scholar]
- Simpson LA, Eng JJ, Hsieh JT, Wolfe DL. The health and life priorities of individuals with spinal cord injury: a systematic review. J Neurotrauma. 2012;29:1548–1555. doi: 10.1089/neu.2011.2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steers WD, Kolbeck S, Creedon D, Tuttle JB. Nerve growth factor in the urinary bladder of the adult regulates neuronal form and function. J Clin Invest. 1991;88:1709–1715. doi: 10.1172/JCI115488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steers WD, Creedon DJ, Tuttle JB. Immunity to nerve growth factor prevents afferent plasticity following urinary bladder hypertrophy. J Urol. 1996;155:379–385. doi: 10.1016/S0022-5347(01)66664-3. [DOI] [PubMed] [Google Scholar]
- Thompson SW, Bennett DL, Kerr BJ, Bradbury EJ, McMahon SB. Brain-derived neurotrophic factor is an endogenous modulator of nociceptive responses in the spinal cord. Proc Natl Acad Sci U S A. 1999;96:7714–7718. doi: 10.1073/pnas.96.14.7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle JB, Steers WD. Nerve growth factor responsiveness of cultured major pelvic ganglion neurons from the adult rat. Brain Res. 1992;588:29–40. doi: 10.1016/0006-8993(92)91341-B. [DOI] [PubMed] [Google Scholar]
- Vizzard MA. Alterations in growth-associated protein (GAP-43) expression in lower urinary tract pathways following chronic spinal cord injury. Somatosens Mot Res. 1999;16:369–381. doi: 10.1080/08990229970429. [DOI] [PubMed] [Google Scholar]
- Vizzard MA. Neurochemical plasticity and the role of neurotrophic factors in bladder reflex pathways after spinal cord injury. Prog Brain Res. 2006;152:97–115. doi: 10.1016/S0079-6123(05)52007-7. [DOI] [PubMed] [Google Scholar]
- Weaver LC, Verghese P, Bruce JC, Fehlings MG, Krenz NR, Marsh DR. Autonomic dysreflexia and primary afferent sprouting after clip-compression injury of the rat spinal cord. J Neurotrauma. 2001;18:1107–1119. doi: 10.1089/08977150152693782. [DOI] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- Yoshiyama M, Nezu FM, Yokoyama O, Chancellor MB, de Groat WC. Influence of glutamate receptor antagonists on micturition in rats with spinal cord injury. Exp Neurol. 1999;159:250–257. doi: 10.1006/exnr.1999.7124. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Zinck ND, Rafuse VF, Downie JW. Sprouting of CGRP primary afferents in lumbosacral spinal cord precedes emergence of bladder activity after spinal injury. Exp Neurol. 2007;204:777–790. doi: 10.1016/j.expneurol.2007.01.011. [DOI] [PubMed] [Google Scholar]