

Abstract
The extracellular matrix (ECM) is a highly dynamic structure that is present in all tissues and continuously undergoes controlled remodelling. This process involves quantitative and qualitative changes in the ECM, mediated by specific enzymes that are responsible for ECM degradation, such as metalloproteinases. The ECM interacts with cells to regulate diverse functions, including proliferation, migration and differentiation. ECM remodelling is crucial for regulating the morphogenesis of the intestine and lungs, as well as of the mammary and submandibular glands. Dysregulation of ECM composition, structure, stiffness and abundance contributes to several pathological conditions, such as fibrosis and invasive cancer. A better understanding of how the ECM regulates organ structure and function and of how ECM remodelling affects disease progression will contribute to the development of new therapeutics.
The extracellular matrix (ECM) is a three-dimensional, non-cellular structure that is present in all tissues and is essential for life. Every organ has an ECM with unique composition that is generated in early embryonic stages. The function of the ECM goes beyond providing physical support for tissue integrity and elasticity: it is a dynamic structure that is constantly remodelled to control tissue homeostasis1. The functional importance of the ECM is illustrated by the wide range of tissue defects or, in severe cases, the embryonic lethality caused by mutations in genes that encode components of the ECM2,3. Loss-of-function studies have also shown the importance of ECM proteins in developmental processes, as genetic deletion of specific ECM proteins such as fibronectin and collagens are often embryonic lethal (reviewed in REF. 4).
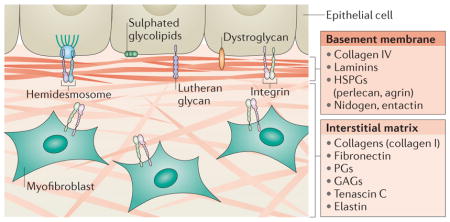
In mammals, the ECM is composed of around 300 proteins, known as the core matrisome, and includes proteins such as collagen, proteoglycans (PGs) and glycoproteins (reviewed in REF. 5). There are two main types of ECM that differ with regard to their location and composition: the interstitial connective tissue matrix, which surrounds cells and provides structural scaffolding for tissues; and the basement membrane, which is a specialized form of ECM that separates the epithelium from the surrounding stroma (BOX 1).
Box 1. The mammalian matrisome.
Using different proteomic techniques and analysing the human and mouse genomes, Hynes and colleagues reported what is so far the most comprehensive list of proteins that define the matrisome in mammals. Among these, ~300 proteins constitute the core matrisome, which consists of 43 collagen subunits, 36 proteoglycans (PCs) and ~200 complex glycoproteins5.
Collagens are the main structural proteins of the extracellular matrix (ECM) and are classified into both fibrillar (collagens I–III, V and XI) and non-fibrillar forms. Collagen fibrils provide tensile strength to the ECM, limiting the distensibility of tissues.
PGs, such as aggrecan, versican, perlecan and decorin, are core proteins with attached glycosaminoglycan (GAG) side chains and are interspersed among collagen fibrils. PGs fill the extracellular interstitial space and confer hydration functions by sequestering water within the tissue. GAGs, especially heparin sulphates, also bind many growth factors, which sequester them in the ECM.
Glycoproteins, such as laminins, elastin, fibronectins, thrombospondins, tenascins and nidogen, have diverse functions. In addition to their role in ECM assembly, they are also involved in ECM–cell interaction by acting as ligands for cell surface receptors such as integrins. Glycoproteins also function as a reservoir of growth factors, which are bound to the ECM and can be released after proteolysis. Cleavage of glycoproteins can generate fragments with different functions than in their original full-length protein.
In addition, there are many ECM-associated proteins that are not part of the matrisome but are nonetheless important in ECM remodelling. These proteins are growth factors and cytokines, mucins, secreted C-type lectins, galectins, semaphorins, plexins and ECM-modifying enzymes that are involved in crosslinking (for example, transglutaminase, lysyl oxidase and hydroxylase).
There are two main types of ECM: the interstitial connective tissue matrix and the basement membrane, a specialized form of ECM separating epithelium from the surrounding stroma and controlling cell organization and differentiation through interactions with cell surface receptors and ECM proteins (see the figure). The interstitial matrix surrounds cells and is mainly composed of collagen I and fibronectin, which provide structural scaffolding for tissues. By contrast, the basement membrane is more compact than the interstitial matrix and mainly consists of collagen IV, laminins, heparan sulphate proteoglycans (HSPGs) and proteins, such as nidogen and entactin, that are synthesized and secreted by epithelial cells, endothelial cells and underlying integrin-expressing myofibroblasts95. Basement membrane express different receptors, such as integrins and hemidesmosomes, that bind to ECM proteins. Hemidesmosomes include two transmembrane proteins, α6β4 integrin and BP180 (180 kDa bullous pemphigoid antigen 2) and two cytoplasmic proteins, BP230 and plectin, that are related to the cytoskeleton. ECM proteins can bind other receptors such as dystroglycan, the Lutheran glycoprotein and sulphated glycolipids such as sulphatides136 (see the figure).
Components of the ECM constantly interact with epithelial cells by serving as ligands for cell receptors such as integrins, thereby transmitting signals that regulate adhesion, migration, proliferation, apoptosis, survival or differentiation. The ECM can also sequester and locally release growth factors, such as epidermal growth factor (EGF), fibroblast growth factor (FGF) and other signalling molecules (such as WNTs, transforming growth factor-β (TGFβ) and amphiregulin). ECM components released through ECM cleavage also regulate ECM architecture and influence cell behaviour1. Moreover, cells are constantly rebuilding and remodelling the ECM through synthesis, degradation, reassembly and chemical modification6. These processes are complex and need to be tightly regulated to maintain tissue homeostasis, especially in response to injury. Indeed, dysregulated ECM remodelling is associated with pathological conditions and can exacerbate disease progression. For example, abnormal ECM deposition and stiffness are observed in fibrosis and cancer7, and excessive ECM degradation is linked to osteoarthritis8.
Here, we review recent advances in our understanding of ECM dynamics in development and tissue homeostasis and discuss how dysregulation of ECM structure and composition leads to disease. We first describe the endopeptidases that degrade and cleave ECM components, which can also generate fragments with biological functions that differ from their full-length counterparts. We then summarize progress in elucidating the molecular mechanisms underlying the role of the ECM during normal morphogenesis, focusing on the best known examples: the development of the intestine, the lungs and the mammary and submandibular glands (also known as salivary glands). Finally, we discuss how dysregulated ECM remodelling leads to diseases such as fibrosis and cancer and highlight some ECM-related targets with therapeutic potential that are currently being tested in clinical trials. These conceptual advances in understanding the ECM in physiological and pathological processes will guide us in developing better therapies for disease.
ECM breakdown by proteases
Cleavage of ECM components is the main process during ECM remodelling and is important for regulating ECM abundance, composition and structure, as well as for releasing biologically active molecules (such as growth factors). The ECM can be cleaved by different families of proteases.
Matrix metalloproteinases
Matrix metalloproteinases (MMPs) are the main enzymes involved in ECM degradation. Their activity is low in normal conditions but increased during repair or remodelling processes and in diseased or inflamed tissue. MMPs are produced either as soluble or cell membrane-anchored proteinases and cleave ECM components with wide substrate specificities.
MMPs were discovered in 1962 in a study of collagen remodelling during tadpole tail metamorphosis9. So far, 23 human MMPs have been identified (FIG. 1). Most MMPs are secreted as zymogens and are subsequently activated in the extracellular space. MMP activation primarily occurs via proteolytic cleavage (by Ser proteases or other MMPs) or by modifying the thiol group by oxidation (for example, through reactive oxygen species generated by leukocytes). Collectively, MMPs can degrade all ECM proteins6 (FIG. 1) and their proteolytic actions on the ECM have crucial roles in organogenesis and branching morphogenesis.
Figure 1. Structure and targets substrates of metalloproteinases.

Metalloproteinases belong to the metzincin enzyme family, which includes matrix metalloproteinases(MMPs), adamalysins (which includes ADAMs (a disintegrin and metalloproteinases) and ADAMTS (ADAMs with a thrombospondin motif)) and astacins (including meprins). They are multidomain enzymes that contain the highly conserved motif HEXXHXXGXXH (where X is any amino acid), in which three His residues chelate a zinc ion in the catalytic site. Metalloproteinases are produced either as soluble or membrane-anchored enzymes that cleave components of the extracellular matrix (ECM). MMPs are composed of several shared functional domains: signal peptide domain, propeptide domain, catalytic domain and haemopexin-like domain (except MMP7, MMP23 and MMP26). The amino-terminal signal peptide domain is required for the secretion of MMPs. The propeptide domain contains the Cys-switch motif PRCGXPD. The catalytic domain (which has proteolytic activity) contains the zinc-binding motif; the Cys residue in this motif interacts with the zinc ion that keeps pro-MMPs inactive until the propeptide domain is removed. The carboxy-terminal haemopexin-like domain, which is present in almost all MMPs, is involved in substrate specificity and in the non-proteolytic functions of MMPs160. Membrane-type MMPs (MTMMPs) such as MMP14 are anchored to the cell surface by either a transmembrane domain followed by a short cytoplasmic tail or a glycosylphosphatidylinositol (GPI) sequence. Some MMPs, including MTMMPs, MMP11, MMP17, MMP21, MMP23, MMP25 and MMP28 can be activated by the furin convertase, which cleaves the propeptide of inactive precursors in the Golgi apparatus, to release functional proteins. ADAMs are transmembrane proteins that are structurally similar to MTMMPs, except that they lack the haemopexin domain and instead have three other domains: the Cys-rich domain, the epidermal growth factor (EGF)-like repeat domain (except ADAM10 and ADAM17) and the disintegrin domain. Only ADAM9, ADAM10, ADAM12 and ADAM15 are shown, as the other ADAMs do not have known ECM protein substrates. ADAMTSs are secreted proteinases and have thrombospondin type I-like repeats in their C-terminal sequence. In addition to the metalloproteinase domains, the meprins also have an astacin-like catalytic domain (Ast-like Cat), a MAM (meprin A5 protein Tyr phosphatase) domain, a TRAF (TNFR-associated factor) domain and a C-terminal cytosolic tail. Meprin-α also contains a furin cleavage domain, cleavage of which results in the loss of the EGF-like transmembrane domain and the cytosolic domain and release of the enzyme into the extracellular space. MMP23 contains an immunoglobulin (Ig) domain that is unique among the MMPs. This Ig domain facilitates protein–protein or lipid–protein interactions similar to the haemopexin domain of other MMPs.
