

Abstract
Alzheimer's disease (AD) is a neurodegenerative disease exhibiting amyloid beta (Aβ) peptide accumulation as a key characteristic. Autophagy, which is dysregulated in AD, participates in the metabolism of Aβ. Unexpectedly, we recently found that autophagy, in addition to its degradative function, also mediates the secretion of Aβ. This finding adds Aβ to an increasing number of biomolecules, the secretion of which is mediated by autophagy. We also showed that inhibition of Aβ secretion through genetic deletion of autophagy leads to intracellular Aβ accumulation, which enhanced neurodegeneration induced by autophagy deficiency. Hence, autophagy may play a central role in two pathological hallmarks of AD: Aβ amyloidosis and neurodegeneration. Herein, we summarize the role of autophagy in AD with focus on Aβ metabolism in light of the recently established role of autophagy in protein secretion. We discuss potential routes for autophagy-mediated Aβ secretion and suggest experimental approaches to further elucidate its mechanisms.
Keywords: Alzheimer's disease, Aβ secretion, autophagy, neurodegeneration
Introduction
Alzheimer's disease is the main cause of dementia in the elderly. More than 30 million people are affected worldwide. Additionally, because the main risk factor for AD is age, the number of AD patients is rapidly growing with increased life expectancy. Current AD medications ameliorate the symptoms either by maintaining and enhancing the cholinergic signaling system in the brain [1] or by lowering excitotoxicity by antagonizing the NMDA receptor [2]. However, no disease-modifying treatment targeting the well-characterized pathologies of AD, amyloid beta (Aβ) plaque, and tau-containing neurofibrillary tangles (NFT), are yet available, though intensive research in this direction is ongoing.
The AD brain manifests increased levels of the aggregation-prone Aβ peptide. This increase in Aβ gives rise to intraneuronal Aβ accumulation already in early stage AD, the extent of which is influenced by the AD risk factor allele ApoE4, and later leads to the formation of extracellular Aβ plaques [3–8]. Aβ is generated from Aβ precursor protein (APP) through proteolytic cleavage by β- and γ-secretase, and is secreted at the PM [9]. In addition to Aβ, hyperphosphorylated microtubule-associated tau protein aggregates intracellularly into the NFTs. The extent of NFT formation clinically correlates with the cognitive dysfunction of AD to a greater degree than does Aβ plaque load [10]. On the other hand, the mutations identified in APP and γ-secretase-associated presenilin 1 and 2, which cause aggressive early onset familial AD (FAD), strongly link Aβ to AD [11]. Although these FAD-linked mutations account for only a small percentage of all AD cases, they severely affect Aβ metabolism, which leads to increased levels of Aβ, especially the hydrophobic Aβ42 and Aβ43 [12]. This strong causative relationship between Aβ and AD has led to the amyloid cascade hypothesis, which predicts that Aβ accumulation precedes the NFT formation that causes the cognitive dysfunction observed in AD [13]. However, the mechanistic link between Aβ and NFTs has not yet been fully clarified. It also remains to be completely resolved how these two pathologies – separately or together – induce the dramatic synaptic loss and neurodegeneration that occurs in AD brains.
In addition to the long known Aβ plaques and NFTs, there is increased attention on autophagy and its role in AD development. Autophagy is an important clearance system for cellular waste, including toxic protein aggregates. Autophagy is impaired in AD as shown by the accumulation of autophagosomes in the dystrophic neurites [14]. Moreover, autophagy has been implicated in Aβ metabolism and is therefore a potential therapeutic target in AD treatment. Unexpectedly, we recently found that autophagy, in addition to its well-established degradative function, mediates the secretion of Aβ into the extracellular space [15]. Autophagy therefore directly influences and contributes to extracellular Aβ plaque formation. This finding creates a new perspective on the role of autophagy in AD and Aβ metabolism, which should be considered during the design of AD therapeutics targeting autophagy. Here, we summarize the role of autophagy in AD with special focus on Aβ metabolism, and discuss the implication of the recently established function of autophagy in Aβ secretion in relation to previous findings on autophagy-mediated secretion.
Autophagy is impaired in Alzheimer's disease
Autophagy regulates protein homeostasis in the cell, in parallel and sometimes in concert with the proteasome [16]. Hence, autophagy plays a vital role in cellular quality control. Autophagy maintains the cell by degrading cellular waste, which in turn generates free amino acids that are used for the synthesis of novel proteins. Waste material in the cytoplasm that is degraded by autophagy includes dysfunctional organelles, e.g. depolarized mitochondria and potentially toxic protein aggregates, including those exceeding the size limitation for proteasome-mediated degradation. The balance between proteasome- and autophagy-mediated degradation and protein synthesis is greatly influenced by physiological conditions. For example, initiation of autophagy is tightly controlled by network signaling that responds not only to starvation but also different nutrients, hypoxia, and reactive oxygen species (ROS) [17, 18].
The autophagic process
One of the main signaling pathways that control autophagy is the mTOR1 complex (TOR1C) [19]. Inhibition of TOR1C initiates autophagy by activating a cascade of phosphorylation events, which leads to the formation of a cup shaped membrane – termed the “isolation membrane” – in the vicinity of the targets termed the “phagosome assembly site” (PAS) (Fig. 1). The membrane source of the autophagosome is still debated, but it may include the endoplasmatic reticulum (ER), mitochondria, the PM, and Golgi [20]; and the contact site between ER and mitochondria may serve as the PAS [21]. One of the key reactions following the formation of the isolation membrane is the conjugation of Atg5 to Atg12. These two factors (of 36 that have been identified so far) belong to the autophagy (Atg) protein family [22]. Atg5-Atg12 conjugation is mediated by Atg7, which upon interaction with LC3, closes the autophagosome and thereby sequesters the substrate inside the double membrane autophagosome [23]. The autophagosomes move by kinesin-mediated transport along the microtubules, and their content is subsequently degraded upon fusion of the autophagosome with a lysosome to form an autolysosome (Fig. 1) [24]. Alternatively, the autophagosome may also fuse with a late endosome/multivesicular body (MVB) to form an amphisome on the route toward final lysosomal degradation, thereby linking the endosomal, autophagosomal, and lysosomal systems (Fig. 1) [25]. Autophagy – which is evolutionarily conserved from yeast to mammals – was initially regarded as a bulk degradation system, but it is now evident that different forms of autophagy exist, which utilize distinct mechanisms, i.e. macroautophagy (herein referred to simply as autophagy), microautophagy and chaperone-mediated autophagy [26].
Figure 1.
