

Abstract
DNA methylation acts as an epigenetic modification in vertebrate DNA. Recently it has become clear that the DNA and histone lysine methylation systems are highly interrelated and rely mechanistically on each other for normal chromatin function in vivo. Here we examine some of the functional links between these systems, with a particular focus on several recent discoveries suggesting how lysine methylation may help to target DNA methylation during development, and vice versa. In addition, the emerging role of non-methylated DNA found in CpG islands in defining histone lysine methylation profiles at gene regulatory elements will be discussed in the context of gene regulation. This article is part of a Special Issue entitled: Methylation: A Multifaceted Modification — looking at transcription and beyond.
Keywords: DNA methylation, Histone lysine methylation, Epigenetics, CpG island, Embryonic development
Graphical abstract
Highlights
-
•
There is an emerging realisation that DNA and histone lysine methylation in mammals are highly interrelated.
-
•
Targeting of DNA methylation is mechanistically linked to H3K9 methylation.
-
•
Uhrf1 acts as a link between H3K9 methylation and maintenance methylation during DNA replication.
-
•
Targeting of Dnmt3a/b is influenced by H3K4 and H3K36 methylation.
-
•
Non-methylated DNA at CpG islands influences histone methylation through ZF-CxxC proteins.
1. Introduction
DNA can be modified by the addition of a methyl group to the 5-position of cytosine. In most vertebrate cell types, cytosine methylation is found in the context of CpG dinucleotides, while recently a much smaller proportion of 5-methylcytosine has been shown to be enzymatically converted to its hydroxymethylated, formylated or carboxylated derivatives [1], [2], [3], [4]. By virtue of the fact that CpG dinucleotides are symmetrically methylated, this functions as an elegant system during semiconservative DNA replication to faithfully copy pre-existing CpG dinucleotide DNA methylation patterns to the daughter strand. This has established DNA methylation as an archetypal example of an epigenetic modification. In general, DNA methylation is thought to contribute to the formation of heterochromatic regions in the genome and transcriptional silencing. In contrast, regions of the genome that are enriched for non-methylated CpGs, called CpG islands, are associated with gene promoters and other regulatory features, where they appear to contribute to a transcriptionally permissive environment [5], [6], [7], [8], [9]. Methylation of these CpG island regions is associated with gene silencing and is a prevalent feature of abnormally silenced genes in cancers.
Within the context of chromatin, DNA methylation does not function in isolation. Instead, there is a complex interplay between DNA methylation and histone modifications, including acetylation, methylation and ubiquitylation. The relationship between DNA methylation and histone lysine methylation is particularly interesting as the two appear to be highly interrelated. For example, there is now emerging evidence that the modification state and sequence of DNA can affect the methylation states on accompanying histones in chromatin, while the histone lysine methylation state of chromatin can in turn influence modification of the DNA itself. Although histone lysine methylation is conserved among eukaryotes, the evolution of DNA methylation is more complex (reviewed in detail by Iyer et al. [8]), being found in some fungi (not including yeast), heterolobosea and stramenopiles, most plants, and some metazoans (including vertebrates, sea urchins, sea anemones and some insects, but not Drosophila or nematodes). Here we will focus mainly on DNA and histone methylation in mammals, though the absence of DNA methylation in many species raises interesting questions about alternative mechanisms governing histone lysine methylation and its interactions with DNA sequence. More specifically, we will explore the relationship between these epigenetic and chromatin based systems in mammals, highlighting some of the most recent discoveries that have provided fascinating and revealing insight into how these processes occur at the molecular and cellular level.
2. A direct link between the readers of DNA methylation and the histone lysine methylation system
5-methylcytosine (5mC) is widespread throughout the mammalian genome and found on up to 80% of CpG dinucleotides. In human and mouse, DNA methylation is best known for the roles it plays in heterochromatin formation at pericentromeric regions, transcriptional repression on the inactive X chromosome in females, and gene regulatory functions at imprinting control regions in a parent-of-origin specific manner. DNA methylation is generally thought to elicit effects that result in changes to chromatin structure, including histone deacetylation, methylation, and local chromatin compaction. Some, if not most, of these effects are likely mediated by one of two families of methylated CpG DNA binding proteins, comprised of the MBD family (MeCP2 and Mbd1–4) and the BTB/POZ family (Kaiso and ZBTB4/38) (reviewed in Ref. [10]).
Biochemical work studying methyl-CpG binding proteins over the past twenty years has revealed that in addition to specifically associating with methylated CpG dinucleotides, these proteins also bind to a multitude of different chromatin modifying enzymes, including histone deacetylases and histone lysine methyltransferases. For example, MeCP2 associates with the Suv39h1/2 histone methyltransferases, which modify histone H3 on lysine at position 9 (H3K9me) [11], [12]. Like DNA methylation, H3K9me is generally associated with transcriptionally repressed gene regulatory elements and heterochromatic regions of the genome. Fittingly, MeCP2 and H3K9 trimethylation (H3K9me3) appear concentrated at pericentromeric regions of heterochromatin in cell-based immunostaining experiments.
Mutations in MeCP2 cause the debilitating neurological disease known as Rett syndrome. Somewhat surprisingly, despite its localization to pericentromeric regions of chromatin and its association with H3K9 methyltransferase activity, cells from a mouse model of Rett Syndrome lacking MeCP2 do not show obvious defects in H3K9me3 deposition at pericentromeric regions [13]. This indicates that although MeCP2 associates with H3K9 methyltransferase activity, this interaction is not essential for H3K9me3 at major satellite pericentromeric heterochromatin and suggests that the H3K9me3 modification state must be achieved through redundant or mechanistically distinct processes.
One possibility is that multiple MBD proteins may function in a redundant manner to drive H3K9me to regions containing DNA methylation. In keeping with this idea, the Mbd1 protein can also associate with H3K9 methyltransferase activity. In the case of Mbd1, the associated activity was attributed to the Setdb1 methyltransferase and this interaction was proposed to play an essential role in the correct maintenance of H3K9 methylation in heterochromatin during DNA replication. This function is mediated by a protein complex consisting of Mbd1, Setdb1 and the chromatin assembly factor Caf1 that assembles during S-phase of the cell cycle to ensure correct placement of H3K9 methylation during replication-coupled chromatin assembly [14]. Mbd1 is also reported to associate with Suv39h1/HP1 to coordinate DNA methylation and H3K9 methylation [15], suggesting that it may play multiple roles in H3K9me deposition. However, deletion of the Mbd1 protein in mouse does not lead to catastrophic developmental defects and instead the mice display only mild neurological defects, though some retrotransposon reactivation was observed [16]. This suggests that the replication-dependent activity leading to deposition of H3K9me by Mbd1 is not essential in mouse.
Together, these observations suggest that either the MBD proteins play partially redundant roles in specifying H3K9me deposition at regions with DNA methylation during development, or that other parallel targeting mechanisms can compensate in their absence in knockout mouse models. Nevertheless, when DNA methylation is lost in cancer cells lacking the major cellular DNA methyltransferase, Dnmt1, there are decreases in H3K9me3/me2 levels at heterochromatic regions [17]. However, when all three mammalian DNMTs are lost in mouse embryonic stem cells, H3K9 methylation distribution appears broadly unchanged in sub-nuclear localisation, and proviral sequences which are normally silenced by H3K9 methylation are not reactivated [18], [19]. This suggests that DNA methylation may play an important role in specifying how and where H3K9me is placed, but that this is likely context-dependent. Interestingly, it is also now becoming clear that H3K9 methylation plays important roles in the maintenance of DNA methylation (see Subsection 3.5). Therefore, it appears clear that the interplay between DNA and histone methylation cannot solely be accounted for by a simple linear pathway whereby MBD proteins recruit histone methyltransferases to chromatin.
3. Getting DNA methylation where it needs to go
Controlling the timing and placement of DNA methylation in the genome is essential for normal development and cellular function. DNA methylation is catalysed by three active DNA methyltransferases (DNMTs), none of which has significant inherent sequence specificity beyond the CpG dinucleotide, implying that their targeting to particular genomic loci must be achieved by other means. These may include the exclusion of DNA methylation by DNA-bound transcription factors, or interference by the transcriptional machinery itself, or the effects of nucleosome positioning and histone modifications. Recently, it has become clear that the histone methylation state of chromatin may have more profound roles than previously realised in the targeting of DNMTs in mammalian genomes.
Of the three DNMTs found in mammals, Dnmt1 is generally considered to act as a ‘maintenance’ methyltransferase. It recognises hemimethylated CpG sites resulting from the synthesis of the daughter strand following semi-conservative DNA replication and modifies these to effectively reinstate the methylation patterns that originated in the parental strands. Dnmt3a and Dnmt3b are highly homologous de novo methyltransferases (though several splice variants of each enzyme exist, some of which are catalytically inactive) that are thought to catalyse the methylation of CpG at previously non-methylated sites. The most profound examples of de novo methylation by Dnmt3a/b occur in two phases in early mouse development (recently reviewed in Ref. [20]). The first occurs following the global demethylation event that precedes embryo implantation, and the second following a similar wave of demethylation in developing primordial germ cells (PGCs). In the zygote, the paternal pronucleus appears to undergo a phase of rapid global demethylation (with the exception of paternal imprinted loci) in a process dependent on Tet3 [21], [22] and the base excision repair (BER) machinery [23]. In contrast, the maternal pronucleus is demethylated more slowly, which may occur via a passive mechanism related to DNA replication [24]. DNA methylation is then re-established by Dnmt3a/b, beginning in the inner cell mass of the developing embryo. Interestingly, the latter wave of de novo methylation in PGCs also depends on the catalytically inactive Dnmt3 homologue, Dnmt3L (see Subsection 3.3). The capacity of Dnmt3a/b to recognise and methylate the appropriate regions of the genome is thus a critical mechanism in establishing the mammalian epigenome that will be faithfully maintained by the maintenance methyltransferase Dnmt1. Perhaps unsurprisingly, given the challenging nature of studying molecular mechanisms in the early developing mouse embryo, there remains very little mechanistic understanding of how targeting of de novo methylation occurs. Nevertheless, there has recently been a series of advances that suggest that de novo methylation may rely, at least in part, on pre-existing histone lysine methylation, and in some cases on the enzymes that catalyse lysine methylation or demethylation. Furthermore, lysine methylation also appears to play a role in protecting DNA from active demethylation.
3.1. The H3K9 methylation system and de novo methylation
Studies aimed at understanding how DNA methylation is specified in lower eukaryotes have been instrumental in identifying general targeting mechanisms that might be shared across species. This has revealed that targeting of DNA methylation to heterochromatin by the de novo methyltransferases Dnmt3a/b is dependent, in some contexts, on pre-existing H3K9 methylation. For example, in the filamentous fungus Neurospora crassa all DNA methylation depends on H3K9 methylation [25], [26], while a similar requirement has been observed in Arabidopsis thaliana [27], [28] for CpNpG methylation deposited by CHROMOMETHYLASE3. Several instances of direct interactions between Dnmt3a/b and the histone H3K9 methyltransferase enzymes have been reported in mammals, though in most cases, the details of how these interactions mediate DNA methylation are yet to be elucidated. For example, the Suv39h1/2 H3K9 methyltransferases are required for the establishment of DNA methylation at pericentric heterochromatin through their enzymatic placement of H3K9me3 [29]. Accordingly, Dnmt3a/b are recruited to H3K9-methylated heterochromatin by direct interactions with heterochromatin protein 1 (HP1), which binds to H3K9me3 through its chromodomain [29] (though one report has suggested that Dnmt3a can also displace HP1 from chromatin by competition for binding to the histone H3 tail [30]). This suggests, at least in heterochromatin, that H3K9me3 plays a role in targeting DNA methylation.