Adamalysins
This protein family includes ADAMs (a disintegrin and metalloproteinases)10 and ADAMTS (ADAMs with a thrombospondin motif)11. So far, 22 ADAM genes have been identified in humans but only 12 encode active proteinases. ADAMs are ‘sheddases’: they can cleave transmembrane protein ectodomains that are adjacent to the cell membrane, thus releasing the complete ectodomain of cytokines, growth factors, receptors and adhesion molecules (reviewed in REF. 12). The disi ntegrin domains mediate cell–ECM interactions by binding integrins, and the Cys-rich domains interact with heparan sulphate proteoglycans (HSPGs; reviewed in REF. 13). ADAM10, ADAM12 and ADAM15 can also cleave ECM proteins such as collagens (FIG. 1).
In contrast to ADAMs, ADAMTS are secreted proteinases with thrombospondin type I-like repeats in their carboxy-terminal sequences. The aggrecanases (ADAMTS1, ADAMTS4, ADAMTS5, ADAMTS8, ADAMTS9, ADAMTS15 and ADAMTS20) are proteo-glycanolytic. ADAMTS2, ADAMTS3 and ADAMTS14 are pro-collagen N-propeptidases that process pro-collagens I, II and III and are important for depositing normal collagen fibrils onto the ECM in a tissue-specific manner (reviewed in REF. 14). ADAMTS13, which cleaves von Willebrand factor, is involved in coagulation and thrombotic thrombocytopenic purpura (TTP). The functions of ADAMTS6, ADAMTS7, ADAMTS10, ADAMTS12, ADAMTS16, ADAMTS17, ADAMTS18 and ADAMTS19 are still unclear.
Meprins
The meprins belong to the astacin family and are composed of two subunits (α and β) that are encoded by two different genes15. The subunits can form heterocomplexes and homocomplexes linked by disulphide bridges16. In contrast to meprin-α (which is a secreted protein as it loses its transmembrane domain by cleavage during biosynthesis), meprin-β is predominantly expressed on the cell surface but can be released from the membrane by shedding via ADAM10 (REF. 17). Meprins can cleave ECM proteins such as collagen IV, nidogen and fibronectin18 (FIG. 1). In addition, meprins may be necessary for the generation of mature collagen molecules by cleaving pro-collagen I that is assembled into collagen fibrils, which are important for skin tensile strength19. Meprins can also indirectly regulate ECM remodelling by activating the other metalloproteinases. For example, ADAM10 is cleaved by meprin-β20, and both meprin-α and meprin-β promote the cleavage of pro-MMP9 by MMP3, thus accelerating the activation of MMP9 (REF. 21). Compared with the other metallo-proteinases, the roles of meprins in ECM remodelling are poorly understood.
Metalloproteinase inhibitors
ECM proteolysis requires tight regulation to avoid excessive and deleterious tissue degradation. The activity of endogenous inhibitors that inactivate ECM proteinases is thus important for tissue integrity. The tissue inhibitor of metalloproteinases (TIMP) family consists of four members (TIMP1–TIMP4) that reversibly inhibit the activity of MMPs, ADAMs and ADAMTS, but not of meprins. MMP-to-TIMP ratios determine the overall proteolytic activity, and each TIMP displays preferential MMP-binding specificity (reviewed in REF. 22). TIMP3 is sequestered in the ECM, whereas the other TIMPs are present in soluble form in vivo. Although TIMP3 is the main inhibitor of ADAMs and ADAMTS, the membrane-associated RECK (reversion-inducing Cys-rich protein with Kazal motifs) also regulates the activity of MMPs and ADAMs23. Cystatin C, elafin and fetuin A have been identified as natural inhibitors of meprins20.
Other enzymes important in ECM remodelling
Ser proteases can also target many ECM proteins. The two plasminogen activators urokinase and tissue plasminogen activator target plasminogen to generate plasmin, a protein that degrades fibrin, fibronectin and laminin24. Moreover, the Ser protease elastase is released by neutrophils and promotes the breakdown of fibronectin and elastin25, and the membrane-anchored Ser protease matriptase, which is expressed by epithelial cells, is important in maintaining the intestinal barrier (reviewed in REF. 26).
In addition, cathepsins are found both extracellularly and intracellularly in lysosomes. Secreted cathepsins degrade extracellular ECM proteins, but many cells can also internalize ECM components such as collagen through endocytosis and degrade them in the lysosomes27. Families of cathepsins include the Ser cathepsins (cathepsins A and G), Asp cathepsins (cathepsins D and E) and Cys cathepsins28.
Finally, heparanases and sulphatases can alter the properties of ECM PGs (reviewed in REF. 6). Heparanase, an endoglucuronidase responsible for heparan sulphate (HS) cleavage, regulates the structure and function of HSPGs. This results in structural alterations of the ECM and the release of bioactive saccharide fragments and HS-bound growth factors and cytokines. Suphatase 1 and sulphatase 2 are secreted endosulphatases that remove 6-O-sulphate residues from HS and modulate HS binding to many cytokines and growth factors, including FGF1 and vascular endothelial growth factor (VEGF)29.
ECM dynamics in intestinal development
In vertebrates, the intestine is formed from the embryonic endoderm layer, which undergoes multiple morphological changes to give rise to an adult epithelium with finger-like luminal projections called villi and epithelial invaginations called intestinal crypts. Within these crypts reside intestinal stem cells, which express Leu-rich-repeat-containing G protein-coupled receptor 5 (LGR5)30. To illustrate the roles of the ECM in development, we discuss our current understanding of how the ECM regulates intestinal morphogenesis in anuran (for example, frog and toad) tadpoles and in other vertebrates.
Intestinal morphogenesis in anurans
The anuran tadpole intestine is composed of a simple tubular organ made of a monolayer of epithelial cells (FIG. 2a). Metamorphosis from tadpole to the adult anuran involves the death of these cells, which undergo apoptosis induced by thyroid hormone; meanwhile, the adult epithelial cells proliferate rapidly (reviewed in REF. 31). During intestinal metamorphosis, the basement membrane also thickens, which suggests that the ECM might have a role in intestinal morphogenesis. Moreover, the addition of the ECM proteins laminin, collagen and fibronectin in vitro inhibits tadpole epithelial cell apoptosis induced by thyroid hormone32, which supports the idea that ECM remodelling influences cell fate during intestinal morphogenesis.
Figure 2. Extracellular matrix remodelling during intestinal development.

a | Tadpole-to-adult intestinal epithelium remodelling during Xenopus laevis morphogenesis. In the pre-metamorphosis tadpole, the small intestine consists of a single layer of larval epithelium (also known as typhlosole), connective tissue and a thin muscle layer. During metamorphosis, thyroid hormone (TH) is produced in high levels, inducing the release of matrix metalloproteinase 11 (MMP11) by stromal cells to trigger apoptosis of larval epithelial cells. At the same time, proliferating stem and progenitor cells give rise to new adult epithelial cells that replace the larval epithelium. During metamorphosis, the basement membrane and the muscle layer are thicker. The levels of other MMPs, such as MMP2 and/or MMP9 and MMP14, increase during tadpole metamorphosis after epithelial cell death, suggesting that they may have a role post-apoptosis. At the end of metamorphosis, the differentiated adult intestine becomes capable of self-renewal and forms a multiply folded epithelium, similar to the mammalian adult intestine.
b–d | Intestinal epithelium remodelling in other vertebrates. Laminin distribution during mammalian intestine development determines small intestine and colon architecture. The basement membrane of villi in the intestine is composed mainly of laminin 511 α5 chain. In mice, a lack of laminin 511 in the intestinal basement membrane leads to a compensatory deposition of colonic laminins (laminin 111 and laminin 411), which results in the transformation of the small intestinal to a tissue with a colon-like mucosal structure that shows high levels of cell proliferation, low levels of the cell cycle inhibitor cyclin-dependent kinase inhibitor B (CDKN1B), and higher numbers of goblet cells (part b). RGD-dependent substrates such as fibronectin can bind to α8β1 integrin. This anchorage prevents anoikis in undifferentiated human intestinal epithelial crypt (HIEC) cells through the recruitment of vinculin and the activation of the PI3K–AKT signalling pathway (part c). Collagen VI is produced by HIEC cells and regulates fibronectin assembly by restraining cell–fibronectin interactions, which influences cell functions such as migration. A lack of collagen VI leads to recruitment of tensin at the fibrillar adhesion points via the activation of myosin light chain kinases (MLCKs), which mediate actomyosin contractility, extensive fibrillogenesis and cell migration (part d). FAK, focal adhesion kinase. Figure part a modified from Establishment of intestinal stem cell niche during amphibian metamorphosis. Curr. Top. Dev. Biol Volume 103. Chapter 11. Pages 305–327. 2013. With permission from Elsevier.161
Several MMPs induced by thyroid hormone during metamorphosis have a pivotal role in remodelling the intestine. For example, MMP11 (also known as stromelysin 3) is released and activated by fibroblasts after thyroid hormone stimulation33 and is essential for epithelial cell apoptosis and invasion of the connective tissue during metamorphosis34 (FIG. 2a). Although the exact role of MMP11 is unknown, MMP11 can cleave laminin receptor and may therefore affect cell fate and migration during intestinal development35. Other MMPs, such as MMP2, MMP9 and MMP14 (also known as membrane type 1 MP; MT1MMP)36, are also upregulated during tadpole metamorphosis in response to thyroid hormone. The levels of MMP2 and MMP9 increase following epithelial cell death, suggesting that, in contrast to MMP11, these MMPs may have a role in post-apoptosis ECM remodelling at later stages of intestinal morphogenesis. Despite the fact that both of these MMPs function cooperatively during metamorphosis by contributing to the MMP14-mediated activation of pro-MMP2, their exact biological role in intestinal development has not been elucidated37.
Intestinal morphogenesis in other vertebrates
In mammals, the involvement of ECM remodelling in intestinal development was first suggested by changes in the levels and localization of ECM proteins along the intestinal villus–crypt axis (reviewed in REF. 38). For example, in the perinatal rat intestine, there is a transient disappearance of fibronectin and pro-collagen III at the top of the outgrowing villus, whereas the basement membrane proteins are restricted to the base of the villi when crypts develop39. In addition, the levels of collagen IV mRNA synthesized by the mesenchyme are high during intestinal morphogenesis, suggesting an important role for this type of collagen in gut development40.
Laminins also play an important part in cell–ECM interactions during intestinal morphogenesis and differentiation (reviewed in REF. 41). For example, mice lacking the laminin 511 α5 chain in the small intestinal basement membrane compensate by depositing colonic laminins (laminin 111 and laminin 411), which results in the transformation of the small intestine to a tissue with a colon-like mucosal architecture characterized by high levels of cell proliferation, probably owing to decreases levels of the cell cycle regulator cyclin-dependent kinase inhibitor B (CDKN1B)42 (FIG. 2b). These studies show that ECM content remodelling has a crucial role in guiding tissue architecture during intestinal development.