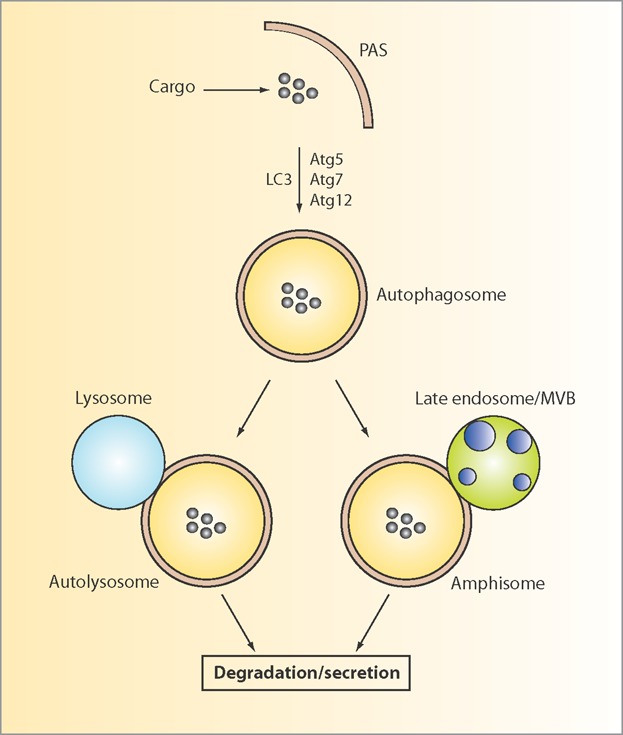
Schematic overview of the general autophagy pathways. Autophagy is initiated at the phagosome assembly site (PAS). Upon elongation of the membrane, which involves Atg5, Atg7, Atg12, and LC3 among others, the selected cargo is enclosed in a double membrane vesicle termed the autophagosome. Depending on the path, the autophagosome can either fuse with a lysosome to form an autolysosome, or with an endosome/MVB to generate an amphisome. The mechanistic details of the balance between degradation and secretion remain to be clarified. PAS, phagosome assembly site; MVB, multivesicular body.
Autophagy is important for brain function
Autophagy is crucial for the physiological regulation of the cell, e.g. to assure sufficient levels of amino acids, to supply energy but also plays a central role in pathological states such as infection, inflammation, cancer, and neurodegenerative diseases [27–30]. In the brain, neurons with complex axonal and dendritic structures are dependent on intense transport and efficient proteostasis to accommodate a dynamic microenvironment during both brain development and aging. This is orchestrated to a large extent by autophagy; measurements have revealed a highly efficient autophagic turnover in neurons [31]. The neuronal dependence on autophagy is further highlighted by the neurodegeneration, including axonal loss and cell death that occurs in the absence of autophagy [32–35]. Hence, well-functioning autophagy is a requirement for a healthy brain. Neurodegenerative diseases, including AD, Parkinson's, Huntington's, and amyotrophic lateral sclerosis (ALS) are all disorders of pathological depositions of potentially toxic protein aggregates and they display impaired autophagy [27].
Autophagy is impaired in AD
In AD, the presence of both key pathologies (Aβ plaques and NFTs) is a prerequisite for disease penetrance. However, the AD brain also exhibits dystrophic neurites, which are not found in healthy brains. These neurites are swollen axons, which exhibit aggregated phosphorylated tau and Aβ, and electron microscopy studies have identified an accumulation of autophagosomes in different stages [14]. This pathology clearly shows that the autophagic system is disturbed in AD. The accumulation of autophagosomes may be due to either increased induction of autophagy or decreased lysosomal clearance or a combination thereof. Indeed inhibition of lysosomal proteolysis in cultured neurons causes an AD-like accumulation of autophagosomes and axonal dystrophy, indicating that impaired lysosomal activity may cause the autophagosomal accumulation [36]. This hypothesis is further supported by the observation that the lysosomal hydrolases are increased and abnormally distributed in AD brains, which indicate compromised lysosomes [37]. The same impairment is also observed in AD mouse models and may be caused by an overloaded lysosome system [38]. Importantly, FAD-linked mutations in PS1 disrupt lysosomal proteolysis by impairing the acidification of the lysosome [39]. In addition, the autophagy-initiating Beclin 1 protein is reduced in early AD, which indicates that initiation of autophagy is decreased in AD [40]. However, transcriptional data indicate an upregulation of autophagy-activating factors in AD brains [17], implying increased autophagic activity. These slightly contradictory results may reflect a compensatory effect at the transcriptional level induced by impaired proteostasis, or that the level of autophagy initiation varies with disease progression. Further studies are required to clarify the level of autophagic activity during the different stages of AD.
Secretion of proteins by autophagy
Autophagy has been known for more than 50 years and its degradative function has been extensively studied. However, only recently has a role for autophagy in the transport and secretion of biomolecules emerged. These recent studies revealed that autophagy contributes to both conventional and unconventional protein secretion (UPS). Below, we briefly summarize the latest findings on the role of autophagy in protein secretion. For a detailed description, we refer the reader to some excellent recent reviews on the topic [41–44]. It should be noted that the terminology “secretion” has been used in the description of the different autophagy-mediated pathways that lead to the release of proteins to the extracellular space. However, some of these pathways may also include excretory mechanisms that transport cellular waste out of the cell, which are likely distinct from the regulated secretion of proteins with a biological function, see Table 1.
Table 1.
Potential Aβ secretory and excretory pathways influenced by autophagy
| Pathway (origin of route) | Mechanism |
|---|---|
| Conventional constitutive (Golgi) | Secretory |
| Conventional constitutive (TASCC) | Secretory/excretory |
| Regulated (lysosome, endosome/MVBs) | Secretory |
| UPS (omegasome) | Secretory/excretory |
Autophagy plays a role in UPS
Proteins to be secreted via the conventional secretory pathway are routed to the ER after their translation and transported through the Golgi-network toward the PM where the proteins are secreted into the extracellular space. However, this mode of trafficking is dependent on the presence of a signal peptide (SP) sequence within the protein, which correctly directs the protein to its destination. Nevertheless, a number of non-SP-containing proteins and peptides are present in the extracellular space, hence indicating the existence of alternative secretory pathways.