Interestingly, it also appears that Dnmt3a/b may take some guidance in recognising chromatin substrates through directly interacting with histone methyltransferases themselves [31]. Dnmt3a/b have been shown to interact with Suv39h1 [32] and Setdb1 [33] via their ADD domain. In the case of Setdb1, this association was essential for methylation and repression of certain CpG-methylated promoters in cancer cells. Furthermore, Dnmt3a interacts with the euchromatin-associated H3K9 methyltransferases G9a/GLP, probably via the chromodomain protein MPP8 [34]. In this specific instance the interaction appears to be Dnmt3a-specific, as neither Dnmt3a2 nor any of the Dnmt3b splice variants contain the protein sequence shown to interact with MPP8. Also, a direct interaction between the C-terminal catalytic domains of Dnmt3a/b and the ankyrin repeat domains of G9a [35], which are themselves able to bind to H3K9 methylation, has been shown to play roles in de novo methylation [36]. The association of the DNMT enzymes with histone methyltransferases is also reported to extend to the maintenance methyltransferase Dnmt1 which associates with G9a [37].
Although a multitude of observations suggest that either the presence of H3K9 methylation or direct association of the DNMTs with H3K9 methyltransferases might play a central role in targeting de novo DNA methylation at heterochromatic regions, the precise molecular details of these relationships remain poorly understood (Fig. 1). For example, do the interactions of Dnmt3a/b with histone methyltransferases contribute to (i) the allosteric activation of Dnmt3a/b by interactions with the lysine methyltransferases, (ii) recruitment to correct genomic loci by interactions with H3K9 methylation, either through HP1 or the SET-domain methyltransferases themselves, or (iii) methylation of Dnmt3a/b themselves, either to activate or to promote interactions with histone methyltransferases? As it stands now, some of these questions appear to have contradictory answers with the requirement for G9a–Dnmt3a/b interactions in establishing DNA methylation in some cases not requiring the catalytic activity of G9a [35]. However, in other cases it has been reported that the G9a–Dnmt3a/b interaction itself is mediated by G9a-catalysed methylation of lysine residues in Dnmt3a [34]. The situation is further complicated by the complex alternative splicing that gives rise to variants of the Dnmt3a/b proteins. Studies linking Dnmt3a/b to H3K9 methylation have, in general, not drawn distinctions between Dnmt3 splice variants, and their possible roles in interactions with H3K9 methyltransferases, HP1 and H3K9 methylation itself. In order to address these questions, a more defined understanding of the biochemical nature of DNMT3 protein complexes is required, coupled to dissection of how these activities function in vivo (Fig. 1).
Fig. 1.

Targeting of DNA methylation and H3K9 methylation to (a) heterochromatin, where it interacts with the H3K9 methylation system, and (b) gene bodies, where it may interact with H3K36 methylation to target DNA methylation to these regions. Several alternative but not mutually exclusive models are shown here.
3.2. The H3K36 methylation system and de novo methylation
Several studies have suggested a link between H3K36me3 and de novo DNA methylation. H3K36 trimethylation (H3K36me3) is catalysed by Setd2 [38], which associates with the C-terminal domain of elongating RNA PolII through a phosphorylation-dependent interaction, and is targeted to the body of actively transcribed genes [39]. Within gene bodies, H3K36 methylation is enriched in exons relative to introns [40], [41], [42], [43], and correlates with enrichment of DNA methylation and depletion of histone acetylation [44]. A combination of DNA methylation and histone methylation in gene bodies seems to work synergistically to regulate the splicing machinery (see Ref. [45] for review). This potential relationship between DNA methylation and H3K36me3 is further supported by the observation that the bodies of actively transcribed genes in the mouse oocyte attract high levels of DNA methylation [46]. This is in contrast to the rest of the oocyte genome, which differs from most somatic cells in that it is largely hypomethylated. This interesting correlation between H3K36 methylation and DNA methylation suggests that mechanisms might exist whereby H3K36 methylation can play a role in recruiting Dnmt3a/b to gene bodies, or vice versa. In support of this, Dnmt3a/b contain, in addition to the histone-binding ADD domain, a PWWP domain, which preferentially binds to H3K36me3 [47]. Mutation of the PWWP domain inhibits its DNA methyltransferase activity on nucleosomal substrates in vitro [47] and leads to reduced affinity of Dnmt3a/b for nucleosomes [48]. Somewhat surprisingly, based on its proposed H3K36me3 binding activity, loss of the PWWP domain also abrogated the ability of Dnmt3a/b to bind to pericentric heterochromatin and its ability to methylate major satellite repeats in pericentric heterochromatic regions of the genome [49], [50]. This was unexpected, given that these regions of the genome are not generally thought to contain significant amounts of H3K36me3 methylation. This suggests that the PWWP domain may play more complex roles in regulating DNMT3 enzyme association with chromatin than simply recognising H3K36me3.
Nevertheless, the importance of the PWWP domain for Dnmt3 function is highlighted by the fact that mutations of the PWWP domain of Dnmt3b have been associated with ICF syndrome (Immunodeficiency, Centromere instability and Facial anomalies syndrome), a severe autosomal recessive disease in humans. ICF syndrome patients carrying the PWWP domain mutation lose DNA methylation in the classical satellite repeat II, consistent with PWWP domain mutations affecting methyltransferase activity in vitro [51]. However, these studies were carried out before the PWWP interaction with H3K36me3 was characterised, so it remains unknown if these outcomes are due to failure to bind H3K36me3 or are the result of other less well-characterised chromatin interactions. It would be interesting to examine ICF syndrome patients with PWWP mutations for alterations in gene body DNA methylation at actively transcribed genes using modern genome-wide methylation profiling techniques, as has recently been done for another ICF patient cell line [52]. However, the relationship between H3K36me3 and DNMT3 recruitment might not be so simple, as reduced H3K36me3 mediated by knockdown of the histone lysine methyltransferase Setd2 in a cancer cell line showed no effect on DNA methylation in gene bodies despite the loss of H3K36me3 in these regions [53]. If H3K36me3 does have a role in recruiting DNA methylation, it likely functions in specific genomic contexts, and further work is needed to elucidate the mechanisms responsible. It is also possible that the PWWP domain interacts with other lysine methylation marks in a context-dependent mechanism to recruit Dnmt3a/b to heterochromatin.
3.3. A role for H3K4 methylation in blocking DNA methylation
Several observations suggest that histone methylation may play a role not only in recruiting DNA methylation to certain genomic regions, but also in excluding it from others. One such modification, H3K4me3, appears to be mutually exclusive with DNA methylation and therefore may be a candidate DNA methylation-blocking histone modification [54]. A hint as to why H3K4me3 might block de novo methylation came from the discovery that the Dnmt3-associated protein Dnmt3L contains an ADD domain that specifically interacts with unmodified H3K4, and which is blocked from binding the H3 tail when H3K4 is methylated [55]. In support of a potential role for Dnmt3L in regulating DNA methylation by the de novo DNMTs, Dnmt3a/b appear to rely in some contexts, particularly in the wave of DNA methylation in developing PGCs, on the function of Dnmt3L. Indeed, the phenotype of a conditional Dnmt3a knockout is very similar to that of Dnmt3L knockout mice in that Dnmt3L−/− males are sterile, and completely lack germ cells, while the heterozygous offspring of Dnmt3L−/− females die in mid-gestation due to abnormal expression of imprinted genes [56], [57], [58], [59]. The observation that Dnmt3L is able to bind to non-methylated H3K4 but cannot bind to H3K4me3 suggested that H3K4 methylation may play a role in blocking de novo DNA methylation at some genomic loci [30], [55]. Interestingly, the antagonistic effects of H3K4me3 on DNA methylation are phenocopied when Dnmt3a is ectopically expressed in budding yeast, a fungus that lacks endogenous DNA methylation. In these heterologous experiments both the presence of Dnmt3L and the histone H3 tail containing residues 1–4 were required for this effect [60]. Furthermore, mutation of the ADD domain of Dnmt3L reduced DNA methylation levels, while mutation of the sole H3K4 methyltransferase in budding yeast, Set1, resulted in a global increase in ectopic DNA methylation. The mechanisms by which Dnmt3L might stimulate Dnmt3a/b activity are not clear; Dnmt3L forms a tetrameric complex with Dnmt3a/b, which might promote Dnmt3a/b activity by stabilising the conformation of the active site loop of Dnmt3a/b [61] or by preventing the formation of Dnmt3 protein aggregates [62]. However, this does not explain why binding to H3K4me0 is important for DNMT3 activity. Further, Dnmt3a/b also contain ADD domains which can bind H3K4me0 (which has an allosteric activating effect on catalytic activity in vitro [63]). Thus, while these observations suggest that H3K4me3 antagonises DNA methylation by the Dnmt3 methyltransferases, and H3K4me0 stimulates it, the mechanisms governing this effect are still unclear.
Consistent with the role of H3K4me0 in facilitating DNA methylation by Dnmt3a/b/L is the observation that Lsd2-deficient female mice show a maternal-effect lethal phenotype, with major disruption of DNA methylation at some imprinted genes (Mest, Grb10, Zac1 and Impact) [64]. Lsd2/Kdm1b is a histone lysine demethylase that removes H3K4me2/me1, and this observation suggests that removal of histone H3K4 methylation may be required for efficient DNA methylation at certain imprinted loci. While Lsd2/Kdm1b is not required for embryonic development, its paralogue Lsd1/Kdm1a is, and embryos lacking Lsd1 fail to progress through gastrulation [65]. Significant reductions in DNA methylation are observed in the Lsd1 mutant mice, though it is unclear whether the effects of Lsd1 deficiency are mediated through an inability of Dnmt3a/b/L to catalyse 5mC, or via direct effects on the maintenance methyltransferase Dnmt1, which has been reported to be a substrate for Lsd1 and whose stability may be reduced in the absence of Lsd1 (though this effect appears to be cell-type specific [169]) [65]. It is also likely that the effects of H3K4 methylation in excluding the DNA methyltransferases could be mediated through multiple mechanisms, including its capacity to act as a docking site for components of the transcription machinery (e.g. TAF3/TFIID [66]) and the H3K4 lysine methyltransferase complexes themselves [67]. Interestingly, H3K4me3 can also act as a binding site for H3K9me2 demethylases [68]. As Dnmt3a/b appear to rely in some contexts on HP1-mediated interactions with H3K9 methylation for their activity and targeting (see Subsection 3.1), this suggests another means whereby interplay between different histone lysine methylation sites may influence DNA methylation.
Finally, the requirement for Dnmt3L-facilitated methylation by Dnmt3a/b does not seem to be universal. The expression of Dnmt3L is limited to germ cells and early developmental stages ([69], [70], [71], and reviewed in Ref. [72]), whereas Dnmt3a/b are active in many other genomic and developmental contexts, implying that their catalytic activity must be amenable to regulation by other proteins apart from Dnmt3L. Indeed, recent studies have shown that catalytically inactive splice variants of Dnmt3b (Dnmt3b3 and Dnmt3b4) are able to modulate the activity of the Dnmt3a/b in a manner analogous to Dnmt3L [72], [73]. The mechanisms by which this is achieved have not yet been elucidated, and it is unclear whether this relies on interactions with histone lysine methylation. It is worth noting, however, that the modulatory effects of Dnmt3b3/4 rely on the presence of intact PWWP domains.