Integrins are differentially expressed during intestinal development (reviewed in REF. 43). Interestingly, in vitro studies show that intestinal epithelial cells express RGD-dependent integrins, which recognize the RGD motif found in many ECM molecules, such as fibronectin, to anchor to the ECM and control their position and fate. In human intestinal epithelial crypt (HIEC) cells, which share features with intestinal epithelial stem cells, the interaction of α8β1 integrin with the ECM regulates adhesion, migration and cell proliferation via a ROCK (RHO-associated protein kinase)-dependent mechanism44. This serves as a checkpoint to regulate anoikis sensitivity, with adhesion preventing anoikis through the recruitment of vinculin and the activation of phosphoinositide 3-kinase (PI3K)–AKT signalling45 (FIG. 2c). In addition, another study showed that the amount of collagen VI in the basement membrane regulates fibronectin assembly by restraining cell–fibronectin interactions, which influences HIEC cell migration via the activation of myosin light chain kinases (MLCKs)46 (FIG. 2d). However, further in vivo or ex vivo studies are required to understand whether these phenomena occur only in the crypt area, where the LGR5-expressing intestinal stem cells are localized or in other more differentiated areas of the intestine.
How metalloproteinases are involved in ECM remodelling during intestinal development in vertebrates has not been well studied. Although meprin-α and meprin-β are differentially expressed during small intestine development in rats47, it is not clear how this affects ECM organization during this process.
So, there is evidence showing that ECM remodelling is essential for intestinal development and maturation; however, our understanding of the molecular mechanisms involved is very limited and thus calls for further studies. Recent advances in ex vivo culture systems, such as the development of intestinal organoids48, will allow us to further study the ECM dynamics that govern intestinal development and differentiation. Characterizing ECM components and their role in the adult intestinal stem cell niche will also have implications in understanding stem cell self-renewal mechanisms.
ECM remodelling in branching morphogenesis
Many organs, including the lungs and the mammary and submandibular glands, are formed during embryonic development by epithelial branching, which establishes the architecture of these organs. Branching involves repetitive formation of epithelial clefts and buds that invade surrounding embryonic ECM, which changes in composition and distribution over time. Although they share some common structural features, each organ is characterized by a specific structure and uses distinct molecular mechanisms for branching (reviewed in REF. 49). ECM remodelling plays a crucial part in governing organ branching by providing structural integrity and regulating diverse cellular processes such as cell shape, cell motility and cell growth.
Deposition of ECM proteins controls morphogenesis
Many ECM components are locally synthesized and reorganized to modulate cell behaviour and promote branching morphogenesis. For example, collagen I and III accumulate at the cleft points of the branching submandibular glands, and treatment with collagenase completely inhibits cleft formation and branching50,51. In mammary glands, collagen I is predominantly localized around the duct52, and collagen I fibres are oriented before branching initiation. Collagen I fibres direct epithelial branching by promoting an intracellular reorganization of epithelial cell actomyosin network. This process is mediated by the activation of the RHOA–ROCK pathway, which triggers cell contraction53 (FIG. 3Aa).
Figure 3. Extracellular matrix remodelling during branching morphogenesis.

A| Ductal elongation and branching of the mammary and submandibular glands. Aa | Collagen is locally synthesized and aligned to increase extracellular matrix (ECM) stiffness and create a mechanical anisotropy that will drive branching. Collagen synthesis is mediated by the activation of the RHOA–RHO-associated protein kinase (ROCK) signalling pathway. The ECM at the end bud tip is much thinner than in cleft region and around the duct. Matrix metalloproteinase 2 (MMP2) and MMP14 are expressed and active at the end bud tip, whereas MMP3 is involved inside branching. Ab | Cleft formation and deepening in the submandibular glands. Fibronectin is locally assembled in the basement membrane and induces BTBD7 at the base of forming clefts, which in turn upregulates the transcription factor SNAIL2 and downregulates the adhesion molecule E-cadherin. These molecular events promote alterations in cell shape, decreasing cell–cell adhesion and promoting a motile phenotype to promote cleft progression. Fibronectin assembly requires focal adhesion kinase (FAK) activation and RHOA–ROCK-mediated actomyosin contraction. B | Role of elastin and collagen deposition in alveolar branching in the lung. Elastin and collagen deposition promotes ECM stiffness in the neonatal lung and facilitates signalling through the endothelial lipoprotein receptor-related protein 5 (LRP5)–TIE2 (also known as angiopoietin 1 receptor) pathway, which is required for normal lung development. Consistent with this, disrupting lung collagen I, III and VI and elastin expression and localization through treatment with β-aminopropionitrile, an inhibitor of the collagen crosslinking enzyme lysyl oxidase (LOX), softens neonatal mouse lung tissue and downregulates the expression of LRP5 and TIE2, which leads to an inhibition of vascular and alveolar morphogenesis in neonatal mice. Ca | Heparan sulphate proteoglycans (HSPGs) bind fibroblast growth factors (FGFs) with different affinity and help to create a concentration gradient that can control cell fate in submandibular glands. In contrast to FGF10, FGF7 binds HSPG with low affinity and diffuses broadly, promoting branching in the submandibular gland. Cb| HSPGs such as perlecan bind FGF10 with high affinity. Following cleavage by heparanase, perlecan releases FGF10, which can then diffuse locally and promote duct elongation. FGFR2, FGF receptor 2; MLC, myocin light chain; MYPT, myosin phosphatase. Figure part Ca modified from Differential interactions of FGFs with heparin sulfate control gradient formation and branching morphogenesis. Sci. Signal. Volume 2. Issue 88. Page ra55. 2009. Reprinted with permission from AAAS.
Fibronectin is also essential for cleft formation during the initiation of epithelial branching in the submandibular gland54. The local accumulation of fibronectin rapidly induces the expression of the transcriptional regulator BTBD7, which in turn induces local expression of the epithelial–mesenchymal transition (EMT)-promoting factor SNAIL2 and suppresses E-cadherin levels, thus altering cell morphology and reducing cell–cell adhesion to promote cleft progression55 (FIG. 3Ab). This cascade is mediated by integrins, which, through the action of focal adhesion kinase (FAK), activate RHOA–ROCK-mediated actomyosin contraction to stabilize the newly formed cleft56,57. RHOA–ROCK inhibit the myosin phosphatase MYPT1 (also known as PPP1R12A), stimulating the phosphorylation of the myosin regulatory light chain (MLC), tipping the balance towards contractility and enhancing α5β1 integrin expression on the cell surface. This modulates fibronectin assembly involved in cell migration and branching morphogenesis in submandibular glands58. It will be interesting to investigate whether these signalling events also happen in other branching organs such as mammary glands, in which fibronectin levels are increased during development59.
Finally, elastin is the most abundant ECM protein in the lungs, and its deposition and remodelling at the leading edge of a growing secondary alveolar septal ridge is crucial for the formation of alveoli in mice60. The molecular mechanisms of how elastin deposition controls lung morphogenesis have not been investigated. However, it has been proposed that elastin and collagen deposition increases ECM stiffness in the neonatal lung by facilitating signalling through the endothelial lipoprotein receptor-related protein 5 (LRP5)–TIE2 (also known as angiopoietin 1 receptor) pathway, which is required for normal lung development.[Au: OK?] Indeed, inhibition of collagen III and elastin expression through treatment with β-aminopropionitrile (which inhibits the collagen crosslinking enzyme lysyl oxidase (LOX)) results in softening of neonatal mouse lung tissue and downregulation of the expression of LRP5 and TIE2, which leads to inhibition of the vascular and alveolar morphogenesis in neonatal mice61 (FIG. 3B).
Taken together, these results demonstrate that local deposition of specific ECM proteins is crucial for branching organs, not only by giving a structural stability to guide tissue expansion in a certain direction but also by inducing signalling pathways to change cell shape and migration and favouring other events such as vascularization, which are important for a normal organ development.
ECM cleavage facilitates branching morphogenesis
The ECM at the end bud tip is much thinner than in cleft region and around ducts62, suggesting a need to cleave the ECM at the invasive front of the epithelium, giving the cells an opportunity to proliferate, migrate and invade the surrounding mesochyme (FIG. 3A). The importance of ECM cleavage in this process is evident from the phenotype of mice lacking metalloproteinases: a lack of MMP11 leads to a decreased periductal collagen content and decreased mammary gland morphogenesis63. MMP14 is highly expressed at the invading edges of terminal end buds, and its expression increases during branching, indicating that it has a role in the branching process64. MMP3 and MMP2 are also involved mammary gland branching morphogenesis: Mmp2-null mice have defects in primary branching, and mice lacking Mmp3 have a defect in side branching but not in primary branching65 (FIG. 3A).
In addition to their proteolytic actions, recent studies show that several MMPs, such as MMP3 (REFS 66, 67) and MMP14 (REF. 64), regulate invasion and branching in the mammary gland independently of their proteolytic activity. For example, MMP3 uses its haemopexin domain to bind WNT5B and increase the number of mammary stem cells and branching morphogenesis66, whereas MMP14 uses its transmembrane and cytoplasmic domains to regulate the expression and activity of β1 integrin to control cell invasion and mammary gland branching. These data show that MMPs function both proteolytically and non-proteolytically to regulate the extracellular microenvironment during branching morphogenesis.
ECM remodelling releases growth factors
The ECM functions as a ligand ‘reservoir’ by binding numerous growth factors. In this way, ECM can retain some growth factors that can be proteolytically released and can locally induce proliferation and branching morphogenesis1.
For example, HSPGs are essential for biological processes that are regulated by FGFs (reviewed in REF. 68). The affinity of FGFs for HSPGs varies and regulates their gradient and effects on submandibular gland branching. As FGF10 binds HSPGs with high affinity, only a portion of the epithelium is exposed to this growth factor, resulting in directional outgrowth of elongating ducts in submandibular glands. By contrast, FGF7 binds HSPG with low affinity, and its gradient is broader, inducing the formation of many buds in multiple directions69 (FIG. 3Ca). In addition, heparanase endogenously colocalizes with perlecan in the basement membrane and in submandibular gland cleft. The addition of heparanase to submandibular gland cultures increases morphogenesis owing to the release of FGF10 from perlecan and the activation of mitogen-activated protein kinase (MAPK) signalling69,70 (FIG. 3Cb) These data show that ECM remodelling through proteolytic degradation can release growth factors from the ECM reservoir, which can affect epithelial cell proliferation and migration and regulate organ morphogenesis.
Aberrant ECM remodelling in disease
Given the crucial importance of the ECM during development and for the maintenance of tissue homeostasis, it is not surprising that dysregulation of ECM components can lead to disease. For example, mutations in genes that encode factors that affect the composition and architecture of the ECM, such as COL1A1 (α-chain of collagen I), result in severe defects in bone formation. However, aberrant ECM remodelling can also lead to various pathological states in more subtle ways such as in osteoarthritis, fibrosis and cancer.