Recently, it has been shown that the secretion of several leaderless proteins lacking a SP is mediated by autophagy via a pathway that is independent of conventional secretion, and which is sometimes referred to as autosecretion. One such example is the secretion of acyl-coenzyme A-binding protein (Acb1 in yeast, ACBP in mammals), which is a 10 kDa cytoplasmic protein lacking a SP. The secretion of Acb1 is dependent on several Atg proteins, including Atg1, Atg6, and Atg8, and it is induced upon starvation or induction of autophagy by rapamycin [45, 46]. UPS involves the formation of an omegasome, a membranous structure found on the ER, which may possibly be related to the compartment for UPS (CUPS) in yeast [47]. Although the mechanistic details remain to be further investigated, the formation of the omegasome shares some of the factors required for the formation of PAS. Further investigation of the secretory pathway of Acb1 revealed that it relies on the Golgi reassembly and stacking protein (GRASP), whose function was initially ascribed to the Golgi cisternae, and which potentially associates the Golgi to the UPS [46]. Interestingly, Acb1 secretion is also dependent on factors required for the fusion of autophagosomes with endosomes and formation of MVBs [47]. This indicates that the UPS pathway involves the fusion of the autophagic vacuole with late endosome/MVBs to form an amphisome on its route toward the PM. It remains to be resolved whether the released Acb1 is contained within an exosome (exophagy) that disrupts extracellularly, or whether it is directly released as a free protein into the extracellular space. In a similar way, α-synuclein is secreted by UPS, mediated by the amphisome via exophagy [48]. Yet another protein lacking an SP and that is secreted – at least to some extent – by UPS is interleukin-1β (IL-1β) [49].
Autophagy-mediated non-secretory transport of membrane proteins
Not only cytoplasmic proteins but also integral membrane proteins (though not secreted) are transported to the PM by autophagy. One such example is the cystic fibrosis transmembrane conductance regulator (CFTR), an ion channel that causes cystic fibrosis when mutated [50]. In a detailed analysis, it was found that blocking the conventional ER-Golgi-PM secretory pathway enhanced the transport of CFTR in a GRASP55-dependent manner. Notably, autophagy-mediated trafficking of CFTR leads to the correct insertion of the protein into the PM, and strikingly, transgene expression in mice of GRASP rescues the phenotype of the mutant ΔF508-CFTR mice [50].
Autophagy interferes with conventional constitutive secretion
In addition to a role for autophagy in UPS, autophagy also intersects with the conventional secretory pathway, as exemplified by the transport of IL-6 and IL-8 to the extracellular space. The role of autophagy in the secretion of IL-6 and IL-8 starts with a structure termed TOR-autophagy spatial coupling compartment (TASCC), which is located closely to the Golgi apparatus [51]. This type of secretion is sensitive to brefeldin A, which suggests a close interaction with the conventional ER-Golgi-PM secretory pathway. TASCC resembles the lysosome in that it contains degrading organelles positive for the autophagy adaptor protein p62. By largely unknown mechanisms, this autophagic structure selectively mediates the degradation of certain cargoes while others remain intact and get secreted.
Autophagy mediates regulated secretion from lysosomes
In yet another pathway, autophagy contributes to the secretion of proteins stored in secretory lysosomes, so called regulated secretion. Several proteins from a wide range of cell types are secreted from secretory lysosomes or granules by autophagy. These proteins include antimicrobial peptides from Paneth cells in Crohn's disease in which single nucleotide polymorphisms in Atg16L1 is a risk factor [52, 53]; insulin from pancreatic cells [54]; cathepsin K from osteoclasts in bone resorption [55]; and von Willebrand factor from endothelial cells [56]. The mechanisms behind autophagy-mediated regulated secretion are little known and need further investigation. A key question again is how certain proteins can escape lysosomal proteolysis for secretion while others are degraded. In summary, a growing number of proteins are secreted by autophagy-related mechanisms through at least three different pathways (constitutive, unconventional, and regulated secretion). One crucial aspect of autophagy-mediated secretion to be elucidated is whether the pathways are dependent on the whole autophagy machinery, including the autophagosome, or if the secretory pathways share a restricted number of factors with degradative autophagy.
Autophagy participates in Aβ metabolism
Increased levels of Aβ leading to the accumulation and aggregation of Aβ into plaques, is one of the most extensively studied pathologies in AD. In autosomal dominant FAD, the overproduction of Aβ – especially the highly aggregation-prone Aβ42 and Aβ43 – is due to mutations in APP, PS1, or PS2. In addition, around 20 other risk genes associated with AD with diverse functions in the immune system, synaptic activity, and endocytosis have been identified by meta-analysis [57, 58]. However, the associated risk of each of these loci is relatively low, hence indicating a complex genetic background in sporadic AD (SAD). In SAD, which corresponds to up to 98% of all AD cases, increased Aβ levels may be caused by decreased catabolism of Aβ. In fact, levels of one of the major Aβ degrading enzymes, neprilysin, decline with age and may contribute to increased Aβ levels in SAD [59].
In addition to the classical AD pathologies Aβ plaques and NFTs, a pronounced accumulation of autophagosomes occurs in the dystrophic neurites within the AD brain, suggesting a dysregulated or impaired autophagic system [14]. Significantly, the autophagosomes contain the Aβ-generating γ-secretase component PS1 along with Aβ, indicating that autophagy plays a role in Aβ metabolism [60]. In addition, the adaptor complex AP2/PICALM has been identified as an autophagic cargo receptor that concomitantly interacts with APP-CTF and LC3. This interaction directly recruits APP-CTF from endosomes to autophagosomes, where it can be further processed to Aβ [61]. The close association of autophagy with Aβ has led to a number of studies aimed at elucidating the precise role of autophagy in Aβ metabolism. Various experimental approaches to manipulating autophagy have been applied, including pharmacological and genetical means. Pharmacological inhibition of mTor signaling by rapamycin or other activators of autophagy [62–64] induces autophagy, which clears intracellular Aβ accumulation, reduces extracellular Aβ plaque load, and improves cognitive function in 3 × Tg and PDAPP AD mouse models [65–67]. Furthermore, genetic deletion of the endogenous cathepsin D inhibitor, cystatin B, in TgCRND8 mice rescues autophagic-lysosomal dysfunction and decreases Aβ levels [38]. In contrast, heterozygous deletion of autophagy-initiating Beclin 1 increases intracellular and extracellular Aβ depositions [40]. Mouse models of lysosomal storage disorders that mimic impaired end stage autophagic lysosomal clearance accumulate autophagosomes, as determined by LC3 metabolism, and manifest increased APP-CTF and Aβ levels [68]. In addition to starvation, autophagy is also induced by oxidative stress or proteasome inhibition, which increases the generation and lysosomal accumulation of Aβ [69–71]. These studies together imply autophagy in Aβ metabolism. Considering that autophagy is dysregulated in AD it is possible that autophagy impairment contributes to the Aβ pathology of AD.
