

Abstract
AIM: To examine the effects of Helicobacter pylori (H pylori) infection on the invasiveness of gastric cancer cells, and to elucidate its mechanism.
METHODS: Gastric carcinoma cells, MKN-45, were incubated with CagA-positive H pylori, and cell invasion was determined by Matrigel analysis. The expression of matrix metallopr-oteinase-9 (MMP-9), vascular endothelial growth factor (VEGF), and cyclooxygenase-2 (COX-2) were assessed by Western-blot analysis, and transcriptional activation of the COX-2 promoter was examined by measuring luciferase and β-galactosidase activities. Lastly, the protein-DNA interaction was confirmed by an electrophoretic mobility shift assay.
RESULTS: The current studies showed that: (1) incubation of CagA-positive H pylori with MKN-45 cells significantly promotes gastric cancer cells invasion, and this effect is attenuated by pre-treatment with NS-398, a COX-2 inhibitor, or PDTC, a nuclear factor κB (NF-κB) inhibitor; (2) the induction of MKN-45 cells invasion by H pylori is associated with increases in COX-2, MMP-9, and VEGF protein expression, and co-incubation of NS-398 or PDTC significantly reduces these effects; (3) H pylori infection transactivates COX-2 promoter activity and increases the binding of NF-κB to this promoter.
CONCLUSION: Our data demonstrate that H pylori infection promotes gastric epithelial cells invasion by activating MMP-9 and VEGF expression. These effects appear to be mediated through a NF-κB and COX-2 mediated pathway, as COX-2 or NF-κB inhibitor significantly attenuate the invasiveness of gastric cancer cells and the expressions of MMP-9 and VEGF protein.
Keywords: H pylori, Gastric cancer, Invasion, MMP-9, VEGF, COX-2, NF-κB
INTRODUCTION
Helicobacter pylori (H pylori) is a spiral, microaerophilic, neutralophilic gram-negative bacterium that colonizes the gastric mucosa in 25-50% and 70-90% of the population in the developed and developing countries, respectively[1]. H pylori is believed to be the major contributing factor to the development of chronic gastritis and peptic ulcer diseases in human, and epidemiological and interventional studies in human as well as in experimental animals strongly suggest that H pylori infection increases the risk of adenocarcinoma in the distal stomach[2,3]. Although H pylori has been demonstrated to be associated with gastric cancer occurrence, whether H pylori promote gastric cancer cells invasion is still unknown.
Upon bacterial infection, host effectors induced by H pylori are likely to contribute to gastric carcinogenesis and tumor invasion. Matrix metalloproteinases (MMPs), a family of closely related enzymes that degrade extracellular matrix (ECM), are considered to be important factors in facilitating tumor invasion and spread[4]. MMPs displays broad and overlapping substrate specificity and collectively and they are capable of degrading the major components of ECM. Furthermore, MMPs are found to play major roles in connective tissue remodeling during pathologic conditions, such as cancer and inflammatory disease. Among these MMPs, matrix metalloproteinase-9 (MMP-9) has been considered to be an important factor in facilitating lymphatic invasion and metastases in early gastric carcinoma[5], and the level of tissue MMP-9 has been shown to be related to the overall survival of patients with gastric carcinoma[6]. Recently, MMP-9 has been reported to be induced by H pylori through activation of nuclear factor κB (NF-κB)[7]. Whether H pylori can promote gastric cancer cell invasion through MMP-9 is unknown.
Vascular endothelial growth factor (VEGF), the most well-characterized angiogenic factor, is known to play a major role in the multistep process leading to the reconstruction of normal mucosa architecture. This process is believed to be mediated through angiogenesis, ensuring an adequate supply of nutrients to the healing tissue[8]. Moreover, VEGF also plays a vital role in tumor-associated microvascular invasion[9]. In human gastric cancers, VEGF has been found to be over-expressed[10,11], and in a recent study, VEGF expression has been reported to be upregulated by H pylori through a cyclooxygenase-2 (COX-2) dependent mechanism[12]. Whether VEGF contributes to gastric cancer invasion induced by H pylori infection remains unknown.
Cyclooxygenase (COX), the rate-limiting enzyme in the conversion of arachidonic acid to prostaglandin H2, is the main target of non-steroid anti-inflammatory drugs (NSAIDs). Two isoforms of this enzyme have been identified: COX-1 is constitutively expressed in most tissues and is involved in the production of prostaglandins to maintain normal physiological functions; and COX-2 is involved in inflammation and has been shown to be induced by mitogens, cytokines, hormones, and growth factors. Several recent studies suggested that COX-2 might be an important factor in carcinogenesis, and COX-2 inhibitors were shown to possess anticancer effects. These properties were mediated through the inhibition of prostaglandins production by COX-2, leading to decreases in angiogenic factors, and changes in MMP activity[13]. In human gastric cancer cells, NF-κB mediated COX-2 expression is associated with cell proliferation[14]. Furthermore, H pylori activates NF-κB expression in gastric cancer cells[15].