Increased ECM breakdown causes tissue destruction
The importance of ECM homeostasis is clear from diseases characterized by abnormal ECM breakdown, a process primarily mediated by proteinases such as MMPs and ADAMs. For example, abnormally high levels of heart-specific MMP1 expression result in collagen loss and diminished contractility that leads to cardiomyopathy71. In osteoarthritis, ADAMTS4 and ADAMTS5 are abnormally overexpressed and are partially responsible for the pathological destruction of cartilage ECM72.
The mechanisms mediating metalloproteinase upregulation remain unclear but may involve receptors involved in cell–ECM interactions, such as syndecan 4, which activate MMP3 and ADAMTS5 to promote ECM breakdown73 (FIG. 4A). Interestingly, loss-of-function mutations in MMP2 also cause a severe osteolytic and arthritic syndrome74. This counterintuitive discovery that deficiency of a protease results in osteolysis and arthritis demonstrates that normal bone and cartilage homeostasis depends on balanced ECM breakdown and synthesis. Taken together, these studies highlight the importance of an intact ECM network in maintaining tissue architecture, support and homeostasis.
Figure 4. Aberrant extracellular matrix remodelling leads to numerous human diseases.

A | Over-degradation of the extracellular matrix (ECM), mediated primarily by matrix metalloproteinases (MMPs) and ADAMTS (ADAMs with a thrombospondin motif), results in osteoarthritis and increased breakdown of the connective tissue. B | As a result of chronic inflammation or tissue injury, transforming growth factor-β (TGFβ), connective tissue growth factor (CTGF), interleukin-13 (IL-13) and other factors stimulate fibroblasts and myofibroblasts (the main ECM producers) to produce more ECM, resulting in pathological fibrosis. The excess ECM further stimulates fibroblasts to continue making ECM, forming a positive feedback loop. Fibrosis is a major risk factor for developing cancer, including hepatocellular carcinoma and breast cancer. C | The ECM contributes to cancer pathogenesis by several mechanisms: functioning as a barrier to chemotherapy, to monoclonal antibodies such as cetuximab and to immune therapy mediated, for example, by cytotoxic T cells (CTLs) (part Ca); forming migration track ‘highways’ that regulate the interaction of immune cells with cancer cells (part Cb); stimulating integrin signalling through increased ECM stiffness, which promotes ECM synthesis, invasion and proliferation (lysyl oxidase (LOX) and LOX-like 2 (LOXL2) are enzymes that crosslink collagen and are the main enzymes responsible for increasing ECM stiffness) (part Cc); forming a niche for new metastatic cells and providing survival and proliferative signals (part Cd); generating novel bioactive ECM fragments from native ECM chains, and stimulating cell migration or immune cell recruitment (part Ce); activating cell–ECM receptors such as discoidin domain-containing receptor 1 (DDR1) and DDR2, which bind directly to collagen and regulate transcriptional pathways to increase MMP and epithelial–mesenchymal transition (EMT) marker expression (part Cf); and sequestering growth factors that can be released by proteolytic cleavage, which then diffuse to bind receptors to stimulate cell growth, EMT or angiogenesis (part Cg).
Excess ECM leads to pathological fibrosis
Chronic or severe tissue injuries leading to excessive ECM production and deposition without reciprocally balanced degradation can result in fibrosis (FIG. 4B). This aberrant healing process can be so severe that it leads to organ failure, such as cirrhosis in the liver or myelofibrosis in the bone marrow. Fibrosis also increases the risk of cancer; for example, liver cirrhosis increases the risk of hepatocellular carcinoma by 20–30%75, and increased mammographic density, which reflects the amount of collagen in the breast, correlates with increased risk of breast cancer76.
Fibrosis is mediated primarily by fibroblasts or myofibroblasts, as well as other stromal cells. The TGFβ pathway is perhaps the most well-studied and potent stimulator of fibrosis77. TGFβ induces the translocation of the SMAD2–SMAD3 complex of transcription factors into the nucleus, where it directly promotes the expression of ECM genes such as COL1A1, COL3A1 and TIMP1, as well as about 60 other ECM-related genes78. Owing to space limitations, the reader is referred to several excellent recent reviews regarding additional molecular signals that regulate fibrosis79–81.
Some cytokines, such as interleukin-33 (IL-33), promote fibrosis by activating immune cells82. In the liver, IL-33 promotes the expansion of resident innate lymphoid cells, which produce IL-13 to activate hepatic stellate cells, the main ECM producers in the liver. IL-13 promotes fibrosis by stimulating collagen accumulation, downregulating MMPs and recruiting pro-fibrotic innate immune cells83. IL-13 also promotes the differentiation of fibroblasts into myofibroblasts and increases TGFβ expression84. How these cytokines and growth factors are upregulated in different tissues remains to be elucidated. In the case of the intestinal tract, recent studies have suggested that the micro-biome is an important pro-inflammatory component that stimulates fibrosis85.
Interestingly, it was recently shown that a fibrotic ECM can stimulate fibroblasts to further increase ECM production86. Experiments in which ECM and fibro-blasts were isolated from patients with or without idiopathic pulmonary fibrosis (IPF) and then co-cultured indicated that the origin of the ECM had a greater effect on gene expression than the origin of the fibro-blasts. ECM derived from patients with IPF increased ECM production in both non-IPF and IPF fibroblasts by inducing downregulation of the microRNA miR-29, which regulates the expression of many ECM gene products86. In addition, fibronectin extra domain A (EDA) fragments, which are increased in patients with scleroderma, stimulate collagen production via Toll-like receptor 4 (TLR4) signalling and induce myofibroblast differentiation and ECM stiffness. This ultimately leads to chronic cutaneous fibrosis87. Taken together, these studies demonstrate that the ECM actively participates in stimulating further ECM synthesis. These ECM-driven pathways that further exacerbate fibrosis may be amenable to therapeutic intervention (BOX 2).
Box 2. Extracellular matrix remodelling as a potential therapeutic target.
Targeting the extracellular matrix (ECM), the enzymes that remodel it and the receptors that transduce their signals offers promising therapeutic opportunities for many diseases. For example, one study found that injecting ECM scaffolds derived from decellularized porcine myocardial tissue into porcine models of myocardial infarction can improve cardiac function137. These ECM scaffolds seemed to be haemocompatible and to not induce a pathological pro-inflammatory reaction. Although the number of pigs tested in this study was small, the findings support advancing this strategy into human trials. Other ECM molecules such as perlecan have been shown to be neuroprotective after acute stroke by promoting the secretion of vascular endothelial growth factor (VEGF) and angiogenesis, which ultimately promotes motor function recovery in rodent models138. In addition, strategies to increase growth factor affinity to the ECM (via a domain derived from placental growth factor 2 (PIGF2)) may improve repair of chronic wounds and bone defects139, but these studies are still in the preclinical stages.
However, some ECM proteins compromise wound repair and regeneration. For example, chondroitin sulphate proteoglycans (CSPGs) generated after trauma or spinal cord injury inhibit neuronal repair140,141. Treatment with chondroitinase ABC enzymes improves nerve regeneration in mouse models142. In addition, hyaluronan, which accumulates in patients with multiple sclerosis, prevents remyelination and inhibits oligodendrocyte maturation. Consistent with this, degrading hyaluronan promotes oligodendrocyte maturation in vitro, which may improve nerve regrowth143. Therefore, therapeutics that degrade the abnormal ECM may be beneficial in these conditions.
Strategies to target the enzymes involved in remodelling the ECM are also being pursued. For example, inhibiting lysyl oxidase-like 2 (LOXL2) using the antibody simtuzumab showed promising results in mouse models of fibrosis144. Simtuzumab is currently being evaluated in Phase 2 and Phase 3 clinical trials for scleroderma, idiopathic pulmonary fibrosis (IPF) and cancer. Matrix metalloproteinase (MMP) inhibitors (such as marimastat and prinomastat), which were initially thought to be promising anticancer agents, have unfortunately not demonstrated efficacy in the clinic. This is due to their lack of specificity and also their poor tolerability145. Therefore, altering individual MMPs using monoclonal antibodies may be more promising146. In addition, MMPs have non-proteolytic functions and so inhibiting these other domains (for example, the haemopexin domain) needs to be considered, as well as the timing of MMP inhibitor administration as MMPs can have opposing functions at different stages of disease147. Furthermore, given the importance of transforming growth factor-β (TGFβ) in regulating ECM gene expression, several trials are currently evaluating a TGFβ-inhibitory antibody, fresolimumab, in systemic sclerosis, IPF and myelofibrosis.
ECM stiffness can influence responses to anticancer agents by regulating access to chemotherapy148 and potentially forming a physical barrier to promote resistance149 (FIG. 4c). In addition, lung cancer cells that express high levels of fibronectin, laminin and collagen IV are protected against chemotherapy-induced apoptosis150. Notably, recent work has shown that the epidermal growth factor receptor (EGFR) inhibitor cetuximab actually self-attenuates its cytotoxic effects by inducing the synthesis of fibronectin and promoting radioresistance151. Therefore, targeting the ECM may be an important strategy to improve tumour responses to systemic and radiation therapy.
Finally, several drugs targeting integrins, including natalizumab (which targets α4 integrin) for multiple sclerosis and Crohn’s disease and abciximab (which targets glycoprotein IIbx–IIIa) for thrombotic disorders, are being investigated. Targeting α5 integrin may also be promising for various fibrotic diseases152. Further work is clearly required to translate preclinical studies to the clinic. However, taken together, these studies illustrate several exciting advances in perturbing the ECM to treat several human diseases.
Mechanisms that limit fibrosis
Signalling mediators such as interferon-γ (IFNγ) and peroxisome proliferator-activated receptor-γ (PPARγ) exert their anti-fibrotic effects by antagonizing TGFβ signalling. Vitamin D receptor (VDR) ligands, for example, decrease liver fibrosis by redirecting VDR to SMAD3 sites on the DNA, thereby reducing SMAD3 occupancy. This molecular competition for binding sites inhibits SMAD3-mediated transcription of ECM targets and prevents hepatic stellate cell activation in the liver88.
In addition, MMPs have also been shown to be anti-fibrotic in specific tissue microenvironments80. For example, MMP12 limits fibrosis during corneal injury by altering the expression of CC-chemokine ligand 2 (CCL2) and CXC-chemokine ligand 1 (CXCL1) and therefore interfering with the infiltration of immune cells89. Similarly, the transmembrane HSPG syndecan 4 inhibits fibrosis by binding the chemokine CXCL10 and preventing fibroblast migration to areas of injury, so that new ECM cannot be deposited at these sites90. Of note, recent studies highlight the importance of divergent chemokine signals in the liver that control the balance between fibrosis and tissue repair91. Perturbing chemokine expression and gradients (such that different cell types with varying propensities to deposit ECM are recruited) is an important way to indirectly affect the local ECM.