Autophagy influences Aβ secretion
Genetic deletion of autophagy inhibits extracellular delivery of Aβ
As summarized above, results from both in vitro and in vivo experiments have implicated autophagy in Aβ metabolism. However, the effects on Aβ metabolism of genetic ablation of autophagy in a mouse model of AD had not been investigated. Therefore, we created an autophagy-deficient APP transgenic mouse by conditionally knocking down Atg7 in excitatory neurons in the mouse forebrain [15]. Remarkably – and contrary to our expectations – taking the degradative function of autophagy into account, the autophagy-deficient mouse exhibited drastically reduced Aβ plaque load (Fig. 2A). Consistent with these results, Aβ ELISA measurements revealed significantly decreased Aβ levels (Fig. 2B). After thoroughly investigating the properties of the mutant mice, we found that autophagy-deficiency induced intracellular Aβ accumulation prior to Aβ plaque formation. Similar intracellular Aβ accumulation is observed in AD brain and appears prior to extracellular plaque formation [72–74]. The intracellular Aβ accumulation induced by deficiency of autophagy, which is subsequently followed by a reduced extracellular Aβ plaque load, indicated that the neuronal lack of autophagy impairs Aβ secretion. Indeed, measurement of Aβ release from primary neurons derived from autophagy-deficient mice revealed that the secretion was reduced by 90%; supplementing the neurons with lentivirus-expressed Atg7 restored Aβ secretion levels back to normal. In an additional approach, we treated primary neurons from wildtype mice with pharmacological compounds that either enhanced or inhibited autophagy. Exposing the neurons to rapamycin, an inhibitor of mTor, increased autophagy and induced Aβ secretion. Conversely, inhibition of autophagy by spautin-1, which inhibits the deubiquitinating enzymes USP10/13 and promotes VPS34 PI 3-kinase complex degradation, decreased Aβ secretion. Similarly, inhibiting microtubule-dependent transport of autophagy by vinblastine significantly decreased Aβ secretion. Furthermore, tracing Aβ in the autophagy-deficient neurons by immunofluorescence revealed a build-up of Aβ in the perinuclear region of the neuron, which supports the notion that transport of Aβ inside the cell is affected by the lack of autophagy. These data show that autophagy influences the release of Aβ to the extracellular space. Moreover, the decreased total Aβ levels, as determined by ELISA measurements, support previous findings that Aβ is generated in the autophagosomes (Fig. 2B) [60].
Figure 2.
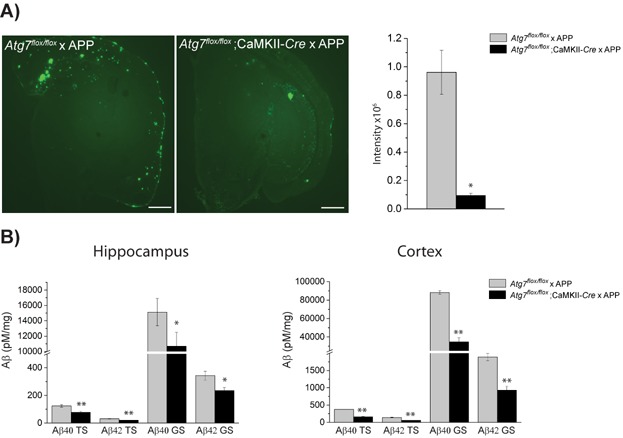
Autophagy mediates the secretion of Aβ in a mouse model of Alzheimer's disease and contributes to extracellular Aβ plaque formation. A: Aβ plaque staining of 15-month-old Atg7flox/flox × APP and Atg7flox/flox; CamKII-Cre × APP mouse brain. The fluorescence intensities were quantified. Note the drastic decrease in Aβ plaque load upon deletion of Atg7. B: Aβ ELISA measurement of brain homogenate of hippocampus and cortex of 15-month-old Atg7flox/flox × APP and Atg7flox/flox; CamKII-Cre × APP mice. *p < 0.01 **p < 0.05. TS and GS denote tris-soluble and guanidine-HCl-soluble, respectively. Scale bar represents 500 µm.
Classical secretory routes of Aβ involve constitutive secretion and release from endosomes
In addition to a potential autophagosomal source of Aβ [60], Aβ is generally believed to be produced and secreted by two major pathways: (i) translated APP is translocated to the ER and further to Golgi, where APP is processed by β- and γ-secretase within the Golgi-apparatus and post-Golgi vesicles, which subsequently deliver Aβ to the PM for extracellular release; (ii) alternatively, APP located at the PM is endocytozed, and Aβ is generated within the endosomes, which bring Aβ to the PM for secretion upon fusion of the endosome with the PM. However, the contribution of each of these two pathways to total Aβ generation is a subject of debate. One important aspect of Aβ generation is the requirement of an acidic environment for β-secretase, which is fulfilled in both the post-Golgi vesicles and the endosomes. Indeed, the presence of Aβ has been experimentally verified in both Golgi and endosomes, where Aβ is known to accumulate in AD brains [73, 75, 76]. Aβ has also been found in MVBs [77, 78] where Aβ impairs the multivesicular sorting system, which causes abnormal synaptic morphology [77, 79]. In addition, the MVBs may also fuse with late endosomes during vesicle sorting.
What is the pathway of autophagy-mediated extracellular delivery of Aβ?
The interference of autophagy with Aβ metabolism is thought to occur within the endosomal-lysosomal network that destines Aβ for lysosomal degradation. However, our recent finding that autophagy influences Aβ secretion indicates alternative fates of Aβ in the cell. This finding raises several questions: (i) does the autophagy-mediated delivery of Aβ to the extracellular space follow a regulated secretory pathway or is it a secondary effect linked to the excretion of cellular waste, (ii) is the autophagosome solely responsible for the delivery of Aβ to the PM or does the secretion involve other cellular compartments, (iii) does autophagy-mediated Aβ secretion interfere with the classical Aβ secretory routes or is it an independent pathway (see below), (iv) what determines whether autophagy transports Aβ for degradation versus secretion? Degradative autophagy and secretory autophagy may either share common molecular determinants and may not diverge until the late stage, or the pathways may be distinctly separate.
As summarized above, autophagy mediates the secretion of proteins through conventional, unconventional, and regulated secretion, which originate at the TASCC (in close proximity of the Golgi), the omegasome and the lysosome, respectively (Fig. 3). Potentially, Aβ-containing autophagosomes originating from the omegasome (which fuses with endosomes/MVBs to form amphisomes) could deliver Aβ to the PM for extracellular release, similar to the secretory pathway of Acb1 [46]. However, Acb1 is a cytoplasmic protein, while Aβ is membrane bound. If such a scenario occurs, APP, contained within the autophagosomal membrane, could be processed to Aβ within the autophagosome, which indeed contains β-cleaved APP-CTF and the Aβ cleaving enzyme PS1 [60]. By analogy with the autophagy-dependent translocation of CFTR from ER to PM [50], APP could be recruited directly from the ER or the Golgi along with the membrane for the autophagosome and Aβ generated within autophagosomes and secreted at the PM. Alternatively, autophagy may control the secretion of Aβ generated from endocytozed PM-located APP that is processed within endosomes, and released from amphisomes at the PM. Secretion through amphisomes involves the release of exosomes and, indeed, Aβ-containing exosomes have been experimentally verified [80, 81]. The observation of endosomal accumulation of Aβ in the AD brain is in agreement with this hypothesized pathway.