The present study was undertaken to examine the effect of H pylori infection on gastric cancer cells invasiveness and to elucidate its mechanism. Our results suggest that H pylori may induce the expression of MMP-9 and VEGF and promote gastric cell invasion through a NF-κB-and COX-2-mediated pathway.
MATERIALS AND METHODS
Cell line
Human gastric carcinoma cell line, MKN-45, was obtained from American Type Culture Collection (Manassas, VA, USA). MKN-45 cells were maintained in DMEM medium containing 10% fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin. On the day of experiment, cells were refed with fresh medium and co-cultured with H pylori.
Bacterial strain
Cag pathogenicity island-positive H pylori (ATCC 43504) strain was used in experiments described in this study. Stock cultures were maintained at -70 °C in brucella broth supplemented with 30% glycerol. The bacteria were grown at 37 °C in 5% horse blood agar plates and in a microaerobic condition. Cultures were routinely screened for urease activity. For co-infection studies, H pylori were harvested between 48 and 72 h after inoculation of agar plates, resuspended in sterile phosphate buffered saline (PBS), and enumerated by absorbance at 600 nm (1 optical density (A) at 600 nm = 2.4×108 colony-forming units/mL). MKN-45 cells were seeded into 65 mm dishes, and H pylori at a multiplicity of infection 80 were added in the culture.
Cell migration assays
The effect of H pylori infection on cell migration was examined using the Matrigel Invasion chamber, as suggested by the manufacturer (BD Bioscience). The lower surface of the chamber contained a transwell filter (8-μm pores), coated with fibronectin, and vitronectin. MKN 45 cells (1×105) and H pylori were added to the upper chamber, in the presence or absence of NS-398, and incubated overnight in a humidified tissue culture incubator, at 37 °C, 50 mL/L CO2 atmosphere. The next day, a cotton tipped swab was inserted into chambers to remove non-invading cells by applying gentle but firm pressure while moving the tip around the membrane surface. The cells on the lower surface of insert chambers were stained with hematoxylin for 10 min, and the cell number was counted under a microscope (40 to 200×magnifications). The extent of cell invasion was expressed as fold increases of total number of cells on the lower surface of chambers in treated over untreated samples.
Western blot analysis
To obtain whole-cell extracts, cells were washed twice with ice-cold phosphate-buffer saline (PBS), and pelleted by centrifugation (200 r/min). Cell pellets were then lysed in a standard RIPA buffer (50 mmol/L Tris-HCl, pH 7.5, 150 mmol/L NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS), containing protease inhibitors. Protein concentrations were determined by Bio-Rad assays. Protein samples were dissolved in the loading buffer (60 mmol/L Tris-HCl, pH 6.8, 2% SDS, 100 mmol/L dithiothreitol, and 0.01% bromophenol blue), heated to 100 °C for 3 min, and loaded onto the gel in an electrophoresis buffer containing 25 mmol/L Tris-HCl, pH 8.3, 250 mmol/L glycine, and 0.1% SDS. At the completion of electrophoresis, protein was transferred to a nitrocellulose membrane (Hybond-ECL, Amersham Life Science). The membrane was incubated in the blocking buffer (10 mmol/L Tris, pH 7.5, 100 mmol/L NaCl, 0.1% Tween 20), containing 5% nonfat powdered milk for 2 h. The membrane was immunoblotted with COX-2, MMP-9, or VEGF antiserum (obtained from Santa Cruz Biotech, Santa Cruz, CA, USA). After incubation with the secondary antibody, the membrane was visualized with Enhanced Chemiluminescence kit from Amersham.
Transfection and luciferase assay
To examine transcriptional regulation of COX-2 promoters by NF-κB, MKN-45 cells were transiently transfected with pMT2-LacZ and COX-2-Luc or phPES2 (KBM)-Luc DNAs, in the presence of NF-κB p65, p50, or control pMT2 plasmid (kindly supplied by Dr. Gail Sonenshein, Boston Medical Center, Boston, MA, USA). The phPES2 (KBM) plasmid contains a full-length COX-2 promoter in which a putative NF-κB binding site is mutated, as described previously[16]. For luciferase assays, cells were washed twice with PBS and then lysed in 500 μL of lysis buffer following the manufacturer’s instructions (Analytical Luminescence, San Diego, CA, USA). To assay luciferase activity, cell lysate (100 μL) was mixed with 100 μL of luciferase substrate solution A (Analytical Luminescence). Using a luminometer with automatic injection, 100 μL of luciferase solution B was added (Analytical Luminescence) and luciferase activity was detected as the light emission over a 30-s period.
The β-galactosidase activity in 40 μL of the cell lysate was determined after a 5-30-min incubation at 37 °C with 2 mmol/L chlorophenol red β-galactopyranoside (Boehringer Mannheim) in 2 mmol/L MgCl2, 0.1 mmol/L MnCl2, 45 mmol/L 2-mercaptoethanol, and 100 mmol/L NaHPO4, pH 8.0. The reactions were terminated by adding 500 μL of 0.5 mol/L EDTA, pH 8.0, and the absorbance at 570 nm was measured using a spectrophotometer. With each experiment, the luciferase activity was determined in duplicate and normalized to the β-galactosidase activity for each dish.
