

Abstract
IL-1β production is critically regulated by cytosolic molecular complexes, termed inflammasomes. Different inflammasome complexes have been described to date.
While all inflammasomes recognize certain pathogens, it is the distinctive feature of NLRP3 inflammasome to be activated by many and diverse stimuli making NLRP3 the most versatile, and importantly also the most clinically implicated inflammasome. However, NLRP3 activation has remained the most enigmatic. It is not plausible that the intracellular NLRP3 receptor is able to detect all of its many and diverse triggers through direct interactions; instead, it is discussed that NLRP3 is responding to certain generic cellular stress-signals induced by the multitude of molecules that trigger its activation.
An ever increasing number of studies link the sensing of cellular stress signals to a direct pathophysiological role of NLRP3 activation in a wide range of autoinflammatory and autoimmune disorders, and thus provide a novel mechanistic rational, on how molecules trigger and support sterile inflammatory diseases. A vast interest has created to unravel how NLRP3 becomes activated, since mechanistic insight is the prerequisite for a knowledge-based development of therapeutic intervention strategies that specifically target the NLRP3 triggered IL-1β production. In this review, we have updated knowledge on NLRP3 inflammasome assembly and activation and on the pyrin domain in NLRP3 that could represent a drug target to treat sterile inflammatory diseases. We have reported mutations in NLRP3 that were found to be associated with certain diseases. In addition, we have reviewed the functional link between NLRP3 inflammasome, the regulator of cellular redox status Trx/TXNIP complex, endoplasmic reticulum stress and the pathogenesis of diseases such as type 2 diabetes. Finally, we have provided data on NLRP3 inflammasome, as a critical regulator involved in the pathogenesis of obesity and cardiovascular diseases.
Keywords: NLRP3 inflammasome (PDB id: Q96P20); IL-1β (PDB id: P01584, P10749); IL-18 (PDB id: Q14116); Obesity; Cardiovascular diseases
Abbreviations: Alum, aluminum hydroxide; ATMs, adipose tissue macrophages; ATP, adenosine triphosphate; ApoE, murin apolipoprotein E; ASC, Apoptosis-associated Speck-like protein containing a Caspase-recruitment domain; CARD, caspase recruitment domain; CAPS, cryopyrin-associated periodic syndrome; CAP1, caspase-1; CINCA, chronic infantile neurological cutaneous articular syndrome; CPPD, calcium pyrophosphate dihydrate; CVD, cardiovascular diseases; DAMPs, danger-associated molecular patterns; DD, death domains; FCAS, familial cold autoinflammatory syndrome; FCU, familial cold urticaria; HFD, high fat Western-type diet; HPFs, hereditary periodic fevers; IAPP, amyloid-containing amylin—islet amyloid polypeptide; IL-1β, Interleukin-1β; IL-1 R1, IL-1 receptor 1; IL-18, interleukin-18; IR, insulin receptor; IRS-1, insulin receptor substrate-1; LDL, low density lipoprotein; LDLR, LDL receptor; MDP, muramyl dipeptide; mmLDL, minimally modified low-density lipoprotein; MSU, uric acid crystals; MWS, Muckle–Wells syndrome; NBD, nucleotide-binding domain; NLRP3, nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3; Nlrp3−/−, Nlrp3-deficient mice3; NLRs, receptors; NOMID, neonatal-onset multisystem inflammatory disease; Nrf2, NF-E2-related 2; LPS, lipopolysaccharide; LRR, leucine-rich-repeat domain; oxLDL, oxidized LDL; PAMPs, pathogen-associated molecular patterns; PMNL, polymorphonuclear leukocytes; POP, PYD-only proteins; PRRs, pattern-recognition receptors; PYD, pyrin domain; RLRs, RIG-1-like helicases; ROS, reactive oxygen species; SMCs, smooth muscle cells; TLRs, toll-like receptors; TNF, tumor necrosis factor; Trx-1, thioredoxin-1; TXNIP, thioredoxin-interacting P; T2DM, type 2 diabetes mellitus; UPR, unfolded protein response
Highlights
-
•
We review recently described knowledge that have been proposed to be involved in NLRP3 inflammasome assembly and activation.
-
•
We have updated knowledge on the pyrin domain in NLRP3 that could represent a drug target to treat sterile inflammatory diseases.
-
•
We have reported mutations in NLRP3 that were found to be associated with certain diseases.
-
•
We reviewed the functional link between NLRP3 inflammasome, the regulator of cellular redox status Trx/Txnip complex, endoplasmic reticulum stress and the pathogenesis of diseases such as type 2 diabetes.
-
•
We have provided data on NLRP3, as a critical regulator, involved in the pathogenesis of obesity, type 2 diabetes and cardiovascular diseases.
Introduction
Vertebrates evolved two different systems to recognize and eliminate pathogens: the innate and the adaptive immune systems. The innate immune system is the first one to be activated and can sense a wide range of pathogenic microbes through a limited number of receptors, called pattern-recognition receptors (PRRs), by recognizing conserved microbial signatures, named pathogen-associated molecular patterns (PAMPs) [1]. PRRs are expressed by many cell types (macrophages, monocytes, neutrophils, and others), allowing the detection of pathogens to take place directly at the site of infection. Once activated, the innate immune system initiates the inflammatory response by secreting cytokines and chemokines. This leads to the expression of adhesion and co-stimulatory molecules able to recruit immune cells and to stimulate the adaptive immune response. Because of the need to distinguish between pathogenic and non-pathogenic or commensal microbes, it has been proposed that the innate immune system is activated by the recognition of an antigen, but only in presence of danger signals released by cells (danger-associated molecular patterns or DAMPs) [2].
The NOD-like receptors (NLRs) are a family of PRRs mostly expressed in the cytosol and able therefore to detect signs of intracellular invaders [3]. Some of the NLRs can also sense non-microbial danger signals and form large cytoplasmic complexes called inflammasomes, responsible for the activation of caspase-1 and -5, which ultimately leads to the proteolytic activation of the proinflammatory cytokines interleukin-1β (IL-1β) and interleukin-18 (IL-18) [4].
Secretion of the key inflammatory cytokine IL-1β (and its family member IL-18) is a consequence of phagocyte activation and promotes a multitude of metabolic, physiologic, inflammatory, hematologic and immunologic effects. Excessive or prolonged IL-1β generation can cause widespread tissue damage and is a well-documented phenomenon of, and is associated with numerous acute and chronic inflammatory human diseases, many of which are autoinflammatory or autoimmune pathologies (for detailed reviews see [5–7]). IL-1β production is critically regulated by cytosolic molecular complexes, termed inflammasomes [8]. Several different inflammasome complexes have been described to date. All of them specialize in pattern recognition of danger signals, and subsequently instruct general defense mechanisms of the human immune system.
While all inflammasomes recognize certain PAMPs or DAMPs, it is the distinctive feature of NLRP3 (NLRP3: nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 OR Nod-like receptor protein 3) to be activated by unusually many and diverse stimuli making NLRP3 the most versatile, and importantly also the most clinically implicated inflammasome. At the same time NLRP3 activation has remained the most enigmatic. It is not plausible that the intracellular NLRP3 receptor is able to detect all of its many and diverse triggers through direct interactions; instead, it is discussed that NLRP3 is responding to certain generic cellular stress-signals induced by the multitude of PAMPs and DAMPs that trigger its activation.
An ever increasing number of studies link the sensing of cellular stress signals to a direct pathophysiological role of NLRP3 activation in a wide range of autoinflammatory and autoimmune disorders, and thus provide a novel mechanistic rational, on how DAMPs trigger and support sterile inflammatory diseases. Several of these pathologies related to undue NLRP3 activation, like, e.g. gout and pseudogout, obesity, atherosclerosis, Alzheimer's disease or type 2 diabetes mellitus (T2DM) [6], have an immense impact on our society. This has created vast interest to unravel how NLRP3 becomes activated, since mechanistic insight is the prerequisite for a knowledge-based development of therapeutic intervention strategies that specifically target the NLRP3 triggered IL-1β production.
To this end, many studies have contributed very valuable insights. It is known that only stimulated cells activate NLRP3, which has a tripartite structure consisting of a pyrin domain (PYD), a nucleotide-binding domain (NBD) and a leucine-rich-repeat (LRR) domain [9]. Upon activation, NLRP3 associates with the adaptor protein ASC, which comprises a caspase recruitment domain (CARD) and a pyrin domain, that are held together solely by a semi-flexible linker [10] allowing both domains to engage freely with other partners. The NLRP3:ASC complex oligomerizes and binds the enzyme caspase 1, thus forming active inflammasome complexes (NLRP3, ASC, and caspase-1) that produce IL1-β [11] (Fig. 1).
Fig. 1.
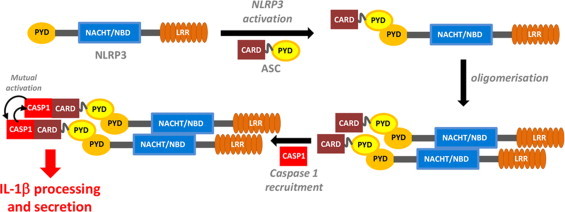
NLRP3 inflammasome assembly. CARD, caspase recruitment domain; LRR, leucine-rich repeat; NACHT/NBD, nucleotide binding domain; PYD, pyrin domain; CAP1, caspase-1.
It is well established that the recruitment of ASC by NLRP3 is an absolute prerequisite in this process. If ASC recruitment fails, the NLRP3 inflammasome complex does not assemble. Formation of the ASC:NLRP3 complex is established between two pyrin domains, one each in ASC and NLRP3. The presence of both pyrin domains is sufficient and necessary to promote the bimolecular interaction between ASC and NLRP3 [12]. To this end, it is also worth to note that both, host cells (to regulate) and pathogens (to evade immune response) produce PYD-only proteins (POP) that interfere with PYD-driven assembly of inflammasomes indicating the central role of PYD interactions in regulating IL-1β production. In contrast to this pivotal role that the pyrin domain association plays in the formation of the NLRP3 inflammasome, the mechanisms that prevent this binding event during quiescent cellular states are not known (Fig. 2A). However, it has been elucidated that over 90 disease-associated mutations in human NLRP3, which map mainly to the NBD and its vicinity, render the protein constitutively active [7] and allow continuous inflammasome activation, e.g. by association of the NLRP3's pyrin domain with its adaptor protein ASC, despite the absence of PAMP or DAMP stimuli [12] (Fig. 2B). Furthermore, the deletion of the LRR domain promotes NLRP3's association with its adaptor ASC via their pyrin domains [12] (Fig. 2C).
Fig. 2.
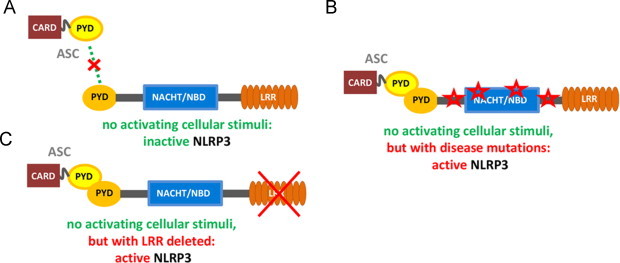
NLRP3 domain structure and its influence on interactions of its pyrin domain. (A) It is unresolved how the pyrin domains shielded from engaging the readily available pyrin domain in ASC in non-activated NLRP3. (B) Disease-associated mutations in NLRP3 were shown to promote pyrin–pyrin domain engagement between NLRP3 and ASC. (C) Biochemical deletion of the LRR-domain in NLRP3 unmasks the pyrin domain and thus facilitates its binding to ASC's pyrin domain.