Figure 3.
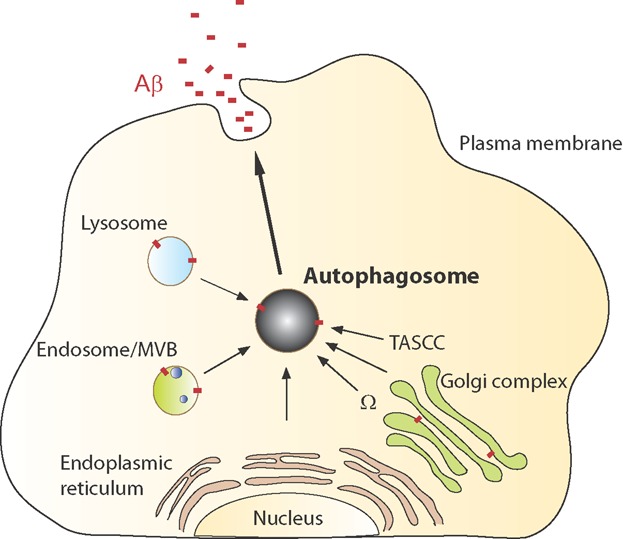
Schematic overview of potential autophagy-mediated Aβ-secretory routes. Compartments/organelles in the cell that could be part of autophagy-mediated delivery of Aβ to the extracellular space include the Golgi apparatus, endosome/MVBs, lysosome, and the autophagosome, all known to harbor Aβ. The endoplasmatic reticulum may contribute to the autophagosomal membrane. It remains to be resolved through which pathway autophagy mediates Aβ secretion. MVB, multivesicular body; Ω, omegasome; TASCC, TOR-autophagy spatial coupling compartment.
On the other hand, lysosomes, or possibly secretory lysosomes, contain substantial amounts of Aβ, which could potentially be released to the extracellular space by autophagy-mediated mechanisms similar to the regulated secretion of a number of proteins (as described above). Increased amounts of lysosomal Aβ have also been observed upon autophagy induction or inhibition of secretion [69, 82]. Further support for Aβ secretion from lysosomes comes from a recent study where the loss of the lysosomal sialidase NEU1 both increases the processing of over-sialylated APP and enhances Aβ exocytosis from lysosomes [83]. Taken together, there are several potential intersections of autophagy and Aβ generation that could lead to the secretion of Aβ (Fig. 3). More research is warranted to elucidate the mechanisms underlying autophagy-mediated secretion of Aβ, including the origin of the Aβ-secretory autophagosome, and the site of Aβ generation for autophagy-mediated secretion (Golgi vs. endosomal vs. autophagosomal). Furthermore, it needs to be clarified whether other cellular compartments are involved in the pathway (secretory vesicles, endosome/MVBs, and lysosome) and if the delivery is a regulated process or a consequence of disposal of cellular waste by an excretory mechanism.
Aβ accelerates autophagy-deficiency-induced neurodegeneration
Massive neurodegeneration and cell death occurs in the AD brain, which leads to up to 10% loss of the total brain weight. However, mechanistic insights into the neuronal loss and how it is linked to Aβ and tau are limited. Autophagy may be involved in this process because autophagy is impaired in AD and lack of autophagy in the mouse brain causes neurodegeneration [33, 34]. Similarly, several other neurodegenerative diseases such as ALS and Parkinson's disease exhibit neuronal cell death linked to autophagosomal-lysosomal impairments [27, 84]. We found that the autophagy-deficient APP mice exhibited enhanced neurodegeneration compared to autophagy-deficient mice without Aβ amyloidosis, which indicates that Aβ exacerbates the degeneration caused by impaired autophagy [15]. The enhanced neurodegeneration was coupled to increased caspase 3 activation, which indicates an onset of apoptosis, seemingly independent of tau. Interestingly, a recent report showed that unconventional secretion from endothelial cells is regulated by caspase 3 [85]. Thus, it is possible that the enhanced accumulation of cleaved caspase 3 in the autophagy-deficient APP mice may be associated with the secretory impairment to some extent. This finding indicates a possible link between impaired secretion and apoptosis, although further investigations are warranted to establish such connection. Moreover, decreased secretion of Aβ has been observed over time in cultured primary neurons derived from AD model mice, leading to intracellular Aβ accumulation [86]. The accumulation of intracellular Aβ is, along with other molecules such as ROS, toxic to lysosomes and mitochondria and can induce cell death by lysosomal membrane permeabilization and subsequent release of cathepsins [69, 87–91]. Aβ interacts with cyclofilin D [92], which causes mitochondrial swelling and permeability transition while the interaction of Aβ with Aβ-binding alcohol dehydrogenase induces apoptosis [93]. Thus, it is possible that impaired autophagy inhibits the secretion of Aβ, which leads to intracellular Aβ accumulation that induces neurodegeneration potentially including lysosomal cell death. Simultaneously, endocytozed Aβ inhibits autophagy, which then may initiate a vicious cycle [65]. The association of intracellular Aβ with neurodegeneration could therefore play a pivotal role in the neurodegeneration observed in AD. However, further research is needed to establish such a connection and resolve the question of the relative toxicity of intracellular versus extracellular Aβ.
Conclusions and outlook
Autophagy is dysregulated in AD, plays a central role in Aβ metabolism and therefore potentially contributes to AD pathology. In addition to its well-established degradative role of Aβ, we found that autophagy also influences the secretion of Aβ. Therefore, the role of autophagy in Aβ metabolism is bi-functional.
