

Abstract
In an effort to explore the possible causes of human radiosensitivity and identify more rapid assays for cellular radiosensitivity, we interrogated a set of assays that evaluate cellular functions involved in recognition and repair of DNA double-strand breaks: (1) neutral comet assay, (2) radiation-induced γ-H2AX focus formation, (3) the temporal kinetics of structural maintenance of chromosomes 1 phosphorylation, (4) intra-S-phase checkpoint integrity, and (5) mitochondrial respiration. We characterized a unique panel of 19 “radiosensitive” human lymphoblastoid cell lines from individuals with undiagnosed diseases suggestive of a DNA repair disorder. Radiosensitivity was defined by reduced cellular survival using a clonogenic survival assay. Each assay identified cell lines with defects in DNA damage response functions. The highest concordance rate observed, 89% (17/19), was between an abnormal neutral comet assay and reduced survival by the colony survival assay. Our data also suggested that the neutral comet assay would be a more rapid surrogate for analyzing DNA repair/processing disorders.
INTRODUCTION
An integral relationship has been noted between clinical radiosensitivity, cellular radiosensitivity measured by the colony survival assay, and inefficient repair of double-strand breaks (DSBs) after exposure to ionizing radiation. In an effort to further sort out this relationship, we examined various DNA damage response-related end points such as (1) clonogenic survival, (2) intra-S-phase checkpoint, (3) the kinetics of ATM-dependent substrate phosphorylation, (4) neutral comet assay, and (5) mitochondrial respiratory function using a panel of 19 lymphoblastoid cell lines (LCLs) established from individuals with clinical phenotypes suggestive of a DNA repair disorder/syndrome (1–5). All 19 LCLs were radiosensitive when assessed by the colony survival assay (5). We also sought independent confirmation of the DSB repair deficiency suggested by the clonogenic survival results and interrogated whether any single assay or group of assays might correlate with the impaired colony survival assay, thereby providing an additional diagnostic marker of radiosensitivity for future studies.
The use of cells (fibroblasts or lymphoblasts) from AT (ataxia telangiectasia) patients as controls in studies of radiation-induced DNA damage stems from the seminal observation that AT patients are clinically radiosensitive (6–9). The colony survival assay was also used as an inclusion criterion for AT in family linkage studies that led to the positional cloning of the disease-causing ATM gene (10–12). Furthermore, when AT LCLs were transfected with wild-type full-length ATM cDNA, the reduced clonogenic survival was corrected (13, 14).
The comet assay is a technique that is widely used for the rapid analysis of DNA fragmentation after DNA damage. To study DNA single-strand breaks, alkaline conditions are most appropriate, whereas the neutral comet assay reflects recognition and recovery of DNA DSBs, as assessed by various parameters related to the comet assay profiles. These are obtained from measurements of the amount DNA in the comet head (head DNA), the amount of DNA in the comet tail (tail DNA), the length of the comet tail (tail length), length of the entire comet (comet length), or the percentage tail DNA multiplied by the tail length (tail moment). In time course studies, these parameters, in particular the tail moment and the tail length, are those most frequently used to obtain information on the repair of DNA damage. One can also use these parameters to calculate the percentage of DNA damage that returned to baseline levels after the induction of DSBs by radiation. The latter parameters reflect not only recognition of DNA damage but also repair of that damage.
Cell cycle checkpoints such as the intra-S phase that we measured here allow time for DNA repair before DNA replication and cell division (18–22). A typical checkpoint response involves sensor proteins (RAD50, BRCA1, NBS1), transducer proteins (ATM, CHK1, CHK2), and effector proteins (p53, p21, CDK). ATM protein appears early at a DSB and coordinately regulates cell cycle checkpoints by activating many of these proteins almost simultaneously, such that deficiencies in ATM or its ATM substrates manifest cell cycle checkpoint defects. Analyzing the inhibition of DNA synthesis once DSB damage has been “sensed” or recognized can further assess S-phase checkpoint defects. Failure to properly inhibit DNA synthesis after radiation damage is a hallmark of ATM-deficient cells (23).
The phosphorylation of SMC1 (structural maintenance of chromosomes 1) by the ATM kinase reflects the radiation-induced DNA DSB response network that functions in the ATM/NBS1-dependent S-phase checkpoint pathway (24–26). Phosphorylation of SMC1 is completely absent in AT LCLs when measured shortly after irradiation; minimal phosphorylation at serines 966 and 957 occurs at later times from redundant phosphorylation by other kinases (27). SMC1 phosphorylation has recently been used in the diagnosis of AT and the identification of AT carriers (28, 29).
DSBs are induced either by endogenous metabolic processes leading to the formation of free radicals or by exogenous sources such as radiation. DSBs represent the most lethal of DNA lesions. Early in the response to repair of DSBs, H2AX is phosphorylated at residue serine 139, in the highly conserved C-terminal SQEY motif forming γ-H2AX (30, 31). This triggers a myriad of chromatin modifications and spreads to adjacent areas of chromatin, affecting approximately 0.03% of total cellular γ-H2AX per DSB (31). Repair is thought to be complete when γ-H2AX is dephosphorylated. Thus the quantitative analysis of γ-H2AX foci has led to a wide range of applications in radiation research.
Endogenously induced free radicals also cause DNA damage (32, 33). Free radicals include both reactive oxygen (ROS) and nitrogen (RNS) species. ROS and RNS react with macromolecules like DNA, RNA, proteins and lipids, which can lead to severe and prolonged cellular toxicity. When the production of ROS exceeds the cell’s ability to metabolize and detoxify them, a cell is said to be in a state of oxidative stress, and this state can be self-perpetuating and ultimately fatal to the cell(s). Defects in endogenous mitochondrial oxidative metabolism can also perpetuate ROS production. AT LCLs have been reported to be in a chronic state of oxidative stress, suggesting that the role of DNA repair enzymes in regulating cellular redox homeostasis can affect a cell’s ability to respond to the genotoxic insult caused by radiation (32). In addition, a recent study demonstrated that the induction of oxidative stress induces ATM autophosphorylation and transactivation phosphorylation of the tumor suppressor p53 and protein kinase Chk2. Moreover, the activation of the oxidized form of ATM (ATM as a disulfide-crosslinked dimer) by ROS-inducing agents can occur in the absence of DNA DSBs and the MRE11/RAD50/NBS1 complex (33).
When the colony survival assay is included in diagnostic testing to distinguish AT from other types of neurological conditions, a small but consistent subset of non-AT patients (i.e., those clearly without AT) show reduced clonogenic survival (5). Immunoblotting confirms the presence of ATM, NBS and other proteins associated with known DNA repair disorders. In this report, we show that each radiosensitive LCL manifests additional DNA repair abnormalities; the strongest concordance with impaired colony survival was with the neutral comet assay. We conclude that most of the 19 patients from whom these LCLs were derived carry a DSB repair disorder, most likely autosomal recessive, and would likely manifest some degree of clinical radiosensitivity, cancer susceptibility and possibly a progressive neurological deterioration (4).