Interestingly, other mechanisms besides protease-mediated degradation of the ECM can also limit fibrosis. One such mechanism is cellular re-uptake of ECM proteins. For example, milk fat globule EGF 8 (MFGE8; also known as lactadherin) inhibits pulmonary fibrosis by binding collagen and promoting its uptake by macrophages92; collagen can then be degraded by lysosomal pathways. This illustrates an important alternative process to facilitate the removal of collagen from the extracellular space. Additional receptors that promote endocytosis-mediated degradation of other ECM components and the basis for the specific uptake of individual components will be important to identify and may yield new strategies for attenuating fibrosis.
ECM stiffness and remodelling in cancer progression
The ECM provides essential signals to maintain tissue architecture and polarity and to regulate cell growth and apoptosis. In the appropriate context, the ECM is sufficient to restrain malignant tumour progression93. For example, a high molecular mass hyaluronan (that is, more than five times larger than human or mouse hyaluronan) protects against cancer in naked mole rats94. HA accumulates abundantly in naked mole-rat tissues owing to the decreased activity of HA-degrading enzymes and a unique sequence of hyaluronan synthase 2 (HAS2).
However, the ECM can also promote tumour progression (reviewed in REFS 1,6,95). For example, collagen IV overexpression enhances cell survival and provides a growth advantage for lung cancer cells in the liver96. In fact, ECM gene signatures can stratify breast cancer into subclasses that predict patient outcome. Tumours with high expression of protease inhibitors correlate with good prognosis, whereas those with high MMPs correlate with poor prognosis and increased risk of recurrence97. In addition, high MMP1 expression stratifies atypical ductal hyperplasia into benign versus pre-malignant lesions that are at risk for becoming cancer98. Several recent studies also point to the importance of the ECM in facilitating metastatic tumour cell growth in the metastatic niche (BOX 3).
Box 3. The extracellular matrix is a crucial component of the metastatic niche.
Successful metastasis requires not only a local niche to support primary tumour growth but also a metastatic niche to enable disseminated cancer cells to survive, colonize and proliferate at a distant site153. This niche consists of various extracellular matrix (ECM) components and the enzymes that remodel them (for example, matrix metalloproteinases (MMPs)), as well as other cell types such as bone marrow-derived cells (BMDCs), endothelial cells and fibroblasts. For example, before the arrival of tumour cells, Lewis lung carcinoma (LLC) cells upregulate fibronectin at sites of future metastasis, which promotes BMDC clustering through α4β1 integrin154. These BMDCs are recruited to the metastatic site by tumour-derived factors and express MMP9 to facilitate breakdown of the basement membrane. Thus, ECM remodelling occurs as an early step in metastasis to help circulating tumour cells colonize and thrive in distant organs.
In addition to fibronectin, other ECM components support the metastatic niche. Tenascin C, which is normally enriched in stem cell niches, is initially expressed by breast cancer cells and later by stromal cells to promote survival and outgrowth of lung metastases155. Periostin, another ECM component that is important in bone and tooth development, promotes metastatic colonization by recruiting WNT ligands, thereby increasing WNT signalling in cancer stem cells156. Periostin expression by fibroblasts is induced by infiltrating tumour cells via transforming growth factor-β (TGFβ), and is required for cancer stem cell maintenance. Interestingly, endothelial tip cells within new vascular sprouts secrete both periostin and TGFβ, which promotes angiogenesis and micrometastatic outgrowth157. Furthermore, LLC-conditioned medium contains versican, which is a large chondroitin sulphate proteoglycan that is upregulated by many human tumours. Versican activates macrophages through Toll-like receptor 2 (TLR2) to produce interleukin-6 (IL-6) and tumour necrosis factor (TNF), thereby establishing a pro-inflammatory microenvironment that is conducive for metastatic growth123. Osteopontin in gliomas also promotes stem cell-like properties and radioresistance via CD44 signalling158. A systematic approach to identify responses to ECM has further uncovered ECM molecules that promote metastasis159. Taken together, these studies illustrate how the ECM regulates metastasis and suggest that inhibiting the ECM niche may be therapeutically beneficial in cancer.
Insights into ECM elasticity and stiffness, which is determined by ECM organization, orientation and chemical modification, have challenged our understanding of how cells interact with, and respond to, the micro-environment. For example, ECM stiffening, induced by increased collagen deposition and crosslinking, disrupts tissue morphogenesis and contributes to malignant progression99. Collagen crosslinking is mediated primarily by LOX and LOX-like (LOXLs) enzymes, which are frequently overexpressed in many cancers and at meta-static sites, and patients with high LOX expression have poor survival100. In breast cancer, LOX-induced collagen crosslinking increases stiffness, β1 integrin clustering, PI3K signalling and focal adhesion formation to drive invasion and tumour progression (FIG. 4C). Conversely, inhibiting LOX suppresses fibrosis and increases tumour latency101. ECM stiffening also facilitates metastatic colonization and infiltration of tumour-promoting immune cells102, and induces the expression of miR-18a, which is pro-tumorigenic by targeting the tumour suppressor phosphatase and tensin homologue (PTEN)103.
Collagen also signals to cells through the receptor Tyr kinases discoidin domain-containing receptor 1 (DDR1) and DDR2, both of which have been implicated in cancer (reviewed in REF. 104) (FIG. 4C). DDR1 is required for collective cell migration105, and increased expression of DDR2 has been observed in invasive human breast tumours106. Notably, DDR2 activation stabilizes the transcription factor SNAIL1, which induces collagen I and MMP14, and promotes EMT and metastasis106. The extracellular collagen network can therefore alter cell-intrinsic properties such as epithelial plasticity through these cell receptors. Together, these data demon strate that collagen crosslinking activates signalling pathways and miRNA networks that promote malignant progression.
Several studies have implicated miRNAs as potent regulators of the ECM. The miR-29 family, for example, controls the expression of a network of genes involved in ECM remodelling, including several collagen chains, LOX and LOXLs, as well as MMP2 and MMP9 (REFS 107,108). In breast cancer, miR-29 expression inversely correlates with cell adhesion and ECM gene expression109, and miR-29b overexpression alters the tumour microenvironment and suppresses metastasis108. Additional studies are required to identify other miRNAs that are important in regulating the ECM and to determine whether other non-coding RNAs, such as long non-coding RNAs, might also regulate the chromatin state, and therefore transcription, of ECM genes. Factors upstream of these non-coding RNAs will also be important to identify. This exciting area of biology should yield additional insights into how ECM remodelling is controlled.
The ECM promotes angiogenesis
The ECM also modulates angiogenesis to promote tumour growth110. For example, in a pancreatic β-cell tumour model, tenascin C not only promotes tumour cell survival and proliferation but also induces angiogenesis and blood vessel permeability by downregulating Dickkopf 1 (DKK1) and increasing WNT signalling111.
In addition, ECM fragments generated by cleavage of the full-length ECM protein also have pro- and anti-angiogenic functions. These include endostatin, tumstatin, canstatin, arresten and hexastatin, all of which are derived from collagen IV and XVIII (reviewed in REF. 112) and can bind to cell receptors such as integrins and EGFR113. For example, arresten, which is generated from the NC1 domain of the collagen a1(IV) chain, binds to α1β1 integrin and inhibits angiogenesis by antagonizing MAPK signalling114. Together, these studies demonstrate that the ECM also promotes non-cell autonomous mechanisms to enhance tumour growth (FIG. 4C).
The ECM regulates immune and cancer cell migration
Immune responses occur in the context of integrin-mediated adhesive interactions with the ECM. For example, α1β1 integrin, which binds collagen I and IV, is expressed by peripheral CD8+ T cells during influenza infection and mediates retention of influenza-specific memory T cells in the lung after viral clearance, which is important for secondary immunity115. Furthermore, the ECM niche within the spleen, which consists of laminin and agrin, promotes marginal zone B cell differentiation and survival to support antibody production116. These studies highlight the importance of the ECM in mediating immune responses.
Many ECM proteins also contain cryptic domains with structures that are similar to chemokines and cytokines117. These domains can be exposed by proteolysis and elicit biological responses that are distinct from those of the full length ECM component (FIG. 4C). These matrikines regulate many processes, including migration, adhesion and differentiation, and can affect immune and pro-inflammatory cell behaviour118. For example, N-acetyl Pro-Gly-Pro (PGP) is a bioactive collagen I fragment generated by MMP8 and MMP9. PGP shares structural homology with CXC chemokines and signals through CXC-chemokine receptor 1 (CXCR1) and CXCR2 to attract neutrophils to sites of inflammation119. Interestingly, these PGP collagen derivatives are found in patients with chronic inflammatory airway disorders, which illustrates their relevance in human disease120. Other ECM fragments, such those generated by overexpressing hyaluronidase 1, promote dendritic cell egress from the skin to facilitate antigen presentation. This migration is mediated by TLR4, as mice lacking this receptor do not exhibit hyaluronan-associated phenotypes121. These studies demonstrate the importance of ECM fragments in controlling immune cell migration, which may be exploited therapeutically to treat pro-inflammatory diseases.
The density and orientation of the ECM fibres also controls immune cell migration. Loose areas of fibronectin and collagen facilitate T cell motility, whereas dense ECM areas impede migration. These ECM fibres also govern cell migratory tracks, which can restrict the inter action of immune cells with cancer cells122 (FIG. 4C). Notably, treatment with collagenase enhances the contact of T cells with cancer cells, suggesting that modulating ECM architecture might be an important way to improve immunotherapy access. Other ECM components, such as versican, recruit and activate pro-tumorigenic immune cells such as macrophages, thereby promoting inflammation and metastasis123.
Interestingly, ECM geometry dictates the mode of cell migration, as cells can use both proteolytic and non-proteolytic mechanisms to migrate and squeeze through the ECM124. Confined ECM geometries alter cell deformability and shift the relationship between adhesion, contractility and velocity to promote bleb migration patterns at tumour margins125. Together, these studies illustrate how the ECM regulates immune and cancer cell migration.
Abnormal release of growth factors during pathogenesis
The ECM is a rich reservoir of growth factors and other bioactive molecules that can be released by proteolysis via MMPs (FIG. 4C). For example, in pancreatic neuro-endocrine tumours, VEGF is abundant in the ECM, even in non-angiogenic islets. The switch from vascular quiescence to active angiogenesis involves upregulation of MMP9, which releases sequestered VEGF from the ECM126. In addition to MMPs127, the Ser protease plasmin can also release ECM-bound VEGF into soluble forms128. The multiple isoforms of VEGF generated by alternative splicing have unique C-terminal domains, which allow some isoforms to bind collagen and fibrinogen129. In addition, HSPGs interact with platelet-derived growth factor (PDGF) in a sulphation-dependent manner. Inhibition of sulphatase 2 decreases growth in glioblastomas by attenuating PDGF receptor signalling, indicating that the ECM and the enzymes that modify it control the bioavailability of growth factors130.