3.4. The relationship between H3K27 methylation and DNA methylation
Histone lysine methylation on position 27 of H3 (H3K27me) is associated with regions of the genome that are silenced by the polycomb group of transcriptional repressors. This modification is catalysed by the Ezh1/2 components of the polycomb repressive complex 2 (PRC2). The relationship between H3K27 methylation and DNA methylation remains poorly defined. In embryonic stem cells, H3K27me3 is located in discrete, punctate regions coinciding almost exclusively with CpG islands, which are generally devoid of DNA methylation. At face value this might suggest that H3K27me3 and DNA methylation are mutually exclusive. However, in somatic cell types and cancer cell lines H3K27me3 is much less restricted to CpG islands and there is extensive overlap between DNA methylation and H3K27me3 methylation, suggesting that the two are not incompatible [74], [75]. Interestingly, promoters that are marked with H3K27me3 in embryonic stem cells are more likely to gain DNA methylation during differentiation and carcinogenesis than those lacking H3K27me3 [76], [77], [78]. One possible explanation for this observation is that silencing of these genes is initiated by the polycomb repressive complexes in early development, and these genes then subsequently designated for long term silencing by the acquisition of DNA methylation in tissues where the polycomb complexes are not expressed. In support of this possibility, it has been suggested that PRC2 may recruit DNMTs [79], though a more recent report indicates that Dnmt3L may inhibit this interaction, thus preventing DNA methylation at regions where PRC2 is present [80]. Alternatively, a recent report suggested that PRC2 may also associate with Tet1 [81], which catalyses hydroxylation of 5mC and may act to enforce the exclusion of DNA methylation from CpG island regions that are actively targeted by polycomb mediated repression in embryonic stem cells. This could explain why the subset of CpG islands that are heavily occupied by polycomb group proteins in embryonic stem cells are not subject to encroachment of DNA methylation. Nevertheless, these interesting relationships between DNA and H3K27 methylation clearly warrant more careful molecular examination in vivo.
3.5. A complex relationship between maintenance DNA methylation, histone lysine methylation, and other chromatin modifications
During DNA replication the maintenance DNA methyltransferase Dnmt1 functions to specifically recognise hemimethylated DNA and reinstate symmetrical CpG methylation on the daughter DNA strands, while ignoring CpG dinucleotides that that lack methylation. Somewhat surprisingly, on naked DNA substrates in vitro Dnmt1 alone has robust methyltransferase activity toward non-methylated CpG dinucleotides and, according to some reports, sometimes only weak biochemical preference for hemimethylated CpGs over non-methylated CpGs in vitro (reviewed in Ref. [82]). This suggests that additional mechanisms must contribute to Dnmt1's elegant specificity for hemimethylated CpG dinucleotides in vivo and inability to de novo-methylate non-methylated CpG island regions. During DNA replication Dnmt1 associates with PCNA and a second protein called Uhrf1 at replication forks. Uhrf1 is an interesting multidomain protein that contains several chromatin binding domains, including a tandem Tudor and PHD domain that together bind H3K9me3/me2/H3K4me0 [83], [84], and an SRA domain that binds to hemimethylated DNA [85], [86], [87], [88]. Importantly, Uhrf1 is absolutely required for efficient maintenance methylation, suggesting it may play an essential role in the Dnmt1 catalytic cycle [89], [90].
Several models have been proposed to explain the substrate selectivity and recruitment of Dnmt1. One model involves autoinhibition of DNA methylation by binding to non-methylated DNA through the ZF-CxxC domain of Dnmt1 [91], [92], though this has been largely discounted [93]. A second model (Fig. 2a) invokes the binding of Uhrf1 to the methylated DNA base in a hemimethylated CpG dinucleotide via its SRA domain allowing Dnmt1 to specifically methylate the unmodified cytosine base on the opposite strand [94], [95]. In the latter case, modelling of the available crystal structures of Dnmt1 and the Uhrf1 SRA domain bound to CpG dinucleotides suggests that it is unlikely that both molecules can simultaneously engage the same site due to steric clashes between the two proteins [87], [91]. This suggests that Uhrf1 may act to first engage a hemimethylated site which is then subsequently bound by Dnmt1, displacing Uhrf1, before the methylation reaction can occur [95]. A displacement type mechanism may be mediated through the replication foci targeting sequence (RFTS) domain in Dnmt1 that normally functions to inhibit catalytic activity [96], [97]. This is supported by reports that Uhrf1 interacts with Dnmt1 through the RFTS domain [98], and this interaction may be instrumental in relieving the autoinhibitory effect on Dnmt1, making it competent for DNA methylation. However, a more recent report showed that Uhrf1, rather than interacting directly with Dnmt1, ubiquitylates histone H3K23 using its C-terminal Ring domain as an E3 ligase [99]. H3K23ub then recruits Dnmt1 to replication foci through interaction with the RFTS domain. It is possible that this recruitment by H3K23ub then alleviates the autoinhibition of Dnmt1. According to this model (Fig. 2b), Uhrf1 engages hemimethylated sites through its SRA domain, whereupon it ubiquitylates H3K23, recruits Dnmt1, and leads to methylation of the hemimethylated CpG. Consistent with this recruitment model is the finding that a Dnmt1–PCNA fusion protein was able to rescue DNA methylation defects in Uhrf1−/− cells [100], although this doesn't explain the autoinhibition effects observed for the RFTS domain.
Fig. 2.

Maintenance of DNA methylation and histone methylation at replication forks by Dnmt1. Two alternative models are shown here. (a) Model I: Uhrf1 localises to replication forks through interactions with PCNA, H3K9me3/me2 and hemimethylated DNA, where it recruits Dnmt1 and H3K9 methyltransferases to replace DNA methylation and histone methylation on newly synthesised chromatin. Mbd1 also recruits Setdb1 to newly synthesised chromatin by interactions with Caf1 and methylated DNA, where it assists in maintaining H3K9 methylation.
(b) Model II: Uhrf1 is recruited to replication forks by interactions with PCNA, hemimethylated DNA and H3K9me3/me2, where it ubiquitylates H3K23, which recruits Dnmt1 to methylated hemimethylated DNA.
It was recently shown that the recruitment of Dnmt1 via Uhrf1 still occurs with SRA domain mutants that lack base-flipping activity [100]. Interestingly, the ability of Uhrf1 to target Dnmt1 activity to replication foci was lost when Uhrf1 lost both its base-flipping and H3K9me3/me2 binding capacity [100], or its E3 ligase activity [99]. This suggests that histone methylation at H3K9me3/me2, in addition to recognition of hemimethylated DNA by Uhrf1, may function synergistically to ensure correct recruitment of Uhrf1 to the appropriate genomic loci. Following on from this, Uhrf1-dependent H3K23 ubiquitylation would then function to recruit Dnmt1 and maintain DNA methylation during S-phase [100], [101]. Another report indicates that Uhrf1, through its PHD domain, contributes to the decompaction of chromatin, suggesting that this effect on chromatin structure may play a role in targeting Dnmt1 activity to heterochromatic regions [102]. Interestingly, the ability to target Dnmt1 to replication foci is limited to Uhrf1 but not its paralogue Uhrf2. Although Uhrf2 can associate with Dnmt1, Dnmt3a/b and G9a, and has similar base-flipping and H3K9me3-binding properties to Uhrf1, it cannot associate with replication foci or target Dnmt1 there [103], [104], suggesting that the ability to ubiquitylate H3 may be limited to Uhrf1. Uhrf1 knockdown, or mutation of its H3K9me3/me2-binding tandem Tudor domain, also reduced the stability of Dnmt1 during mitosis, suggesting another mechanism whereby DNA maintenance methylation at heterochromatic regions might rely on recognition of histone methylation [83], [105]. It has also been shown that the stability of Dnmt1 is regulated through the cell cycle by a phospho/methyl switch; phosphorylation of Ser143 by AKT1 stabilises Dnmt1 by antagonising methylation of Lys142 by Set7, which targets Dnmt1 for proteasomal degradation [106], [107]. This leads to accumulation of Dnmt1 during S-phase. It is possible that some of the cell-cycle specific effects of Uhrf1 in recruiting Dnmt1 to chromatin are in part a result of cell-cycle dependent stability of Dnmt1.
Finally, while the maintenance of DNA methylation during replication is fairly well understood, many questions remain regarding the maintenance of histone methylation on newly-synthesised chromatin. H3K4 and H3K27 methylation appear to be maintained on newly-synthesised DNA by association of the respective methyltransferases (TrxG and PcG complexes) with DNA through the replication fork [169], [170]. By contrast, it seems that Uhrf1 and Dnmt1 contribute to the maintenance of H3K9 methylation through interactions with H3K9 methyltransferases [108], [109], [110]. There is thus emerging evidence for complex interactions between histone and DNA methylation in the maintenance of heterochromatin during DNA replication. To this end, there remains a need for structural information that describes the molecular relationships and physical interactions between the central players in this system, including Dnmt1 and H3K23ub, Uhrf1, PCNA, the nucleosome and the histone methyltransferases. Furthermore, detailed kinetic studies to examine the fidelity of DNA methylation and H3K9 methylation maintenance are required to understand how these systems function together.
3.6. H3K9me2 methylation protects DNA from demethylation
Histone modifications also appear to function in ways that protect DNA from demethylation. It is proposed that, following fertilisation in the mouse oocyte, DNA methylation may be counteracted via Tet3-mediated hydroxylation (and possibly demethylation) in the male pronucleus, with the maternal genome being protected from this activity. Emerging evidence shows that the maternal genome, and certain paternal imprinted loci, are protected from Tet3-mediated hydroxylation by the binding of PGC7 to H3K9me2 at these loci [111]. The mechanism by which PGC7 recognises H3K9me2 and prevents hydroxylation of 5mC to 5hmC has yet to be determined. Histone methylation at H3K9 may also influence imprinting at other loci by interactions with Zfp57 and Trim28, though the mechanisms governing these interactions are still to be determined. Initial evidence indicates that they seem to function by facilitating heterochromatinisation and DNA re-methylation (rather than protecting from Tet3-mediated hydroxylation) (reviewed in [112]). To this end, Zfp57 and Trim28 co-exist on chromatin with H3K9me3, Setdb1, HP1 and Uhrf1, and recruit the DNMTs [113], [114], [115]. These observations together suggest that H3K9 methylation plays an important role in the establishment and maintenance of parent-of-origin-specific imprints through a variety of mechanisms.
4. Direct links between non-methylated DNA readers and histone lysine methylation
While most genomic CpG dinucleotides are methylated, regions of the genome known as CpG islands generally remain free of DNA methylation. CpG islands are associated with approximately 70% of mammalian gene promoters and are also found at many other gene regulatory elements including enhancers [5], [6], [7]. These non-methylated regions are usually associated with particular histone methylation signatures, including methylation of H3K4 and H3K27 and the absence of H3K36 methylation.
Much like the MBD family of proteins that recognise methylated CpG dinucleotides, a family of proteins that bind to non-methylated CpG dinucleotides has also been discovered [116]. These proteins recognise non-methylated CpG dinucleotides via a highly conserved ZF-CxxC DNA binding domain. ZF-CxxC domain-containing proteins are found in a number of proteins/protein complexes which have the ability to modify histones or DNA, including Mll1/2, Cfp1, Kdm2a/b, Dnmt1, Tet1/3 and Mbd1. While these proteins have recently been reviewed in detail [117], we highlight here the functional links between ZF-CxxC proteins and histone lysine methylation (Fig. 3).
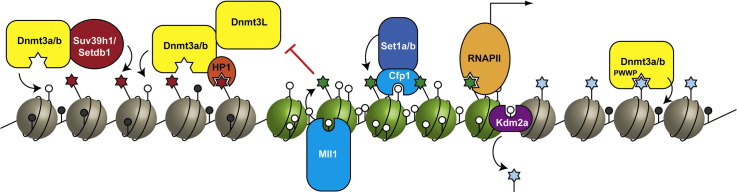
Fig. 3.