As described above, the NLRP3 inflammasome is formed by NLRP3 (NACHT, LRR and PYD domains-containing protein 3, also called cryopyrin, NLRP3, PYPAF1, CIAS1 and CLR1.1), the adaptor protein Apoptosis-associated Speck-like protein containing a CARD (ASC) and the inflammatory caspase-1 (cysteine-dependent aspartate-directed protease-1).
The current model of inflammasome activation has been developed in analogy to mammalian toll-like receptors (TLRs) and plant disease resistance gene products (R proteins). When the LRR domain recognizes an activating stimulus, the autorepression state of NLRP3 (probably mediated by the LRR domain itself) is relieved. As a consequence, the PYD and NBD domains are exposed, thus allowing for NLRP3 oligomerization by homotypic NBD domain interaction (most likely upon ATP binding) and recruitment of the adaptor protein ASC, which in turn recruits caspase-1 (see Fig. 1 above). Finally, caspase-1 clustering leads to its activation via autoprocessing, allowing the proteolytic cleavage of pro-IL-1β and other cellular targets [11].
ASC is composed of the two death domains (DD), PYD and caspase recruitment domain (CARD), and acts as an adaptor molecule not only in inflammation [4], but also in apoptosis [12,13]. ASC binds to other pyrin-containing proteins via its own pyrin domain [14] and recruits members of the caspase family via CARD/CARD interaction [15–17]. It has been shown that ASC can also oligomerize into functional complexes, like the pyroptosome, a potent caspase-1 activator responsible for pyroptosis [18], and serves as scaffold for other supramolecular platforms involved in caspase activation [4].
Caspases are a family of cysteine proteases that play essential roles in apoptosis, necrosis and inflammation. They are formed by a CARD domain followed by a domain containing a catalytic cysteine and are synthesized as inactive zymogens that are activated upon proteolytic processing [19]. Inflammasomes activate a class of caspases known as inflammatory caspases [20,21], that are so called because the main substrates of their representative member, caspase-1, are cytokines (IL-1β, IL-18 and possibly IL-33), which are crucial mediators of the inflammatory response. In mammals, inflammatory caspases include human and murine caspase-1 and -12, murine caspase-11, and the two caspase-1-related human caspase-4 and -5 [22,23]. In order to be activated, pro-caspase-1 needs to undergo autoproteolysis, which requires clustering of pro-caspase-1 on oligomerized NLRP3.
The pyrin domain in NLRP3 assembly
As detailed above, the NLRP3 inflammasome assembly critically relies on the PYD/PYD interaction between NLRP3 and ASC. Both pyrin domains belong to the DD superfamily and share a common structural feature (i.e. a six-helical bundle fold). Almost all known protein–protein interactions in the DD superfamily are formed either by self-association or homotypic interactions with other members of the same subfamily (e.g. the PYD/PYD interaction between NLRP3 and ASC). However the members of the DD superfamily often display entirely different surface features from other proteins of the same fold, which may account for their specificity in protein–protein interactions [24]. In contrast to the CARD/CARD interaction, which has already been characterized structurally [25], the structural binding mode of PYD/PYD interactions is currently still unknown. Recently, Vajjhala et al. used a mutagenesis approach combined with coimmunoprecipitation to investigate residues in ASC that govern PYD/PYD mediated self-association or NLRP3-binding of ASC [26]. Available high resolution structures of ASC [10], ASC-PYD [27] and NLRP3-PYD [28] exist and were used as a template for the structural mapping of the analyzed mutants as well as for building a model of the oligomerization of NLRP3's adaptor protein, ASC. Five specific amino acids were shown to be responsible for both, ASC self-association and NLRP3 recruitment. However, self-association of ASC did not compete with NLRP3 binding, but to the contrary promoted binding of NLRP3-PYD. Vajjhala et al. reconciled these data by constructing a model that describes how an ASC-PYD homodimer can interact with two NLRP3-PYDs. They further propose that ASC combines its ability to self-associate with simultaneous interactions with NLRP3-PYD to form higher order structures, which build the basis for caspase-1 recruitment and activation. The detailed mapping of interaction sites on NLRP3-PYD is necessary to critically evaluate this model, since in contrast to ASC, critical amino acids in NLRP3's pyrin domain responsible for its interaction with ASC remain undiscovered. Moreover, it is not understood how the NLRP3's PYD is masked in non-activated cellular states (see above). Therefore, the structure–function relationship which mediates the PYD-triggered NLRP3 inflammasome assembly is unavailable, which impedes the knowledge-based design of inhibitors that specifically target the NLRP3 inflammasome.
The pyrin domain in NLRP3 as a drug target for sterile inflammatory diseases
Successful strategies of inhibiting PYD/PYD interactions are already in place in nature. Since prolonged inflammation beyond eradication of endogenous DAMPs or foreign PAMPs can lead to excessive tissue damage, host cells have an arsenal of inflammasome inhibitors available, which cells produce to conclude a successful inflammatory insult [29,30]. One example of these inflammasome regulators are the POPs, which function as endogenous dominant negative proteins that modulate inflammasome activity. POP1 shares a sequence identity of 64% with ASC-PYD. It interacts with ASC in a PYD-dependent manner, thus interfering with the recruitment of several NLRPs by ASC [31]. A recent study, however, raises some questions in regard to the solidity of this mechanism [26]. From a biological viewpoint, a broad shutdown of signals from all NLRPs, which utilize the adaptor protein ASC, seems favorable in cases when cells aim to conclude an inflammatory insult. Contrary to POP1, POP2 interacts only weakly with ASC-PYD, but binds more specifically to the PYD of NLRP2. This way, POP2 is more selective and mainly affects NLRP2-specific danger sensing [32] without interfering much with the pattern-recognition activities of other members of the NLR family. Certain pathogens have copied some of the host's own strategies to control inflammasome assembly in order to evade the host's immune surveillance system. Members of the family of poxviruses encode viral POP proteins, which target the PYD of ASC [29], thus interfering with the host's ability to produce IL-1β via any PYD-containing pattern recognition receptors [33].
While it is useful for pathogens to broadly block the host's immune mechanisms, for therapeutic intervention, selective inhibition of the specific pathway in question is superior. In regards to treatment strategies for the sterile inflammatory immunopathologies that are associated with NLRP3 activation, specific targeting of the PYD in NLRP3 (as opposed to the PYD in ASC) promises a better safety profile. By specific targeting of NLRP3-PYD pattern recognition and signaling, functions of all other inflammasome complexes that rely on recruitment of the adaptor protein ASC (i.>e. AIM2 and 14, NLRP3 inflammasomes [9,34]) remain in place to support the host immune surveillance.
Elusive mechanism of NLRP3 activation
Overall, the mechanism leading to NLRP3 inflammasome activation is poorly understood. Differently from other inflammasomes, which only respond to few specific PAMPs, the NLRP3 inflammasome can be activated by an increasing number of physically and chemically diverse triggers. Among them: (i) exogenous microbial stimuli, including lipopolysaccharide (LPS) [35–37], lipooligosaccharide [38], nucleic acids [35,36,39,40], muramyl dipeptide (MDP) [41], and certain pore-forming toxins, like pneumolysin [42], nigericin and maitotoxin [43]; (ii) environmental large inorganic crystalline structures, such as asbestos and silica [44,45], nanoparticles [46], adjuvants, like aluminum hydroxide (alum) [45,47], commonly used to boost vaccine responses and ultraviolet irradiation [48]; (iii) endogenous danger signals such as extracellular adenosine triphosphate (ATP) [43], uric acid crystals (MSU) [49], hyaluronan and heparan sulfate [50], and amyloid-β fibrils [51]. The NLRP3 inflammasome is also activated by necrotic, but not apoptotic cells, leading to release of IL-1β and IL-18, which contribute to the so-called sterile inflammation response [52]. Sensing of necrosis is mediated via Ca-sensing mechanism of NLRP3 [53,54].
Given the high number of very different NLRP3 activators, it is unlikely that each of them binds directly to the inflammasome. Therefore it has been suggested that a common molecule or pathway must be responsible for the activation of the inflammasome. Several studies have indicated three possible pathways. The first hypothesis is that reactive oxygen species (ROS) are proximal signals for NLRP3 inflammasome activation. ROS are ancient and highly evolutionarily conserved danger signals and elevated ROS production is observed upon treatment with many NLRP3 activators tested to date [44,55,56]. The second model suggests that a drop in intracellular potassium (K+) concentration, through endogenous ion channels or pore-forming bacterial toxins, causes inflammasome activation [57]. Consistently with these two hypotheses, both sequestration of ROS and blockade of K+ efflux induced by NLRP3 activators suppress inflammasome activation, with a consequent reduction in caspase-1 activation and IL-1β maturation [44,55–59]. Furthermore, ROS generation is often associated with K+ efflux [60], although the interplay between these two pathways is currently unclear. A third model proposes that the disruption of the lysosomal membrane, as a consequence of the phagocytosis of particulate or live pathogens, causes the release of a putative NLRP3-activating molecule into the cytosol, most likely cathepsin B or a protein modified by cathepsin B [45,51]. This hypothesis is supported by the efficacy of phagocytosis inhibitors in blocking inflammasome activation by particulates. Interestingly, Shimada et al. propose a unifying theory of the three potential NLRP3 activating mechanisms, where potassium efflux, lysosomal damage, and ROS signals all converge to induce release of oxidized mitochondrial DNA that then activates NLRP3 [61].
Analyzing all of the triggers that induce inflammasome activation to date (including those causing channel formation and lysosome rupture), the production of ROS certainly seems to be the most prominent trigger. However, ROS-triggered NLRP3 activation has also an impressive track record of controversies in the field, as reviewed by Rubartelli et al. [62]. These arise from the study design, which relies on differently cultured sets (or subsets) of primary cells or various immortalized cell cultures. Both, cell origin and culture conditions have a tremendous impact on the delicate cellular redox balance and thus, variation in experimental assays may result in starkly different or even opposing outcomes [62]. A further reason is that the vast majority of interaction studies are restricted to a single experimental design: overexpression of desired analytes in cell cultures followed by interaction analysis of cell lysates by the use of coimmunoprecipitation.
NLRP3 activation by redox-proteins – the controversy around TXNIP and potential involvement of another intracellular redox-protein
In a recent study, Zhou et al. demonstrate that an increase in ROS concentration following cellular stress leads to dissociation of thioredoxin-interacting protein (TXNIP also called VDUP1) from oxidized thioredoxin-1 (Trx-1), subsequent association of TXNIP with NLRP3 (via its NBD and/or LRR domain) and NLRP3 activation [63]. It is interesting to note that TXNIP knockout or knockdown impairs caspase-1 activation, yet not completely, thus indicating that other regulators of the inflammasome activity or other pathways might function together with ROS production to initiate the inflammatory response. Furthermore, many stimuli that induce generation of ROS are not able to activate the NLRP3 inflammasome (such as tumor necrosis factor, TNF), suggesting either that a specific type or subcellular location of ROS is required, or that ROS are necessary, but not sufficient, for NLRP3 inflammasome activation. However, the direct involvement of TXNIP in NLRP3 binding and activation has been challenged by another report by Masters et al. [64], which was unable to reproduce a selection of experiments, which involve TXNIP-triggered NLRP3 activation. Recently, Lunov et al. reported that specifically functionalized nanoparticles activate NLRP3 [46]. These findings demonstrate that all of the following processes, accumulation of ROS, thioredoxin oxidation and TXNIP recruitment of NLRP3, happen in the same time frame as the secretion of IL-1β.