Electrophoretic mobility shift assay
Nuclear extracts were prepared using Nuclear Extract kit (Active Motif, Carlsbad, CA, USA). A double-strand oligonucleotide (5’-AGTTGAGGGGACTTTCCCAGGC-3’), corresponding to the putative NF-κB binding domain on the COX-2 promoter was synthesized and was labeled with [γ-32P] ATP (3000 Ci/mmol at 10 mCi/mL) using a T4 polynucleotide kinase. Nuclear protein (1 μg) was incubated in a buffer containing 20% glycerol, 5 mmol/L MgCl2, 2.5 mmol/L EDTA, 2.5 mmol/L DTT, 250 mmol/L NaCl, 50 mmol/L Tris-HCl (pH 7.5), 0.25 mg/mL poly (dl-dC)·poly (dl-dC), for 10 min at room temperature, and 32P-labeled NF-κB oligo was added to each reaction and incubated for additional 20 min. Samples were subjected to electrophoresis at room temperature on a 4% acrylamide gel at 25 mA using 0.5×TBE buffer. The gels were dried at 80 °C for 2 h and exposed to radiography film at -70 °C.
Immunohistochemical studies
To further investigate the role of MMP-9 and VEGF in gastric cancer invasion, the expression of these two proteins were examined in H pylori-positive gastric cancer tissues. Tissue sections from gastric cancer were de-paraffinized, rehydrated, and immersed in 3% hydrogen peroxide–methanol solution for 10 min at room temperature to inhibit endogenous peroxidase activity. The sections were then incubated with unmasking solution (0.01 mol/L citrate buffer pH 6.0), heated for 10 min. The sections were allowed to cool down to room temperature and washed twice in PBS buffer for 5 min. The sections were pre-incubated with diluted normal serum for 10 min, and then incubated with 1:40 mouse anti-MMP-9 monoclonal antibody (Novocastra Lab. Ltd, Newcastle, UK), 1:100 rabbit anti-VEGF polyclonal antibody (Zymed Lab. Inc., South San Francisco, CA, USA), or 1:40 mouse anti-COX-2 monoclonal antibody (Cayman Chemical Co., Ann Arbor, MI, USA) for 1 h at room temperature. The sections were washed in PBS for 5 min twice, and incubated with appropriate biotinylated secondary antibody. After washing in PBS for 5 min twice, the slides were incubated with ABC reagents, followed with DAB or other suitable peroxidase substrates. The sections were counterstained with hematoxylin for 30 s, washed, dried, and mounted.
RESULTS
H pylori infection promotes gastric cancer cells invasion
In vivo tumor invasion includes not only migration process, but also adhesion, proliferation and angiogenesis, etc., dissociation of in vitro assay for tumor cell invasion (migration) with in vivo tumor invasion might exist. However, similar migration assay to assess tumor cell invasion has been used by several articles published recently[17].
We used Matrigel invasion chamber to examine whether H pylori infection induces gastric cancer cells invasion and to determine the effect of COX-2 inhibitor on this process. MKN-45 cells were incubated with H pylori in the presence or absence of COX-2 inhibitor, NS-398 (100 ng/mL). As illustrated in Figure 1A co-infection with H pylori induced a 2.5 fold increase in MKN-45 cells migrated through Matrigel-coated filters, indicating that H pylori promoted gastric cancer cells invasion. The COX-2 inhibitor, NS398, significantly reduced cell invasion in H pylori stimulated, but not in untreated cells. This result suggested that H pylori induced gastric cancer cells invasion was, in part, mediated through a COX-2-dependent mechanism.
Figure 1.
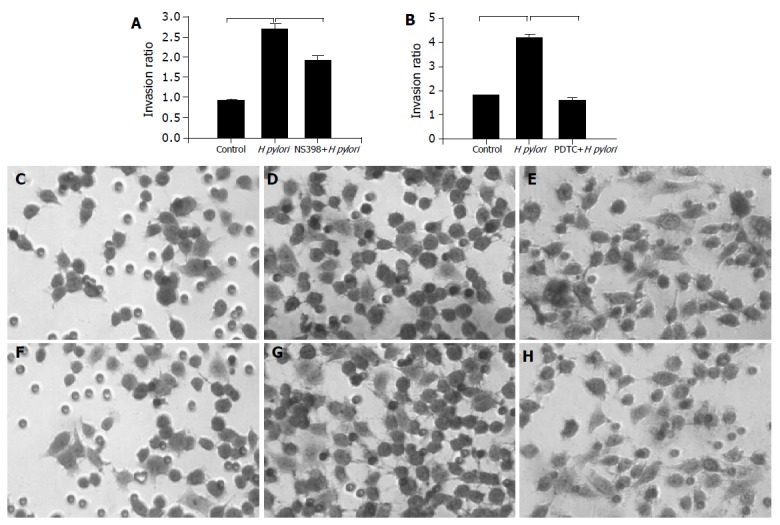
Effects of H pylori infection, a cox-2 (NS398), or a NF-κB inhibitor (PDTC) on gastric cancer cell invasion. A: MKN-45 cells were treated with H pylori in the presence or absence of NS398. Cells on the lower surface of insert chamber were stained with hematoxylin for 10 min and counted under microscope with 200× magnifications. Data are presented as mean±SD of three separate experiments (P<0.05); B: MKN-45 cells were treated with H pylori in the presence or absence of PDTC (P<0.05); microscopic photos of stained migration cells: C: control; D: with H pylori; E: with H pylori and NS-398; F: control; G: with H pylori; H: with H pylori and PDTC.