TGFβ is also sequestered and bound to PGs in the ECM. In osteogenesis imperfecta, impaired collagen formation leads to reduced binding to decorin, a known regulator of TGFβ activity, which results in over-active TGFβ signalling131. Excessive TGFβ signalling is a common driving mechanism in many mouse models of osteogenesis imperfecta, and treatment with TGFβ-specific antibodies partially corrects the bone phenotype in mice. In addition, the fibrinogen-like domain of tenascin X interacts with the small latent TGFβ complex and regulates the bioavailability of mature TGFβ, which controls EMT in mammary epithelial cells132. These studies establish a role for the ECM as an important reservoir of growth factors and as an indirect regulator of signal transduction in multiple diseases.
Summary and conclusions
The ECM is a vital structure that has a dynamic and complex organization and can trigger multiple biological activities that are essential for a normal organ development and tissue homeostasis. Dysregulated ECM remodelling leads to many diseases, including fibrosis and cancer.
In this Review, we describe how ECM remodelling affects organ morphogenesis, using the intestine, the lung, and the mammary and submandibular glands as examples. In all four cases, appropriate development requires the tight control and regulation of ECM assembly, modification and degradation. However, unique mechanisms must be at play for each organ, given their distinct structure and function. The exact nature of these organ-specific mechanisms remains elusive. Is the molecular mechanism involved in submandibular gland cleft formation (which involves deposition of fibronectin at the cleft site driven by BTBD7) specific to this organ, or is it generalizable to other branched organs? What is the role of the microenvironment, which includes immune cells, endothelial cells and fibroblasts, in ECM remodelling, and how does this influence the branched pattern of each organ? It will be interesting to investigate the interactions between the ECM, epithelial cells and microenvironment in organogenesis.
In addition, the role of the ECM in regulating the stem cell niche needs to be more extensively explored. For example, a single mammary stem cell can reproduce an entire organ when transplanted into a cleared fat pad. This suggests the presence of a niche in the mammary gland that contains all the signals required to programme stem cells. The ECM has a crucial role in the release of the niche signals that are essential for stem cell fate133, which probably has implication for diseases such as cancer, in which cancer stem cells might also be using such ECM signals to promote their survival and growth. A better understanding of the niche signals that regulate stem cell behaviour might also have therapeutic potential in regenerative medicine.
Ex vivo three-dimensional organoids cultured in Matrigel has improved our understanding of the development of branching organs. However, these models do not recapitulate the complex microenvironment of the developing organ, nor do they reproduce the exact composition and stiffness of the native ECM. Organotypic tissue culture systems that incorporate native ECM and stromal cells will continue to improve our understanding of cancer134 and enable us to study developmental and disease processes in more physiologic environments that better model human disease135.
The identification of the matrisome and key ECM remodelling effects that promote certain diseases opens several exciting possibilities for therapeutic intervention. Attention must be given to targeting specific individual ECM components as well as to timing the therapy correctly, given that the ECM is actively remodelled. Current clinical trials using inhibitors of ECM-related targets are ongoing and promising (BOX 2). However, a deeper understanding of the diverse biologic activities and properties of the ECM must be attained to uncover new targets for future therapy.
Acknowledgments
This study was supported by funds from the National Cancer Institute (grant numbers CA057621 and CA138818), a Department of Defense (DOD) Era of Hope Scholar Expansion grant (BC122990) and Institut National de la Santé et de la Recherche Médicale (INSERM).
Glossary
- Basement membrane
The ECM layer that is located basolaterally to all epithelium and endothelium in the body. It provides structural support to the tissue and modulates epithelial and endothelial cell functions. It is mostly composed of collagen IV and laminins
- TGFβ
(Transforming growth factor-β). The TGFβ isoforms (TGFβ1–TGFβ3) are synthesized as latent precursors in complex with both latency-associated peptide (LAP) and latent TGFβ-binding proteins (LTBPs). Proteinases such as plasmin and MMPs catalyse the release of active TGFβ from the complex
- Zymogens
Proenzymes that are inactive enzyme precursors. Zymogens need to be biochemically modified to become active enzymes
- Integrins
Heterodimeric cell surface receptors that mediate cell–cell and cell–ECM interactions and orchestrate cell attachment, movement, growth, differentiation and survival
- von Willebrand factor
A blood glycoprotein that is involved in haemostasis and is defective in von Willebrand disease. This factor is also increased in the plasma of cardiovascular, neoplastic and connective tissue diseases and can contribute to an increased risk of thrombosis
- Villi
A finger-like projection that protrudes from the epithelium of the intestinal wall. Intestinal villi increase the epithelium surface area to promote the absorption of nutrients
- Intestinal crypts
(Also known as crypts of Lieberkühn). Proliferative compartments found in the small intestine and the colon; they contain intestinal stem cells and other specialized cells such as Paneth cells (only in the small intestine) and goblet cells
- Intestinal stem cells
Undifferentiated cells that can self-renew and differentiate into all of the specialized cell types of the tissue or organ. The main role of stem cells is to maintain and repair the tissue in which they reside
- RGD-dependent integrins
A group of integrins that specifically recognize the RGD motif, a sequence of three amino acids (Arg-Gly-Asp) found in many ECM molecules such as fibronectin and osteopontin. Collectively, these interactions are termed the RGD-dependent adhesion system
- Anoikis
A programmed cell death that is induced by lack of correct cell–ECM interactions. Invasive tumour cells may escape from anoikis to target different metastatic organs
- Organoids
Organ epithelial fragments that resemble the whole organ in structure and function during three-dimensional culture
- Stem cell niche
A specialized microenvironment that interacts with cells such as stem cells or tumour cells to regulate their fate
- E-cadherin
A calcium-dependent cell–cell adhesion molecule with pivotal roles in epithelial cell behaviour, tissue formation and suppression of cancer
- Matrikines
ECM fragments released from the ECM by proteolysis or by cryptic site exposurel; they have biological activities that are different from those of the full-length protein
- Matrigel
A gelatinous protein mixture secreted by Engelbreth–Holm–Swarm (EHS) mouse sarcoma cells. It contains structural proteins such as laminin, entactin, collagen and HSPGs. Growth factors such as TGFβ and EGF are also present in Matrigel, although growth factor-reduced formulations are also available
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jarvelainen H, Sainio A, Koulu M, Wight TN, Penttinen R. Extracellular matrix molecules: potential targets in pharmacotherapy. Pharmacol Rev. 2009;61:198–223. doi: 10.1124/pr.109.001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bateman JF, Boot-Handford RP, Lamande SR. Genetic diseases of connective tissues: cellular and extracellular effects of ECM mutations. Nature Rev Genet. 2009;10:173–183. doi: 10.1038/nrg2520. [DOI] [PubMed] [Google Scholar]
- 4.Rozario T, DeSimone DW. The extracellular matrix in development and morphogenesis: a dynamic view. Dev Biol. 2010;341:126–140. doi: 10.1016/j.ydbio.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hynes RO, Naba A. Overview of the matrisome—an inventory of extracellular matrix constituents and functions. Cold Spring Harb Perspect Biol. 2012;4:a004903. doi: 10.1101/cshperspect.a004903. This review gives a complete list of ECM proteins that are part of the matrisome, and describes the ECM structure and function modifiers and the evolution of the matrisome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol. 2011;3:a005058. doi: 10.1101/cshperspect.a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123:4195–4200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhen G, Cao X. Targeting TGFβ signaling in subchondral bone and articular cartilage homeostasis. Trends Pharmacol Sci. 2014;35:227–236. doi: 10.1016/j.tips.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gross J, Lapiere CM. Collagenolytic activity in amphibian tissues: a tissue culture assay. Proc Natl Acad Sci USA. 1962;48:1014–1022. doi: 10.1073/pnas.48.6.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hite LA, Shannon JD, Bjarnason JB, Fox JW. Sequence of a cDNA clone encoding the zinc metalloproteinase hemorrhagic toxine from Crotalus atrox: evidence for signal, zymogen, and disintegrin-like structures. Biochemistry. 1992;31:6203–6211. doi: 10.1021/bi00142a005. [DOI] [PubMed] [Google Scholar]
- 11.Kuno K, et al. Molecular cloning of a gene encoding a new type of metalloproteinase-disintegrin family protein with thrombospondin motifs as an inflammation associated gene. J Biol Chem. 1997;272:556–562. doi: 10.1074/jbc.272.1.556. [DOI] [PubMed] [Google Scholar]
- 12.Murphy G. The ADAMs: signalling scissors in the tumour microenvironment. Nature Rev Cancer. 2008;8:929–941. doi: 10.1038/nrc2459. [DOI] [PubMed] [Google Scholar]
- 13.White JM. ADAMs: modulators of cell-cell and cell–matrix interactions. Curr Opin Cell Biol. 2003;15:598–606. doi: 10.1016/j.ceb.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Apte SS. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J Biol Chem. 2009;284:31493–31497. doi: 10.1074/jbc.R109.052340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bond JS, Rojas K, Overhauser J, Zoghbi HY, Jiang W. The structural genes, MEP1A and MEP1B, for the α and β subunits of the metalloendopeptidase meprin map to human chromosomes 6p and 18q, respectively. Genomics. 1995;25:300–303. doi: 10.1016/0888-7543(95)80142-9. [DOI] [PubMed] [Google Scholar]
- 16.Bertenshaw GP, Norcum MT, Bond JS. Structure of homo- and hetero-oligomeric meprin metalloproteases. Dimers, tetramers, and high molecular mass multimers. J Biol Chem. 2003;278:2522–2532. doi: 10.1074/jbc.M208808200. [DOI] [PubMed] [Google Scholar]