Disease-associated mutations in NLRP3
Mutations in the gene coding for NLRP3 have been associated with several disorders belonging to the family of so-called autoinflammatory diseases. Hereditary periodic fevers (HPFs), or cryopyrin-associated periodic syndrome (CAPS) [65], are heritable diseases characterized by unexplained and recurrent episodes of fever and severe inflammation. These cryopyrinopathies were once thought to be distinct conditions but considering the overlapping characteristics and the fact that they form a clinical continuum, the different HPFs are now used to describe disease severity [66], which seems to correlate well with the total amount of IL-1β produced [66]. Patients suffering from familial cold autoinflammatory syndrome (FCAS), also called familial cold urticaria (FCU), have less severe symptoms that generally include fever (often triggered by exposure to cold), arthralgia and recurrent urticaria. Muckle–Wells syndrome (MWS) patients display the same symptoms and may, in addition, develop renal amyloidosis together with deafness [67]. Patients affected by chronic infantile neurological cutaneous articular syndrome (CINCA), also termed neonatal-onset multisystem inflammatory disease (NOMID), show the most severe symptoms. These include arthropathy, chronic urticaria, and central nervous system involvement ranging from hearing loss to chronic aseptic meningitis and mental retardation [68].
HPFs are all caused by mutations in the third exon of NLRP3 [69,70]. The disease-associated mutations in FCAS, MWS and NOMID cause hyperactivity of the inflammasome and constitutive IL-1β production [71,72], suggesting that these mutations lead to a constitutively active NLRP3. This model is supported by clinical studies showing that treatment of patients with an inhibitor of IL-1β leads to a very striking and dramatic improvement of symptoms in all three conditions [73–75].
An aberrant activation of the NLRP3 inflammasome has also been observed in the case of other autoinflammatory diseases such as gout, pseudogout, silicosis, and asbestosis. In these cases, the aberrant activation is not caused by an inherited mutation but by chronic exposure to inflammasome activators such as MSU (causing gout), calcium pyrophosphate dihydrate (CPPD, responsible for pseudogout), or inflammation-inducing dust [76].
The majority of the over 90 disease-associated mutations causing HPFs reside in the NBD domain of NLRP3, but not in residues predicted to directly interact with nucleotides. By using the structure of the N-terminal ATPase domain of Cdc6 (PDB code: 1FNN) [77] as template to model the NBD domain of NLRP3, Albrecht et al. showed that almost all of the disease-associated mutations comprised in the aligned sequence (V200-L371) cluster in the proximity of the nucleotide-binding region. Although none of the mutations maps to residues considered essential for the activity of most NTPases, such as the so-called Walker A-motif (corresponding to residues G226, G231, K232, and T233 in NLRP3) or the Mg2+-anchoring aspartate in the Walker B-motif (D302 in NLRP3), the different variants could still disturb the NTP-binding and -hydrolysis activity by causing unfavorable structural changes near the active site.
Alternatively, the disease-associated mutations could interfere with domain–domain interactions within the same protein or between two proteins or in dimer formation [78]. Both ATP-binding and NLRP3 oligomerization are essential for the NLRP3 inflammasome activation and loss of one of these functions could explain constitutive NLRP3 activation [79]. Additionally, another report shows that cAMP negatively regulates the NLRP3 activation and that selected disease-associated mutations have less sensitivity for cAMP regulation, which also could explain the constitutive activity [54]. To complicate things further, yet, another report could not detect a dependency between cAMP levels and inflammasome activation [53]. Some of the disease-associated mutations might very well interfere with ATP or cAMP binding. However, it is difficult to imagine that all 90 plus mutations shown to cause constitutive NLRP3 activation, can be explained by altering the relatively small binding pockets for ATP or cAMP.
A more likely explanation is that these mutations disrupt the interaction between NLRP3 and an unknown regulator or between two domains, which normally keeps the whole protein in an inactive form, potentially by shielding NLRP3's PYD from binding ASC. In support of this is the finding that the deletion of the LRR domain renders NLRP3 constitutively active. Similarly, in NLRP1, the oligomerization of the NBD becomes independent of the activation stimulus (muramyl dipeptide) once the LRR domain has been removed [80].
Treatment of the CAPS, is currently restricted to targeting the extracellular IL-1β by three different biopharmaceutical drugs (reviewed in 66): anakinra, a recombinant form of a naturally occurring IL-1β receptor antagonist [81], rilonacept, a dimeric fusion protein consisting of the extracellular domain of human interleukin-1β receptor and the Fc domain of human IgG1 [82], and canakinumab, an anti-IL-1β monoclonal antibody [83]. The successful use of anti-IL-1β treatments in the afore-mentioned diseases and ongoing clinical trials have boosted enormous efforts to discover and develop small-molecule approaches to specifically inhibit inflammasome activation [9].
NLRP3 inflammasome in diabetes and obesity
T2DM and obesity represent major public health problems with steadily increasing prevalence worldwide. Chronic metaflammation ‘metabolic inflammation’ [84] is the hallmark of obesity causing insulin resistance and T2DM [85]. Activation of inflammasome proteins and caspase-1 are drastically increased in cellular compartments of adipose and liver tissue of obese humans and mice. Total body weight loss in obese T2DM individuals is associated with diminished NLRP3 and IL-1β expression in subcutaneous adipose tissue. Hence, expression rates of inflammasome components and caspase-1 are significantly correlated with disease severity of T2DM-obese subjects [86,87]. The influx of macrophages, T and B cells into adipose tissue, adipocyte hypertrophy, total body weight gain and circulating adipokine levels are dependent on the NLRP3 inflammasome activation [88]. Many authors investigated the role of the inflammasome complex proteins such as caspase-1 and ASC protein in the development of high-fat diet (HFD)-induced obesity. Deficiency in NLRP3 inflammasome and adaptor ASC protein in obese mice improves glucose tolerance and ameliorates insulin signaling pathways [89]. Nlrp3−/− mice fed an HFD for a long period are protected from pancreatic β-cells apoptosis as well as a significant enlargement of Langerhans islets [89]. Compared with wild-type, ASC−/− mice are protected from liver steatosis, adipocyte hypertrophy and the HFD-induced insulin resistance [88]. These findings underline the importance of the NLRP3 inflammasome in the development of insulin resistance and T2DM progression. More interestingly, NLRP3 in adipocytes may play important roles in the context of obesity [9]. A high activity of inflammasome proteins especially caspase-1, IL-1β and IL-18 has been detected in white adipose tissue of obese mice [87]. Compared with wild-type precursor cells, caspase-1-deficient precursor cells differentiate more efficiently into mature adipocytes with a higher level of oxidation [87]. This suggests a direct role of inflammasome in the control of adipocyte developmental programs [90], with adipocyte-specific proteins possibly serving as substrates for the caspase-1 enzyme [9]. Thus, leakage of caspase-1 is well correlated with decreased insulin resistance, presence of smaller adipocytes in adipose tissue and lower percentage of total fat mass [87]. Nutritional excess of HFD leads to the infiltration of M1 macrophages into the adipose tissue [91]. Free fatty acids are elevated in plasma and probably scavenged by adipose tissue macrophages (ATMs) leading to generation of lipotoxic ceramide molecules composed from sphingoside and fatty acid [92,93]. At higher concentration, ceramide and palmitate act as endogenous danger signals responsible for activation of adipose-tissue infiltrating macrophages that in addition to proinflammatory cytokines, preferentially express Nlrp3, ASC, and caspase-1 proteins. In the case of obesity, the NLRP3 inflammasome is activated through a pathway involving reduction of the AMP-protein kinase (AMPK) activity leading to defective autophagy of mitochondria, the so-called mitophagy. This leads to greater ROS generation by mitochondria. ROS has been suggested to activate NLRP3 and hence could increase the release of the active form of IL1-β and IL-18 [86,94]. The increased circulating levels of the potent proinflammatory cytokine IL1-β directly inhibits the insulin signaling cascade via serine phosphorylation of the insulin receptor substrate 1 and indirectly promotes production of tumor necrosis-factor-α, known as an inducer of insulin-resistance. The IL1-β- and IL-18-mediated promotion of the infiltration of the effector adipose type 1 CD4 T helper cells infiltrate into adipose tissues, is the second deleterious consequence of inflammasome activation [86]. Evidence showing that the deficiency of the NLRP3 inflammasome reduces but does not abrogate the caspase-1 pathway signaling in the pathophysiology of obesity [9], leaves room for numerous questions and speculations about the role of other metabolic sensor subtypes, the nature of their stimuli and mechanisms required for caspase-1 activation in liver and adipose tissue. The current view on the role of NLRP3 activation in insulin resistance and T2DM development is illustrated in Fig. 3.
Fig. 3.
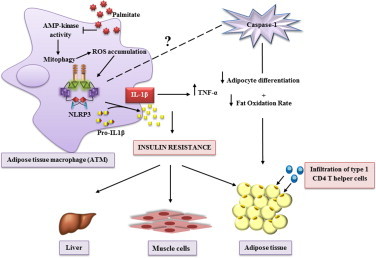
The NLRP3 inflammasome is a key mediator of metabolic inflammation and disorder. In ATM, saturated free fatty acids, but not unsaturated free fatty acids, inhibit regulation of energy storage and cell lipid metabolism by decreasing AMP-activated kinase activity, which normally leads to degradation and recycling of mitochondrial components. When mitophagy is inhibited, the accumulation of dysfunctional mitochondria promotes, (i) mitochondrial generation of ROS and (ii) mitochondrial release of DNA into the cytosol, which combined activate the NLRP3 inflammasome and cleave pro-IL-1β into the active form IL1-1β. This proinflammatory state leads to deterioration of the metabolism. IL1-β enhances insulin-resistance through serine phosphorylation of insulin receptor substrate-1 (IRS-1) that impairs engagement of the insulin receptor (IR) with IRS-1. It also triggers a direct-insulin resistance by promoting expression of TNF-α. Absence of caspase-1 improves adipogenesis and increases fat oxidation rate. Other unknown inflammasome sensors might regulate adipocyte differenciation/maturation and control the cellular energy metabolism through the enzyme caspase-1.
Elevated levels of IL-1β in the circulation and in pancreatic islets are associated with an increased risk for developing T2DM [95]. Chronic hyperglycemia induced upon peripheral insulin resistance inhibits β-cell insulin secretion and contributes to islet dysfunction and destruction in an IL-1β-dependent manner [96,97]. Chronic exposure to elevated glucose triggers, mitochondrial dysfunction and oxidative stress in rat pancreatic islets and a β-cell line [98]. Upon increased glycolysis, ROS are generated by increasing the activity of the mitochondrial electron transport chain [99]. Elevated intracellular ROS may dissociate the Trx-1/TXNIP complex. Free Trx-1 and TXNIP act respectively as a ROS scavenger and NLRP3 inflammasome activator, leading to the subsequent secretion of IL-1β [100]. In fact, TXNIP exerts multiple deleterious effects on β-cells and glucose metabolism. In addition to the inhibition of the anti-oxidative effect of Trx-1; TXNIP represses cellular glucose uptake directly by binding to the glucose transporter, Glut1, and indirectly by reducing the level of Glut1 mRNA. The TXNIP knocking down, increases glucose uptake in peripheral tissues in both insulin-dependent and independent manners [101]. It has been shown that chronic hyperglycemia of rat pancreatic islets induced ER stress leading to cellular oxidative stress. ER stress causes activation of the NLRP3 inflammasome and IL-1β induction by cleavage of caspase-1. The resultant release of IL-1β is necessary for the initiation of inflammation and mitochondrial cell death. ER stress activates NLRP3 inflammasome, independently of the classical unfolded protein response (UPR) but required the production of ROS and K+ efflux. Genetic deletion of TXNIP suppressed IL-1β release from islet cells and prevented ER stress-induced β-cell death. These findings highlight the role of TXNIP as a functional link between ER stress, NLRP3 inflammasome activation and inflammation related to T2DM [102].