Further mechanistic insights into how autophagy influences Aβ secretion and how the balance is obtained between Aβ degradation and Aβ secretion may increase the chances of developing a therapeutic AD drug involving autophagy. It is therefore important to elucidate the precise pathway of autophagy-mediated Aβ secretion. This includes identification of indispensible factors that initiate and participate in the autophagy-mediated Aβ secretion. Specifically, it is crucial to determine if Aβ, or even APP, is recruited to autophagosomes from any of the organelles/compartments known to harbor APP/Aβ including Golgi, MVBs/late endosomes, and lysosomes or if the delivery of Aβ to the extracellular space is a result of an exocytic activity that clears cellular waste. Since Aβ is mainly associated with membranes, elucidation of the membrane source of the autophagosome may give hints as to the origin of APP/Aβ. By meticulously tracing Aβ upon manipulation of autophagy by genetic or pharmacological means in vivo in AD mouse models that exhibit Aβ pathology, or in vitro in neuronal cells, it will be possible to identify proteins that are important for autophagy-mediated Aβ secretion. It is also necessary to determine to what extent autophagy-mediated Aβ secretion contributes to total Aβ secretion. This will in turn open up for the investigation of the drugability of autophagy in AD, taking into account the dual function of autophagy in Aβ metabolism; degradation and secretion. Augmenting autophagy could clear potentially toxic intracellular Aβ at the expense of increasing Aβ secretion. However, co-administration of an Aβ-lowering compound could be a complement to such treatment.
Acknowledgments
We would like to thank all the members in the PNS laboratory for fruitful discussions during the completion of this study. We especially thank at RIKEN Brain Science Institute the people involved in the finding of autophagy-mediated Aβ secretion, Takashi Saito, Nobuhisa Iwata, Misaki Sekiguchi, Yukio Matsuba, Satoshi Tsubuki, Kelvin Hui, Motomasa Tanaka, and Krishnapriya Loganathan. We are grateful to Masaaki Komatsu for providing the Atg7flox/flox mice. This project was financially supported by research grants from the Swedish Research Council, Sweden; RIKEN Brain Science Institute, Japan; Ministry of Education, Sports, Science and Technology, Japan; and Ministry of Health, Labour and Welfare, Japan.
The authors have declared no conflict of interest.
Glossary
- Aβ
amyloid beta
- Acb1/ACBP
acetyl coenzyme A-binding protein 1
- AD
Alzheimer's disease
- AP2
adaptor protein 2
- APP
Aβ precursor protein
- APP-CTF
APP carboxy-terminal fragment
- Atg
autophagy related protein
- CFTR
cystic fibrosis transmembrane conductance regulator
- CUPS
compartment of UPS
- ER
endoplasmatic reticulum
- FAD
familial AD
- GRASP
Golgi reassembly and stacking protein
- IL
interleukin
- LC3
microtubule-associated protein 1A/1B-light chain 3
- mTOR1
mammalian target of rapamycin 1
- MVB
multivesicular body
- NFT
neurofibrillary tangles
- NMDAr
N-methyl-d-aspartate receptor
- PAS
phagosome assembly site
- PDAPP
APP transgenic mouse with Indiana mutation
- PICALM
phosphatidylinositol binding clathrin assembly protein
- PM
plasma membrane
- PS1/2
presenilin 1/2
- ROS
reactive oxygen species
- SAD
sporadic AD
- SP
signal peptide
- TASCC
TOR-autophagy spatial coupling compartment
- TgCRND8
transgenic APP mouse with FAD-linked Swedish and Indiana mutation
- TOR1C
mammalian target of rapamycin 1 complex
- UPS
unconventional protein secretion
- USP10/13
ubiquitin-specific protease 10/30
- VPS34 PI
vacuolar protein sorting 34 phosphatidylinositol
References
- 1.Di Santo SG, Prinelli F, Adorni F, Caltagirone C. A meta-analysis of the efficacy of donepezil, rivastigmine, galantamine, and memantine in relation to severity of Alzheimer's disease. J Alzheimers Dis. 2013;35:349–61. doi: 10.3233/JAD-122140. [DOI] [PubMed] [Google Scholar]
- 2.Winblad B. Donepezil in severe Alzheimer's disease. Am J Alzheimers Dis Other Demen. 2009;24:185–92. doi: 10.1177/1533317509332094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gouras GK, Tsai J, Naslund J, Vincent B. Intraneuronal Aβ42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagele RG, D'Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of β-amyloid 1-42 in neurons is facilitated by the α7 nicotinic acetylcholine receptor in Alzheimer's disease. Neuroscience. 2002;110:199–211. doi: 10.1016/s0306-4522(01)00460-2. [DOI] [PubMed] [Google Scholar]
- 5.D'Andrea MR, Nagele RG, Wang HY, Peterson PA. Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer's disease. Histopathology. 2001;38:120–34. doi: 10.1046/j.1365-2559.2001.01082.x. [DOI] [PubMed] [Google Scholar]
- 6.D'Andrea MR, Nagele RG, Wang H-Y, Lee DHS. Consistent immunohistochemical detection of intracellular β-amyloid42 in pyramidal neurons of Alzheimer's disease entorhinal cortex. Neurosci Lett. 2002;333:163–6. doi: 10.1016/s0304-3940(02)00875-3. [DOI] [PubMed] [Google Scholar]
- 7.Mochizuki A, Tamaoka A, Shimohata A, Komatsuzaki Y. Aβ42-positive non-pyramidal neurons around amyloid plaques in Alzheimer's disease. Lancet. 2000;355:42–3. doi: 10.1016/S0140-6736(99)04937-5. [DOI] [PubMed] [Google Scholar]
- 8.Zhao W, Dumanis SB, Tamboli IY, Rodriguez GA. Human APOE genotype affects intraneuronal Aβ1-42 accumulation in a lentiviral gene transfer model. Hum Mol Genet. 2013;23:1365–75. doi: 10.1093/hmg/ddt525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haass C, Kaether C, Thinakaran G, Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med. 2012;2:a006270. doi: 10.1101/cshperspect.a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giacobini E, Gold G. Alzheimer disease therapy – moving from amyloidbeta to tau. Nat Rev Neurol. 2013;9:677–86. doi: 10.1038/nrneurol.2013.223. [DOI] [PubMed] [Google Scholar]
- 11.Kim S, Kim J. Sequence analyses of presenilin mutations linked to familial Alzheimer's disease. Cell Stress Chaperones. 2008;13:401–12. doi: 10.1007/s12192-008-0046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saito T, Suemoto T, Brouwers N, Sleegers K. Potent amyloidogenicity and pathogenicity of Ab43. Nat Neurosci. 2011;14:1023–32. doi: 10.1038/nn.2858. [DOI] [PubMed] [Google Scholar]
- 13.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 14.Nixon RA, Wegiel J, Kumar A, Yu WH. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–22. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 15.Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013;5:61–9. doi: 10.1016/j.celrep.2013.08.042. [DOI] [PubMed] [Google Scholar]
- 16.Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol. 2012;8:108–17. doi: 10.1038/nrneurol.2011.200. [DOI] [PubMed] [Google Scholar]
- 17.Lipinski MM, Zheng B, Lu T, Yan Z. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc Natl Acad Sci USA. 2010;107:14164–9. doi: 10.1073/pnas.1009485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lipinski MM. Towards the global understanding of the autophagy regulatory network. Autophagy. 2010;6:1218–20. doi: 10.4161/auto.6.8.13772. [DOI] [PubMed] [Google Scholar]