CpG islands are associated with distinctive chromatin environments. (a) CpG islands associated with actively transcribed genes recruit H3K4 methyltransferases (Mll1/2, Set1a/b) through interactions between ZF-CxxC domains and non-methylated CpG dinucleotides. RNA PolII associates with H3K4me3, while Kdm2a/b remove H3K36me2 from CpG islands. CpG islands also block DNA methylation by preventing the binding of Dnmt3L/Dnmt3a/b to histone tails that are methylated at H3K4. CpG islands may also recruit Mll translocations in leukaemia, where they recruit elongation complexes and Dot1L, which catalyses H3K79 methylation. (b) CpG island associated genes may be repressed by the polycomb complexes PRC1 and PRC2. PRC1 can be recruited to CpG islands via interaction with Kdm2b to ubiquitylate H2AK119. PRC2 is targeted via unknown mechanisms to methylate H3K27 at CpG islands, though it may be excluded from non-CpG island regions by its reduced ability to methylate substrates that have H3K36 methylation.
4.1. CpG islands and the placement of H3K4 methylation
The association of H3K4me3 with active gene promoters is well documented [118], although the precise mechanisms that lead to its recruitment to these genomic regions, and its function in regulating transcription, still remain unclear. Recent work suggests that H3K4me3 aids in the association of RNA PolII with promoters via interactions with the general transcription factor TFIID via the TAF3 subunit [66], [119]. Interestingly, most CpG island-associated genes, regardless of their transcriptional state, are also modified by H3K4me3. This suggests that the underlying non-methylated DNA state may in some way contribute to this modification profile. Fittingly, most of the mammalian H3K4me3 methyltransferase complexes contain ZF-CxxC domain proteins which may act as the functional link between the activity of these protein complexes and the specification of H3K4me3 at CpG islands [120].
In support of this possibility, recent work has demonstrated that the ZF-CxxC domain protein Cfp1 recruits the Set1a/b H3K4 methyltransferase complexes to CpG islands, where they catalyse H3K4me3 [121]. Unexpectedly, in Cfp1−/− cells the most dramatic defects in H3K4me3 placement were not observed at lowly expressed CpG island associated genes, but instead a the most highly expressed subset of genes. This suggests a role for Cfp1 in the amplification of H3K4me3 at highly expressed genes as opposed to the more ubiquitous placement of H3K4me3 at CpG islands. Furthermore, new peaks of H3K4me3 appeared at other regulatory regions such as enhancers in the Cfp1 null cells. When Cfp1 null cells were reconstituted with Cfp1 mutant protein lacking a functional ZF-CxxC domain, Cfp1 rescued the defects in H3K4me3 at transcribed genes but the intact ZF-CxxC domain was required to prevent the appearance of ectopic H3K4me3 at other regions [122]. These observations suggest that DNA sequence and methylation state work together with transcriptional state to ensure correct targeting of histone lysine methylation marks, rather than a more simplistic model where DNA sequence alone influences the placement of histone marks. Consistent with this model is the observation that, while the Kdm2b ZF-CxxC domain protein is able to recruit the polycomb repressive complex 1 (PRC1) to most CpG island promoters, a repressive polycomb state (characterised by H3K27me3 and H2AK119ub) is only established at a small subset of CpG island promoters (see Subsection 4.2), indicating that other factors also influence the ability of DNA sequences to affect the attendant histone marks [123], [124], [125].
Although the Set1 complexes are responsible for most H3K4 methylation in mammalian cells [120], [126], the Mll1/2 methyltransferase complexes are also thought to contribute to H3K4me3 at specific regions of the genome. For example, Mll1/2 methylate less than 5% of H3K4me3 in mammals, but were found to be essential for the methylation of Hox loci [5], [127], [128], [129] and in mouse embryonic stem cells Mll2 contributes to H3K4me3 at bivalent (H3K27me3/H3K4me3 positive) gene promoters [129]. Like Cfp1, the Mll1/2 proteins encode a ZF-CxxC domain that can bind to non-methylated DNA and in some instances this DNA binding activity is required for the functionality of the Mll protein. For example, Mll1 translocations which are involved in many instances of leukaemogenesis also rely on the ZF-CxxC domain and binding to CpG islands for the establishment and maintenance of aberrant Hoxa9 gene expression through the recruitment of transcriptional elongation complexes [130], [131], [132]. The recruitment of Mll1 fusion proteins also results in the recruitment of Dot1L, leading to elevated levels of H3K79me2 at these loci [133], [134], [135]. TET proteins, which contain or interact with ZF-CxxC domains, may also play a role in recruiting H3K4me3 to CpG islands. TET proteins are reported to recruit OGT (O-GlcNac transferase) to CpG islands, which in turn interacts with and glycosylates HCF1, a component of the Set1a/b and MLL1/2 complexes, suggesting that this may function as another means to recruit H3K4 methylation to CpG island regions [136], [137], [138], [139], [140]. Thus there are several possible pathways by which non-methylated DNA in CpG islands may function to recruit H3K4 methylation.
4.2. CpG islands, H3K27 methylation and Polycomb
In contrast to H3K4me3, H3K27me3 is associated with repression of transcription, although the mechanisms by which this is achieved are not clear [141]. H3K27me3 seems to promote chromatin compaction [142], which may be associated with repression, while there is also evidence that it may be involved in excluding Mediator from gene promoters, thus hindering transcriptional activation [143], [144]. H3K27me3 in embryonic stem cells correlates strongly with CpG islands [145], and anticorrelates with DNA methylation at these regions [74], [146]. Nevertheless, the mechanisms governing the placement of H3K27me3 by polycomb repressive complex 2 (PRC2) in vertebrates are still being elucidated. The PRC2 complex consists of a core complex of Ezh2, the active methyltransferase component, Eed, Suz12 and Rbap46/48, and a number of ancillary components including Jarid2, Aebp2 and Phc1–3 [147], [148], [149], [150], [151], [152], [153], [154], [155]. None of these proteins has been identified as having specific non-methylated CpG-binding capability, though Jarid2 has some preference for binding to GC-rich sequences through its C-terminal region, which contains an ARID domain and a C5HC2 Zn-finger domain [147]. However, a number of lines of evidence – most notably (i) the acquisition of H3K27me3 by bacterial non-methylated GC-rich sequences integrated into mouse genomes [156], (ii) the acquisition of H3K27me3 by CpG islands that have activating sequences removed [156], and (iii) the anticorrelation of H3K27me3 with DNA methylation [74], [146] – point to the requirement for CpG islands in establishing H3K27me3/polycomb repressed domains in mammals. In the absence of an obvious DNA-binding candidate for this role, indirect effects have been suggested, including the ability of H3K36me2 (which is removed from CpG islands through interactions with the ZF-CxxC-domain containing proteins Kdm2a/b, among other mechanisms) to inhibit PRC2 activity [157], [158]. However, it should be noted that, although beyond the scope of this review, other mechanisms including transcription factor-specific mechanisms and interactions with non-coding RNAs have been posited to account for the recruitment of PRC2 to its target regions of the genome (see Ref. [159] for a recent review), and it is likely that a combination of factors, including but not limited to the underlying DNA sequence and DNA methylation state, is responsible.
4.3. CpG islands and the removal of H3K36 methylation
While H3K4me3 and H3K27me3 are associated with CpG islands, trimethylation of H3K36 is associated with the bodies of actively transcribed genes, where it associates with DNA methylation (see Subsection 3.2). Most of our mechanistic understanding of the H3K36 methylation comes from experiments done in budding yeast that suggest that it may function to suppress histone exchange in actively transcribed genes through recruitment of the Rpd3s histone deacetylase corepressor complex [160], thus reducing cryptic transcriptional initiation in gene bodies [161]. If similar mechanisms are at play in vertebrate gene bodies, this activity may be further stabilised through the recruitment of de novo DNA methyltransferases Dnmt3a/b and increased levels of DNA methylation via recognition of H3K36me3 [47] (discussed in Subsection 3.2). In contrast to H3K36me3, dimethylation of H3K36 is abundant throughout the genome (30–50% of histone H3 is dimethylated at H3K36), but targeting and functions of this modification are less well-understood. It remains possible that this modification also contributes to transcriptional quelling, supported by the observation that, in budding yeast, H3K36me2 is sufficient to suppress cryptic initiation and elicit Rpd3s mediated effects [162], [163]. Recently it was demonstrated [123], [124], [125], [164] that the Kdm2a/b histone demethylases are recruited via their ZF-CxxC domains to CpG island chromatin, where they lead to removal of H3K36me2. If H3K36me2 acts at the genome scale in mammals to quell spurious transcriptional initiation, then the concerted activity of Kdm2a/b at CpG islands may liberate these regions from this generalized repression and make them more amenable to transcription. Given the broad overlap of CpG islands with gene promoters and other regulatory features, this may be a means to highlight regulatory regions in large and complex vertebrate genomes. In addition to Kdm2b acting as an H3K36me2 demethylase, it is also able to recruit a variant PRC1 complex to CpG islands. This may function as a sampling mechanism which provides CpG islands with the opportunity to become occupied by the polycomb group repressive proteins should they lack counteracting transcriptional activities [123], [124], [125] (see Ref. [159] for a detailed discussion). Interestingly, the ability of the PRC2 complex to methylate H3K27me3 is inhibited by nucleosomes that have H3K36 methylation, suggesting that Kdm2b may also function at PRC2-occupied CpG islands to ensure efficient deposition of H3K27me3. H3K36 methylation may also be excluded from CpG islands by H2AK119ub, which is catalysed at these regions by the PRC1 complexes [165]. Clearly there is much that remains to be understood about the functionality of the H3K36 methylation system in mammals, but nevertheless it appears, much like other methylation marks, to be highly interconnected with the DNA methylation and CpG island systems.
5. Outlook
Our understanding of the interactions between DNA methylation and histone modifications is becoming clearer, as we understand the molecular mechanisms that lead to their deposition and the precise biochemistry that underpins their catalysis. We now understand that non-methylated DNA in CpG islands can act as part of a genomic signature to recruit H3K4 and H3K27 trimethylation, and to exclude H3K36 methylation, possibly creating chromatin environments unique to gene regulatory elements that are able to modulate transcriptional states. Similarly, the maintenance of DNA methylation through replication is becoming more clearly understood, particularly through understanding the role of Uhrf1 in targeting and coordinating this activity through histone modifications. Nevertheless, understanding how de novo DNA methylation is targeted during development remains poorly understood. Although there are indications that unmodified H3K4, H3K9me3 and H3K36me3 are involved in these processes, in many cases these features appear to be context-specific, stressing that additional effort is needed to elucidate the molecular detail underpinning these systems and the generality of their usage. Finally, it seems likely that functional links between 5-hydroxymethylcytosine and histone lysine methylation will be identified, as exemplified by the discovery that Uhrf1 and MeCP2 can also bind to 5hmC [166], [167]. With the molecular components linking the DNA and histone methylation systems being rapidly identified, we are well placed to reveal how these fascinating systems contribute to genome function.
Acknowledgements
Work in the Klose laboratory is supported by the Wellcome Trust, the Lister Institute of Preventive Medicine, the European Molecular Biology Organization (EMBO) and Cancer Research UK. N.R.R. is a Junior Research Fellow at St John's College, Oxford. We thank Anca Farcas, Hamish King and David Brown for the critical reading of the manuscript.
Footnotes
This article is part of a Special Issue entitled: Methylation: A Multifaceted Modification — looking at transcription and beyond.