AMPK is an energy sensor characterized by numerous protective metabolic effects including anti-inflammatory activity and maintenance of metabolic homeostasis. A low cellular energy status activates AMPK, leading to the phosphorylation of a host of key cellular proteins in order to suppress ATP consumption and increase ATP production to restore energy homeostasis [103,104]. It has been shown that, AMPK regulates chREBP/Mlx activity by phosphorylation-dependent nuclear translocation and indirectly regulates TXNIP protein level [105]. Indeed, AMPK, phosphorylates TXNIP leading to its rapid degradation [101].
While extensive study regarding the important role of TXNIP during hyperglycemia was reported; the role of TXNIP in inflammasome NLRP3 activation and IL-1β release requires further investigations to clearly highlight its implication. It has been shown that IL-1β signals in an autocrine and/or paracrine manner triggering β-cells dysfunction. Furthermore, it aggravates the local inflammatory environment through the secretion of other proinflammatory cytokines and chemotactic factors that drive infiltration of immune cells into pancreatic islets [106]. Elevated levels of glucose induce expression of TXNIP leading to an enhanced production of IL-1β in pancreatic islets [100] as well as human and mouse adipose tissue [107], but not in macrophages [100]. The role of the NLRP3 inflammasome in β-cell dysfunction is illustrated in Fig. 4.
Fig. 4.
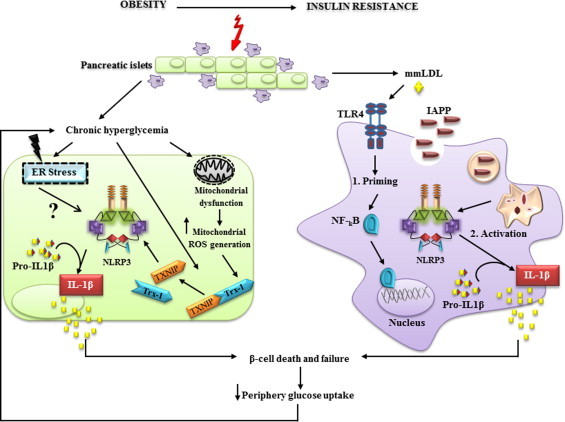
Inflammasome is a central player in the induction of β-cell death and T2DM progression. Chronic exposure of pancreatic β-cells to elevated concentrations of glucose promotes TXNIP expression, endoplasmic reticulum stress and accumulation of dysfunctional mitochondria leading to intracellular ROS accumulation. ROS generation drives NLRP3 activation in a TXNIP-dependent manner. However, ER activation pathway(s) remain poorly understood. IAPP is deposited in pancreas and quickly taken up by macrophages. 1. mmLDL primes cells through TLR4 signaling and 2. IAPP specifically activates NLRP3 inflammasome and pro-IL-1β cleavage. IL1-β signaling induces a local proinflammatory environment through activation of other chemotactic factors and immune cell infiltration that aggravates β-cell failure.
Islet amyloid polypeptide (IAPP) also known as amylin, is deposited in the islet interstitium of T2DM individuals and is tightly correlated with the disease severity and β-cell function [108]. The 37-amino-acid polypeptide amylin is a hormone co-secreted with insulin [108], which requires ROS to trigger β-cell apoptosis [109]. It has recently been reported that IAPP triggers the activation of the NLRP3 inflammasome when mouse pancreatic macrophages were primed by minimally oxidized low-density lipoprotein (mmLDL) [64], which warrants the NF-κB-dependent induction of pro-IL-1β. Under those circumstances, the activation of the inflammasome and the subsequent cleavage of pro-IL-1β into its active form, IL-1β, is based on the phagolysosomal disruption by IAPP amyloid, provided sufficient glucose was present during the priming phase [110].
Inflammasome in cardiovascular diseases
Over the last two decades, the incidence of cardiovascular diseases (CVD) has greatly increased and CVD remain the leading cause of morbidity and mortality [111] with more than 80% of deaths occurring in low- and middle-income countries [112,113]. The cardiovascular system, especially the endothelial tissue, is highly exposed to pathogens and is the first barrier to react with PAMPs through activation of cytokines, chemokines and dilator hormones [114]. Vascular smooth muscle cells [115], cardiomyocytes and heart resident fibroblasts play also a major role in controlling PAMPs leading to injury and cardiovascular dysfunction [116] via TLRs and/or nucleotide-binding domain and NLRs [117,118].
Hypertension is the most common risk factor for the development of cerebrovascular and cardiovascular diseases. In the year 2000, 26% of all adults worldwide (972 million) were suffering from hypertension and the disease is expected to hit 29.2% of the adult population, i.e. 1.56 billion, in 2025 [119]. Chronic inflammation and oxidative stress are crucial factors in vascular remodeling of large and small arteries [120–123], which is important for both the development and the subsequent complications of hypertension [124,125]. As to oxidative stress, polymorphonuclear leukocytes (PMNL) have been identified as the main producers of ROS in both, hypertensive human subjects as well as in animal models of hypertension [126,127]. As a complex disease, hypertension may have different reasons, but it is nevertheless the predominant underlying risk factor for the metabolic syndrome [128]. Indeed, in patients suffering from hypertension, the NLRP3 gene is frequently mutated, which could explain the high correlation of hypertension with the development of insulin resistance in T2DM [129].
Fibrinogen is essential for the maintenance of hemostasis [130] and high plasma fibrinogen concentrations are a risk factor for cardiovascular disease. Of note, single nucleotide polymorphisms in the inflammatory NLRP3 gene locus are highly concordant with the fibrinogen phenotype [131]. IL-1β and IL-18 are major mediators of ischemia/reperfusion (I/R)-induced human myocardial injury [132]. Their levels are drastically increased in response to several mediators relevant to injury such as host-derived DNA, RNA or particles and under activation of NLRP3 [132]. Therefore, the NLRP3 inflammasome is considered as an initial sensor for danger signal(s) in myocardial I/R injury. Indeed, the inflammasome activator ASC is markedly overexpressed in mononuclear cells and neutrophils infiltrated into myocardial tissues of patients that have developed myocardial infarction. As compared to wild-type controls, ASC−/− and caspase-1−/− mice exhibited a significantly lower heart/body weight ratio, less inflammatory responses, and less cardiac myopathies such as myocardial fibrosis, and less dysfunction after myocardial ischemia and reperfusion (I/R) injury as well as development of myocardial infarction. A pivotal role in the early injuries occurring after myocardial I/R has been established for infiltrating leukocytes, but likewise for the activation of the NLRP3 inflammasome in cardiac fibroblasts. The activation of the NLRP3 inflammasome in this setting is dependent on ROS production [133]. In analogy to the situation after myocardial I/R, Nlrp3−/− mice subjected to renal I/R are similarly protected from injury and retain their renal function independent from ASC and caspase-1 proteins [134]. Besides myocardial and kidney I/R injury, NLRP3 signaling is causally involved in hepatic I/R damage and NLRP3 gene silencing prevents this injury based on down-regulation of caspase-1 activation and NF-κB activity [135].
The majority of cardiovascular diseases results from complications of atherosclerosis [136,137], a complex and multifactorial disease implicated in 50% of deaths occurring in developed countries [138]. For a long time, atherosclerosis has been merely considered as a result of lipid accumulation that obstructs arterial vessel wall [139]. Two centuries ago, Virchow has highlighted inflammation as a central cause of atherosclerosis that has been described as endarteritis deformans [140]. After that and through the discovery of various inflammatory markers, Ross has proposed the concept of inflammation as a response to vascular injury [136,141]. Hence, atherosclerosis is now defined as a chronic inflammatory disorder [142], in which inflammation characterizes all phases of the pathogenic process including formation, progression and rupture of atherosclerotic plaques [143,144]. Immune cells (macrophages and T cells), foam cells, vascular endothelial cells, smooth muscle cells (SMCs), platelets, extracellular matrix and a lipid rich core with extensive necrosis and fibrosis of surrounding tissues constitute the basic elements of atherosclerotic plaques [136,137].
Oxidation of low density lipoprotein (oxLDL) is a central step contributing to the progression of atherosclerosis and endothelial dysfunction [145] through an array of pro-atherogenic and proinflammatory properties [146,147]. Endothelial cells, SMCs and macrophages are the sources of oxidants implicated in the oxidative modification of phospholipids [148]. The atherogenic process is initiated by recruitment of LDL cholesterol into the arterial wall leading to inflammatory injury and the excess of cholesterol is partially deposited as cholesterol crystals [149,150]. Crystallographic studies revealed that cholesterol monohydrate is the predominant cholesterol species in human atherosclerotic plaques [151]. An abundance of large extracellular cholesterol crystals can be detected as ‘cholesterol crystal clefts’ in advanced atherosclerotic lesions. In addition to these tissue clefts, smaller cholesterol crystals are frequently observed inside immune cells and in the extracellular space [152,153]. By using confocal reflection microscopy, Rajamaki et al. showed that, in vitro, human macrophages are able to fully internalize the crystals by phagocytosis despite their relatively large size. This phagocytosis leads to accumulation of cellular cholesteryl esters in macrophages [154]. Due to the intimate relationship between IL-18 [155,156] and IL-1β [157,158] levels and atherosclerosis severity, cholesterol crystals activate the NLRP3 inflammasome through a mechanism that involves potassium efflux and phago-lysosomal damage [154]. In human [154] and murine immune cells [159], the activation of the NLRP3 inflammasome requires a priming step probably provided by a modified form of LDL [64,159], which stimulates an NF-κB dependent proinflammatory cascade through a receptor complex involving either TLR4/6 homodimer and CD14 [159], or numerous scavenger receptors like CD36 and scavenger receptor A [160,161].
Myeloid differentiation primary response protein 88 and the kinase activity of IL-1β receptor-associated kinase-4 have also been implicated in the NLRP3 activation mechanism [162,163]. Cholesterol crystals phagocytosed by macrophages induce lysosomal damage [154] leading to translocation of phago-lysosomal content into the cytosol. The mechanism of NLRP3 activation involves leakage of cathepsin B and L into the cytoplasm [159], lowering intracellular potassium concentrations [154] and/or ROS formation [44]. These intermediate steps seem to be important to activate the NLRP3 receptor by as yet unknown processes [164,165]. In murine primary macrophages, Menu et al. have shown that 7-ketocholesterol (oxidative cholesterol derivative) is the major component of oxLDL present in atherosclerotic plaques that enhances expression of the processed form of caspase-1 in an NLRP3 inflammasome-dependent manner.