To examine the effect of NF-κB on the migration of MKN-45 cells, cells were incubated with H pylori in the presence or absence of NF-κB inhibitor, (pyrrolidine dithiocarbamate (PDTC), 0.1 μmol/L, purchased from Sigma Chem, St. Louis, MO, USA). As illustrated in Figure 1B the effect of H pylori infection on gastric cancer cells migration was completely abolished by NF-κB inhibitor, suggesting a potential involvement NF-κB in this process. The representative microscopic photos of stained cells treated with H pylori in the presence or absence of COX-2 inhibitor were shown in Figures 1C-1E.
H pylori infection induces MMP-9 and VEGF expression
Several proteins, including MMP-9 and VEGF, have been reported to play an important role in tumor invasion. The effects of H pylori on the expression level of MMP-9 and VEGF were examined in MKN 45 cells after co-infection with H pylori for a different period of time. The induction of VEGF protein expression was noticeable within an hour after H pylori infection and reached the highest level in 24 h (Figure 2A). Although the increment of MMP-9 level after H pylori infection was smaller than that of VEGF, a significant increase was observed at 24 h (Figure 2A).
Figure 2.
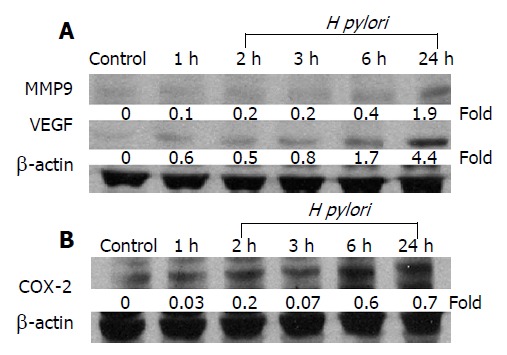
H pylori infection increases MMP-9, VEGF, and COX-2 expressions in gastric epithelial cell. MKN-45 gastric cancer cells were incubated with H pylori for 0-24 h and total cellular protein was extracted for Western blot analysis for the expression of A: MMP-9 and VEGF and B: COX-2 proteins. The blots were stripped and probed with β-actin to document equal protein loading. The experiment was performed for thrice with similar results.
COX-2 and NF-κB inhibitors reduce MMP-9 and VEGF expressions
Recent studies showed that COX-2 inhibitor reduced the release of MMP and COX-2-induced MMP-9 expression[18,19]. Therefore, we examined whether the activation of MMP-9 and VEGF by H pylori infection was also dependent on COX-2 expression. The MKN-45 cells were co-cultured with H pylori for 24 h in the presence or absence of a COX-2 inhibitor, NS-398, and our results showed that co-culture with H pylori resulted in a time-dependent increase in COX-2 protein concentration in MKN-45 cells (Figure 2B). Moreover, NS398 significantly reduced the expression levels of COX-2, MMP-9, and VEGF, induced by H pylori in MKN-45 cells at 24 h (Figure 3A). These data suggest that the induction of MMP-9 and VEGF by H pylori is mediated through a COX-2-dependent mechanism.
Figure 3.
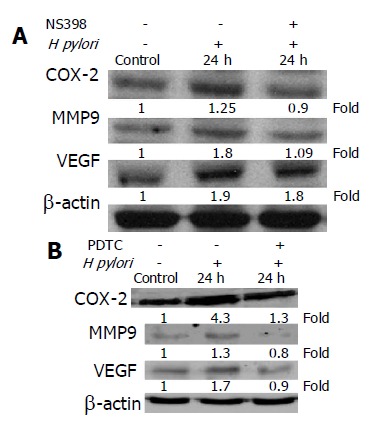
Effect of COX-2 or NF-κB inhibitor on COX-2, MMP-9, or VEGF protein level in MKN-45 cells. A: Cells were cultured in the presence or absence of H pylori and NS398 for 24 h, and cellular protein was extracted and subjected to Western blot analysis; B: MKN-45 cells were treated with or without PDTC and in the presence or absence of H pylori for 24 h. The experiments were repeated on at least three occasions, and the results were identical to these presented here. All blots were stripped and probed with β-actin to check for equal protein loading.
To examine whether activations of MMP-9 and VEGF by H pylori infection were also dependent on NF-κB expression, MKN-45 cells were co-cultured with H pylori for 24 h in the presence or absence of a NF-κB inhibitor, PDTC. As demonstrated in Figure 3B, the effects of H pylori on COX-2, MMP-9, and VEGF expression were significantly reduced by PDTC. These results suggest that the induction of COX-2, MMP-9, and VEGF by H pylori is also NF-κB-dependent.