References
- 1.Kriaucionis S., Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tahiliani M., Koh K.P., Shen Y., Pastor W.A., Bandukwala H., Brudno Y., Agarwal S., Iyer L.M., Liu D.R., Aravind L., Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He Y.F., Li B.Z., Li Z., Liu P., Wang Y., Tang Q., Ding J., Jia Y., Chen Z., Li L., Sun Y., Li X., Dai Q., Song C.X., Zhang K., He C., Xu G.L. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito S., Shen L., Dai Q., Wu S.C., Collins L.B., Swenberg J.A., He C., Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Long H.K., Sims D., Heger A., Blackledge N.P., Kutter C., Wright M.L., Grutzner F., Odom D.T., Patient R., Ponting C.P., Klose R.J. Epigenetic conservation at gene regulatory elements revealed by non-methylated DNA profiling in seven vertebrates. eLife. 2013;2:e00348. doi: 10.7554/eLife.00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Illingworth R.S., Gruenewald-Schneider U., Webb S., Kerr A.R., James K.D., Turner D.J., Smith C., Harrison D.J., Andrews R., Bird A.P. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 2010;6:e1001134. doi: 10.1371/journal.pgen.1001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Illingworth R., Kerr A., Desousa D., Jorgensen H., Ellis P., Stalker J., Jackson D., Clee C., Plumb R., Rogers J., Humphray S., Cox T., Langford C., Bird A. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008;6:e22. doi: 10.1371/journal.pbio.0060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iyer L.M., Abhiman S., Aravind L. Natural history of eukaryotic DNA methylation systems. Progress in Molecular Biology and Translational Science. 2011;101:25–104. doi: 10.1016/B978-0-12-387685-0.00002-0. [DOI] [PubMed] [Google Scholar]
- 9.Deaton A.M., Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bogdanovic O., Veenstra G.J. DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma. 2009;118:549–565. doi: 10.1007/s00412-009-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fuks F., Hurd P.J., Wolf D., Nan X., Bird A.P., Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 2003;278:4035–4040. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
- 12.Lunyak V.V., Burgess R., Prefontaine G.G., Nelson C., Sze S.H., Chenoweth J., Schwartz P., Pevzner P.A., Glass C., Mandel G., Rosenfeld M.G. Corepressor-dependent silencing of chromosomal regions encoding neuronal genes. Science. 2002;298:1747–1752. doi: 10.1126/science.1076469. [DOI] [PubMed] [Google Scholar]
- 13.Jiang Y., Matevossian A., Guo Y., Akbarian S. Setdb1-mediated histone H3K9 hypermethylation in neurons worsens the neurological phenotype of Mecp2-deficient mice. Neuropharmacology. 2011;60:1088–1097. doi: 10.1016/j.neuropharm.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarraf S.A., Stancheva I. Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol. Cell. 2004;15:595–605. doi: 10.1016/j.molcel.2004.06.043. [DOI] [PubMed] [Google Scholar]
- 15.Fujita N., Watanabe S., Ichimura T., Tsuruzoe S., Shinkai Y., Tachibana M., Chiba T., Nakao M. Methyl-CpG binding domain 1 (MBD1) interacts with the Suv39h1–HP1 heterochromatic complex for DNA methylation-based transcriptional repression. J. Biol. Chem. 2003;278:24132–24138. doi: 10.1074/jbc.M302283200. [DOI] [PubMed] [Google Scholar]
- 16.Zhao X., Ueba T., Christie B.R., Barkho B., McConnell M.J., Nakashima K., Lein E.S., Eadie B.D., Willhoite A.R., Muotri A.R., Summers R.G., Chun J., Lee K.F., Gage F.H. Mice lacking methyl-CpG binding protein 1 have deficits in adult neurogenesis and hippocampal function. Proc. Natl. Acad. Sci. U. S. A. 2003;100:6777–6782. doi: 10.1073/pnas.1131928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Espada J., Ballestar E., Fraga M.F., Villar-Garea A., Juarranz A., Stockert J.C., Robertson K.D., Fuks F., Esteller M. Human DNA methyltransferase 1 is required for maintenance of the histone H3 modification pattern. J. Biol. Chem. 2004;279:37175–37184. doi: 10.1074/jbc.M404842200. [DOI] [PubMed] [Google Scholar]
- 18.Matsui T., Leung D., Miyashita H., Maksakova I.A., Miyachi H., Kimura H., Tachibana M., Lorincz M.C., Shinkai Y. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature. 2010;464:927–931. doi: 10.1038/nature08858. [DOI] [PubMed] [Google Scholar]
- 19.Tsumura A., Hayakawa T., Kumaki Y., Takebayashi S., Sakaue M., Matsuoka C., Shimotohno K., Ishikawa F., Li E., Ueda H.R., Nakayama J., Okano M. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells. 2006;11:805–814. doi: 10.1111/j.1365-2443.2006.00984.x. [DOI] [PubMed] [Google Scholar]
- 20.Seisenberger S., Peat J.R., Hore T.A., Santos F., Dean W., Reik W. Reprogramming DNA methylation in the mammalian life cycle: building and breaking epigenetic barriers. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2013;368:20110330. doi: 10.1098/rstb.2011.0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gu T.P., Guo F., Yang H., Wu H.P., Xu G.F., Liu W., Xie Z.G., Shi L., He X., Jin S.G., Iqbal K., Shi Y.G., Deng Z., Szabo P.E., Pfeifer G.P., Li J., Xu G.L. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477:606–610. doi: 10.1038/nature10443. [DOI] [PubMed] [Google Scholar]
- 22.Wossidlo M., Nakamura T., Lepikhov K., Marques C.J., Zakhartchenko V., Boiani M., Arand J., Nakano T., Reik W., Walter J. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat. Commun. 2011;2:241. doi: 10.1038/ncomms1240. [DOI] [PubMed] [Google Scholar]
- 23.Hajkova P., Jeffries S.J., Lee C., Miller N., Jackson S.P., Surani M.A. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science. 2010;329:78–82. doi: 10.1126/science.1187945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Howell C.Y., Bestor T.H., Ding F., Latham K.E., Mertineit C., Trasler J.M., Chaillet J.R. Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell. 2001;104:829–838. doi: 10.1016/s0092-8674(01)00280-x. [DOI] [PubMed] [Google Scholar]
- 25.Tamaru H., Zhang X., McMillen D., Singh P.B., Nakayama J., Grewal S.I., Allis C.D., Cheng X., Selker E.U. Trimethylated lysine 9 of histone H3 is a mark for DNA methylation in Neurospora crassa. Nat. Genet. 2003;34:75–79. doi: 10.1038/ng1143. [DOI] [PubMed] [Google Scholar]
- 26.Tamaru H., Selker E.U. A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature. 2001;414:277–283. doi: 10.1038/35104508. [DOI] [PubMed] [Google Scholar]
- 27.Jackson J.P., Lindroth A.M., Cao X., Jacobsen S.E. Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature. 2002;416:556–560. doi: 10.1038/nature731. [DOI] [PubMed] [Google Scholar]
- 28.Lindroth A.M., Shultis D., Jasencakova Z., Fuchs J., Johnson L., Schubert D., Patnaik D., Pradhan S., Goodrich J., Schubert I., Jenuwein T., Khorasanizadeh S., Jacobsen S.E. Dual histone H3 methylation marks at lysines 9 and 27 required for interaction with CHROMOMETHYLASE3. EMBO J. 2004;23:4286–4296. doi: 10.1038/sj.emboj.7600430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lehnertz B., Ueda Y., Derijck A.A., Braunschweig U., Perez-Burgos L., Kubicek S., Chen T., Li E., Jenuwein T., Peters A.H. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 2003;13:1192–1200. doi: 10.1016/s0960-9822(03)00432-9. [DOI] [PubMed] [Google Scholar]
- 30.Otani J., Nankumo T., Arita K., Inamoto S., Ariyoshi M., Shirakawa M. Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX–DNMT3–DNMT3L domain. EMBO Rep. 2009;10:1235–1241. doi: 10.1038/embor.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muramatsu D., Singh P.B., Kimura H., Tachibana M., Shinkai Y. Pericentric heterochromatin generated by HP1 interaction-defective histone methyltransferase Suv39h1. J. Biol. Chem. 2013;288:25285–25296. doi: 10.1074/jbc.M113.470724. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Fuks F., Hurd P.J., Deplus R., Kouzarides T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003;31:2305–2312. doi: 10.1093/nar/gkg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H., Rauch T., Chen Z.X., Szabo P.E., Riggs A.D., Pfeifer G.P. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J. Biol. Chem. 2006;281:19489–19500. doi: 10.1074/jbc.M513249200. [DOI] [PubMed] [Google Scholar]
- 34.Chang Y., Sun L., Kokura K., Horton J.R., Fukuda M., Espejo A., Izumi V., Koomen J.M., Bedford M.T., Zhang X., Shinkai Y., Fang J., Cheng X. MPP8 mediates the interactions between DNA methyltransferase Dnmt3a and H3K9 methyltransferase GLP/G9a. Nat. Commun. 2011;2:533. doi: 10.1038/ncomms1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Epsztejn-Litman S., Feldman N., Abu-Remaileh M., Shufaro Y., Gerson A., Ueda J., Deplus R., Fuks F., Shinkai Y., Cedar H., Bergman Y. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat. Struct. Mol. Biol. 2008;15:1176–1183. doi: 10.1038/nsmb.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collins R.E., Northrop J.P., Horton J.R., Lee D.Y., Zhang X., Stallcup M.R., Cheng X. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nat. Struct. Mol. Biol. 2008;15:245–250. doi: 10.1038/nsmb.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smallwood A., Esteve P.O., Pradhan S., Carey M. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev. 2007;21:1169–1178. doi: 10.1101/gad.1536807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edmunds J.W., Mahadevan L.C., Clayton A.L. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008;27:406–420. doi: 10.1038/sj.emboj.7601967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun X.J., Wei J., Wu X.Y., Hu M., Wang L., Wang H.H., Zhang Q.H., Chen S.J., Huang Q.H., Chen Z. Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J. Biol. Chem. 2005;280:35261–35271. doi: 10.1074/jbc.M504012200. [DOI] [PubMed] [Google Scholar]
- 40.Kolasinska-Zwierz P., Down T., Latorre I., Liu T., Liu X.S., Ahringer J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat. Genet. 2009;41:376–381. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spies N., Nielsen C.B., Padgett R.A., Burge C.B. Biased chromatin signatures around polyadenylation sites and exons. Mol. Cell. 2009;36:245–254. doi: 10.1016/j.molcel.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andersson R., Enroth S., Rada-Iglesias A., Wadelius C., Komorowski J. Nucleosomes are well positioned in exons and carry characteristic histone modifications. Genome Res. 2009;19:1732–1741. doi: 10.1101/gr.092353.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huff J.T., Plocik A.M., Guthrie C., Yamamoto K.R. Reciprocal intronic and exonic histone modification regions in humans. Nat. Struct. Mol. Biol. 2010;17:1495–1499. doi: 10.1038/nsmb.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lorincz M.C., Dickerson D.R., Schmitt M., Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat. Struct. Mol. Biol. 2004;11:1068–1075. doi: 10.1038/nsmb840. [DOI] [PubMed] [Google Scholar]