MATERIALS AND METHODS
Cell Panel
The radiosensitive LCLs used in this study were derived mainly from patients suspected of having hereditary ataxia, primary immunodeficient clinical radiosensitivity, or some other related genetic disorder. They were coded for anonymity in accordance with an approved protocol for study of human subjects (Exemption no. 4). No human subjects were recruited for this study. Peripheral blood lymphocytes (PBLs) were isolated and transformed with Epstein-Barr Virus (EBV) as described previously (5). Wild-type control LCLs from healthy individuals exhibited no apparent DNA repair defects or reduced clonogenic survival compared to a previously determined normal range (5). No significant differences were observed (P > 0.1) within the group of wild-type or AT LCLs used in these studies (Supplementary Tables 1 and 2; http://dx.doi.org/10.1667/RR2580.1.S1). Prior to irradiation, the majority of cells were in the G1 phase of the cell cycle as determined by FACS analysis of PI-stained cells. The times and doses evaluated for each functional assay were selected after extensive laboratory optimizations to maximize signal-to-noise ratios at selected end points.
Colony Survival Assay
Survival was assessed by the colony survival assay (5). Transformed cells were maintained in RPMI 1640 medium, 15% fetal bovine serum (Hyclone, Logan, UT), and 1% penicillin/streptomycin (Gibco BRL, Grand Island, NY) at 37°C in 95% air/5% CO2. Cells were grown to log phase (1 × 106 cells/ml) and harvested by centrifugation at 1000 rpm for 5 min. Supernatants were filtered and reused as conditioned medium. The cells were resuspended in a 1:1 mixture of fresh and conditioned medium at concentrations of 2–14 × 102 cells/ml, depending on the estimated plating efficiency of that particular LCL. The cells were then plated in duplicate 96-well plates at 100 or 50 cells per well. One plate was exposed to 1.0 Gy radiation at a dose rate of 4.5 Gy/min (Mark 1 137Cs) and the other was kept as a control. Both plates were returned to 37°C. The cells were incubated for 10 to 13 days, at which time they were stained with MTT dye (tetrazolium-based colorimetric assay, Sigma, St Louis, MO). After a 2- to 4-h incubation, each well was checked under a microscope; viable cells were dark blue. The presence of a colony of >32 LCLs was scored as a positive well (i.e., five cell divisions) (5). Colony-forming efficiency (CFE) was calculated as CFE = (−ln F)/W, where F is the fraction of negative wells and W is the number of cells seeded per well. Cell survival after irradiation was obtained by dividing the CFEi (the CFE of irradiated plate) by the CFEc (the CFE of control plate): Survival = (CFEi/CFEc) × 100. Based on earlier studies comparing 104 bona fide AT LCLs and 29 wild-type LCLs (i.e., phenotypically healthy), LCLs with a survival <21% (13 ± 7%) were interpreted as radiosensitive and those with a survival >36% were considered as normal (5). Normal and bona fide AT LCLs were included as negative and positive controls in all experiments but were not used to generate additional data points; each LCL was tested in a minimum of three independent colony survival assay experiments.
Neutral Comet Assay
When the comet assay is performed under neutral pH conditions, repair of DSBs can be assessed (15, 16). Neutral comet assay experiments were performed according to the manufacturer’s protocol (Trevigen Inc., Gaithersburg, MD). Wild-type and AT controls were included in each experiment. To define normal positive and negative ranges, a total of five independent wild-type (WT1, 2, 3, 4 and 5) and four independent AT (AT1, 2, 3 and 4) LCLs were either shamirradiated or irradiated with 15 Gy followed by incubation at 37°C for 30 min and 5 h postirradiation to allow repair (see Supplementary Tables 1 and 2 for data comparisons among controls; http://dx.doi.org/10.1667/RR2580.1.S1). A total of 1 × 106 LCLs were centrifuged at 1000 rpm and resuspended in 1 ml PBS. Low-melting agarose (maintained at 37°C) was added to the cell mixture at a 1:10 dilution just before 2,500 cells were pipetted onto each well of a 20-well slide. Once the agarose solidified, slides were incubated at 4°C in cell lysis solution for 30 min, then electrophoresed at 20 V for 10 min. After electrophoresis, slides were dried and stained with SyberGold for 10 min at 4°C. Slides were then visualized using a Zeiss fluorescence microscope equipped with AxioVision camera and acquisition software.
The images acquired were analyzed with Comet Score™ software (TriTek Corp, Sumerduck, VA). Image analysis was optimized using the densest region of fluorescence (i.e., the greatest concentration of DNA, using a heat-map imaging program) as a reference point for the best signal-to-noise ratio. In cases where no single region of DNA density was apparent, the cursor was placed in the center of the comet head (according to TriTek Corp. protocol). Approximately 100 cells were analyzed per cell line/experiment.
Tail moment was used to compute the percentage of DNA damage that returned to baseline (time zero) levels (i.e. %RTB) by 5 h after 15 Gy. Thus %RTB was the metric used to indicate adequate DNA repair. A mean tail moment value was obtained for each time/cell line. The tail moment values had minimal variation or near-complete return to basal levels at 5 h in pilot experiments comparing 5- and 24-h tail moment values for wild-type cells. No appreciable differences were observed in tail moment or %RTB in the ATM-deficient controls at 5 or 24 h postirradiation (data not shown). Thus all subsequent experiments used 5 h postirradiation as the end point. The %RTB is the ratio of irradiated to nontreated tail moments. The error bars for all LCLs tested represent means ± SD from three experiments normalized to the mean %RTB of five different wild-type LCLs (WT1, 2, 3, 4 and 5). The average %RTB for the five wild-type LCLs tested was 98.2% 6 1 ± 16.0%. The average %RTB for the four AT LCLs was 54.1 ± 10.2 (see Supplementary Tables 1 and 2 for a comparison among the controls; http://dx.doi.org/10.1667/RR2580.1.S1). The average %RTB of the AT LCLs was normalized to the wild-type controls. Normalized %RTBs from the four AT LCLs (AT 1, 2, 3 and 4) provide the experimental expression of defective DNA repair: 55 ± 10 (wild-type = 100). The same calculations were done for each radiosensitive LCL. Comparable tail moments for wild-type and AT LCLs 30 min postirradiation were observed. Therefore, the tail moment values 30 min postirradiation were used as an internal experimental control to qualitatively indicate induction of DNA damage by radiation (data not shown).