The role of ER stress in cardiovascular diseases has not been extensively studied. However, there is increasing evidence suggesting that ER stress impairs endothelial dysfunction through initiation of oxidative stress, inflammation and endothelium apoptosis. Therefore, ER stress appears to be important in the pathogenesis of atherosclerosis and myocardium ischemia–reperfusion [166]. Recent data suggest that TXNIP is required for NLRP3 inflammasome activation and the release of IL-1β in endothelial cells in mice fed an HFD causing oxidative and inflammatory stress, obesity and retinal microvascular degeneration [167]. Although TXNIP/NLRP3 inflammasome activation implicated in the endothelial dysfunction, is regulated by AMPK activity. This anti-inflammatory factor is highly implicated in the maintenance of endothelial homeostasis, the activation of endothelial nitric oxide synthase, the induction of endothelial NO production and the protection of endothelial function against oxidative stress [168]. It has been shown that AMPK acts as an ER stress suppressor and its resulting inflammation in vascular endothelium. Indeed, AMPK inhibits TXNIP activity by promoting its degradation [101] leading to the maintenance of endothelial homeostasis and the setting of ER stress. Regarding these findings, TXNIP appears to play a key molecule linking ER stress with endothelium dysfunction event, leading to inflammatory cells infiltration and atherosclerosis initiation. In vivo studies based on double deficient-mice ApoE−/−/Nlrp3−/−, ApoE−/−/Asc−/− and ApoE−/−/caspase-1−/−, have shown no differences between double knockouts and wild-type mice in the abundance of cholesterol crystals in plaques, plaque macrophage infiltration, atherosclerosis progression and plaque stability [169]. Thus, no influence has been detected of Nlrp3, ASC or caspase-1 deficiency on the atherogenic process in the ApoE mouse model. Compared with ApoE−/−/IL-1β+/+, ApoE−/−/IL-1β−/− mice showed a significant decrease in the size of atherosclerotic lesions in the aortic sinus and in the percentage of the atherosclerotic area to total aortic area up to a 30% [170]. In addition, lacking both ApoE and IL-18 in atherosclerosis mouse models reduced the lesion size assessed in the aortic root of the offspring [171] and exogenous IL-18 administration to ApoE−/− mice promoted a 2-fold increase in the lesion size in both the ascending aorta and the aortic arch [172]. These data point to an important role of the NLRP3 activation for the development of atherosclerosis.
Compared to wild-type, in C57BL/6 IL-1α−/− and C57BL/6 IL-1β−/− mice, the lesion area in aortic sinus was decreased on average by 56% and 50%, respectively. In addition, aortic sinus lesion area in mice transplanted with IL-1α−/− bone marrow cells was 59% lower than IL-1α/β+/+ transplanted mice. However the difference in lesion area between IL-1β−/− and IL-1α/β+/+ transplanted mice was not statistically significant [173]. Thus, the active IL-1α form, which is synthetized through an NLRP3 inflammasome-independent manner via a calpain-mediated mechanism contributed mostly to the pathogenesis of atherosclerosis in ApoE−/− mice [174].
According to Duewell et al., the NLRP3 inflammasome activation by bone marrow derived cells presents the major contributor to murine atherosclerosis. Indeed, irradiated LDL Receptor (LDLR)−/− mice transplanted with bone marrow cells from Nlrp3−/−, Asc−/− or IL-1β−/−/IL-1α−/− mice displayed significantly reduced plasma IL-18 levels as well as decreased total lesion size at the aortic sinus (69%) compared to LDLR−/− mice reconstituted with wild type bone marrow [159]. The discrepant results obtained in these two studies, are not quite surprising. In fact, it has been reported that ApoE−/− mice are more hypercholesterolemic than LDLR−/− mice, even when given the same atherogenic diet [169]. Thus, besides the specific genetic modification of the mouse model, the development of atherosclerosis is also influenced by the choice and the duration of the high-fat diet [175]. Another difference between the two studies is deficiency of the NLRP3 inflammasome components only in bone marrow cells versus whole body double knockout of the inflammasome components [159].
Another study implicated the oxidative stress-responsive transcription factor NF-E2-related 2 (Nrf2) in NRLP3 activation and atherosclerosis development in ApoE−/− mice [176]. In this model, atherosclerotic lesion size was about 50% lower in Nrf2−/−/ApoE−/− mice than in heterogzygous Nrf2+/−/ApoE−/− mice. In addition, expression of bioactive IL-1α and IL-1β pro-inflammatory cytokines was completely abrogated after exposure of Nrf2-deficient macrophages to cholesterol crystals. Taken together, cholesterol crystals act as an endogenous pro-atherogenic danger signal that triggers and sustains vascular inflammation in Nrf2-dependent pathway. Induction of the Nrf2 signaling is required to initiate the atherogenic effects of IL-1β and IL-1α in an NLRP3/caspase-1-dependent as well as -independent manner [176].
Thus, although the NLRP3 inflammasome is an important source of the active form of IL-1β and IL-18, other NLRP3-independent processes yielding those cytokines have also been implicated in the pathogenesis of atherosclerosis [169]. Evidently, further researches are needed to clarify the exact implications of the inflammasome activation and the associated production of the proinflammatory cytokines IL-1β/IL-18 for the atherogenic process.
In summary, despite the increased knowledge gained over the past decade, many aspects of the biology of the host response to danger signals via the NLRP3 inflammasome are still not well defined or even completely unknown. Therefore, a thorough investigation of the molecular mechanisms underlying the NLRP3 inflammasome assembly and activation will be crucial to elucidate these aspects. In addition, given the implications of NLRP3 in several sterile immunopathologies, design and development of novel anti-inflammatory drugs specifically targeting the NLRP3 inflammasome is highly desirable and anxiously awaited.
References
- 1.Janeway C.A., Jr. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunology Today. 1992;13(1):11–16. doi: 10.1016/0167-5699(92)90198-G. 1739426 [DOI] [PubMed] [Google Scholar]
- 2.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296(5566):301–305. doi: 10.1126/science.1071059. 11951032 [DOI] [PubMed] [Google Scholar]
- 3.Martinon F., Tschopp J. NLRs join TLRs as innate sensors of pathogens. Trends in Immunology. 2005;26(8):447–454. doi: 10.1016/j.it.2005.06.004. 15967716 [DOI] [PubMed] [Google Scholar]
- 4.Martinon F., Burns K., Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular Cell. 2002;10(2):417–426. doi: 10.1016/s1097-2765(02)00599-3. 12191486 [DOI] [PubMed] [Google Scholar]
- 5.Dinarello C.A. A clinical perspective of IL-1beta as the gatekeeper of inflammation. European Journal of Immunology. 2011;41(5):1203–1217. doi: 10.1002/eji.201141550. 21523780 [DOI] [PubMed] [Google Scholar]
- 6.Leemans J.C., Cassel S.L., Sutterwala F.S. Sensing damage by the NLRP3 inflammasome. Immunological Reviews. 2011;243(1):152–162. doi: 10.1111/j.1600-065X.2011.01043.x. 21884174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masters S.L., Simon A., Aksentijevich I. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*) Annual Review of Immunology. 2009;27:621–668. doi: 10.1146/annurev.immunol.25.022106.141627. 19302049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dinarello C.A. IL-1: discoveries, controversies and future directions. European Journal of Immunology. 2010;40(3):599–606. doi: 10.1002/eji.201040319. 20201008 [DOI] [PubMed] [Google Scholar]
- 9.Strowig T., Henao-Mejia J., Elinav E. Inflammasomes in health and disease. Nature. 2012;481(7381):278–286. doi: 10.1038/nature10759. 22258606 [DOI] [PubMed] [Google Scholar]
- 10.De Alba E. Structure and interdomain dynamics of apoptosis-associated speck-like protein containing a CARD (ASC) Journal of Biological Chemistry. 2009;284(47):32932–32941. doi: 10.1074/jbc.M109.024273. 19759015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gross O., Thomas C.J., Guarda G. The inflammasome: an integrated view. Immunological Reviews. 2011;243(1):136–151. doi: 10.1111/j.1600-065X.2011.01046.x. 21884173 [DOI] [PubMed] [Google Scholar]
- 12.Masumoto J., Dowds T.A., Schaner P. ASC is an activating adaptor for NF-kappa B and caspase-8-dependent apoptosis. Biochemical and Biophysical Research Communications. 2003;303(1):69–73. doi: 10.1016/s0006-291x(03)00309-7. 12646168 [DOI] [PubMed] [Google Scholar]
- 13.Ohtsuka T., Ryu H., Minamishima Y.A. ASC is a Bax adaptor and regulates the p53-Bax mitochondrial apoptosis pathway. Nature Cell Biology. 2004;6(2):121–128. doi: 10.1038/ncb1087. 14730312 [DOI] [PubMed] [Google Scholar]
- 14.Richards N., Schaner P., Diaz A. Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. Journal of Biological Chemistry. 2001;276(42):39320–39329. doi: 10.1074/jbc.M104730200. 11498534 [DOI] [PubMed] [Google Scholar]
- 15.Masumoto J., Taniguchi S., Ayukawa K. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. Journal of Biological Chemistry. 1999;274(48):33835–33838. doi: 10.1074/jbc.274.48.33835. 10567338 [DOI] [PubMed] [Google Scholar]
- 16.Masumoto J., Taniguchi S., Nakayama J. Expression of apoptosis-associated speck-like protein containing a caspase recruitment domain, a pyrin N-terminal homology domain-containing protein, in normal human tissues. Journal of Histochemistry and Cytochemistry. 2001;49(10):1269–1275. doi: 10.1177/002215540104901009. 11561011 [DOI] [PubMed] [Google Scholar]
- 17.Srinivasula S.M., Poyet J.L., Razmara M. The pyrin-CARD protein ASC is an activating adaptor for caspase-1. Journal of Biological Chemistry. 2002;277(24):21119–21122. doi: 10.1074/jbc.C200179200. 11967258 [DOI] [PubMed] [Google Scholar]
- 18.Fernandes-Alnemri T., Wu J., Yu J.W. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death and Differentiation. 2007;14(9):1590–1604. doi: 10.1038/sj.cdd.4402194. 17599095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen G.M. Caspases: the executioners of apoptosis. Biochemical Journal. 1997;326(1):1–16. doi: 10.1042/bj3260001. 9337844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinon F., Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117(5):561–574. doi: 10.1016/j.cell.2004.05.004. 15163405 [DOI] [PubMed] [Google Scholar]
- 21.Nadiri A., Wolinski M.K., Saleh M. The inflammatory caspases: key players in the host response to pathogenic invasion and sepsis. Journal of Immunology. 2006;177(7):4239–4245. doi: 10.4049/jimmunol.177.7.4239. 16982854 [DOI] [PubMed] [Google Scholar]
- 22.Martinon F., Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death and Differentiation. 2007;14(1):10–22. doi: 10.1038/sj.cdd.4402038. 16977329 [DOI] [PubMed] [Google Scholar]
- 23.Saleh M., Vaillancourt J.P., Graham R.K. Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature. 2004;429(6987):75–79. doi: 10.1038/nature02451. 15129283 [DOI] [PubMed] [Google Scholar]