- 45.Brown S.J., Stoilov P., Xing Y. Chromatin and epigenetic regulation of pre-mRNA processing. Hum. Mol. Genet. 2012;21:R90–96. doi: 10.1093/hmg/dds353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kobayashi H., Sakurai T., Imai M., Takahashi N., Fukuda A., Yayoi O., Sato S., Nakabayashi K., Hata K., Sotomaru Y., Suzuki Y., Kono T. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012;8:e1002440. doi: 10.1371/journal.pgen.1002440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dhayalan A., Rajavelu A., Rathert P., Tamas R., Jurkowska R.Z., Ragozin S., Jeltsch A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 2010;285:26114–26120. doi: 10.1074/jbc.M109.089433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jeong S., Liang G., Sharma S., Lin J.C., Choi S.H., Han H., Yoo C.B., Egger G., Yang A.S., Jones P.A. Selective anchoring of DNA methyltransferases 3A and 3B to nucleosomes containing methylated DNA. Mol. Cell. Biol. 2009;29:5366–5376. doi: 10.1128/MCB.00484-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ge Y.Z., Pu M.T., Gowher H., Wu H.P., Ding J.P., Jeltsch A., Xu G.L. Chromatin targeting of de novo DNA methyltransferases by the PWWP domain. J. Biol. Chem. 2004;279:25447–25454. doi: 10.1074/jbc.M312296200. [DOI] [PubMed] [Google Scholar]
- 50.Chen T., Tsujimoto N., Li E. The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol. Cell. Biol. 2004;24:9048–9058. doi: 10.1128/MCB.24.20.9048-9058.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shirohzu H., Kubota T., Kumazawa A., Sado T., Chijiwa T., Inagaki K., Suetake I., Tajima S., Wakui K., Miki Y., Hayashi M., Fukushima Y., Sasaki H. Three novel DNMT3B mutations in Japanese patients with ICF syndrome. Am. J. Med. Genet. 2002;112:31–37. doi: 10.1002/ajmg.10658. [DOI] [PubMed] [Google Scholar]
- 52.Heyn H., Vidal E., Sayols S., Sanchez-Mut J.V., Moran S., Medina I., Sandoval J., Simo-Riudalbas L., Szczesna K., Huertas D., Gatto S., Matarazzo M.R., Dopazo J., Esteller M. Whole-genome bisulfite DNA sequencing of a DNMT3B mutant patient. Epigenetics. 2012;7:542–550. doi: 10.4161/epi.20523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hahn M.A., Wu X., Li A.X., Hahn T., Pfeifer G.P. Relationship between gene body DNA methylation and intragenic H3K9me3 and H3K36me3 chromatin marks. PLoS One. 2011;6:e18844. doi: 10.1371/journal.pone.0018844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weber M., Hellmann I., Stadler M.B., Ramos L., Paabo S., Rebhan M., Schubeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 55.Ooi S.K., Qiu C., Bernstein E., Li K., Jia D., Yang Z., Erdjument-Bromage H., Tempst P., Lin S.P., Allis C.D., Cheng X., Bestor T.H. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bourc'his D., Bestor T.H. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature. 2004;431:96–99. doi: 10.1038/nature02886. [DOI] [PubMed] [Google Scholar]
- 57.Bourc'his D., Xu G.L., Lin C.S., Bollman B., Bestor T.H. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–2539. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- 58.Kaneda M., Okano M., Hata K., Sado T., Tsujimoto N., Li E., Sasaki H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–903. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- 59.Hata K., Okano M., Lei H., Li E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development. 2002;129:1983–1993. doi: 10.1242/dev.129.8.1983. [DOI] [PubMed] [Google Scholar]
- 60.Hu J.L., Zhou B.O., Zhang R.R., Zhang K.L., Zhou J.Q., Xu G.L. The N-terminus of histone H3 is required for de novo DNA methylation in chromatin. Proc. Natl. Acad. Sci. U. S. A. 2009;106:22187–22192. doi: 10.1073/pnas.0905767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jia D., Jurkowska R.Z., Zhang X., Jeltsch A., Cheng X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature. 2007;449:248–251. doi: 10.1038/nature06146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kareta M.S., Botello Z.M., Ennis J.J., Chou C., Chedin F. Reconstitution and mechanism of the stimulation of de novo methylation by human DNMT3L. J. Biol. Chem. 2006;281:25893–25902. doi: 10.1074/jbc.M603140200. [DOI] [PubMed] [Google Scholar]
- 63.Li B.Z., Huang Z., Cui Q.Y., Song X.H., Du L., Jeltsch A., Chen P., Li G., Li E., Xu G.L. Histone tails regulate DNA methylation by allosterically activating de novo methyltransferase. Cell Res. 2011;21:1172–1181. doi: 10.1038/cr.2011.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ciccone D.N., Su H., Hevi S., Gay F., Lei H., Bajko J., Xu G., Li E., Chen T. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature. 2009;461:415–418. doi: 10.1038/nature08315. [DOI] [PubMed] [Google Scholar]
- 65.Wang J., Hevi S., Kurash J.K., Lei H., Gay F., Bajko J., Su H., Sun W., Chang H., Xu G., Gaudet F., Li E., Chen T. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 2009;41:125–129. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- 66.Vermeulen M., Mulder K.W., Denissov S., Pijnappel W.W., van Schaik F.M., Varier R.A., Baltissen M.P., Stunnenberg H.G., Mann M., Timmers H.T. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 67.Eberl H.C., Spruijt C.G., Kelstrup C.D., Vermeulen M., Mann M. A map of general and specialized chromatin readers in mouse tissues generated by label-free interaction proteomics. Mol. Cell. 2013;49:368–378. doi: 10.1016/j.molcel.2012.10.026. [DOI] [PubMed] [Google Scholar]
- 68.Horton J.R., Upadhyay A.K., Qi H.H., Zhang X., Shi Y., Cheng X. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat. Struct. Mol. Biol. 2010;17:38–43. doi: 10.1038/nsmb.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.La Salle S., Mertineit C., Taketo T., Moens P.B., Bestor T.H., Trasler J.M. Windows for sex-specific methylation marked by DNA methyltransferase expression profiles in mouse germ cells. Dev. Biol. 2004;268:403–415. doi: 10.1016/j.ydbio.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 70.Webster K.E., O'Bryan M.K., Fletcher S., Crewther P.E., Aapola U., Craig J., Harrison D.K., Aung H., Phutikanit N., Lyle R., Meachem S.J., Antonarakis S.E., de Kretser D.M., Hedger M.P., Peterson P., Carroll B.J., Scott H.S. Meiotic and epigenetic defects in Dnmt3L-knockout mouse spermatogenesis. Proc. Natl. Acad. Sci. U. S. A. 2005;102:4068–4073. doi: 10.1073/pnas.0500702102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liao H.F., Tai K.Y., Chen W.S., Cheng L.C., Ho H.N., Lin S.P. Functions of DNA methyltransferase 3-like in germ cells and beyond. Biol. Cell. 2012;104:571–587. doi: 10.1111/boc.201100109. [DOI] [PubMed] [Google Scholar]
- 72.Gordon C.A., Hartono S.R., Chedin F. Inactive DNMT3B splice variants modulate de novo DNA methylation. PLoS One. 2013;8:e69486. doi: 10.1371/journal.pone.0069486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Van Emburgh B.O., Robertson K.D. Modulation of Dnmt3b function in vitro by interactions with Dnmt3L, Dnmt3a and Dnmt3b splice variants. Nucleic Acids Res. 2011;39:4984–5002. doi: 10.1093/nar/gkr116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Statham A.L., Robinson M.D., Song J.Z., Coolen M.W., Stirzaker C., Clark S.J. Bisulfite sequencing of chromatin immunoprecipitated DNA (BisChIP-seq) directly informs methylation status of histone-modified DNA. Genome Res. 2012;22:1120–1127. doi: 10.1101/gr.132076.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brinkman A.B., Gu H., Bartels S.J., Zhang Y., Matarese F., Simmer F., Marks H., Bock C., Gnirke A., Meissner A., Stunnenberg H.G. Sequential ChIP–bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res. 2012;22:1128–1138. doi: 10.1101/gr.133728.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mohn F., Weber M., Rebhan M., Roloff T.C., Richter J., Stadler M.B., Bibel M., Schubeler D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell. 2008;30:755–766. doi: 10.1016/j.molcel.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 77.Schlesinger Y., Straussman R., Keshet I., Farkash S., Hecht M., Zimmerman J., Eden E., Yakhini Z., Ben-Shushan E., Reubinoff B.E., Bergman Y., Simon I., Cedar H. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007;39:232–236. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- 78.Ohm J.E., McGarvey K.M., Yu X., Cheng L., Schuebel K.E., Cope L., Mohammad H.P., Chen W., Daniel V.C., Yu W., Berman D.M., Jenuwein T., Pruitt K., Sharkis S.J., Watkins D.N., Herman J.G., Baylin S.B. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 2007;39:237–242. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vire E., Brenner C., Deplus R., Blanchon L., Fraga M., Didelot C., Morey L., Van Eynde A., Bernard D., Vanderwinden J.M., Bollen M., Esteller M., Di Croce L., de Launoit Y., Fuks F. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 80.Neri F., Krepelova A., Incarnato D., Maldotti M., Parlato C., Galvagni F., Matarese F., Stunnenberg H.G., Oliviero S. Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs. Cell. 2013;155:121–134. doi: 10.1016/j.cell.2013.08.056. [DOI] [PubMed] [Google Scholar]
- 81.Neri F., Incarnato D., Krepelova A., Rapelli S., Pagnani A., Zecchina R., Parlato C., Oliviero S. Genome-wide analysis identifies a functional association of Tet1 and Polycomb PRC2 in mouse embryonic stem cells. Genome Biol. 2013;14:R91. doi: 10.1186/gb-2013-14-8-r91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jeltsch A. On the enzymatic properties of Dnmt1: specificity, processivity, mechanism of linear diffusion and allosteric regulation of the enzyme. Epigenetics. 2006;1:63–66. doi: 10.4161/epi.1.2.2767. [DOI] [PubMed] [Google Scholar]
- 83.Rothbart S.B., Dickson B.M., Ong M.S., Krajewski K., Houliston S., Kireev D.B., Arrowsmith C.H., Strahl B.D. Multivalent histone engagement by the linked tandem Tudor and PHD domains of UHRF1 is required for the epigenetic inheritance of DNA methylation. Genes Dev. 2013;27:1288–1298. doi: 10.1101/gad.220467.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nady N., Lemak A., Walker J.R., Avvakumov G.V., Kareta M.S., Achour M., Xue S., Duan S., Allali-Hassani A., Zuo X., Wang Y.X., Bronner C., Chedin F., Arrowsmith C.H., Dhe-Paganon S. Recognition of multivalent histone states associated with heterochromatin by UHRF1 protein. J. Biol. Chem. 2011;286:24300–24311. doi: 10.1074/jbc.M111.234104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hashimoto H., Horton J.R., Zhang X., Bostick M., Jacobsen S.E., Cheng X. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature. 2008;455:826–829. doi: 10.1038/nature07280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Qian C., Li S., Jakoncic J., Zeng L., Walsh M.J., Zhou M.M. Structure and hemimethylated CpG binding of the SRA domain from human UHRF1. J. Biol. Chem. 2008;283:34490–34494. doi: 10.1074/jbc.C800169200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Arita K., Ariyoshi M., Tochio H., Nakamura Y., Shirakawa M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008;455:818–821. doi: 10.1038/nature07249. [DOI] [PubMed] [Google Scholar]
- 88.Avvakumov G.V., Walker J.R., Xue S., Li Y., Duan S., Bronner C., Arrowsmith C.H., Dhe-Paganon S. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature. 2008;455:822–825. doi: 10.1038/nature07273. [DOI] [PubMed] [Google Scholar]
- 89.Bostick M., Kim J.K., Esteve P.O., Clark A., Pradhan S., Jacobsen S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317:1760–1764. doi: 10.1126/science.1147939. [DOI] [PubMed] [Google Scholar]
- 90.Sharif J., Muto M., Takebayashi S., Suetake I., Iwamatsu A., Endo T.A., Shinga J., Mizutani-Koseki Y., Toyoda T., Okamura K., Tajima S., Mitsuya K., Okano M., Koseki H. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450:908–912. doi: 10.1038/nature06397. [DOI] [PubMed] [Google Scholar]