Immunofluorescence Detection of γ-H2AX
Immunofluorescence detection of γ-H2AX by IRIF has been described previously (30, 31). Briefly, LCLs were harvested 30 min and 24 h after irradiation with 1 Gy. Fixed cells were placed on cover slips with 3% buffered formaldehyde solution for 15 min, and washed with 1× PBS. They were then permeabilized for 5 min with 0.2% Triton X-100 and washed. The primary antibody, a mouse monoclonal γ-H2AX (Upstate, MA) antibody, was applied at a 1:500 dilution for 1 h. After primary incubation and washing, a secondary antibody, Alexa Fluor 488 goat anti-mouse (Invitrogen, Carlsbad, CA), was applied for 1 h. After washing, cover slips were mounted in Vectashield with DAPI. A cell was considered positive with the observation of four or more γ-H2AX foci per nucleus. A total of 100 nuclei per sample were scored for foci. Three independent experiments were performed for each cell line. The average percentage of cells positive for foci 24 h postirradiation and preirradiation were subsequently calculated and plotted. The ratios of γ-H2AX foci 24 h postirradiation to spontaneous γ-H2AX focus formation (i.e., not induced by radiation) were averaged over three independent experiments and used to establish a γ-H2AX focus formation profile of the radiosensitive LCLs and controls. WT1 and 5 and AT3 and 4 cells were used as controls. Thirty minutes postirradiation was used as a control in each experiment for radiation-induced DNA damage focus formation (data not shown).
pSMC1 Kinetics Immunoblotting
Nuclear extracts from 5 × 106 cells were prepared as described previously (34). Cells were harvested 1, 4 and 24 h after 10 Gy or mock treatment. Twenty-five micrograms of nuclear lysate was loaded and electrophoresed in a 7.5% SDS-polyacrylamide gel (PAGE), transferred onto PVDF membranes (Bio-Rad, Hercules, CA), and blocked with 5% dry milk. Blots were incubated overnight at 4°C with the following antibodies: anti-SMC1ser966 and SMC1 (both from Novus, Littleton, CO). A horseradish peroxidase-conjugated anti-rabbit secondary antibody was incubated at 20°C for 40 min for detection by enhanced chemiluminescence (ECL) (Amersham Pharmacia, Piscataway, NJ). All immunoblots included WT3 and AT4 controls (not shown) and were tested in a minimum of three independent experiments.
Radioresistant DNA Synthesis (RDS)
To analyze the intra-S-phase checkpoint, RDS using LCLs was performed as described previously (23, 34). Asynchronous radiosensitive cells were incubated with [14C]dThd for 24 h to establish background synthesis of DNA. All cells were tested 1 day after feeding. The cells were exposed to varying doses of radiation (0, 2, 5, 10 and 25 Gy) and then returned to the incubator for 60 min and pulse-labeled in medium containing [3H]dThd for an additional 60 min (23). The samples were harvested and counted in a Packard 2900TR scintillation counter. The ratio of incorporated [3H]dThd to [14C]dThd was used as a measure of the amount of DNA synthesis after irradiation. “WT1 and 5 and AT3 and 4 cells were used as experimental controls to establish control profiles of a “wild-type-like” reduced ratio of [3H]dThd/[14C]dThd incorporation and a sustained “AT-like” incorporation postirradiation. Radiosensitivity cells with a percentage [3H]dThd/[14C]dThd incorporation at 10 Gy similar to that of AT cells were considered AT-like. The experiments were performed in triplicate in a minimum of three independent experiments to obtain the average percentage ratios. No significant differences were observed between the wild-type controls, WT1 and 5 (P > 0.05), and the AT controls, AT3 and 4 (P > 0.05) (see Supplementary Tables 1 and 2; http://dx.doi.org/10.1667/RR2580.1.1.S1).
Mitochondrial Respiration Assay
Resazurin (also known as Alamar Blue) is a fluorogenic dye that is commonly used to detect production of cellular reactive oxygen species and mitochondrial function/respiration. The resazurin fluorescence intensity was measured at 530 nm excitation and 590 nm emission every 60 min over 3 h using a FLX-800 microplate fluorescence reader (Bio-Tek Instruments, Inc.). When reduced by the mitochondria, resazurin is reduced to resorufin and is measured by an increase in fluorescence intensity (32). Radiosensitive WT4 and AT1 cells (1 × 105 cells/well) were incubated with resazurin (3 mM) in a 96-well microtiter plate format under exponential growth conditions. Each experimental condition was repeated in six replicates, averaged and repeated in three independent experiments (see Supplementary Tables 1 and 2; http://dx.doi.org/10.1667/RR2580.1.S1). Wells containing only resazurin and medium served as additional controls. No significant differences were observed when the averages of six replicates and the three independent experiments were compared between experiments using WT4 and AT1 cells (P > 0.05) (see Supplementary Tables 1 and 2; http://dx.doi.org/10.1667/RR2580.1.S1).
RESULTS
Radiosensitive Cell Panel
Table 1 summarizes all findings in this report. Of the 19 radiosensitive LCLs, which were identified on the basis of having abnormal colony survival, 14/19 had an average survival within a previously determined radiosensitivity range of <21%, and 5/19 fell within an intermediate range of 21–37% (5) (Fig. 1). The abnormal colony survival results prompted us to perform immunoblotting to determine the protein levels for candidate protein known to affect radiosensitivity, such as ATM and NBS. None of the 19 radiosensitive LCLs were deficient in these proteins (data not shown). We subsequently abandoned this “candidate protein” approach in favor of using functional analyses to assess each functional assay as a possible surrogate marker for colony survival and to guide follow-up studies that might identify specific pathways with disease-causing genetic defects. Thus a second assay would be 100% concordant with the colony survival assay if all 19 radiosensitive LCLs scored in the abnormal range for that assay. AT and wild-type controls were always included in each experiment and additional data were collected from those controls.
TABLE 1.
Summary of Radiosensitive Cell Testing
| Cell line | CSA | NCA-TM | γ-H2AX | IR/NIR/γ-H2AX | pSMC1s966 | RDS | Mitochondrial respiration |
|---|---|---|---|---|---|---|---|
| WT | 50 ± 13 | 100 | 15 ± 1 | 7 ± 3 | WT | 62 ± 3 | WT |
| A–T | 13 ± 7 | 55 ± 10 | 28 ± 13 | 15 ± 12 | MR | 97 ± 3 | R |
| 7 | 27 ± 15* | 84 ± 6 | 23 ± 14 | 3 ± 0.88 | WT | 97 ± 7 | WT |
| 8 | 24 ± 4* | 42 ± 13 | 18 ± 11 | 2 ± 0.11 | LR | 92 ± 9 | R |
| 12 | 9 ± 4 | 39 ± 14 | 12 ± 13 | 9 ± 5 | LR | 61 ± 7 | R |
| 14 | 19 ± 2 | 59 ± 16 | 21 ± 2 | 6 ± 3 | WT | 99 ± 7 | R |
| 15 | 6 ± 3 | 83 ± 10 | 31 ± 12 | 2 ± 0.58 | WT | 99 ± 1 | WT |
| 17 | 16 ± 6 | 53 ± 6 | 15 ± 6 | 3 ± 0.86 | WT | 67 ± 6 | WT |
| 18 | 16 ± 8 | 53 ± 17 | 17 ± 6 | 18 ± 3 | WT | 54 ± 5 | R |
| 20 | 16 ± 5 | 49 ± 13 | 33 ± 1 | 3 ± 0.88 | WT | 55 ± 9 | R |
| 29 | 30 ± 18* | 56 ± 17 | 11 ± 1 | 3 ± 4 | LR | 97 ± 1 | R |
| 31 | 12 ± 4 | 47 ± 9 | 33 ± 12 | 4 ± 0.75 | WT | 110 ± 3 | R |
| 47 | 10 ± 4 | 60 ± 7 | 65 ± 4 | 2 ± 0.76 | WT | 101 ± 18 | R |
| 49 | 12 ± 2 | 56 ± 18 | 14 ± 2 | 13 ± 9 | IR | 94 ± 4 | R |
| 50 | 25 ± 1* | 53 ± 8 | 52 ± 3 | 29 ± 13 | LR | 95 ± 4 | WT |
| 52 | 19 ± 1 | 28 ± 10 | 14 ± 1 | 9 ± 11 | WT | 96 ± 3 | WT |
| 61 | 13 ± 4 | 42 ± 13 | 14 ± 1 | 4 ± 2 | LR | 96 ± 10 | R |
| 63 | 18 ± 2 | 52 ± 14 | 16 ± 1 | 13 ± 11 | LR | 49 ± 3 | R |
| 64 | 14 ± 3 | 58 ± 4.0 | 16 ± 5 | 4 ± 2 | WT | 97 ± 8 | R |
| 65 | 23 ± 1* | 44 ± 10 | 51 ± 2 | 3 ± 0.28 | LR | 65 ± 5 | WT |
| 68 | 16 ± 4 | 58 ± 9 | 27 ± 1 | 15 ± 10 | IR | 98 ± 2 | R |
| Correlation with CSA (%): | 89 | 53 | 26 | 47 | 68 | 68 | |
Notes. Bold: P < 0.05 compared to wild-type controls. Bold and italicized: RS-LCLs that correlated with results of colony survival assay. Abbreviations used: CSA: colony survival assay;
CSA: intermediate result; NCA: neutral comet assay; IR/NIR γ-H2AX: average % ratio focus formation 24 h postirradiation/nonirradiated; MR: minimal response; LR: late response: IR: intermediate response; WT: wild-type; R: reduced.