- 17.Herzog C, Haun RS, Ludwig A, Shah SV, Kaushal GP. ADAM10 is the major sheddase responsible for the release of membrane-associated meprin A. J Biol Chem. 2014;289:13308–13322. doi: 10.1074/jbc.M114.559088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kruse MN, et al. Human meprin α and β homooligomers: cleavage of basement membrane proteins and sensitivity to metalloprotease inhibitors. Biochem J. 2004;378:383–389. doi: 10.1042/BJ20031163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Broder C, et al. Metalloproteases meprinα and meprinβ are C- and N-procollagen proteinases important for collagen assembly and tensile strength. Proc Natl Acad Sci USA. 2013;110:14219–14224. doi: 10.1073/pnas.1305464110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jefferson T, et al. The substrate degradome of meprin metalloproteases reveals an unexpected proteolytic link between meprin β and ADAM10. Cell Mol Life Sci. 2013;70:309–333. doi: 10.1007/s00018-012-1106-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geurts N, et al. Meprins process matrix metalloproteinase-9 (MMP-9)/gelatinase B and enhance the activation kinetics by MMP-3. FEBS Lett. 2012;586:4264–4269. doi: 10.1016/j.febslet.2012.10.033. [DOI] [PubMed] [Google Scholar]
- 22.Khokha R, Murthy A, Weiss A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nature Rev Immunol. 2013;13:649–665. doi: 10.1038/nri3499. [DOI] [PubMed] [Google Scholar]
- 23.Baker AH, Edwards DR, Murphy G. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci. 2002;115:3719–3727. doi: 10.1242/jcs.00063. [DOI] [PubMed] [Google Scholar]
- 24.Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nature Rev Mol Cell Biol. 2010;11:23–36. doi: 10.1038/nrm2821. [DOI] [PubMed] [Google Scholar]
- 25.Bonnefoy A, Legrand C. Proteolysis of subendothelial adhesive glycoproteins (fibronectin, thrombospondin, and von Willebrand factor) by plasmin, leukocyte cathepsin G, and elastase. Thromb Res. 2000;98:323–332. doi: 10.1016/s0049-3848(99)00242-x. [DOI] [PubMed] [Google Scholar]
- 26.Giuffrida P, Biancheri P, MacDonald TT. Proteases and small intestinal barrier function in health and disease. Curr Opin Gastroenterol. 2014;30:147–153. doi: 10.1097/MOG.0000000000000042. [DOI] [PubMed] [Google Scholar]
- 27.Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nature Rev Cancer. 2006;6:764–775. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- 28.Fonovic M, Turk B. Cysteine cathepsins and extracellular matrix degradation. Biochim Biophys Acta. 2014;1840:2560–2570. doi: 10.1016/j.bbagen.2014.03.017. [DOI] [PubMed] [Google Scholar]
- 29.Uchimura K, et al. HSulf-2, an extracellular endoglucosamine-6-sulfatase, selectively mobilizes heparin-bound growth factors and chemokines: effects on VEGF, FGF-1, and SDF-1. BMC Biochem. 2006;7:2. doi: 10.1186/1471-2091-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barker N, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 31.Hasebe T, et al. Thyroid hormone-induced cell–cell interactions are required for the development of adult intestinal stem cells. Cell Biosci. 2013;3:18. doi: 10.1186/2045-3701-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su Y, Shi Y, Stolow MA, Shi YB. Thyroid hormone induces apoptosis in primary cell cultures of tadpole intestine: cell type specificity and effects of extracellular matrix. J Cell Biol. 1997;139:1533–1543. doi: 10.1083/jcb.139.6.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patterton D, Hayes WP, Shi YB. Transcriptional activation of the matrix metalloproteinase gene stromelysin-3 coincides with thyroid hormone-induced cell death during frog metamorphosis. Dev Biol. 1995;167:252–262. doi: 10.1006/dbio.1995.1021. [DOI] [PubMed] [Google Scholar]
- 34.Ishizuya-Oka A, et al. Requirement for matrix metalloproteinase stromelysin-3 in cell migration and apoptosis during tissue remodeling in Xenopus laevis. J Cell Biol. 2000;150:1177–1188. doi: 10.1083/jcb.150.5.1177. This paper showed that MMP11 is required for cell fate determination and cell migration during morphogenesis, most probably through ECM remodelling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amano T, Kwak O, Fu L, Marshak A, Shi YB. The matrix metalloproteinase stromelysin-3 cleaves laminin receptor at two distinct sites between the transmembrane domain and laminin binding sequence within the extracellular domain. Cell Res. 2005;15:150–159. doi: 10.1038/sj.cr.7290280. [DOI] [PubMed] [Google Scholar]
- 36.Fujimoto K, Nakajima K, Yaoita Y. Expression of matrix metalloproteinase genes in regressing or remodeling organs during amphibian metamorphosis. Dev Growth Differ. 2007;49:131–143. doi: 10.1111/j.1440-169X.2007.00916.x. [DOI] [PubMed] [Google Scholar]
- 37.Hasebe T, Hartman R, Fu L, Amano T, Shi YB. Evidence for a cooperative role of gelatinase A and membrane type-1 matrix metalloproteinase during Xenopus laevis development. Mech Dev. 2007;124:11–22. doi: 10.1016/j.mod.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simon-Assmann P, Kedinger M, De Arcangelis A, Rousseau V, Simo P. Extracellular matrix components in intestinal development. Experientia. 1995;51:883–900. doi: 10.1007/BF01921739. [DOI] [PubMed] [Google Scholar]
- 39.Simon-Assmann P, Kedinger M, Haffen K. Immunocytochemical localization of extracellular-matrix proteins in relation to rat intestinal morphogenesis. Differentiation. 1986;32:59–66. doi: 10.1111/j.1432-0436.1986.tb00556.x. [DOI] [PubMed] [Google Scholar]
- 40.Simon-Assmann P, Bouziges F, Freund JN, Perrin-Schmitt F, Kedinger M. Type IV collagen mRNA accumulates in the mesenchymal compartment at early stages of murine developing intestine. J Cell Biol. 1990;110:849–857. doi: 10.1083/jcb.110.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simon-Assmann P, et al. The laminins: role in intestinal morphogenesis and differentiation. Ann NY Acad Sci. 1998;859:46–64. doi: 10.1111/j.1749-6632.1998.tb11110.x. [DOI] [PubMed] [Google Scholar]
- 42.Mahoney ZX, Stappenbeck TS, Miner JH. Laminin α 5 influences the architecture of the mouse small intestine mucosa. J Cell Sci. 2008;121:2493–2502. doi: 10.1242/jcs.025528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beaulieu JF. Integrins and human intestinal cell functions. Front Biosci. 1999;4:D310–D321. doi: 10.2741/beaulieu. [DOI] [PubMed] [Google Scholar]
- 44.Benoit YD, et al. Integrin α8β1 regulates adhesion, migration and proliferation of human intestinal crypt cells via a predominant RhoA/ROCK-dependent mechanism. Biol Cell. 2009;101:695–708. doi: 10.1042/BC20090060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benoit YD, et al. Integrin α8β1 confers anoikis susceptibility to human intestinal epithelial crypt cells. Biochem Biophys Res Commun. 2010;399:434–439. doi: 10.1016/j.bbrc.2010.07.107. [DOI] [PubMed] [Google Scholar]
- 46.Groulx JF, et al. Collagen VI is a basement membrane component that regulates epithelial cell-fibronectin interactions. Matrix Biol. 2011;30:195–206. doi: 10.1016/j.matbio.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 47.Henning SJ, et al. Meprin mRNA in rat intestine during normal and glucocorticoid-induced maturation: divergent patterns of expression of α and β subunits. FEBS Lett. 1999;462:368–372. doi: 10.1016/s0014-5793(99)01558-6. [DOI] [PubMed] [Google Scholar]
- 48.Sato T, Clevers H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science. 2013;340:1190–1194. doi: 10.1126/science.1234852. [DOI] [PubMed] [Google Scholar]
- 49.Kim HY, Nelson CM. Extracellular matrix and cytoskeletal dynamics during branching morphogenesis. Organogenesis. 2012;8:56–64. doi: 10.4161/org.19813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grobstein C, Cohen J. Collagenase: effect on the morphogenesis of embryonic salivary epithelium in vitro. Science. 1965;150:626–628. doi: 10.1126/science.150.3696.626. [DOI] [PubMed] [Google Scholar]
- 51.Nakanishi Y, Sugiura F, Kishi J, Hayakawa T. Collagenase inhibitor stimulates cleft formation during early morphogenesis of mouse salivary gland. Dev Biol. 1986;113:201–206. doi: 10.1016/0012-1606(86)90122-3. [DOI] [PubMed] [Google Scholar]
- 52.Keely PJ, Wu JE, Santoro SA. The spatial and temporal expression of the α 2 β 1 integrin and its ligands, collagen I, collagen IV, and laminin, suggest important roles in mouse mammary morphogenesis. Differentiation. 1995;59:1–13. doi: 10.1046/j.1432-0436.1995.5910001.x. [DOI] [PubMed] [Google Scholar]
- 53.Brownfield DG, et al. Patterned collagen fibers orient branching mammary epithelium through distinct signaling modules. Curr Biol. 2013;23:703–709. doi: 10.1016/j.cub.2013.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakai T, Larsen M, Yamada KM. Fibronectin requirement in branching morphogenesis. Nature. 2003;423:876–881. doi: 10.1038/nature01712. This paper is the first to show that fibronectin expression is required for cleft formation in branching morphogenesis. [DOI] [PubMed] [Google Scholar]
- 55.Onodera T, et al. Btbd7 regulates epithelial cell dynamics and branching morphogenesis. Science. 2010;329:562–565. doi: 10.1126/science.1191880. This paper is the first to give molecular mechanisms for cleft formation in branching morphogenesis, showing that BTBD7 is a regulatory gene that promotes epithelial tissue remodelling and formation of branched organs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Daley WP, Gulfo KM, Sequeira SJ, Larsen M. Identification of a mechanochemical checkpoint and negative feedback loop regulating branching morphogenesis. Dev Biol. 2009;336:169–182. doi: 10.1016/j.ydbio.2009.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Daley WP, Kohn JM, Larsen M. A focal adhesion protein-based mechanochemical checkpoint regulates cleft progression during branching morphogenesis. Dev Dyn. 2011;240:2069–2083. doi: 10.1002/dvdy.22714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Joo EE, Yamada KM. MYPT1 regulates contractility and microtubule acetylation to modulate integrin adhesions and matrix assembly. Nature Commun. 2014;5:3510. doi: 10.1038/ncomms4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Woodward TL, Mienaltowski AS, Modi RR, Bennett JM, Haslam SZ. Fibronectin and the α5β1 integrin are under developmental and ovarian steroid regulation in the normal mouse mammary gland. Endocrinology. 2001;142:3214–3222. doi: 10.1210/endo.142.7.8273. [DOI] [PubMed] [Google Scholar]
- 60.Liu S, Young SM, Varisco BM. Dynamic expression of chymotrypsin-like elastase 1 over the course of murine lung development. Am J Physiol Lung Cell Mol Physiol. 2014;306:L1104–L1116. doi: 10.1152/ajplung.00126.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mammoto T, Jiang E, Jiang A, Mammoto A. Extracellular matrix structure and tissue stiffness control postnatal lung development through the lipoprotein receptor-related protein 5/Tie2 signaling system. Am J Respir Cell Mol Biol. 2013;49:1009–1018. doi: 10.1165/rcmb.2013-0147OC. [DOI] [PubMed] [Google Scholar]
- 62.Daley WP, Yamada KM. ECM-modulated cellular dynamics as a driving force for tissue morphogenesis. Curr Opin Genet Dev. 2013;23:408–414. doi: 10.1016/j.gde.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tan J, et al. Stromal matrix metalloproteinase-11 is involved in the mammary gland postnatal development. Oncogene. 2013;33:4050–4059. doi: 10.1038/onc.2013.434. [DOI] [PubMed] [Google Scholar]