- 24.Park H.H., Wu H. Crystal structure of RAIDD death domain implicates potential mechanism of PIDDosome assembly. Journal of Molecular Biology. 2006;357(2):358–364. doi: 10.1016/j.jmb.2005.12.082. 16434054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park H.H., Lo Y.C., Lin S.C. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annual Review of Immunology. 2007;25:561–586. doi: 10.1146/annurev.immunol.25.022106.141656. 17201679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vajjhala P.R., Mirams R.E., Hill J.M. Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. Journal of Biological Chemistry. 2012;287(50):41732–41743. doi: 10.1074/jbc.M112.381228. 23066025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liepinsh E., Barbals R., Dahl E. The death-domain fold of the ASC pyrin domain, presenting a basis for pyrin/pyrin recognition. Journal of Molecular Biology. 2003;332(5):1155–1163. doi: 10.1016/j.jmb.2003.07.007. 14499617 [DOI] [PubMed] [Google Scholar]
- 28.Bae J.Y., Park H.H. Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammasome assembly. Journal of Biological Chemistry. 2011;286(45):39528–39536. doi: 10.1074/jbc.M111.278812. 21880711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taxman D.J., Huang M.T., Ting J.P. Inflammasome inhibition as a pathogenic stealth mechanism. Cell Host Microbe. 2010;8(1):7–11. doi: 10.1016/j.chom.2010.06.005. 20638636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stehlik C., Dorfleutner A. COPs and POPs: modulators of inflammasome activity. Journal of Immunology. 2007;179(12):7993–7998. doi: 10.4049/jimmunol.179.12.7993. 18056338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stehlik C., Krajewska M., Welsh K. The PAAD/PYRIN-only protein POP1/ASC2 is a modulator of ASC-mediated nuclear-factor-kappa B and pro-caspase-1 regulation. Biochemical Journal. 2003;373(1):101–113. doi: 10.1042/BJ20030304. 12656673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dorfleutner A., Bryan N.B., Talbott S.J. Cellular pyrin domain-only protein 2 is a candidate regulator of inflammasome activation. Infection and Immunity. 2007;75(3):1484–1492. doi: 10.1128/IAI.01315-06. 17178784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnston J.B., Barrett J.W., Nazarian S.H. A poxvirus-encoded pyrin domain protein interacts with ASC-1 to inhibit host inflammatory and apoptotic responses to infection. Immunity. 2005;23(6):587–598. doi: 10.1016/j.immuni.2005.10.003. 16356857 [DOI] [PubMed] [Google Scholar]
- 34.Schroder K., Tschopp J. The inflammasomes. Cell. 2010;140(6):821–832. doi: 10.1016/j.cell.2010.01.040. 20303873 [DOI] [PubMed] [Google Scholar]
- 35.Kanneganti T.D., Body-Malapel M., Amer A. Critical role for cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. Journal of Biological Chemistry. 2006;281(48):36560–36568. doi: 10.1074/jbc.M607594200. 17008311 [DOI] [PubMed] [Google Scholar]
- 36.Kanneganti T.D., Ozören N., Body-Malapel M. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440(7081):233–236. doi: 10.1038/nature04517. 16407888 [DOI] [PubMed] [Google Scholar]
- 37.Sutterwala F.S., Ogura Y., Szczepanik M. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24(3):317–327. doi: 10.1016/j.immuni.2006.02.004. 16546100 [DOI] [PubMed] [Google Scholar]
- 38.Duncan J.A., Gao X., Huang M.T. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. Journal of Immunology. 2009;182(10):6460–6469. doi: 10.4049/jimmunol.0802696. 19414800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Paolo N.C., Miao E.A., Iwakura Y. Virus binding to a plasma membrane receptor triggers interleukin-1alpha-mediated proinflammatory macrophage response in vivo. Immunity. 2009;31(1):110–121. doi: 10.1016/j.immuni.2009.04.015. 19576795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muruve D.A., Pétrilli V., Zaiss A.K. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008;452(7183):103–107. doi: 10.1038/nature06664. 18288107 [DOI] [PubMed] [Google Scholar]
- 41.Martinon F., Agostini L., Meylan E. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Current Biology. 2004;14(21):1929–1934. doi: 10.1016/j.cub.2004.10.027. [DOI] [PubMed] [Google Scholar]
- 42.Hoegen T., Tremel N., Klein M. The NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis and is activated through ATP-dependent lysosomal cathepsin B release. Journal of Immunology. 2011;187(10):5440–5451. doi: 10.4049/jimmunol.1100790. 22003197 [DOI] [PubMed] [Google Scholar]
- 43.Mariathasan S., Weiss D.S., Newton K. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440(7081):228–232. doi: 10.1038/nature04515. 16407890 [DOI] [PubMed] [Google Scholar]
- 44.Dostert C., Pétrilli V., Van Bruggen R. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320(5876):674–677. doi: 10.1126/science.1156995. 18403674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hornung V., Bauernfeind F., Halle A. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nature Immunology. 2008;9(8):847–856. doi: 10.1038/ni.1631. 18604214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lunov O., Syrovets T., Loos C. Amino-functionalized polystyrene nanoparticles activate the NLRP3 inflammasome in human macrophages. ACS Nano. 2011;5(12):9648–9657. doi: 10.1021/nn203596e. 22111911 [DOI] [PubMed] [Google Scholar]
- 47.Kool M., Pétrilli V., De Smedt T. Cutting edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. Journal of Immunology. 2008;181(6):3755–3759. doi: 10.4049/jimmunol.181.6.3755. 18768827 [DOI] [PubMed] [Google Scholar]
- 48.Feldmeyer L., Keller M., Niklaus G. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Current Biology. 2007;17(13):1140–1145. doi: 10.1016/j.cub.2007.05.074. 17600714 [DOI] [PubMed] [Google Scholar]
- 49.Martinon F., Pétrilli V., Mayor A. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–241. doi: 10.1038/nature04516. 16407889 [DOI] [PubMed] [Google Scholar]
- 50.Yamasaki K., Muto J., Taylor K.R. NLRP3/cryopyrin is necessary for interleukin-1beta (IL-1beta) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. Journal of Biological Chemistry. 2009;284(19):12762–12771. doi: 10.1074/jbc.M806084200. 19258328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Halle A., Hornung V., Petzold G.C. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nature Immunology. 2008;9(8):857–865. doi: 10.1038/ni.1636. 18604209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li H., Ambade A., Re F. Cutting edge: necrosis activates the NLRP3 inflammasome. Journal of Immunology. 2009;183(3):1528–1532. doi: 10.4049/jimmunol.0901080. 19596994 [DOI] [PubMed] [Google Scholar]
- 53.Rossol M., Pierer M., Raulien N. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nature Communications. 2012;3:1329. doi: 10.1038/ncomms2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee G.S., Subramanian N., Kim A.I. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492(7427):123–127. doi: 10.1038/nature11588. 23143333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cassel S.L., Eisenbarth S.C., Iyer S.S. The Nalp3 inflammasome is essential for the development of silicosis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(26):9035–9040. doi: 10.1073/pnas.0803933105. 18577586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cruz C.M., Rinna A., Forman H.J. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. Journal of Biological Chemistry. 2007;282(5):2871–2879. doi: 10.1074/jbc.M608083200. 17132626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pétrilli V., Papin S., Dostert C. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death and Differentiation. 2007;14(9):1583–1589. doi: 10.1038/sj.cdd.4402195. 17599094 [DOI] [PubMed] [Google Scholar]
- 58.Gross O., Poeck H., Bscheider M. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. 2009;459(7245):433–436. doi: 10.1038/nature07965. 19339971 [DOI] [PubMed] [Google Scholar]
- 59.Shio M.T., Eisenbarth S.C., Savaria M. Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLOS Pathogens. 2009;5(8):e1000559. doi: 10.1371/journal.ppat.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kowaltowski A.J., de Souza-Pinto N.C., Castilho R.F. Mitochondria and reactive oxygen species. Free Radical Biology and Medicine. 2009;47(4):333–343. doi: 10.1016/j.freeradbiomed.2009.05.004. 19427899 [DOI] [PubMed] [Google Scholar]
- 61.Shimada K., Crother T.R., Karlin J. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401–414. doi: 10.1016/j.immuni.2012.01.009. 22342844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rubartelli A. Redox control of NLRP3 inflammasome activation in health and disease. Journal of Leukocyte Biology. 2012;92(5):951–958. doi: 10.1189/jlb.0512265. [DOI] [PubMed] [Google Scholar]
- 63.Zhou R., Tardivel A., Thorens B. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nature Immunology. 2010;11(2):136–140. doi: 10.1038/ni.1831. 20023662 [DOI] [PubMed] [Google Scholar]
- 64.Masters S.L., Dunne A., Subramanian S.L. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nature Immunology. 2010;11(10):897–904. doi: 10.1038/ni.1935. 20835230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McGonagle D., Savic S., McDermott M.F. The NLR network and the immunological disease continuum of adaptive and innate immune-mediated inflammation against self. Seminars in Immunopathology. 2007;29(3):303–313. doi: 10.1007/s00281-007-0084-1. 17805542 [DOI] [PubMed] [Google Scholar]
- 66.Yu J.R., Leslie K.S. Cryopyrin-associated periodic syndrome: an update on diagnosis and treatment response. Current Allergy and Asthma Reports. 2011;11(1):12–20. doi: 10.1007/s11882-010-0160-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Muckle T.J., Wellsm Urticaria, deafness, and amyloidosis: a new heredo-familial syndrome. Quarterly Journal of Medicine. 1962;31:235–248. 14476827 [PubMed] [Google Scholar]
- 68.Huttenlocher A., Frieden I.J., Emery H. Neonatal onset multisystem inflammatory disease. Journal of Rheumatology. 1995;22(6):1171–1173. 7674249 [PubMed] [Google Scholar]
- 69.Aksentijevich I., D Putnam C., Remmers E.F. The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis & Rheumatology. 2007;56(4):1273–1285. doi: 10.1002/art.22491. 17393462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shinkai K., McCalmont T.H., Leslie K.S. Cryopyrin-associated periodic syndromes and autoinflammation. Clinical and Experimental Dermatology. 2008;33(1):1–9. doi: 10.1111/j.1365-2230.2007.02540.x. 17927785 [DOI] [PubMed] [Google Scholar]
- 71.Agostini L., Martinon F., Burns K. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle–Wells autoinflammatory disorder. Immunity. 2004;20(3):319–325. doi: 10.1016/s1074-7613(04)00046-9. 15030775 [DOI] [PubMed] [Google Scholar]
- 72.Masters S.L., Lobito A.A., Chae J. Recent advances in the molecular pathogenesis of hereditary recurrent fevers. Current Opinion in Allergy and Clinical Immunology. 2006;6(6):428–433. doi: 10.1097/ACI.0b013e3280109b57. 17088647 [DOI] [PubMed] [Google Scholar]
- 73.Goldbach-Mansky R., Dailey N.J., Canna S.W. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. New England Journal of Medicine. 2006;355(6):581–592. doi: 10.1056/NEJMoa055137. 16899778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hawkins P.N., Lachmann H.J., McDermott M.F. Interleukin-1-receptor antagonist in the Muckle–Wells syndrome. New England Journal of Medicine. 2003;348(25):2583–2584. doi: 10.1056/NEJM200306193482523. 12815153 [DOI] [PubMed] [Google Scholar]
- 75.Hoffman H.M., Rosengren S., Boyle D.L. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet. 2004;364(9447):1779–1785. doi: 10.1016/S0140-6736(04)17401-1. 15541451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McDermott M.F., Tschopp J. From inflammasomes to fevers, crystals and hypertension: how basic research explains inflammatory diseases. Trends in Molecular Medicine. 2007;13(9):381–388. doi: 10.1016/j.molmed.2007.07.005. 17822957 [DOI] [PubMed] [Google Scholar]