FIG. 1.
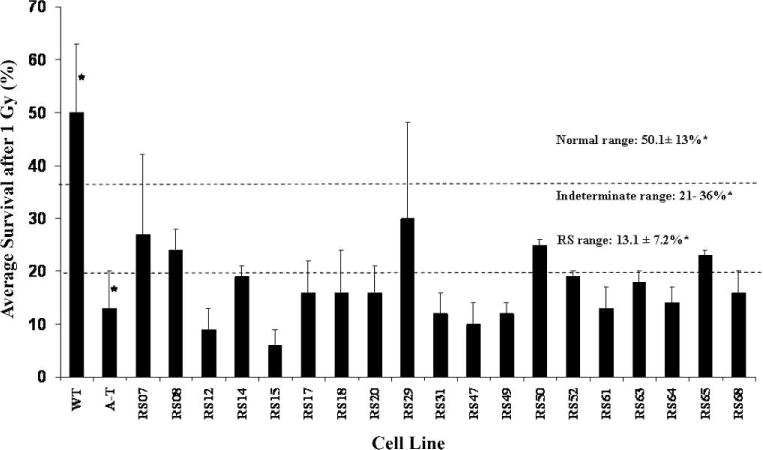
Radiosensitivity of the radiosensitive LCL panel. Radiosensitivity was based on the colony survival assay using previously described ranges (5). Fourteen of the radiosensitive LCLs scored within a radiosensitive range (survival <21%); five scored in an ‘intermediate’ range (21–36%). In all experiments, each radiosensitive LCL was tested with an AT and wild-type LCL internal control. Error bar for each radiosensitive LCL represents the mean ± SD of three experiments. *SE were determined in a previous study (5) using 104 AT and 29 wild-type LCLs.
Neutral Comet Assay
We used neutral comet assay as a second general measure of DNA repair/processing (Fig. 2, Table 1). We scored the %RTB from tail moment values 5 h after 15 Gy as the ratio of nonirradiated/irradiated cells. Five wild-type (WT1–5) and four AT (AT1–4) LCLs were used to establish normal and defective %RTB ranges in these studies. There were no significant differences within the wild-type controls (P = 0.42) or among the AT controls (P = 0.32). The average %RTB of the AT1–4 LCLs normalized to the average tail moment of WT1–5 cells was 55 ± 10 (wild-type = 100). Of the 19 colony survival-defective radiosensitive LCLs, 17/19 (89%) exhibited a %RTB similar to that of ATM-deficient controls by these criteria (P > 0.05); only RS7 (P = 0.16) and RS15 (P = 0.17) did not. We concluded that the neutral comet assay appeared to be a strong potential surrogate assay for colony survival.
FIG. 2.
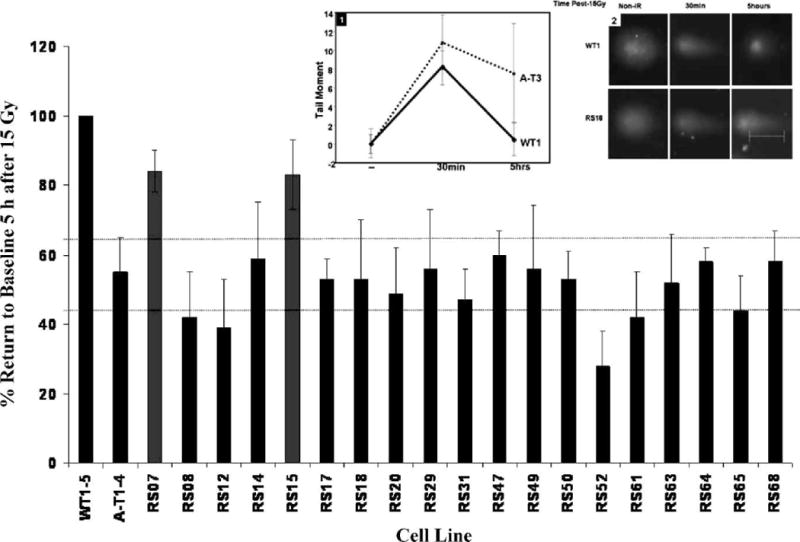
Neutral comet assay measuring the effects of DNA damage 5 h after 15 Gy. Note that all but two radiosensitive LCLs (RS07, RS15) had “AT-like” reduced DNA repair efficiency. Error bars for all LCLs tested represent means ± SD of three experiments, normalized to the average tail moments of five different wild-type LCLs, WT1 to 5 (first bar). Inset 1: Representative data from WT1 and AT3 LCLs demonstrates the kinetics of tail moment 30 min and 5 h postirradiation. Tail moment on the y-axis is the measurement of pixel fluorescence in the form of arbitrary units. The tail moment values of the nonirradiated cells and cells 5 h postirradiation were used to calculate the average percentage return to baseline (%RTB) represented in the bar graph. Inset 2: Representative photomicrographs of comet tails (WT1 and RS18) at 5 h after damage.
γ-H2AX Assay
Previous studies have shown γ-H2AX focus formation in irradiated cells (30, 31, 34). When radiation induces DSBs, γ-H2AX protein complexes rearrange to encompass damaged DNA breakpoints and form microscopically visible foci. The foci dissipate once DSBs are repaired (usually after 1–2 h). We surveyed γ-H2AX focus formation after irradiation. WT1 and 5 and AT3 and 4 cells were again used as internal controls. There were no significant differences among the wild-type control cell lines (P > 0.05) or among the AT control cell lines (P > 0.05) (Supplementary Tables 1 and 2; http://dx.doi.org/10.1667/RR2580.1.S1). We first examined the average spontaneous levels of γ-H2AX focus formation 24 h after irradiation with 1 Gy. Ten of 19 radiosensitive LCLs (53%) exhibited sustained (i.e., 24-h) levels of γ-H2AX focus formation relative to controls (Fig. 3A, Table 1). We also noted a high spontaneous level of γ-H2AX focus formation in some unirradiated LCLs (e.g. RS47, RS65). When we normalized the average γ-H2AX focus formation 24 h postirradiation as a ratio (Fig. 3B), five of the 19 radiosensitive LCLs (26%) exhibited sustained high levels of γ-H2AX foci, especially RS18 and 50. However, the non-normalized data may be more meaningful, reflecting two independent mechanisms: (a) the spontaneous background indicates ongoing unrepaired DNA damage, and (b) the high levels 24 h postirradiation indicate inefficient delayed repair. Thus the ratios in Fig. 3B may be misleading.