Induction of MMP-9 and VEGF by H pylori infection depends on a NF-κB-mediated COX-2 activation
A previous study has shown that NF-κB regulated COX-2 expression and affected cell proliferation in human gastric cancer cells[14]. We hypothesize that the induction of MMP-9 and VEGF by H pylori is associated with NF-κB mediated COX-2 expression. To investigate this hypothesis, we first examined the effect of NF-κB on COX-2 promoter activities in MKN45 cells. As illustrated in Figure 4A co-transfection with NF-κB p65 or p50 plasmid DNA significantly enhanced COX-2 promoter activity, and the induction was completely abolished in phPES2 (KBM) construct where a putative NF-κB binding domain was mutated. Furthermore, the interaction between NF-κB and COX-2 promoter was also enhanced by H pylori infection in MKN-45 cells (Figure 4B).
Figure 4.
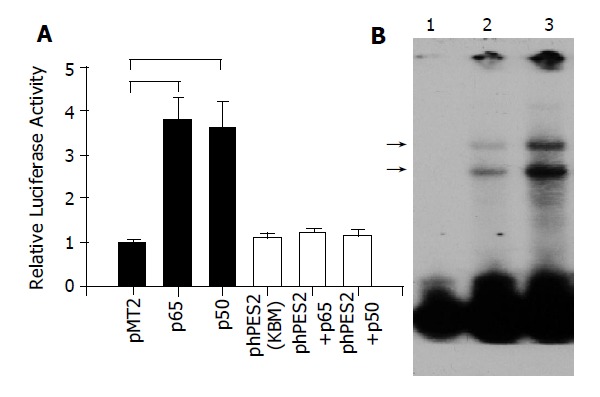
A: Transactivation of COX-2 promoter by NF-κB. MKN-45 cells were transiently transfected with full-length COX-2 promoter (Black bar), or mutated COX-2 construct, phPES2 (KBM), where the putative NF-κB binding domain was mutated (open bar), in the presence of p65, p50, or control vector (pMT2) plasmid. Luciferase and β-galactosidase activities were performed 48 h after transfection. (n = 4, P<0.05); B: Binding of nuclear NF-κB to COX-2 promoter DNA in MKN-45 cells. Nuclear protein was extracted from cells cultured in the absence (lane 2), or presence (lane 3) of H pylori. The protein-DNA interaction bands were shown as (→). Lane 1 showed free probe only.
High level of MMP-9 and VEGF expression in gastric cancer tissues with H pylori infection
To investigate whether these observations were also present in vivo, we randomly selected six gastric cancer patients (three cases with H pylori infection, confirmed by Giemsa stain and CLO test; three cases without H pylori infection) and examined the expression of COX-2, MMP-9, or VEGF protein in the surgical specimens. The immunostain of COX-2 (Figures 5A and 5B), MMP-9 (Figures 5C and 5D), or VEGF (Figures 5E and 5F) was located predominantly on the surface epithelial cells, and the intensity was more abundant in H pylori-positive than H pylori-negative tissue samples (Figure 5).
Figure 5.
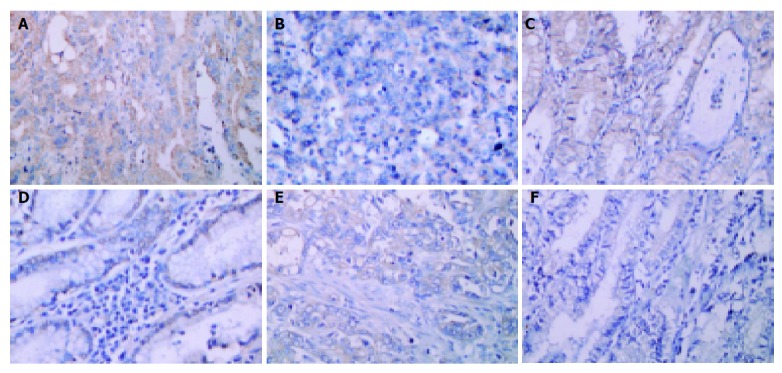
Immunohistochemical detection of COX-2, MMP-9, and VEGF in gastric cancer tissues. The serial sections of gastric cancer surgical specimens were stained with anti-COX-2 (A and B), anti-MMP-9 (C and D), and anti-VEGF (E and F) antibodies. Sections were counterstained with hematoxylin. The immunoreactivities of COX-2, MMP-9 and VEGF are predominantly detected in the cytoplasm of the tumor cell. Tissues in A, C, and E are from H pylori-positive patients, whereas, sections in B, D, and F are from H pylori-negative individuals. (Magnification: A-F: 400×).
DISCUSSION
Gastric cancer is one of the most common malignancies in the world, especially in Eastern Asia. Although the incidence of gastric carcinoma is declining in Western countries, gastric cancer remains the leading cause of cancer death worldwide. Current strategies to reduce mortality from this disease focus on early detection of gastric cancer or its precursor lesions by endoscopic screening. There is an increasing interest in the use of drugs to prevent the occurrence or the invasion of gastric cancers. Epidemiological studies have shown that NSAIDs decrease the risk of gastrointestinal carcinomas[20]. However, the mechanisms by which NSAIDs inhibit neoplastic growth are not fully elucidated[21,22].