- 19.Oddo S. The role of mTOR signaling in Alzheimer disease. Front Biosci. 2012;S4:941–52. doi: 10.2741/s310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rubinsztein David C, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol. 2012;22:R29–34. doi: 10.1016/j.cub.2011.11.034. [DOI] [PubMed] [Google Scholar]
- 21.Hamasaki M, Furuta N, Matsuda A, Nezu A. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–93. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- 22.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 23.Mizushima N, Noda T, Yoshimori T, Tanaka Y. A protein conjugation system essential for autophagy. Nature. 1998;395:395–8. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 24.Mackeh R, Perdiz D, Lorin S, Codogno P. Autophagy and microtubules – new story, old players. J Cell Sci. 2013;126:1071–80. doi: 10.1242/jcs.115626. [DOI] [PubMed] [Google Scholar]
- 25.Lamb CA, Dooley HC, Tooze SA. Endocytosis and autophagy: shared machinery for degradation. BioEssays. 2013;35:34–45. doi: 10.1002/bies.201200130. [DOI] [PubMed] [Google Scholar]
- 26.Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15:713–20. doi: 10.1038/ncb2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–97. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 28.Choi AMK, Ryter SW, Levine B. Autophagy in human health and disease. New Engl J Med. 2013;368:651–62. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 29.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–37. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–10. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boland B, Kumar A, Lee S, Platt FM. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci. 2008;28:6926–37. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Komatsu M, Wang QJ, Holstein GR, Friedrich VL. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci USA. 2007;104:14489–94. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Komatsu M, Waguri S, Chiba T, Murata S. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 34.Hara T, Nakamura K, Matsui M, Yamamoto A. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 35.Inoue K, Rispoli J, Kaphzan H, Klann E. Macroautophagy deficiency mediates age-dependent neurodegeneration through a phospho-tau pathway. Mol Neurodegener. 2012;7:48–61. doi: 10.1186/1750-1326-7-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee S, Sato Y, Nixon RA. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer's-like axonal dystrophy. J Neurosci. 2011;31:7817–30. doi: 10.1523/JNEUROSCI.6412-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cataldo AM, Paskevich PA, Kominami E, Nixon RA. Lysosomal hydrolases of different classes are abnormally distributed in brains of patients with Alzheimer disease. Proc Natl Acad Sci USA. 1991;88:10998–1002. doi: 10.1073/pnas.88.24.10998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang D-S, Stavrides P, Mohan PS, Kaushik S. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain. 2011;134:258–77. doi: 10.1093/brain/awq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee J-H, Yu WH, Kumar A, Lee S. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–58. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pickford F, Masliah E, Britschgi M, Lucin K. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J Clin Invest. 2008;118:2190–9. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deretic V, Jiang S, Dupont N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol. 2012;22:397–406. doi: 10.1016/j.tcb.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Subramani S, Malhotra V. Non-autophagic roles of autophagy-related proteins. EMBO Rep. 2013;14:143–51. doi: 10.1038/embor.2012.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding Y, Wang J, Wang J, Stierhof Y-D. Unconventional protein secretion. Trends Plant Sci. 2012;17:606–15. doi: 10.1016/j.tplants.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 44.Zhang M, Schekman R. Unconventional secretion, unconventional solutions. Science. 2013;340:559–61. doi: 10.1126/science.1234740. [DOI] [PubMed] [Google Scholar]
- 45.Manjithaya R, Anjard C, Loomis WF, Subramani S. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J Cell Biol. 2010;188:537–46. doi: 10.1083/jcb.200911149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duran JM, Anjard C, Stefan C, Loomis WF. Unconventional secretion of Acb1 is mediated by autophagosomes. J Cell Biol. 2010;188:527–36. doi: 10.1083/jcb.200911154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bruns C, McCaffery JM, Curwin AJ, Duran JM. Biogenesis of a novel compartment for autophagosome-mediated unconventional protein secretion. J Cell Biol. 2011;195:979–92. doi: 10.1083/jcb.201106098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ejlerskov P, Rasmussen I, Nielsen TT, Bergström A-L. Tubulin polymerization-promoting protein (TPPP/p25α) promotes unconventional secretion of α-synuclein through exophagy by impairing autophagosome-lysosome fusion. J Biol Chem. 2013;288:17313–35. doi: 10.1074/jbc.M112.401174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dupont N, Jiang S, Pilli M, Ornatowski W. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011;30:4701–11. doi: 10.1038/emboj.2011.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gee Heon Y, Noh Shin H, Tang Bor L, Kim Kyung H. Rescue of ΔF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell. 2011;146:746–60. doi: 10.1016/j.cell.2011.07.021. [DOI] [PubMed] [Google Scholar]
- 51.Narita M, Young ARJ, Arakawa S, Samarajiwa SA. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011;332:966–70. doi: 10.1126/science.1205407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hampe J, Franke A, Rosenstiel P, Till A. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–11. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 53.Cadwell K, Liu JY, Brown SL, Miyoshi H. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–63. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jung HS, Chung KW, Won Kim J, Kim J. Loss of autophagy diminishes pancreatic β cell mass and function with resultant hyperglycemia. Cell Metab. 2008;8:318–24. doi: 10.1016/j.cmet.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 55.DeSelm CJ, Miller BC, Zou W, Beatty WL. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell. 2011;21:966–74. doi: 10.1016/j.devcel.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Torisu T, Torisu K, Lee IH, Liu J. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat Med. 2013;19:1281–7. doi: 10.1038/nm.3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bettens K, Sleegers K, Van Broeckhoven C. Genetic insights in Alzheimer's disease. Lancet Neurol. 2013;12:92–104. doi: 10.1016/S1474-4422(12)70259-4. [DOI] [PubMed] [Google Scholar]