FIG. 3.
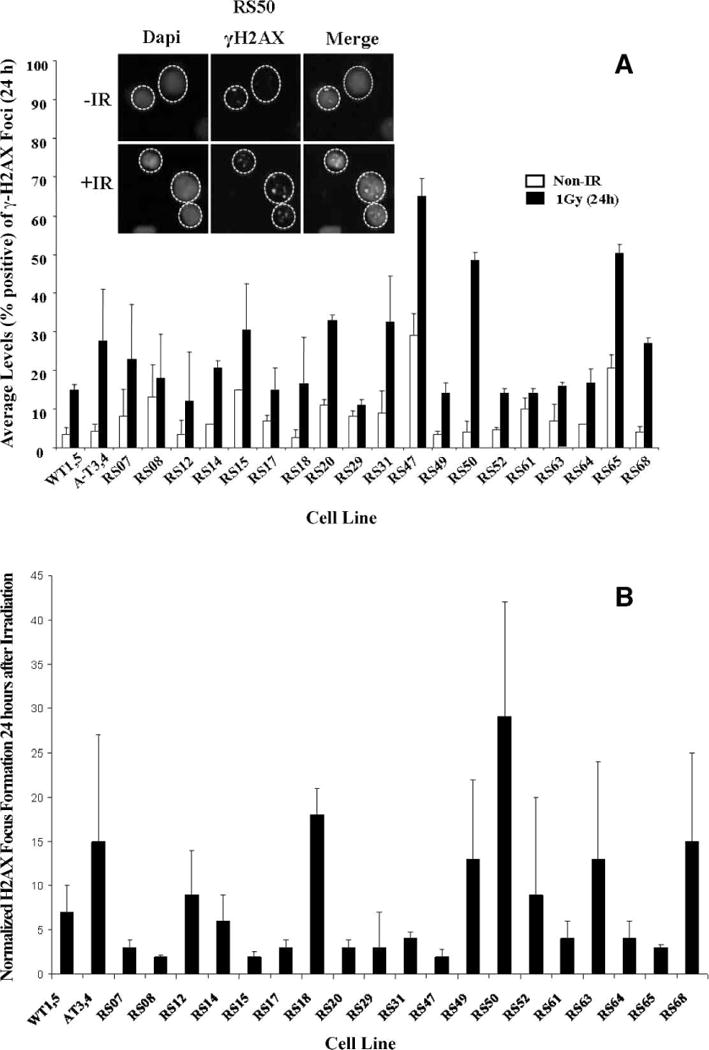
Radiation-induced γ-H2AX focus formation. Panel A: Cells were harvested 24 h after irradiation, stained for γ-H2AX detection (green), counterstained with DAPI (blue), and scored for nuclei with >4 foci. One hundred nuclei were counted per sample (31). WT1 and 5 and AT3 and 4 cells were used as controls. Panel B: Normalization of γ-H2AX focus formation 24 h postirradiation to nonirradiated (spontaneous) focus formation in panel A. Error bars represent means ± SD. N = 3. Inset: representative data for RS50.
pSMC1-s966 Kinetics
By immunoblot analyses, we evaluated the kinetics of ATM-dependent phosphorylation of SMC1-s966 (after 10 Gy) over 24 h. Four patterns (I, II, III, IV) of response were seen (Fig. 4, Table 1), the most striking being pattern IV (see below). In normal LCLs, represented by WT3 cells, phosphorylation of SMC1 peaked at 1 h (pattern I). ATM-deficient cells (AT4) defined a second type of response (pattern II), i.e., no phosphorylation observed at 1 h postirradiation followed by minimally detectable pSMC1 at 4 and 24 h (Fig. 4) as well; the latter times most likely reflect the phosphorylation of SMC1-s966 by other redundant protein kinases (35). A third group of LCLs exhibited a response that peaked at 4 h (rather than at the normal 1-h time) but was generally completed by 24 h postirradiation (pattern III); this pattern was also observed, for example, in Nijmegen breakage syndrome (NBS) LCLs. Such LCLs appeared to be capable of repairing DSBs, but in a somewhat delayed manner. A fourth response, pattern IV, with pSMC1-s966 phosphorylation still continuing at 24 h, most likely represents cells that are not successfully completing DNA repair in a timely manner (Fig. 4); a DNA LigIV-deficient LCL exemplified this pattern. However, many other examples of this pattern were observed, suggesting that this assay may be useful for future translational studies.
FIG. 4.
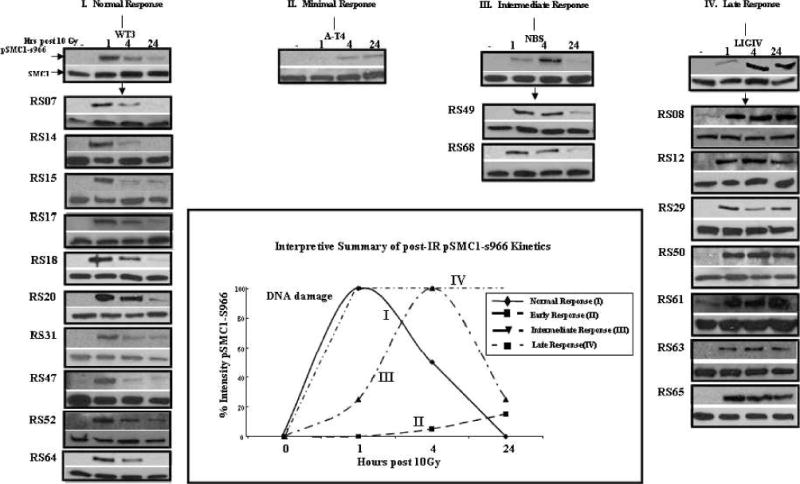
Kinetics of SMC1 phosphorylation after 10 Gy. Normal repair (Pattern I): pSMC1-s966 phosphorylation peaks at 1 h postirradiation; minimal response (pattern II): absence of pSMC1-s966; intermediate response (pattern III): pSMC1-s966 peaks at 4 h postirradiation; late response (pattern IV): pSMC1-s966 peaks at 24 h postirradiation. Native SMC1 was used as a loading control. All experiments were performed three times using WT3 and AT4 controls in each immunoblot. Inset: Qualitative interpretive summary of responses (see text for details).