The involvement of COX-2 in carcinogenesis has been shown in many epidemiological, animal and clinical studies. Individuals who took NSAIDs regularly had a markedly reduced risk of developing colon cancer[13], and COX-2 inhibitors have been proved to be effective in suppressing tumor progression both in vitro and in vivo in nude mice[21]. The anti-tumor effect of COX-2 inhibitors was attributed to their ability to induce apoptosis and inhibit tumor cell proliferation and angiogenesis. COX-2 inhibitors have also been illustrated to prevent tumor invasion in colon cancer, hepatocellular carcinoma[22], and lung cancer[23]. There is little evidence that COX-2 inhibitors may prevent gastric cancer invasion, although gastric cancers have been shown to over-express COX-2 protein[24].
In the present study, we found that H pylori infection promoted gastric cancer cells invasion and a COX-2 specific inhibitor significantly attenuated this process. Furthermore, the induction of gastric cancer cells invasion is associated with an increase in COX-2, MMP-9, or VEGF protein level, and these effects were also attenuated by a COX-2 inhibitor, suggesting a potential role of MMP-9 or VEGF in this process. In MKN-45 cells, H pylori infection enhanced nuclear NF-κB activity and transactivated COX-2 promoter. In addition, the induction of MMP-9 and VEGF by H pylori was suppressed by a NF-κB inhibitor. These data indicate that H pylori-induced MMP-9 and VEGF expressions in MKN-45 cells are mediated through the interaction of NF-κB on the COX-2 promoter. These results also support an important role of COX-2 in gastric cancer cells invasion.
Lim et al[14], have recently shown that inhibition of NF-κB results in inhibition of COX-2 expression and proliferation of gastric cancer cells. These data suggest NF-κB may play an important role in gastric cancer proliferation via COX-2 expression. Recently, Callejas et al[17], reported that COX-2 expression promotes the release of MMP-9 in fetal rat hepatocytes, and Caputo et al[12], also revealed that H pylori induce VEGF expression in MKN-28 gastric epithelial cells through a COX-2 dependent mechanism. Furthermore, Li et al, reported that COX-2 increased the angiogenic and metastatic potential of tumor cells by activation of VEGF in human transitional cell carcinoma cell line, and the effect on invasiveness could be reversed by COX-2 inhibitors[25-27].
Compared with previous studies, we have found several interesting points. In Figure 1 PDTC attenuated cell invasion completely, NS-398 only partially inhibited, suggesting that NF-κB could induce cell invasion not only through COX-2, but also through other pathways. One of the possibilities was through direct activation of VEGF expression. In Figure 3 we found that the inhibition of COX-2 by NS-398 attenuated MMP-9 expression to the control level, but it did not attenuate VEGF expression completely. On the other hand, the inhibition of NF-κB by PDTC inhibited both MMP-9 and VEGF expression to the control levels. These results suggest that MMP-9 expression is dependent on COX-2 pathway, while VEGF expression might be independent of COX-2 pathway. These observations might explain why NS-398 only partially attenuated cell invasion.
The activation of NF-κB by H pylori has been described by several groups[15,28,29]. Mori et al[7], reported that H pylori induced NF-κB activation and stimulated MMP-9 expression. In the present study, we have observed that H pylori infection in gastric cancer cells induced MMP-9 protein level and the increase was attenuated by either a NF-κB inhibitor or a COX-2 inhibitor. These data suggested that the expression of MMP-9 in H pylori-infected cells is mediated by a direct activation NF-κB, or through a COX-2 mediated pathway. This conclusion is supported by a recent report showing that MMP-9 promoter contains several putative NF-κB binding sites, and its transcription requires the activation of NF-κB.
Infection with H pylori affects more than 50% of the world population; some patients exhibit a progression through chronic atrophic gastritis to cancer, others develop peptic ulcer, but most do not exhibit either disease[30]. It is believed that different pathogens, host and environmental factors may lead to variable outcomes. In this study, we suggest that the induction of MMP-9 and VEGF proteins by H pylori can be considered part of a host response to accelerate an oncogenic progression via disruption of epithelial organization or increased invasion. The identification of H pylori-specific signaling pathways leading to the gastric cancer cells invasion will add to our understanding of the mechanism of H pylori-associated gastric carcinogenesis and the potential use of therapeutic agents in preventing H pylori-associated gastric cancer.
In summary, we have demonstrated that H pylori promote gastric epithelial cells invasion by activating the expression of MMP-9 and VEGF, and these effects are attenuated by a COX-2 or a NF-κB inhibitor. Moreover, H pylori infection induces nuclear NF-κB binding activity to the COX-2 promoter, and the activation of COX-2 promoter is abolished when the NF-κB binding site is mutated. These data suggest that the promotion of gastric cancer cells invasion by H pylori infection appears to be mediated through a NF-κB and COX-2 mediated pathway. Therefore, we proposed a model of H pylori-induced gastric cancer cell invasion as shown in Figure 6.
Figure 6.