- 91.Song J., Teplova M., Ishibe-Murakami S., Patel D.J. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science. 2012;335:709–712. doi: 10.1126/science.1214453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Song J., Rechkoblit O., Bestor T.H., Patel D.J. Structure of DNMT1–DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science. 2011;331:1036–1040. doi: 10.1126/science.1195380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bashtrykov P., Jankevicius G., Smarandache A., Jurkowska R.Z., Ragozin S., Jeltsch A. Specificity of Dnmt1 for methylation of hemimethylated CpG sites resides in its catalytic domain. Chem. Biol. 2012;19:572–578. doi: 10.1016/j.chembiol.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 94.Hashimoto H., Horton J.R., Zhang X., Cheng X. UHRF1, a modular multi-domain protein, regulates replication-coupled crosstalk between DNA methylation and histone modifications. Epigenetics. 2009;4:8–14. doi: 10.4161/epi.4.1.7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hashimoto H., Vertino P.M., Cheng X. Molecular coupling of DNA methylation and histone methylation. Epigenomics. 2010;2:657–669. doi: 10.2217/epi.10.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Syeda F., Fagan R.L., Wean M., Avvakumov G.V., Walker J.R., Xue S., Dhe-Paganon S., Brenner C. The replication focus targeting sequence (RFTS) domain is a DNA-competitive inhibitor of Dnmt1. J. Biol. Chem. 2011;286:15344–15351. doi: 10.1074/jbc.M110.209882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Takeshita K., Suetake I., Yamashita E., Suga M., Narita H., Nakagawa A., Tajima S. Structural insight into maintenance methylation by mouse DNA methyltransferase 1 (Dnmt1) Proc. Natl. Acad. Sci. U. S. A. 2011;108:9055–9059. doi: 10.1073/pnas.1019629108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Achour M., Jacq X., Ronde P., Alhosin M., Charlot C., Chataigneau T., Jeanblanc M., Macaluso M., Giordano A., Hughes A.D., Schini-Kerth V.B., Bronner C. The interaction of the SRA domain of ICBP90 with a novel domain of DNMT1 is involved in the regulation of VEGF gene expression. Oncogene. 2008;27:2187–2197. doi: 10.1038/sj.onc.1210855. [DOI] [PubMed] [Google Scholar]
- 99.Nishiyama A., Yamaguchi L., Sharif J., Johmura Y., Kawamura T., Nakanishi K., Shimamura S., Arita K., Kodama T., Ishikawa F., Koseki H., Nakanishi M. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature. 2013;502:249–253. doi: 10.1038/nature12488. [DOI] [PubMed] [Google Scholar]
- 100.Liu X., Gao Q., Li P., Zhao Q., Zhang J., Li J., Koseki H., Wong J. UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat. Commun. 2013;4:1563. doi: 10.1038/ncomms2562. [DOI] [PubMed] [Google Scholar]
- 101.Rottach A., Frauer C., Pichler G., Bonapace I.M., Spada F., Leonhardt H. The multi-domain protein Np95 connects DNA methylation and histone modification. Nucleic Acids Res. 2010;38:1796–1804. doi: 10.1093/nar/gkp1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Papait R., Pistore C., Grazini U., Babbio F., Cogliati S., Pecoraro D., Brino L., Morand A.L., Dechampesme A.M., Spada F., Leonhardt H., McBlane F., Oudet P., Bonapace I.M. The PHD domain of Np95 (mUHRF1) is involved in large-scale reorganization of pericentromeric heterochromatin. Mol. Biol. Cell. 2008;19:3554–3563. doi: 10.1091/mbc.E07-10-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang J., Gao Q., Li P., Liu X., Jia Y., Wu W., Li J., Dong S., Koseki H., Wong J. S phase-dependent interaction with DNMT1 dictates the role of UHRF1 but not UHRF2 in DNA methylation maintenance. Cell Res. 2011;21:1723–1739. doi: 10.1038/cr.2011.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pichler G., Wolf P., Schmidt C.S., Meilinger D., Schneider K., Frauer C., Fellinger K., Rottach A., Leonhardt H. Cooperative DNA and histone binding by Uhrf2 links the two major repressive epigenetic pathways. J. Cell. Biochem. 2011;112:2585–2593. doi: 10.1002/jcb.23185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rothbart S.B., Krajewski K., Nady N., Tempel W., Xue S., Badeaux A.I., Barsyte-Lovejoy D., Martinez J.Y., Bedford M.T., Fuchs S.M., Arrowsmith C.H., Strahl B.D. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat. Struct. Mol. Biol. 2012;19:1155–1160. doi: 10.1038/nsmb.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Esteve P.O., Chang Y., Samaranayake M., Upadhyay A.K., Horton J.R., Feehery G.R., Cheng X., Pradhan S. A methylation and phosphorylation switch between an adjacent lysine and serine determines human DNMT1 stability. Nat. Struct. Mol. Biol. 2011;18:42–48. doi: 10.1038/nsmb.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Esteve P.O., Chin H.G., Benner J., Feehery G.R., Samaranayake M., Horwitz G.A., Jacobsen S.E., Pradhan S. Regulation of DNMT1 stability through SET7-mediated lysine methylation in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 2009;106:5076–5081. doi: 10.1073/pnas.0810362106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kim J.K., Esteve P.O., Jacobsen S.E., Pradhan S. UHRF1 binds G9a and participates in p21 transcriptional regulation in mammalian cells. Nucleic Acids Res. 2009;37:493–505. doi: 10.1093/nar/gkn961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Babbio F., Pistore C., Curti L., Castiglioni I., Kunderfranco P., Brino L., Oudet P., Seiler R., Thalman G.N., Roggero E., Sarti M., Pinton S., Mello-Grand M., Chiorino G., Catapano C.V., Carbone G.M., Bonapace I.M. The SRA protein UHRF1 promotes epigenetic crosstalks and is involved in prostate cancer progression. Oncogene. 2012;31:4878–4887. doi: 10.1038/onc.2011.641. [DOI] [PubMed] [Google Scholar]
- 110.Esteve P.O., Chin H.G., Smallwood A., Feehery G.R., Gangisetty O., Karpf A.R., Carey M.F., Pradhan S. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 2006;20:3089–3103. doi: 10.1101/gad.1463706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nakamura T., Liu Y.J., Nakashima H., Umehara H., Inoue K., Matoba S., Tachibana M., Ogura A., Shinkai Y., Nakano T. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature. 2012;486:415–419. doi: 10.1038/nature11093. [DOI] [PubMed] [Google Scholar]
- 112.Szabo P.E., Pfeifer G.P. H3K9me2 attracts PGC7 in the zygote to prevent Tet3-mediated oxidation of 5-methylcytosine. J. Mol. Cell Biol. 2012;4:427–429. doi: 10.1093/jmcb/mjs038. [DOI] [PubMed] [Google Scholar]
- 113.Quenneville S., Verde G., Corsinotti A., Kapopoulou A., Jakobsson J., Offner S., Baglivo I., Pedone P.V., Grimaldi G., Riccio A., Trono D. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol. Cell. 2011;44:361–372. doi: 10.1016/j.molcel.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zuo X., Sheng J., Lau H.T., McDonald C.M., Andrade M., Cullen D.E., Bell F.T., Iacovino M., Kyba M., Xu G., Li X. Zinc finger protein ZFP57 requires its co-factor to recruit DNA methyltransferases and maintains DNA methylation imprint in embryonic stem cells via its transcriptional repression domain. J. Biol. Chem. 2012;287:2107–2118. doi: 10.1074/jbc.M111.322644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Messerschmidt D.M., de Vries W., Ito M., Solter D., Ferguson-Smith A., Knowles B.B. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science. 2012;335:1499–1502. doi: 10.1126/science.1216154. [DOI] [PubMed] [Google Scholar]
- 116.Voo K.S., Carlone D.L., Jacobsen B.M., Flodin A., Skalnik D.G. Cloning of a mammalian transcriptional activator that binds unmethylated CpG motifs and shares a CXXC domain with DNA methyltransferase, human trithorax, and methyl-CpG binding domain protein 1. Mol. Cell. Biol. 2000;20:2108–2121. doi: 10.1128/mcb.20.6.2108-2121.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Long H.K., Blackledge N.P., Klose R.J. ZF-CxxC domain-containing proteins, CpG islands and the chromatin connection. Biochem. Soc. Trans. 2013;41:727–740. doi: 10.1042/BST20130028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bernstein B.E., Kamal M., Lindblad-Toh K., Bekiranov S., Bailey D.K., Huebert D.J., McMahon S., Karlsson E.K., Kulbokas E.J., III, Gingeras T.R., Schreiber S.L., Lander E.S. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 119.Lauberth S.M., Nakayama T., Wu X., Ferris A.L., Tang Z., Hughes S.H., Roeder R.G. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell. 2013;152:1021–1036. doi: 10.1016/j.cell.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Thomson J.P., Skene P.J., Selfridge J., Clouaire T., Guy J., Webb S., Kerr A.R., Deaton A., Andrews R., James K.D., Turner D.J., Illingworth R., Bird A. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature. 2010;464:1082–1086. doi: 10.1038/nature08924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Clouaire T., Webb S., Skene P., Illingworth R., Kerr A., Andrews R., Lee J.H., Skalnik D., Bird A. Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev. 2012;26:1714–1728. doi: 10.1101/gad.194209.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Farcas A.M., Blackledge N.P., Sudbery I., Long H.K., McGouran J.F., Rose N.R., Lee S., Sims D., Cerase A., Sheahan T.W., Koseki H., Brockdorff N., Ponting C.P., Kessler B.M., Klose R.J. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. eLife. 2012;1:e00205. doi: 10.7554/eLife.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wu X., Johansen J.V., Helin K. Fbxl10/Kdm2b recruits polycomb repressive complex 1 to CpG islands and regulates H2A ubiquitylation. Mol. Cell. 2013;49:1134–1146. doi: 10.1016/j.molcel.2013.01.016. [DOI] [PubMed] [Google Scholar]