As can be seen in Fig. 4, 10/19 radiosensitive LCLs tested (53%) had normal kinetics (I). Thus this assay did not compare well with the “gold standard” colony survival assay. FANCD2-deficient LCLs also showed a Type I normal response pattern (data not shown). No LCLs showed the Type II pattern characteristic of AT LCLs, which was anticipated since ATM deficiency was an exclusion criterion for the radiosensitive LCL panel. Two radiosensitive LCLs exhibited an intermediate (Type III) response, suggesting a common underlying repair defect or pathway; seven LCLs showed a much-delayed (Type IV) response, a pattern similar to the one observed with DNA LigIV-defective LCLs. The general relationships of the SMC1-s966 kinetic response curves observed are portrayed graphically in Fig. 4 (see inset at center).
Taken together, the data in Fig. 4 suggest that temporal kinetic analysis of SMC1-s966 phosphorylation provides a useful general assessment of DNA DSB repair. On the other hand, when one considers that every radiosensitive LCL was categorized as radiosensitive by the colony survival assay, there is a surprising lack of concordance between pSMC1 kinetics, colony survival, and the neutral comet assay (Table 1). Although this assay can identify general underlying defects in DNA repair/processing, it would probably be too laborious for use in high-throughput clinical testing.
RDS Assay
RDS reflects a defect in the intra-S-phase cell cycle checkpoint after increasing doses of radiation, which is characteristic of AT cells (23, 34). RDS with [3H]dThd has been used previously as a highly concordant surrogate marker for a diagnosis of AT (23). Thus it was anticipated that RDS would correlate more strongly with colony survival despite the exclusion of AT LCLs from the study. Surprisingly, only 13 of 19 radiosensitive LCLs tested (68%) exhibited an AT-like RDS response that was significantly different from wild-type LCLs (P < 0.05). Six (32%) exhibited a wild-type pattern of postirradiation DNA synthesis (Fig. 5). RDS results for each cell line are listed in Table 1. These observations highlight that the previously published evidence for a very close correlation of RDS with AT LCLs cannot be extrapolated to other radiosensitive LCLs, strongly suggesting that the former correlation between colony survival and RDS is strongly and uniquely dependent on ATM.
FIG. 5.
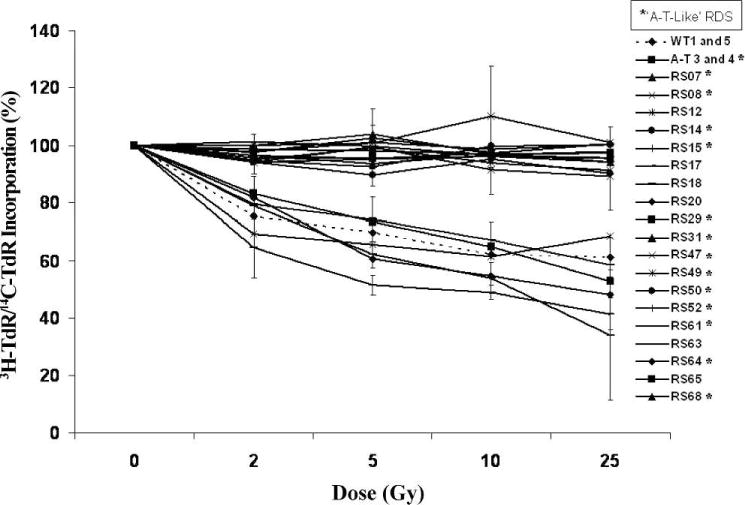
Intra-S-phase cell cycle checkpoint. RDS was performed using cells that were labeled with [14C]dThd for 24 h, subsequently exposed to radiation, and then chased with [3H]dThd for 1 h. Cells were fixed and the [3H]dThd/[14C]dThd ratio was calculated by scintillation (23). Each experiment was performed in triplicate. Error bars represent means ± SD of three experiments. *Represents the radiosensitive LCLs with an AT-like RDS response.
Resazurin Assay
We measured the reduction of resazurin as an indicator of overall mitochondrial respiratory function. Resazurin is blue and nonfluorescent and is reduced by oxidoreductases to resorufin, which is pink and highly fluorescent (32). Of 19 radiosensitive LCLs tested for overall mitochondrial respiratory function, 13 (68%) reduced resazurin at a decreased rate at each time out over the 3-h time course (Fig. 6A, Table 1), while 6 radiosensitive LCLs reduced resazurin at a rate similar to wild-type LCL controls (Fig. 6B, Table 1). These data suggest a fundamental relationship between abnormal mitochondrial respiration and radiosensitivity and are consistent with previously published reports (32, 36).
FIG. 6.
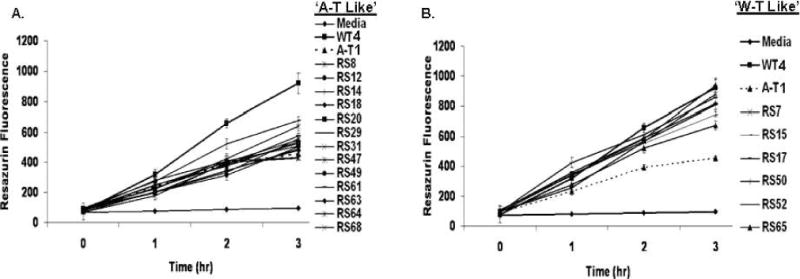
Mitochondrial respiratory function. Resazurin reduction was impaired in 13 of 19 radiosensitive LCLs. Panel A: Thirteen radiosensitive LCLs exhibited a reduced (R) AT-like rate of resazurin reduction. Panel B: Six radiosensitive LCLs exhibited a wild-type (WT)-like rate. Cells were incubated with resazurin (3 μM) in 96-well microtiter plates under constant growth conditions in replicates of six for each condition (32). All LCLs were tested in three independent experiments. Wells containing only resazurin and medium served as experimental controls. Error bars represent means ± SD. N = 3. WT4 and AT1 LCLs were used as further experimental controls. Radiosensitive LCLs exhibited either an AT-like reduced (R) or WT-like (W) response.
DISCUSSION
To identify new surrogate assays for evaluation of individuals with defective DNA DSB sensing/signaling, we analyzed a panel of 19 radiosensitive LCLs that were operationally labeled as radiosensitive based on the colony survival assay. We were attempting to achieve two goals. First, we hoped that functional assays would help us to pinpoint the responsible gene(s) within a repair pathway and would next allow the identification of missing proteins and disease-causing mutation(s). Second, we wanted to establish the degree of concordance between various assays with the “gold standard” colony survival assay. The radiosensitive LCLs were derived from individuals with undiagnosed disorders that included ataxias of unknown etiology, adverse radiation reactions, or symptoms reminiscent of chromosomal instability syndromes (4, 27). Immunoblot analyses had previously excluded known causes of radiosensitivity disorders. Our working model assumed that each radiosensitive LCL has a unique genetic disorder, most likely of an autosomal recessive nature. We found that each LCL precategorized as radiosensitive based on the colony survival assay results was abnormal for at least one additional DNA damage response assay. This was reassuring, because although all known DNA repair disorders tested by us to date have had reduced clonogenic survival and scored similarly to an AT control, this result was not guaranteed a priori in our study. Furthermore, it was entirely possible that the colony survival assay might reflect DSB repair pathways that were not evaluated by some of our assays. Most radiosensitive LCLs were defective for several assays. The low concordance/high variability between the radiosensitive LCLs for each individual assay further illustrates the complexity of the cellular response to radiation. In addition the neutral comet data demonstrate the physical observation of increased DNA breaks and provide a higher concordance with survival than observations of surrogate markers, such as γ-H2AX IRIF. Only RS17 was defective for only one other assay (again, the neutral comet assay). Of the six assays, two involved ATM-dependent functions: pSMC1 and RDS; mitochondrial respiration may be a third. Thus, for example, the data for RS17, 18 and 20 cells most likely represent ATM-independent mechanisms underlying their radiosensitivity and neutral comet assay abnormalities.