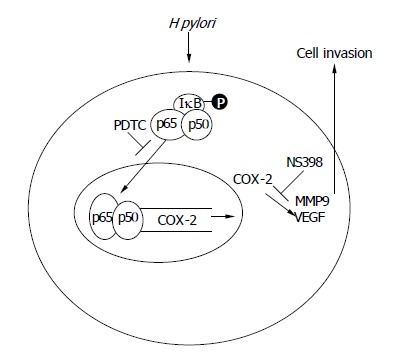
The schematic presentation of proposed mechanism of H pylori promote gastric epithelial cells invasion.
Footnotes
Supported by the Taichung Veterans General Hospital Research Grant: TCVGH-933308C
Co-first-author: Chun-Ying Wu
Co-correspondent: Chi-Chuan Tseng
Science Editor Guo SY Language Editor Elsevier HK
References
- 1.Göõz M, Göõz P, Smolka AJ. Epithelial and bacterial metalloproteinases and their inhibitors in H. pylori infection of human gastric cells. Am J Physiol Gastrointest Liver Physiol. 2001;281:G823–G832. doi: 10.1152/ajpgi.2001.281.3.G823. [DOI] [PubMed] [Google Scholar]
- 2.Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology. 1998;114:1169–1179. doi: 10.1016/s0016-5085(98)70422-6. [DOI] [PubMed] [Google Scholar]
- 3.Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N, Schlemper RJ. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784–789. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- 4.Murray GI, Duncan ME, O'Neil P, Melvin WT, Fothergill JE. Matrix metalloproteinase-1 is associated with poor prognosis in colorectal cancer. Nat Med. 1996;2:461–462. doi: 10.1038/nm0496–461. [DOI] [PubMed] [Google Scholar]
- 5.Kabashima A, Maehara Y, Kakeji Y, Baba H, Koga T, Sugimachi K. Clinicopathological features and overexpression of matrix metalloproteinases in intramucosal gastric carcinoma with lymph node metastasis. Clin Cancer Res. 2000;6:3581–3584. [PubMed] [Google Scholar]
- 6.Sier CF, Kubben FJ, Ganesh S, Heerding MM, Griffioen G, Hanemaaijer R, van Krieken JH, Lamers CB, Verspaget HW. Tissue levels of matrix metalloproteinases MMP-2 and MMP-9 are related to the overall survival of patients with gastric carcinoma. Br J Cancer. 1996;74:413–417. doi: 10.1038/bjc.1996.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mori N, Sato H, Hayashibara T, Senba M, Geleziunas R, Wada A, Hirayama T, Yamamoto N. Helicobacter pylori induces matrix metalloproteinase-9 through activation of nuclear factor kappaB. Gastroenterology. 2003;124:983–992. doi: 10.1053/gast.2003.50152. [DOI] [PubMed] [Google Scholar]
- 8.Jones MK, Tomikawa M, Mohajer B, Tarnawski AS. Gastrointestinal mucosal regeneration: role of growth factors. Front Biosci. 1999;4:D303–D309. doi: 10.2741/a428. [DOI] [PubMed] [Google Scholar]
- 9.Connolly DT, Heuvelman DM, Nelson R, Olander JV, Eppley BL, Delfino JJ, Siegel NR, Leimgruber RM, Feder J. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest. 1989;84:1470–1478. doi: 10.1172/JCI114322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tian X, Song S, Wu J, Meng L, Dong Z, Shou C. Vascular endothelial growth factor: acting as an autocrine growth factor for human gastric adenocarcinoma cell MGC803. Biochem Biophys Res Commun. 2001;286:505–512. doi: 10.1006/bbrc.2001.5409. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto S, Yasui W, Kitadai Y, Yokozaki H, Haruma K, Kajiyama G, Tahara E. Expression of vascular endothelial growth factor in human gastric carcinomas. Pathol Int. 1998;48:499–506. doi: 10.1111/j.1440-1827.1998.tb03940.x. [DOI] [PubMed] [Google Scholar]
- 12.Caputo R, Tuccillo C, Manzo BA, Zarrilli R, Tortora G, Blanco Cdel V, Ricci V, Ciardiello F, Romano M. Helicobacter pylori VacA toxin up-regulates vascular endothelial growth factor expression in MKN 28 gastric cells through an epidermal growth factor receptor-, cyclooxygenase-2-dependent mechanism. Clin Cancer Res. 2003;9:2015–2021. [PubMed] [Google Scholar]
- 13.Church RD, Fleshman JW, McLeod HL. Cyclo-oxygenase 2 inhibition in colorectal cancer therapy. Br J Surg. 2003;90:1055–1067. doi: 10.1002/bjs.4297. [DOI] [PubMed] [Google Scholar]
- 14.Lim JW, Kim H, Kim KH. Nuclear factor-kappaB regulates cyclooxygenase-2 expression and cell proliferation in human gastric cancer cells. Lab Invest. 2001;81:349–360. doi: 10.1038/labinvest.3780243. [DOI] [PubMed] [Google Scholar]