- 58.Lambert J-C, Ibrahim-Verbaas CA, Harold D, Naj AC. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452–8. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iwata N, Higuchi M, Saido TC. Metabolism of amyloid-β peptide and Alzheimer's disease. Pharmacol Ther. 2005;108:129–48. doi: 10.1016/j.pharmthera.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 60.Yu WH, Cuervo AM, Kumar A, Peterhoff CM. Macroautophagy – a novel β-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tian Y, Chang JC, Fan EY, Flajolet M. Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer's APP-CTF for terminal degradation via autophagy. Proc Natl Acad Sci USA. 2013;110:17071–6. doi: 10.1073/pnas.1315110110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tian Y, Bustos V, Flajolet M, Greengard P. A small-molecule enhancer of autophagy decreases levels of Aβ and APP-CTF via Atg5-dependent autophagy pathway. FASEB J. 2011;25:1934–42. doi: 10.1096/fj.10-175158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vingtdeux V, Chandakkar P, Zhao H, d'Abramo C. Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-β peptide degradation. FASEB J. 2011;25:219–31. doi: 10.1096/fj.10-167361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Steele JW, Gandy S. Latrepirdine (Dimebon®), a potential Alzheimer therapeutic, regulates autophagy and neuropathology in an Alzheimer mouse model. Autophagy. 2013;9:617–8. doi: 10.4161/auto.23487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Caccamo A, Majumder S, Richardson A, Strong R. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-β, and Tau. J Biol Chem. 2010;285:13107–20. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Majumder S, Richardson A, Strong R, Oddo S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One. 2011;6:e25416. doi: 10.1371/journal.pone.0025416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spilman P, Podlutskaya N, Hart MJ, Debnath J. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-β levels in a mouse model of Alzheimer's disease. PLoS One. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boland B, Smith DA, Mooney D, Jung SS. Macroautophagy is not directly involved in the metabolism of amyloid precursor protein. J Biol Chem. 2010;285:37415–26. doi: 10.1074/jbc.M110.186411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zheng L, Terman A, Hallbeck M, Dehvari N. Macroautophagy-generated increase of lysosomal amyloid β-protein mediates oxidant-induced apoptosis of cultured neuroblastoma cells. Autophagy. 2011;7:1528–45. doi: 10.4161/auto.7.12.18051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zheng L, Roberg K, Jerhammar F, Marcusson J. Autophagy of amyloid beta-protein in differentiated neuroblastoma cells exposed to oxidative stress. Neurosci Lett. 2006;394:184–9. doi: 10.1016/j.neulet.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 71.Agholme L, Hallbeck M, Benedikz E, Marcusson J. Amyloid-β secretion, generation, and lysosomal sequestration in response to proteasome inhibition: involvement of autophagy. J Alzheimers Dis. 2012;31:343–58. doi: 10.3233/JAD-2012-120001. [DOI] [PubMed] [Google Scholar]
- 72.Oddo S, Caccamo A, Smith IF, Green KN. A dynamic relationship between intracellular and extracellular pools of Aβ. Am. J Pathol. 2006;168:184–94. doi: 10.2353/ajpath.2006.050593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cataldo AM, Petanceska S, Terio NB, Peterhoff CM. Aβ localization in abnormal endosomes: association with earliest Aβ elevations in AD and Down syndrome. Neurobiol Aging. 2004;25:1263–72. doi: 10.1016/j.neurobiolaging.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 74.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-[beta] in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 75.Choy RW-Y, Cheng Z, Schekman R. Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid β (Aβ) production in the trans-Golgi network. Proc Natl Acad Sci USA. 2012;109:2077–82. doi: 10.1073/pnas.1208635109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu H, Sweeney D, Wang R, Thinakaran G. Generation of Alzheimer β-amyloid protein in the trans-Golgi network in the apparent absence of vesicle-formation. Proc Natl Acad Sci USA. 1997;94:3748–52. doi: 10.1073/pnas.94.8.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Takahashi RH, Milner TA, Li F, Nam EE. Intraneuronal Alzheimer Aβ42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–79. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Langui D, Girardot N, El Hachimi KH, Allinquant B. Subcellular topography of neuronal Aβ peptide in APPxPS1 transgenic mice. Am J Pathol. 2004;165:1465–77. doi: 10.1016/s0002-9440(10)63405-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Almeida CG, Takahashi RH, Gouras GK. β-Amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci. 2006;26:4277–88. doi: 10.1523/JNEUROSCI.5078-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Perez-Gonzalez R, Gauthier SA, Kumar A, Levy E. The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. J Biol Chem. 2012;287:43108–15. doi: 10.1074/jbc.M112.404467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rajendran L, Honsho M, Zahn TR, Keller P. Alzheimer's disease β-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci USA. 2006;103:11172–7. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zheng L, Cedazo-Minguez A, Hallbeck M, Jerhammar F. Intracellular distribution of amyloid beta peptide and its relationship to the lysosomal system. Transl Neurodegener. 2012;1:19. doi: 10.1186/2047-9158-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Annunziata I, Patterson A, Helton D, Hu H. Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-β secretion via deregulated lysosomal exocytosis. Nat Commun. 2013;4:2734–47. doi: 10.1038/ncomms3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ghavami S, Shojaei S, Yeganeh B, Ande SR. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24–49. doi: 10.1016/j.pneurobio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 85.Sirois I, Groleau J, Pallet N, Brassard N. Caspase activation regulates the extracellular export of autophagic vacuoles. Autophagy. 2012;8:927–37. doi: 10.4161/auto.19768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tampellini D, Rahman N, Lin MT, Capetillo-Zarate E. Impaired β-amyloid secretion in Alzheimer's disease pathogenesis. J Neurosci. 2011;31:15384–90. doi: 10.1523/JNEUROSCI.2986-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wirths O, Bayer TA. Intraneuronal Aβ accumulation and neurodegeneration: lessons from transgenic models. Life Sci. 2012;91:1148–52. doi: 10.1016/j.lfs.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 88.Friedrich RP, Tepper K, Rönicke R, Soom M. Mechanism of amyloid plaque formation suggests an intracellular basis of Aβ pathogenicity. Proc Natl Acad Sci USA. 2010;107:1942–7. doi: 10.1073/pnas.0904532106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Abramowski D, Rabe S, Upadhaya AR, Reichwald J. Transgenic expression of intraneuronal Aβ42 but not Aβ40 leads to cellular Aβ lesions, degeneration, and functional impairment without typical Alzheimer's disease pathology. J Neurosci. 2012;32:1273–83. doi: 10.1523/JNEUROSCI.4586-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aits S, Jäättelä M. Lysosomal cell death at a glance. J Cell Sci. 1:1905–12. doi: 10.1242/jcs.091181. [DOI] [PubMed] [Google Scholar]
- 91.Hedskog L, Zhang S, Ankarcrona M. Strategic role for mitochondria in Alzheimer's disease and cancer. Antioxid Redox Signal. 2012;16:1476–91. doi: 10.1089/ars.2011.4259. [DOI] [PubMed] [Google Scholar]
- 92.Du H, Guo L, Fang F, Chen D. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008;14:1097–105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lustbader JW, Cirilli M, Lin C, Xu HW. ABAD directly links Aβ to mitochondrial toxicity in Alzheimer's disease. Science. 2004;16:448–52. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]