Attempts have been made to use the comet assay as a measure of clinical radiosensitivity and genotoxicity and in the study of human disease in LCLs and non-EBV-transfected cells (15–17, 21). The assay requires little specialized equipment other than a microscope and under neutral conditions assesses primarily the repair of DSBs. We found an 89% concordance between the neutral comet assay and colony survival assay. Thus the neutral comet assay may potentially serve as a rapid surrogate marker for colony survival. We did not perform the neutral comet assay on untransformed LCLs because this would not be an alternative for the colony survival assay, and thus no comparisons could be made; however, using peripheral blood lymphocytes would greatly shorten the turnaround time.
The phosphorylation and dephosphorylation of γ-H2AX and SMC1, in addition to controlling cell cycle checkpoints, are necessary for DNA damage/processing (30, 31). A recent study compared clonogenic survival to the potential of using residual phosphorylation of γ-H2AX after irradiation as a marker of radiosensitivity in vitro (37). Although concordance was observed, the differences between this study and ours could likely be due to differences in cell types, methods of γ-H2AX scoring, and the doses of radiation used. We observed that approximately half of the radiosensitive LCLs used in our study had sustained γ-H2AX focus formation 24 h postirradiation, comparable to the ATM-deficient control LCLs. The nonconcordant LCLs may have had defects other than DSBs, and further studies would be needed to elucidate the mechanisms used to cope with such defects. In addition, the high spontaneous background levels of γ-H2AX foci consistently observed in some of our radiosensitive LCLs (e.g., RS47 and RS65) could be due to the phase of the cell cycle the cells were in prior to irradiation (i.e. S phase), although in our studies the vast majority of the cells were in the G1 phase of the cell cycle prior to irradiation (data not shown). Moreover, the high spontaneous background levels may provide yet another indicator of endogenous defective DNA repair/processing. Alternatively, a recent review of persistent γ-H2AX focus formation before and after the induction of DNA damage by radiation reflected nuclear markers of permanent structural rearrangements of chromatin rather than the previously held belief of unrepaired DSBs (38). Furthermore, these permanent structural rearrangements of chromatin might also explain the persistent comet tails in the radiosensitive LCLs seen with the neutral comet assay. Further studies will be needed to address these concerns. The low concordance of the SMC1 phosphorylation assay with the colony survival assay was not surprising since LCLs derived from other known radiosensitivity disorders, tested in other recent studies (28), exhibited wild-type patterns of pSMC1 response. Also, because SMC1 is involved in sister chromatid cohesion, the late response observed in the radiosensitive LCLs might be explained by rearranged chromatin rather than persistent unrepaired DNA damage. These assays illustrate the pitfalls of using surrogate markers for DNA damage/repair.
One characteristic of neurodegenerative diseases is the constitutive expression of signaling pathways that respond abnormally to oxidative DNA damage (32, 33). Mitochondrial respiration rates were reduced in the majority of radiosensitive LCLs tested (68%) (Table 1). A recent report using LCLs generated from young lung cancer patients explored the metabolic activity of mitochondria as an indicator of cell survival as a possible diagnostic test for radiosensitivity (36). In addition, Guo et al. (33) reported that ATM-deficient cells cannot activate DNA repair protein substrates after production of ROS by hydrogen peroxide. This finding provides further support for a mechanistic role for DNA repair proteins and mitochondrial function. Moreover, one can infer that the ability to repair DNA damage caused by ROS may lead to cellular carcinogenesis and also to neurodegenerative syndromes, clinical phenotypes that are similar to many of the radiosensitive LCLs from patients in this study. Thus mitochondrial function may be a valuable surrogate marker for assessing the general integrity of DNA repair.
Our studies may further affect the conventional doses of radiation that are delivered today to cancer patients and to patients undergoing bone marrow ablation prior to stem cell transplantation. Our laboratory has consistently identified four or five new radiosensitive cell lines each year (i.e., ~5% of referred samples) that are not caused by any of the known DNA DSB repair disorders. However, none of the assays described herein would be expected to reliably detect individuals carrying heterozygous defects in DNA repair genes. This could encompass 10% of the general population. Although not radiosensitive to the extent seen in the homozygous deficient individuals for these genes, this subset of ostensibly asymptomatic but partially radiosensitive individuals could bias the empirical testing of any cohort for establishing “safe” doses and would lower the radiation doses recommended for the entire group and for subsequent radiation treatment. If cancer patients could be accurately prescreened for radiosensitivity, to exclude these individuals who are genetically predisposed to radiosensitivity, higher “safe” doses of radiation therapy might then be delivered to the remaining patients. The results of the experiments described herein provide a window to better understanding the genetics of radiation hypersensitivity and the assays that might be considered to identify such individuals. Moreover, such individuals are most likely also cancer prone and are likely to appear again in cancer patient cohorts.
Supplementary Material
Acknowledgments
We acknowledge support from USPHS grants NIH NS052528 and AI 067769, the Ataxia-Telangiectasia Medical Research Foundation, the AT Ease Foundation and GM008243 from the NIH, National Institute of General Medical Sciences. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institute of General Medical Sciences. We especially thank the Joseph Drown Foundation for their encouragement and early support of these studies.