- 15.Maeda S, Akanuma M, Mitsuno Y, Hirata Y, Ogura K, Yoshida H, Shiratori Y, Omata M. Distinct mechanism of Helicobacter pylori-mediated NF-kappa B activation between gastric cancer cells and monocytic cells. J Biol Chem. 2001;276:44856–44864. doi: 10.1074/jbc.M105381200. [DOI] [PubMed] [Google Scholar]
- 16.Inoue H, Nanayama T, Hara S, Yokoyama C, Tanabe T. The cyclic AMP response element plays an essential role in the expression of the human prostaglandin-endoperoxide synthase 2 gene in differentiated U937 monocytic cells. FEBS Lett. 1994;350:51–54. doi: 10.1016/0014-5793(94)00731-4. [DOI] [PubMed] [Google Scholar]
- 17.Ueda J, Kajita M, Suenaga N, Fujii K, Seiki M. Sequence-specific silencing of MT1-MMP expression suppresses tumor cell migration and invasion: importance of MT1-MMP as a therapeutic target for invasive tumors. Oncogene. 2003;22:8716–8722. doi: 10.1038/sj.onc.1206962. [DOI] [PubMed] [Google Scholar]
- 18.Attiga FA, Fernandez PM, Weeraratna AT, Manyak MJ, Patierno SR. Inhibitors of prostaglandin synthesis inhibit human prostate tumor cell invasiveness and reduce the release of matrix metalloproteinases. Cancer Res. 2000;60:4629–4637. [PubMed] [Google Scholar]
- 19.Callejas NA, Casado M, Díaz-Guerra MJ, Boscá L, Martín-Sanz P. Expression of cyclooxygenase-2 promotes the release of matrix metalloproteinase-2 and -9 in fetal rat hepatocytes. Hepatology. 2001;33:860–867. doi: 10.1053/jhep.2001.23002. [DOI] [PubMed] [Google Scholar]
- 20.Huls G, Koornstra JJ, Kleibeuker JH. Non-steroidal anti-inflammatory drugs and molecular carcinogenesis of colorectal carcinomas. Lancet. 2003;362:230–232. doi: 10.1016/s0140-6736(03)13915-3. [DOI] [PubMed] [Google Scholar]
- 21.Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst. 2002;94:252–266. doi: 10.1093/jnci/94.4.252. [DOI] [PubMed] [Google Scholar]
- 22.Chan TA. Nonsteroidal anti-inflammatory drugs, apoptosis, and colon-cancer chemoprevention. Lancet Oncol. 2002;3:166–174. doi: 10.1016/s1470-2045(02)00680-0. [DOI] [PubMed] [Google Scholar]
- 23.Barnes CJ, Cameron IL, Hardman WE, Lee M. Non-steroidol anti-inflammatory drug effect on crypt cell proliferation and apoptosis during initiation of rat colon carcinogenesis. Br J Cancer. 1998;77:573–580. doi: 10.1038/bjc.1998.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koga H. Hepatocellular carcinoma: is there a potential for chemoprevention using cyclooxygenase-2 inhibitors? Cancer. 2003;98:661–667. doi: 10.1002/cncr.11576. [DOI] [PubMed] [Google Scholar]
- 25.Dohadwala M, Luo J, Zhu L, Lin Y, Dougherty GJ, Sharma S, Huang M, Pold M, Batra RK, Dubinett SM. Non-small cell lung cancer cyclooxygenase-2-dependent invasion is mediated by CD44. J Biol Chem. 2001;276:20809–20812. doi: 10.1074/jbc.C100140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang XH, Wong BC. Cyclooxygenase-2 inhibition and gastric cancer. Curr Pharm Des. 2003;9:2281–2288. doi: 10.2174/1381612033453983. [DOI] [PubMed] [Google Scholar]
- 27.Li G, Yang T, Yan J. Cyclooxygenase-2 increased the angiogenic and metastatic potential of tumor cells. Biochem Biophys Res Commun. 2002;299:886–890. doi: 10.1016/s0006-291x(02)02707-9. [DOI] [PubMed] [Google Scholar]
- 28.Gupta RA, Polk DB, Krishna U, Israel DA, Yan F, DuBois RN, Peek RM. Activation of peroxisome proliferator-activated receptor gamma suppresses nuclear factor kappa B-mediated apoptosis induced by Helicobacter pylori in gastric epithelial cells. J Biol Chem. 2001;276:31059–31066. doi: 10.1074/jbc.M104141200. [DOI] [PubMed] [Google Scholar]
- 29.Maeda S, Akanuma M, Mitsuno Y, Hirata Y, Ogura K, Yoshida H, Shiratori Y, Omata M. Distinct mechanism of Helicobacter pylori-mediated NF-kappa B activation between gastric cancer cells and monocytic cells. J Biol Chem. 2001;276:44856–44864. doi: 10.1074/jbc.M105381200. [DOI] [PubMed] [Google Scholar]
- 30.Wada A, Ogushi K, Kimura T, Hojo H, Mori N, Suzuki S, Kumatori A, Se M, Nakahara Y, Nakamura M, et al. Helicobacter pylori-mediated transcriptional regulation of the human beta-defensin 2 gene requires NF-kappaB. Cell Microbiol. 2001;3:115–123. doi: 10.1046/j.1462-5822.2001.00096.x. [DOI] [PubMed] [Google Scholar]