- 77.Liu J., Smith C.L., DeRyckere D. Structure and function of Cdc6/Cdc18: implications for origin recognition and checkpoint control. Molecular Cell. 2000;6(3):637–648. doi: 10.1016/s1097-2765(00)00062-9. 11030343 [DOI] [PubMed] [Google Scholar]
- 78.Albrecht M., Domingues F.S., Schreiber S. Structural localization of disease-associated sequence variations in the NACHT and LRR domains of PYPAF1 and NOD2. FEBS Letters. 2003;554(3):520–528. doi: 10.1016/s0014-5793(03)01222-5. 14623123 [DOI] [PubMed] [Google Scholar]
- 79.Duncan J.A., Bergstralh D.T., Wang Y. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(19):8041–8046. doi: 10.1073/pnas.0611496104. 17483456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Faustin B., Lartigue L., Bruey J.M. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Molecular Cell. 2007;25(5):713–724. doi: 10.1016/j.molcel.2007.01.032. 17349957 [DOI] [PubMed] [Google Scholar]
- 81.So A., De Smedt T., Revaz S. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Research & Therapy. 2007;9(2):R28. doi: 10.1186/ar2143. 17352828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stahl N., Radin A., Mellis S. Rilonacept − CAPS and beyond. Annals of the New York Academy of Sciences. 2009;1182:124–134. doi: 10.1111/j.1749-6632.2009.05074.x. 20074281 [DOI] [PubMed] [Google Scholar]
- 83.Lachmann H.J., Kone-Paut I., Kuemmerle-Deschner J.B. Use of canakinumab in the cryopyrin-associated periodic syndrome. New England Journal of Medicine. 2009;360(23):2416–2425. doi: 10.1056/NEJMoa0810787. 19494217 [DOI] [PubMed] [Google Scholar]
- 84.Horng T., Hotamisligil G.S. Linking the inflammasome to obesity-related disease. Nature Medicine. 2011;17(2):164–165. doi: 10.1038/nm0211-164. 21297609 [DOI] [PubMed] [Google Scholar]
- 85.Choi A.M., Nakahira K. Dampening insulin signaling by an NLRP3 ‘meta-flammasome’. Nature Immunology. 2011;12(5):379–380. doi: 10.1038/ni.2028. 21502990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vandanmagsar B., Youm Y.H., Ravussin A. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nature Medicine. 2011;17(2):179–188. doi: 10.1038/nm.2279. 21217695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stienstra R., Joosten L.A., Koenen T. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metabolism. 2010;12(6):593–605. doi: 10.1016/j.cmet.2010.11.011. 21109192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stienstra R., van Diepen J.A., Tack C.J. Inflammasome is a central player in the induction of obesity and insulin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(37):15324–15329. doi: 10.1073/pnas.1100255108. 21876127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Youm Y.H., Adijiang A., Vandanmagsar B. Elimination of the NLRP3-ASC inflammasome protects against chronic obesity-induced pancreatic damage. Endocrinology. 2011;152(11):4039–4045. doi: 10.1210/en.2011-1326. 21862613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Henao-Mejia J., Elinav E., Strowig T. Inflammasomes: far beyond inflammation. Nature Immunology. 2012;13(4):321–324. doi: 10.1038/ni.2257. 22430784 [DOI] [PubMed] [Google Scholar]
- 91.Lumeng C.N., Bodzin J.L., Saltiel A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. Journal of Clinical Investigation. 2007;117(1):175–184. doi: 10.1172/JCI29881. 17200717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shah C., Yang G., Lee I. Protection from high fat diet-induced increase in ceramide in mice lacking plasminogen activator inhibitor 1. Journal of Biological Chemistry. 2008;283(20):13538–13548. doi: 10.1074/jbc.M709950200. 18359942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Boden G. Ceramide: a contributor to insulin resistance or an innocent bystander? Diabetologia. 2008;51(7):1095–1096. doi: 10.1007/s00125-008-1015-y. 18458870 [DOI] [PubMed] [Google Scholar]
- 94.Wen H., Gris D., Lei Y. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nature Immunology. 2011;12(5):408–415. doi: 10.1038/ni.2022. 21478880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Spranger J., Kroke A., Möhlig M. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes. 2003;52(3):812–817. doi: 10.2337/diabetes.52.3.812. 12606524 [DOI] [PubMed] [Google Scholar]
- 96.Maedler K., Sergeev P., Ris F. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. Journal of Clinical Investigation. 2002;110(6):851–860. doi: 10.1172/JCI15318. 12235117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Maedler K., Spinas G.A., Lehmann R. Glucose induces beta-cell apoptosis via upregulation of the Fas receptor in human islets. Diabetes. 2001;50(8):1683–1690. doi: 10.2337/diabetes.50.8.1683. 11473025 [DOI] [PubMed] [Google Scholar]
- 98.Morgan D., Oliveira-Emilio H.R., Keane D. Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia. 2007;50(2):359–369. doi: 10.1007/s00125-006-0462-6. 17151863 [DOI] [PubMed] [Google Scholar]
- 99.Nishikawa T., Araki E. Impact of mitochondrial ROS production in the pathogenesis of diabetes mellitus and its complications. Antioxidants & Redox Signaling. 2007;9(3):343–353. doi: 10.1089/ars.2006.1458. 17184177 [DOI] [PubMed] [Google Scholar]
- 100.Zhou F., Gomi M., Fujimoto M. Attenuation of neuronal degeneration in thioredoxin-1 overexpressing mice after mild focal ischemia. Brain Research. 2009;1272:62–70. doi: 10.1016/j.brainres.2009.03.023. 19328186 [DOI] [PubMed] [Google Scholar]
- 101.Wu N., Zheng B., Shaywitz A. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Molecular Cell. 2013;49(6):1167–1175. doi: 10.1016/j.molcel.2013.01.035. 23453806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Abais J.M., Xia M., Zhang Y. Redox Regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxidants & Redox Signaling. 2014 doi: 10.1089/ars.2014.5994. 25330206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mihaylova M.M., Shaw R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nature Cell Biology. 2011;13(9):1016–1023. doi: 10.1038/ncb2329. 21892142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Viollet B., Athea Y., Mounier R. AMPK: lessons from transgenic and knockout animals. Frontiers in Bioscience (Landmark Edition) 2009;14:19–44. doi: 10.2741/3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kawaguchi T., Osatomi K., Yamashita H. Mechanism for fatty acid “sparing” effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. Journal of Biological Chemistry. 2002;277(6):3829–3835. doi: 10.1074/jbc.M107895200. 11724780 [DOI] [PubMed] [Google Scholar]
- 106.Schroder K., Zhou R., Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 2010;327(5963):296–300. doi: 10.1126/science.1184003. 20075245 [DOI] [PubMed] [Google Scholar]
- 107.Koenen T.B., Stienstra R., van Tits L.J. Hyperglycemia activates caspase-1 and TXNIP-mediated IL-1beta transcription in human adipose tissue. Diabetes. 2011;60(2):517–524. doi: 10.2337/db10-0266. 21270263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Westermark P., Andersson A., Westermark G.T. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiological Reviews. 2011;91(3):795–826. doi: 10.1152/physrev.00042.2009. 21742788 [DOI] [PubMed] [Google Scholar]
- 109.Zraika S., Hull R.L., Udayasankar J. Oxidative stress is induced by islet amyloid formation and time-dependently mediates amyloid-induced beta cell apoptosis. Diabetologia. 2009;52(4):626–635. doi: 10.1007/s00125-008-1255-x. 19148619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wen H., Ting J.P., O’Neill L.A. A role for the NLRP3 inflammasome in metabolic diseases − Did Warburg miss inflammation? Nature Immunology. 2012;13(4):352–357. doi: 10.1038/ni.2228. 22430788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lusis A.J. Atherosclerosis. Nature. 2000;407(6801):233–241. doi: 10.1038/35025203. 11001066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fuster V., Kelly B.B., Vedanthan R. Global cardiovascular health: urgent need for an intersectoral approach. Journal of the American College of Cardiology. 2011;58(12):1208–1210. doi: 10.1016/j.jacc.2011.05.038. 21903051 [DOI] [PubMed] [Google Scholar]
- 113.Reddy K.S. Cardiovascular diseases in the developing countries: dimensions, determinants, dynamics and directions for public health action. Public Health Nutrition. 2002;5(1A):231–237. doi: 10.1079/phn2001298. 12027289 [DOI] [PubMed] [Google Scholar]
- 114.Földes G., Liu A., Badiger R. Innate immunity in human embryonic stem cells: comparison with adult human endothelial cells. PLOS One. 2010;5(5):e10501. doi: 10.1371/journal.pone.0010501. 20463927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schultz K., Murthy V., Tatro J.B. Endogenous interleukin-1alpha promotes a proliferative and proinflammatory phenotype in human vascular smooth muscle cells. American Journal of Physiology – Heart and Circulatory Physiology. 2007;292(6):H2927–H2934. doi: 10.1152/ajpheart.00700.2006. 17293495 [DOI] [PubMed] [Google Scholar]
- 116.Garg N.J. Inflammasomes in cardiovascular diseases. American Journal of Cardiovascular Disease. 2011;1(3):244–254. 22254202 [PMC free article] [PubMed] [Google Scholar]
- 117.Hirao K., Yumoto H., Takahashi K. Roles of TLR2, TLR4, NOD2, and NOD1 in pulp fibroblasts. Journal of Dental Research. 2009;88(8):762–767. doi: 10.1177/0022034509341779. 19734466 [DOI] [PubMed] [Google Scholar]
- 118.Boyd J.H., Mathur S., Wang Y. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-kappaB dependent inflammatory response. Cardiovascular Research. 2006;72(3):384–393. doi: 10.1016/j.cardiores.2006.09.011. 17054926 [DOI] [PubMed] [Google Scholar]
- 119.Kearney P.M., Whelton M., Reynolds K. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365(9455):217–223. doi: 10.1016/S0140-6736(05)17741-1. 15652604 [DOI] [PubMed] [Google Scholar]
- 120.Russo C., Olivieri O., Girelli D. Anti-oxidant status and lipid peroxidation in patients with essential hypertension. Journal of Hypertension. 1998;16(9):1267–1271. doi: 10.1097/00004872-199816090-00007. 9746113 [DOI] [PubMed] [Google Scholar]
- 121.Berry C., Brosnan M.J., Fennell J. Oxidative stress and vascular damage in hypertension. Current Opinion in Nephrology and Hypertension. 2001;10(2):247–255. doi: 10.1097/00041552-200103000-00014. 11224701 [DOI] [PubMed] [Google Scholar]
- 122.Romero J.C., Reckelhoff J.F. State-of-the-art lecture. Role of angiotensin and oxidative stress in essential hypertension. Hypertension. 1999;34(4 Pt 2):943–949. doi: 10.1161/01.hyp.34.4.943. 10523389 [DOI] [PubMed] [Google Scholar]
- 123.Wilcox C.S. Reactive oxygen species: roles in blood pressure and kidney function. Current Hypertension Reports. 2002;4(2):160–166. doi: 10.1007/s11906-002-0041-2. 11884272 [DOI] [PubMed] [Google Scholar]
- 124.Intengan H.D., Schiffrin E.L. Vascular remodeling in hypertension: roles of apoptosis, inflammation, and fibrosis. Hypertension. 2001;38(3 Pt 2):581–587. doi: 10.1161/hy09t1.096249. 11566935 [DOI] [PubMed] [Google Scholar]
- 125.Rodríguez-Iturbe B., Vaziri N.D., Herrera-Acosta J. Oxidative stress, renal infiltration of immune cells, and salt-sensitive hypertension: all for one and one for all. American Journal of Physiology – Renal Physiology. 2004;286(4):F606–F616. doi: 10.1152/ajprenal.00269.2003. 15001451 [DOI] [PubMed] [Google Scholar]
- 126.Moreno M.U., San José G., Orbe J. Preliminary characterisation of the promoter of the human p22(phox) gene: identification of a new polymorphism associated with hypertension. FEBS Letters. 2003;542(1–3):27–31. doi: 10.1016/s0014-5793(03)00331-4. 12729892 [DOI] [PubMed] [Google Scholar]