References
- 1.Zampetti-Bosseler F, Scott D. Cell death, chromosome damage and mitotic delay in normal human, ataxia telangiectasia and retinoblastoma fibroblasts after x-irradiation. Int J Radiat Biol Relat Stud Phys Chem Med. 1981;39:547–58. doi: 10.1080/09553008114550651. [DOI] [PubMed] [Google Scholar]
- 2.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247–54. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 3.Huo YK, Wang Z, Hong JH, Chessa L, McBride WH, Perlman SL, et al. Radiosensitivity of ataxia-telangiectasia, X-linked agammaglobulinemia and related syndromes. Cancer Res. 1994;54:2544–7. [PubMed] [Google Scholar]
- 4.Gatti RA, Boder E, Good RA. Immunodeficiency, radiosensitivity, and the XCIND syndrome. Immunol Res. 2007;38:87–101. doi: 10.1007/s12026-007-0018-y. [DOI] [PubMed] [Google Scholar]
- 5.Sun X, Becker-Catania SG, Chun HH, Hwang MJ, Huo Y, Wang Z, et al. Early diagnosis of ataxia-telangiectasia using radiosensitivity testing. J Pediatr. 2002;140:724–31. doi: 10.1067/mpd.2002.123879. [DOI] [PubMed] [Google Scholar]
- 6.Taylor AM, Harnden DG, Arlett CF, Harcourt SA, Lehmann AR, Stevens S, et al. Ataxia telangiectasia: a human mutation with abnormal radiation sensitivity. Nature. 1975;258:427–9. doi: 10.1038/258427a0. [DOI] [PubMed] [Google Scholar]
- 7.Houldsworth J, Lavin MF. Effect of ionizing radiation on synthesis of sub-replicon size DNA in ataxia telangiectasia LCLs. Biochem Int. 1983;6:349–56. [PubMed] [Google Scholar]
- 8.Gotoff SP, Amirmokri E, Liebner EJ. Ataxia telangiectasia. Neoplasia, untoward response to x-irradiation, and tuberous sclerosis. Am J Dis Child. 1967;114:617–25. doi: 10.1001/archpedi.1967.02090270073006. [DOI] [PubMed] [Google Scholar]
- 9.Pollard JM, Gatti RA. Clinical radiation sensitivity with DNA repair disorders: an overview. Int J Radiat Oncol Biol Phys. 2009;74:1323–31. doi: 10.1016/j.ijrobp.2009.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gatti RA, Berkel I, Boder E, Braedt G, Charmley P, Concannon P, et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22-23. Nature. 1988;336:577–80. doi: 10.1038/336577a0. [DOI] [PubMed] [Google Scholar]
- 11.Lange E, Borresen AL, Chen X, Chessa L, Chiplunkar S, Concannon P, et al. Localization of an ataxia-telangiectasia gene to an approximately 500-kb interval on chromosome 11q23.1: linkage analysis of 176 families by an international consortium. Am J Hum Genet. 1995;57:112–9. [PMC free article] [PubMed] [Google Scholar]
- 12.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–53. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 13.Zhang N, Chen P, Khanna KK, Scott S, Gatei M, Kozlov S, et al. Isolation of full-length ATM cDNA and correction of the ataxia-telangiectasia cellular phenotype. Proc Natl Acad Sci U S A. 1997;94:8021–6. doi: 10.1073/pnas.94.15.8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitui M, Nahas SA, Du LT, Yang Z, Lai CH, Nakamura K, et al. Functional and computational assessment of missense variants in the ataxia-telangiectasia mutated (ATM) gene: mutations with increased cancer risk. Hum Mutat. 2009;30:12–21. doi: 10.1002/humu.20805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olive PL, Banáth JP. The comet assay: a method to measure DNA damage in individual LCLs. Nat Protoc. 2006;1:23–9. doi: 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- 16.Srujana K, Begum SS, Rao KN, Devi GS, Jyothy A, Prasad MH. Application of the comet assay for assessment of oxidative DNA damage in circulating lymphocytes of Tetralogy of Fallot patients. Mutat Res. 2010;688:62–5. doi: 10.1016/j.mrfmmm.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 17.Foray N, Arlett CF, Malaise EP. Underestimation of the small residual damage when measuring DNA double-strand breaks (DSB): is the repair of radiation-induced DSB complete? Int J Radiat Biol. 1999;75:1589–95. doi: 10.1080/095530099139197. [DOI] [PubMed] [Google Scholar]
- 18.Rothkamm K, Kruger I, Thompson LH, Löbrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol. 2003;23:5706–15. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harper W, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 20.Falck J, Mailand N, Syljuåsen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410:842–7. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- 21.Jeggo P. The role of the DNA damage response mechanisms after low-dose radiation exposure and a consideration of potentially sensitive individuals. Radiat Res. 2010;174:825–32. doi: 10.1667/RR1844.1. [DOI] [PubMed] [Google Scholar]
- 22.Deckbar D, Stiff T, Koch B, Reis C, Löbrich M, Jeggo PA. The limitations of the G1-S checkpoint. Cancer Res. 2010;70:4412–21. doi: 10.1158/0008-5472.CAN-09-3198. [DOI] [PubMed] [Google Scholar]
- 23.Painter RB, Young BR. Radiosensitivity in ataxia-telangiectasia: A new explanation. Proc Natl Acad Sci U S A. 2010;70:4412–21. doi: 10.1073/pnas.77.12.7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 25.Kitagawa R, Bakkenist CJ, McKinnon PJ, Kastan MB. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 2004;18:1423–3. doi: 10.1101/gad.1200304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 27.Nahas SA, Gatti RA. DNA double strand break repair defects, primary immunodeficiency disorders, and ‘radiosensitivity’. Curr Opin Allergy Clin Immunol. 2009;9:510–6. doi: 10.1097/ACI.0b013e328332be17. [DOI] [PubMed] [Google Scholar]
- 28.Nahas SA, Butch AW, Du L, Gatti RA. Rapid flow cytometry-based structural maintenance of chromosomes 1 (SMC1) phosphorylation assay for identification of ataxia-telangiectasia homozygotes and heterozygotes. Clin Chem. 2009;55:463–72. doi: 10.1373/clinchem.2008.107128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ivey RG, Moore HD, Voytovich UJ, Thienes CP, Lorentzen TD, Pogosova-Agadjanyan EL, et al. Blood-based detection of radiation exposure in humans based on novel phospho-Smc1 ELISA. Radiat Res. 2011;175:266–81. doi: 10.1667/RR2402.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Löbrich M, Shibata A, Beucher A, Fisher A, Ensminger M, Goodarzi AA, et al. gammaH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle. 2010;9:662–9. doi: 10.4161/cc.9.4.10764. [DOI] [PubMed] [Google Scholar]
- 31.Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435–46. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 32.Ambrose M, Goldstine JV, Gatti RA. Intrinsic mitochondrial dysfunction in ATM-deficient lymphoblastoid LCLs. Hum Mol Genet. 2007;16:2154–64. doi: 10.1093/hmg/ddm166. [DOI] [PubMed] [Google Scholar]
- 33.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330:517–21. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 34.Stewart GS, Stankovic T, Byrd PJ, Wechsler T, Miller ES, Huissoon A, et al. RIDDLE immunodeficiency syndrome is linked to defects in 53BP1-mediated DNA damage signaling. Proc Natl Acad Sci U S A. 2007;104:16910–5. doi: 10.1073/pnas.0708408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tomimatsu N, Mukherjee B, Burma S. Distinct roles of ATR and DNA-PKcs in triggering DNA damage responses in ATM-deficient LCLs. EMBO J. 2009;10:629–35. doi: 10.1038/embor.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guertler A, Kraemer A, Roessler U, Hornhardt S, Kulka U, Moertl S, et al. The WST survival assay: an easy and reliable method to screen radiation-sensitive individuals. Radiat Prot Dosimetry. 2011;143:487–90. doi: 10.1093/rpd/ncq515. [DOI] [PubMed] [Google Scholar]
- 37.Menegakis A, Yaromina A, Eicheler W, Dörfler A, Beuthien-Baumann B, Thames HD, et al. Prediction of clonogenic cell survival curves based on the number of residual DNA double strand breaks measured by gammaH2AX staining. Int J Radiat Biol. 2009;85:1032–41. doi: 10.3109/09553000903242149. [DOI] [PubMed] [Google Scholar]
- 38.Costes SV, Chiolo I, Pluth JM, Barcellos-Hoff MH, Jakob B. Spatiotemporal characterization of ionizing radiation induced DNA damage foci and their relation to chromatin organization. Mutat Res. 2010;704:78–87. doi: 10.1016/j.mrrev.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.