- 125.He J., Shen L., Wan M., Taranova O., Wu H., Zhang Y. Kdm2b maintains murine embryonic stem cell status by recruiting PRC1 complex to CpG islands of developmental genes. Nat. Cell Biol. 2013;15:373–384. doi: 10.1038/ncb2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wu M., Wang P.F., Lee J.S., Martin-Brown S., Florens L., Washburn M., Shilatifard A. Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol. Cell. Biol. 2008;28:7337–7344. doi: 10.1128/MCB.00976-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang P., Lin C., Smith E.R., Guo H., Sanderson B.W., Wu M., Gogol M., Alexander T., Seidel C., Wiedemann L.M., Ge K., Krumlauf R., Shilatifard A. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol. Cell. Biol. 2009;29:6074–6085. doi: 10.1128/MCB.00924-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hu D., Garruss A.S., Gao X., Morgan M.A., Cook M., Smith E.R., Shilatifard A. The Mll2 branch of the COMPASS family regulates bivalent promoters in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 2013;20:1093–1097. doi: 10.1038/nsmb.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hu D., Garruss A.S., Gao X., Morgan M.A., Cook M., Smith E.R., Shilatifard A. The Mll2 branch of the COMPASS family regulates bivalent promoters in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 2013;20:1093–1097. doi: 10.1038/nsmb.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lin C., Smith E.R., Takahashi H., Lai K.C., Martin-Brown S., Florens L., Washburn M.P., Conaway J.W., Conaway R.C., Shilatifard A. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol. Cell. 2010;37:429–437. doi: 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yokoyama A., Lin M., Naresh A., Kitabayashi I., Cleary M.L. A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell. 2010;17:198–212. doi: 10.1016/j.ccr.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Milne T.A., Kim J., Wang G.G., Stadler S.C., Basrur V., Whitcomb S.J., Wang Z., Ruthenburg A.J., Elenitoba-Johnson K.S., Roeder R.G., Allis C.D. Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Mol. Cell. 2010;38:853–863. doi: 10.1016/j.molcel.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Okada Y., Feng Q., Lin Y., Jiang Q., Li Y., Coffield V.M., Su L., Xu G., Zhang Y. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 134.Krivtsov A.V., Feng Z., Lemieux M.E., Faber J., Vempati S., Sinha A.U., Xia X., Jesneck J., Bracken A.P., Silverman L.B., Kutok J.L., Kung A.L., Armstrong S.A. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14:355–368. doi: 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Biswas D., Milne T.A., Basrur V., Kim J., Elenitoba-Johnson K.S., Allis C.D., Roeder R.G. Function of leukemogenic mixed lineage leukemia 1 (MLL) fusion proteins through distinct partner protein complexes. Proc. Natl. Acad. Sci. U. S. A. 2011;108:15751–15756. doi: 10.1073/pnas.1111498108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Voigt P., Tee W.W., Reinberg D. A double take on bivalent promoters. Genes Dev. 2013;27:1318–1338. doi: 10.1101/gad.219626.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Capotosti F., Guernier S., Lammers F., Waridel P., Cai Y., Jin J., Conaway J.W., Conaway R.C., Herr W. O-GlcNAc transferase catalyzes site-specific proteolysis of HCF-1. Cell. 2011;144:376–388. doi: 10.1016/j.cell.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 138.Chen Q., Chen Y., Bian C., Fujiki R., Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature. 2013;493:561–564. doi: 10.1038/nature11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Deplus R., Delatte B., Schwinn M.K., Defrance M., Mendez J., Murphy N., Dawson M.A., Volkmar M., Putmans P., Calonne E., Shih A.H., Levine R.L., Bernard O., Mercher T., Solary E., Urh M., Daniels D.L., Fuks F. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013;32:645–655. doi: 10.1038/emboj.2012.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Vella P., Scelfo A., Jammula S., Chiacchiera F., Williams K., Cuomo A., Roberto A., Christensen J., Bonaldi T., Helin K., Pasini D. Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol. Cell. 2013;49:645–656. doi: 10.1016/j.molcel.2012.12.019. [DOI] [PubMed] [Google Scholar]
- 141.Simon J.A., Kingston R.E. Occupying chromatin: polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol. Cell. 2013;49:808–824. doi: 10.1016/j.molcel.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Grau D.J., Chapman B.A., Garlick J.D., Borowsky M., Francis N.J., Kingston R.E. Compaction of chromatin by diverse Polycomb group proteins requires localized regions of high charge. Genes Dev. 2011;25:2210–2221. doi: 10.1101/gad.17288211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lehmann L., Ferrari R., Vashisht A.A., Wohlschlegel J.A., Kurdistani S.K., Carey M. Polycomb repressive complex 1 (PRC1) disassembles RNA polymerase II preinitiation complexes. J. Biol. Chem. 2012;287:35784–35794. doi: 10.1074/jbc.M112.397430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Min I.M., Waterfall J.J., Core L.J., Munroe R.J., Schimenti J., Lis J.T. Regulating RNA polymerase pausing and transcription elongation in embryonic stem cells. Genes Dev. 2011;25:742–754. doi: 10.1101/gad.2005511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mikkelsen T.S., Ku M., Jaffe D.B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T.K., Koche R.P., Lee W., Mendenhall E., O'Donovan A., Presser A., Russ C., Xie X., Meissner A., Wernig M., Jaenisch R., Nusbaum C., Lander E.S., Bernstein B.E. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hagarman J.A., Motley M.P., Kristjansdottir K., Soloway P.D. Coordinate regulation of DNA methylation and H3K27me3 in mouse embryonic stem cells. PLoS ONE. 2013;8:e53880. doi: 10.1371/journal.pone.0053880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Peng J.C., Valouev A., Swigut T., Zhang J., Zhao Y., Sidow A., Wysocka J. Jarid2/Jumonji coordinates control of PRC2 enzymatic activity and target gene occupancy in pluripotent cells. Cell. 2009;139:1290–1302. doi: 10.1016/j.cell.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Li G., Margueron R., Ku M., Chambon P., Bernstein B.E., Reinberg D. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev. 2010;24:368–380. doi: 10.1101/gad.1886410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kim H., Kang K., Kim J. AEBP2 as a potential targeting protein for Polycomb Repression Complex PRC2. Nucleic Acids Res. 2009;37:2940–2950. doi: 10.1093/nar/gkp149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ciferri C., Lander G.C., Maiolica A., Herzog F., Aebersold R., Nogales E. Molecular architecture of human polycomb repressive complex 2. eLife. 2012;1:e00005. doi: 10.7554/eLife.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cao R., Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED–EZH2 complex. Mol. Cell. 2004;15:57–67. doi: 10.1016/j.molcel.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 152.Brien G.L., Gambero G., O'Connell D.J., Jerman E., Turner S.A., Egan C.M., Dunne E.J., Jurgens M.C., Wynne K., Piao L., Lohan A.J., Ferguson N., Shi X., Sinha K.M., Loftus B.J., Cagney G., Bracken A.P. Polycomb PHF19 binds H3K36me3 and recruits PRC2 and demethylase NO66 to embryonic stem cell genes during differentiation. Nat. Struct. Mol. Biol. 2012;19:1273–1281. doi: 10.1038/nsmb.2449. [DOI] [PubMed] [Google Scholar]
- 153.Ballare C., Lange M., Lapinaite A., Martin G.M., Morey L., Pascual G., Liefke R., Simon B., Shi Y., Gozani O., Carlomagno T., Benitah S.A., Di Croce L. Phf19 links methylated Lys36 of histone H3 to regulation of Polycomb activity. Nat. Struct. Mol. Biol. 2012;19:1257–1265. doi: 10.1038/nsmb.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Musselman C.A., Avvakumov N., Watanabe R., Abraham C.G., Lalonde M.E., Hong Z., Allen C., Roy S., Nunez J.K., Nickoloff J., Kulesza C.A., Yasui A., Cote J., Kutateladze T.G. Molecular basis for H3K36me3 recognition by the Tudor domain of PHF1. Nat. Struct. Mol. Biol. 2012;19:1266–1272. doi: 10.1038/nsmb.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sarma K., Margueron R., Ivanov A., Pirrotta V., Reinberg D. Ezh2 requires PHF1 to efficiently catalyze H3 lysine 27 trimethylation in vivo. Mol. Cell. Biol. 2008;28:2718–2731. doi: 10.1128/MCB.02017-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mendenhall E.M., Koche R.P., Truong T., Zhou V.W., Issac B., Chi A.S., Ku M., Bernstein B.E. GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet. 2010;6:e1001244. doi: 10.1371/journal.pgen.1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Schmitges F.W., Prusty A.B., Faty M., Stutzer A., Lingaraju G.M., Aiwazian J., Sack R., Hess D., Li L., Zhou S., Bunker R.D., Wirth U., Bouwmeester T., Bauer A., Ly-Hartig N., Zhao K., Chan H., Gu J., Gut H., Fischle W., Muller J., Thoma N.H. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell. 2011;42:330–341. doi: 10.1016/j.molcel.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 158.Yuan W., Xu M., Huang C., Liu N., Chen S., Zhu B. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 2011;286:7983–7989. doi: 10.1074/jbc.M110.194027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Klose R.J., Cooper S., Farcas A.M., Blackledge N.P., Brockdorff N. Chromatin sampling—an emerging perspective on targeting polycomb repressor proteins. PLoS Genet. 2013;9:e1003717. doi: 10.1371/journal.pgen.1003717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Carrozza M.J., Li B., Florens L., Suganuma T., Swanson S.K., Lee K.K., Shia W.J., Anderson S., Yates J., Washburn M.P., Workman J.L. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 161.Venkatesh S., Smolle M., Li H., Gogol M.M., Saint M., Kumar S., Natarajan K., Workman J.L. Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature. 2012;489:452–455. doi: 10.1038/nature11326. [DOI] [PubMed] [Google Scholar]
- 162.Youdell M.L., Kizer K.O., Kisseleva-Romanova E., Fuchs S.M., Duro E., Strahl B.D., Mellor J. Roles for Ctk1 and Spt6 in regulating the different methylation states of histone H3 lysine 36. Mol. Cell. Biol. 2008;28:4915–4926. doi: 10.1128/MCB.00001-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Li B., Jackson J., Simon M.D., Fleharty B., Gogol M., Seidel C., Workman J.L., Shilatifard A. Histone H3 lysine 36 dimethylation (H3K36me2) is sufficient to recruit the Rpd3s histone deacetylase complex and to repress spurious transcription. J. Biol. Chem. 2009;284:7970–7976. doi: 10.1074/jbc.M808220200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Blackledge N.P., Zhou J.C., Tolstorukov M.Y., Farcas A.M., Park P.J., Klose R.J. CpG islands recruit a histone H3 lysine 36 demethylase. Mol. Cell. 2010;38:179–190. doi: 10.1016/j.molcel.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yuan G., Ma B., Yuan W., Zhang Z., Chen P., Ding X., Feng L., Shen X., Chen S., Li G., Zhu B. Histone H2A ubiquitination inhibits the enzymatic activity of H3 Lysine 36 methyltransferases. J. Biol. Chem. 2013;288:30832–30842. doi: 10.1074/jbc.M113.475996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Frauer C., Hoffmann T., Bultmann S., Casa V., Cardoso M.C., Antes I., Leonhardt H. Recognition of 5-hydroxymethylcytosine by the Uhrf1 SRA domain. PLoS One. 2011;6:e21306. doi: 10.1371/journal.pone.0021306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Spruijt C.G., Gnerlich F., Smits A.H., Pfaffeneder T., Jansen P.W., Bauer C., Munzel M., Wagner M., Muller M., Khan F., Eberl H.C., Mensinga A., Brinkman A.B., Lephikov K., Muller U., Walter J., Boelens R., van Ingen H., Leonhardt H., Carell T., Vermeulen M. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–1159. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 168.Jin L., Hanigan C.L., Wu Y., Wang W., Park B.H., Woster P.M., Casero R.A. Loss of LSD1 (lysine-specific demethylase 1) suppresses growth and alters gene expression of human colon cancer cells in a p53- and DNMT1(DNA methyltransferase 1)-independent manner. Biochemical Journal. 2013;449:459–468. doi: 10.1042/BJ20121360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Francis N.J., Follmer N.E., Simon M.D., Aghia G., Butler J.D. Polycomb Proteins Remain Bound to Chromatin and DNA during DNA Replication In Vitro. Cell. 2009;137:110–122. doi: 10.1016/j.cell.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Petruk S., Sedkov Y., Johnston Danika M., Hodgson Jacob W., Black Kathryn L., Kovermann Sina K., Beck S., Canaani E., Brock Hugh W., Mazo A. TrxG and PcG Proteins but Not Methylated Histones Remain Associated with DNA through Replication. Cell. 2012;150:922–933. doi: 10.1016/j.cell.2012.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]