- 127.Sela S., Mazor R., Amsalam M. Primed polymorphonuclear leukocytes, oxidative stress, and inflammation antecede hypertension in the Sabra rat. Hypertension. 2004;44(5):764–769. doi: 10.1161/01.HYP.0000144480.10207.34. 15452031 [DOI] [PubMed] [Google Scholar]
- 128.Scrivo R., Vasile M., Bartosiewicz I. Inflammation as “common soil” of the multifactorial diseases. Autoimmunity Reviews. 2011;10(7):369–374. doi: 10.1016/j.autrev.2010.12.006. 21195808 [DOI] [PubMed] [Google Scholar]
- 129.Omi T., Kumada M., Kamesaki T. An intronic variable number of tandem repeat polymorphisms of the cold-induced autoinflammatory syndrome 1 (CIAS1) gene modifies gene expression and is associated with essential hypertension. European Journal of Human Genetics. 2006;14(12):1295–1305. doi: 10.1038/sj.ejhg.5201698. 16868559 [DOI] [PubMed] [Google Scholar]
- 130.Levy J.H., Szlam F., Tanaka K.A. Fibrinogen and hemostasis: a primary hemostatic target for the management of acquired bleeding. Anesthesia & Analgesia. 2012;114(2):261–274. doi: 10.1213/ANE.0b013e31822e1853. 21965371 [DOI] [PubMed] [Google Scholar]
- 131.Wassel C.L., Lange L.A., Keating B.J. Association of genomic loci from a cardiovascular gene SNP array with fibrinogen levels in European Americans and African-Americans from six cohort studies: the Candidate Gene Association Resource (CARe) Blood. 2011;117(1):268–275. doi: 10.1182/blood-2010-06-289546. 20978265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pomerantz B.J., Reznikov L.L., Harken A.H. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1beta. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(5):2871–2876. doi: 10.1073/pnas.041611398. 11226333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kawaguchi M., Takahashi M., Hata T. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123(6):594–604. doi: 10.1161/CIRCULATIONAHA.110.982777. 21282498 [DOI] [PubMed] [Google Scholar]
- 134.Shigeoka A.A., Mueller J.L., Kambo A. An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia–reperfusion injury. Journal of Immunology. 2010;185(10):6277–6285. doi: 10.4049/jimmunol.1002330. 20962258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhu P., Duan L., Chen J. Gene silencing of NALP3 protects against liver ischemia–reperfusion injury in mice. Human Gene Therapy. 2011;22(7):853–864. doi: 10.1089/hum.2010.145. 21128730 [DOI] [PubMed] [Google Scholar]
- 136.Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874. doi: 10.1038/nature01323. 12490960 [DOI] [PubMed] [Google Scholar]
- 137.Madamanchi N.R., Vendrov A., Runge M.S. Oxidative stress and vascular disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(1):29–38. doi: 10.1161/01.ATV.0000150649.39934.13. [DOI] [PubMed] [Google Scholar]
- 138.Hansson G.K., Libby P. The immune response in atherosclerosis: a double-edged sword. Nature Reviews Immunology. 2006;6(7):508–519. doi: 10.1038/nri1882. 16778830 [DOI] [PubMed] [Google Scholar]
- 139.Libby P. Atherosclerosis: the new view. Scientific American. 2002;286(5):46–55. doi: 10.1038/scientificamerican0502-46. 11951331 [DOI] [PubMed] [Google Scholar]
- 140.Virchow R. Cellular pathology. As based upon physiological and pathological histology. lecture XVI − atheromatous affection of arteries. Nutrition Reviews. 1858;1989(47 (1)):23–25. doi: 10.1111/j.1753-4887.1989.tb02747.x. [DOI] [PubMed] [Google Scholar]
- 141.Ross R. Atherosclerosis is an inflammatory disease. American Heart Journal. 1999;138(5 Pt 2):S419–S420. doi: 10.1016/s0002-8703(99)70266-8. 10539839 [DOI] [PubMed] [Google Scholar]
- 142.Small D.M. George Lyman Duff memorial lecture. Progression and regression of atherosclerotic lesions. Insights from lipid physical biochemistry. Arteriosclerosis. 1988;8(2):103–129. doi: 10.1161/01.atv.8.2.103. 3348756 [DOI] [PubMed] [Google Scholar]
- 143.Libby P., Okamoto Y., Rocha V.Z. Inflammation in atherosclerosis: transition from theory to practice. Circulation Journal. 2010;74(2):213–220. doi: 10.1253/circj.cj-09-0706. 20065609 [DOI] [PubMed] [Google Scholar]
- 144.Van Leuven S.I., Franssen R., Kastelein J.J. Systemic inflammation as a risk factor for atherothrombosis. Rheumatology (Oxford) 2008;47(1):3–7. doi: 10.1093/rheumatology/kem202. 17702769 [DOI] [PubMed] [Google Scholar]
- 145.Steinberg D. Low density lipoprotein oxidation and its pathobiological significance. Journal of Biological Chemistry. 1997;272(34):20963–20966. doi: 10.1074/jbc.272.34.20963. 9261091 [DOI] [PubMed] [Google Scholar]
- 146.Berliner J.A., Watson A.D. A role for oxidized phospholipids in atherosclerosis. New England Journal of Medicine. 2005;353(1):9–11. doi: 10.1056/NEJMp058118. 16000351 [DOI] [PubMed] [Google Scholar]
- 147.Bochkov V.N., Oskolkova O.V., Birukov K.G. Generation and biological activities of oxidized phospholipids. Antioxidants & Redox Signaling. 2010;12(8):1009–1059. doi: 10.1089/ars.2009.2597. 19686040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Vora D.K., Fang Z.T., Liva S.M. Induction of p-selectin by oxidized lipoproteins. separate effects on synthesis and surface expression. Circulation Research. 1997;80(6):810–818. doi: 10.1161/01.res.80.6.810. 9168783 [DOI] [PubMed] [Google Scholar]
- 149.Binder C.J., Chang M.K., Shaw P.X. Innate and acquired immunity in atherogenesis. Nature Medicine. 2002;8(11):1218–1226. doi: 10.1038/nm1102-1218. 12411948 [DOI] [PubMed] [Google Scholar]
- 150.Hansson G.K. Inflammation, atherosclerosis, and coronary artery disease. New England Journal of Medicine. 2005;352(16):1685–1695. doi: 10.1056/NEJMra043430. 15843671 [DOI] [PubMed] [Google Scholar]
- 151.Bogren H., Larsson K. An x-ray-diffraction study of crystalline cholesterol in some pathological deposits in man. Biochimica et Biophysica Acta. 1963;75:65–69. doi: 10.1016/0006-3002(63)90580-8. [DOI] [PubMed] [Google Scholar]
- 152.Klinkner A.M., Waites C.R., Kerns W.D. Evidence of foam cell and cholesterol crystal formation in macrophages incubated with oxidized LDL by fluorescence and electron microscopy. Journal of Histochemistry and Cytochemistry. 1995;43(10):1071–1078. doi: 10.1177/43.10.7560885. 7560885 [DOI] [PubMed] [Google Scholar]
- 153.Tangirala R.K., Jerome W.G., Jones N.L. Formation of cholesterol monohydrate crystals in macrophage-derived foam cells. Journal of Lipid Research. 1994;35(1):93–104. 8138726 [PubMed] [Google Scholar]
- 154.Rajamäki K., Lappalainen J., Oörni K. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLOS One. 2010;5(7):e11765. doi: 10.1371/journal.pone.0011765. 20668705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Blankenberg S., Tiret L., Bickel C. Interleukin-18 is a strong predictor of cardiovascular death in stable and unstable angina. Circulation. 2002;106(1):24–30. doi: 10.1161/01.cir.0000020546.30940.92. 12093765 [DOI] [PubMed] [Google Scholar]
- 156.Mallat Z., Corbaz A., Scoazec A. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation. 2001;104(14):1598–1603. doi: 10.1161/hc3901.096721. 11581135 [DOI] [PubMed] [Google Scholar]
- 157.Galea J., Armstrong J., Gadsdon P. Interleukin-1beta in coronary arteries of patients with ischemic heart disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 1996;16(8):1000–1006. doi: 10.1161/01.atv.16.8.1000. 8696938 [DOI] [PubMed] [Google Scholar]
- 158.Moyer C.F., Sajuthi D., Tulli H. Synthesis of IL-1 alpha and IL-1beta by arterial cells in atherosclerosis. American Journal of Pathology. 1991;138(4):951–960. 2012178 [PMC free article] [PubMed] [Google Scholar]
- 159.Duewell P., Kono H., Rayner K.J. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–1361. doi: 10.1038/nature08938. 20428172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Suzuki H., Kurihara Y., Takeya M. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature. 1997;386(6622):292–296. doi: 10.1038/386292a0. 9069289 [DOI] [PubMed] [Google Scholar]
- 161.Febbraio M., Podrez E.A., Smith J.D. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. Journal of Clinical Investigation. 2000;105(8):1049–1056. doi: 10.1172/JCI9259. 10772649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Björkbacka H., Kunjathoor V.V., Moore K.J. Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nature Medicine. 2004;10(4):416–421. doi: 10.1038/nm1008. 15034566 [DOI] [PubMed] [Google Scholar]
- 163.Michelsen K.S., Wong M.H., Shah P.K. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(29):10679–10684. doi: 10.1073/pnas.0403249101. 15249654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tschopp J., Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nature Reviews Immunology. 2010;10(3):210–215. doi: 10.1038/nri2725. 20168318 [DOI] [PubMed] [Google Scholar]
- 165.Latz E. The inflammasomes: mechanisms of activation and function. Current Opinion in Immunology. 2010;22(1):28–33. doi: 10.1016/j.coi.2009.12.004. 20060699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Song J., Li J., Hou F. Mangiferin inhibits endoplasmic reticulum stress-associated thioredoxin-interacting protein/NLRP3 inflammasome activation with regulation of AMPK in endothelial cells. Metabolism. 2014 doi: 10.1016/j.metabol.2014.11.008. 25499441 [DOI] [PubMed] [Google Scholar]
- 167.Mohamed I.N., Hafez S.S., Fairaq A. Thioredoxin-interacting protein is required for endothelial NLRP3 inflammasome activation and cell death in a rat model of high-fat diet. Diabetologia. 2014;57(2):413–423. doi: 10.1007/s00125-013-3101-z. 24201577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wu J., Xu X., Li Y. Quercetin, luteolin and epigallocatechin gallate alleviate TXNIP and NLRP3-mediated inflammation and apoptosis with regulation of AMPK in endothelial cells. European Journal of Pharmacology. 2014;745:59–68. doi: 10.1016/j.ejphar.2014.09.046. 25446924 [DOI] [PubMed] [Google Scholar]
- 169.Menu P., Pellegrin M., Aubert J.F. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death & Disease. 2011;2:e137. doi: 10.1038/cddis.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kirii H., Niwa T., Yamada Y. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23(4):656–660. doi: 10.1161/01.ATV.0000064374.15232.C3. 12615675 [DOI] [PubMed] [Google Scholar]
- 171.Elhage R., Jawien J., Rudling M. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovascular Research. 2003;59(1):234–240. doi: 10.1016/s0008-6363(03)00343-2. 12829194 [DOI] [PubMed] [Google Scholar]
- 172.Whitman S.C., Ravisankar P., Daugherty A. Interleukin-18 enhances atherosclerosis in apolipoprotein E(−/−) mice through release of interferon-gamma. Circulation Research. 2002;90(2):E34–E38. doi: 10.1161/hh0202.105292. 11834721 [DOI] [PubMed] [Google Scholar]
- 173.Kamari Y., Werman-Venkert R., Shaish A. Differential role and tissue specificity of interleukin-1alpha gene expression in atherogenesis and lipid metabolism. Atherosclerosis. 2007;195(1):31–38. doi: 10.1016/j.atherosclerosis.2006.11.026. 17173923 [DOI] [PubMed] [Google Scholar]
- 174.Carruth L.M., Demczuk S., Mizel S.B. Involvement of a calpain-like protease in the processing of the murine interleukin 1alpha precursor. Journal of Biological Chemistry. 1991;266(19):12162–12167. 2061304 [PubMed] [Google Scholar]
- 175.De Nardo D., Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends in Immunology. 2011;32(8):373–379. doi: 10.1016/j.it.2011.05.004. 21733753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Freigang S., Ampenberger F., Spohn G. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. European Journal of Immunology. 2011;41(7):2040–2051. doi: 10.1002/eji.201041316. 21484785 [DOI] [PubMed] [Google Scholar]