- 64.Mori H, et al. Transmembrane/cytoplasmic, rather than catalytic, domains of Mmp14 signal to MAPK activation and mammary branching morphogenesis via binding to integrin β1. Development. 2013;140:343–352. doi: 10.1242/dev.084236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wiseman BS, et al. Site-specific inductive and inhibitory activities of MMP-2 and MMP-3 orchestrate mammary gland branching morphogenesis. J Cell Biol. 2003;162:1123–1133. doi: 10.1083/jcb.200302090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kessenbrock K, et al. A role for matrix metalloproteinases in regulating mammary stem cell function via the Wnt signaling pathway. Cell Stem Cell. 2013;13:300–313. doi: 10.1016/j.stem.2013.06.005. This paper shows that MMP3 is a regulator of WNT signaling and mammary stem cell activity; through its haemopexin domain, MMP3 specifically binds and inactivates WNT5B, a noncanonical WNT ligand that inhibits canonical WNT signalling and mammary epithelial outgrowth in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Correia AL, Mori H, Chen EI, Schmitt FC, Bissell MJ. The hemopexin domain of MMP3 is responsible for mammary epithelial invasion and morphogenesis through extracellular interaction with HSP90β. Genes Dev. 2013;27:805–817. doi: 10.1101/gad.211383.112. The authors show that the haemopexin domain of MMP3, and not its proteolytic activity, is essential for mammary epithelial cells invasion; this domain interacts with the intracellular chaperone heat-shock protein 90β to promote invasion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goetz R, Mohammadi M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nature Rev Mol Cell Biol. 2013;14:166–180. doi: 10.1038/nrm3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Makarenkova HP, et al. Differential interactions of FGFs with heparan sulfate control gradient formation and branching morphogenesis. Sci Signal. 2009;2:ra55. doi: 10.1126/scisignal.2000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patel VN, et al. Heparanase cleavage of perlecan heparan sulfate modulates FGF10 activity during ex vivo submandibular gland branching morphogenesis. Development. 2007;134:4177–4186. doi: 10.1242/dev.011171. [DOI] [PubMed] [Google Scholar]
- 71.Kim HE, et al. Disruption of the myocardial extracellular matrix leads to cardiac dysfunction. J Clin Invest. 2000;106:857–866. doi: 10.1172/JCI8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bondeson J, Wainwright S, Hughes C, Caterson B. The regulation of the ADAMTS4 and ADAMTS5 aggrecanases in osteoarthritis: a review. Clin Exp Rheumatol. 2008;26:139–145. [PubMed] [Google Scholar]
- 73.Echtermeyer F, et al. Syndecan-4 regulates ADAMTS-5 activation and cartilage breakdown in osteoarthritis. Nature Med. 2009;15:1072–1076. doi: 10.1038/nm.1998. [DOI] [PubMed] [Google Scholar]
- 74.Martignetti JA, et al. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nature Genet. 2001;28:261–265. doi: 10.1038/90100. [DOI] [PubMed] [Google Scholar]
- 75.El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 76.Boyd NF, Martin LJ, Yaffe MJ, Minkin S. Mammographic density and breast cancer risk: current understanding and future prospects. Breast Cancer Res. 2011;13:223. doi: 10.1186/bcr2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Biancheri P, et al. The role of transforming growth factor (TGF)-β in modulating the immune response and fibrogenesis in the gut. Cytokine Growth Factor Rev. 2013;25:45–55. doi: 10.1016/j.cytogfr.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 78.Verrecchia F, Chu ML, Mauviel A. Identification of novel TGF-β /Smad gene targets in dermal fibroblasts using a combined cDNA microarray/ promoter transactivation approach. J Biol Chem. 2001;276:17058–17062. doi: 10.1074/jbc.M100754200. [DOI] [PubMed] [Google Scholar]
- 79.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nature Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. This review summarizes our current understanding of the molecular and cellular pathways controlling fibrosis, with special focus on the immune response, as well as the key pathways for therapeutic targeting. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Giannandrea M, Parks WC. Diverse functions of matrix metalloproteinases during fibrosis. Dis Model Mech. 2014;7:193–203. doi: 10.1242/dmm.012062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Duffield JS, Lupher M, Thannickal VJ, Wynn TA. Host responses in tissue repair and fibrosis. Annu Rev Pathol. 2013;8:241–276. doi: 10.1146/annurev-pathol-020712-163930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McHedlidze T, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity. 2013;39:357–371. doi: 10.1016/j.immuni.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bailey JR, et al. IL-13 promotes collagen accumulation in Crohn’s disease fibrosis by down-regulation of fibroblast MMP synthesis: a role for innate lymphoid cells? PLoS ONE. 2013;7:e52332. doi: 10.1371/journal.pone.0052332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Clarke DL, Carruthers AM, Mustelin T, Murray LA. Matrix regulation of idiopathic pulmonary fibrosis: the role of enzymes. Fibrogen Tissue Repair. 2013;6:20. doi: 10.1186/1755-1536-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rieder F. The gut microbiome in intestinal fibrosis: environmental protector or provocateur? Sci Transl Med. 2013;5:190ps10. doi: 10.1126/scitranslmed.3004731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Parker MW, et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest. 2014;124:1622–1635. doi: 10.1172/JCI71386. This paper demonstrates that fibrotic ECM stimulates fibroblasts to produce more ECM through downregulation of miR-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bhattacharyya S, et al. Fibronectin EDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Sci Transl Med. 2014;6:232ra50. doi: 10.1126/scitranslmed.3008264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ding N, et al. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell. 2013;153:601–613. doi: 10.1016/j.cell.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chan MF, et al. Protective effects of matrix metalloproteinase-12 following corneal injury. J Cell Sci. 2013;126:3948–3960. doi: 10.1242/jcs.128033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jiang D, et al. Inhibition of pulmonary fibrosis in mice by CXCL10 requires glycosaminoglycan binding and syndecan-4. J Clin Invest. 2010;120:2049–2057. doi: 10.1172/JCI38644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ding BS, et al. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature. 2014;505:97–102. doi: 10.1038/nature12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Atabai K, et al. Mfge8 diminishes the severity of tissue fibrosis in mice by binding and targeting collagen for uptake by macrophages. J Clin Invest. 2009;119:3713–3722. doi: 10.1172/JCI40053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nature Med. 2011;17:320–329. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tian X, et al. High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature. 2013;499:346–349. doi: 10.1038/nature12234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Burnier JV, et al. Type IV collagen-initiated signals provide survival and growth cues required for liver metastasis. Oncogene. 2011;30:3766–3783. doi: 10.1038/onc.2011.89. [DOI] [PubMed] [Google Scholar]
- 97.Bergamaschi A, et al. Extracellular matrix signature identifies breast cancer subgroups with different clinical outcome. J Pathol. 2008;214:357–367. doi: 10.1002/path.2278. [DOI] [PubMed] [Google Scholar]
- 98.Poola I, et al. Identification of MMP-1 as a putative breast cancer predictive marker by global gene expression analysis. Nature Med. 2005;11:481–483. doi: 10.1038/nm1243. [DOI] [PubMed] [Google Scholar]
- 99.Paszek MJ, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 100.Erler JT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 101.Levental KR, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. This study describes how ECM stiffening through LOX drives tumour progression and shows that inhibiting collagen crosslinking delays tumorigenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Erler JT, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mouw JK, et al. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nature Med. 2014;20:360–367. doi: 10.1038/nm.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Valiathan RR, Marco M, Leitinger B, Kleer CG, Fridman R. Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev. 2012;31:295–321. doi: 10.1007/s10555-012-9346-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hidalgo-Carcedo C, et al. Collective cell migration requires suppression of actomyosin at cell-cell contacts mediated by DDR1 and the cell polarity regulators Par3 and Par6. Nature Cell Biol. 2011;13:49–58. doi: 10.1038/ncb2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang K, et al. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nature Cell Biol. 2013;15:677–687. doi: 10.1038/ncb2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sengupta S, et al. MicroRNA 29c is down-regulated in nasopharyngeal carcinomas, up-regulating mRNAs encoding extracellular matrix proteins. Proc Natl Acad Sci USA. 2008;105:5874–5878. doi: 10.1073/pnas.0801130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chou J, et al. GATA3 suppresses metastasis and modulates the tumour microenvironment by regulating microRNA-29b expression. Nature Cell Biol. 2013;15:201–213. doi: 10.1038/ncb2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Enerly E, et al. miRNA-mRNA integrated analysis reveals roles for miRNAs in primary breast tumors. PLoS ONE. 2011;6:e16915. doi: 10.1371/journal.pone.0016915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cheresh DA, Stupack DG. Regulation of angiogenesis: apoptotic cues from the ECM. Oncogene. 2008;27:6285–6298. doi: 10.1038/onc.2008.304. [DOI] [PubMed] [Google Scholar]
- 111.Saupe F, et al. Tenascin-C downregulates wnt inhibitor dickkopf-1, promoting tumorigenesis in a neuroendocrine tumor model. Cell Rep. 2013;5:482–492. doi: 10.1016/j.celrep.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 112.Mott JD, Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol. 2004;16:558–564. doi: 10.1016/j.ceb.2004.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Burgess JK, Weckmann M. Matrikines and the lungs. Pharmacol Ther. 2012;134:317–337. doi: 10.1016/j.pharmthera.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 114.Sudhakar A, et al. Human α1 type IV collagen NC1 domain exhibits distinct antiangiogenic activity mediated by α1β1 integrin. J Clin Invest. 2005;115:2801–2810. doi: 10.1172/JCI24813. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 115.Ray SJ, et al. The collagen binding α1β1 integrin VLA-1 regulates CD8 T cell-mediated immune protection against heterologous influenza infection. Immunity. 2004;20:167–179. doi: 10.1016/s1074-7613(04)00021-4. [DOI] [PubMed] [Google Scholar]
- 116.Song J, et al. Extracellular matrix of secondary lymphoid organs impacts on B-cell fate and survival. Proc Natl Acad Sci USA. 2013;110:E2915–E2924. doi: 10.1073/pnas.1218131110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sorokin L. The impact of the extracellular matrix on inflammation. Nature Rev Immunol. 2010;10:712–723. doi: 10.1038/nri2852. [DOI] [PubMed] [Google Scholar]
- 118.Monboisse JC, Oudart JB, Ramont L, Brassart-Pasco S, Maquart FX. Matrikines from basement membrane collagens: a new anti-cancer strategy. Biochim Biophys Acta. 2014;1840:2589–2598. doi: 10.1016/j.bbagen.2013.12.029. This review summarizes the involvement of collagen-derived matrikines in cancer, particularly matrikines derived from the NC1 domains of the different constitutive chains of basement membrane-associated collagens, mainly collagen IV. [DOI] [PubMed] [Google Scholar]
- 119.Gaggar A, et al. A novel proteolytic cascade generates an extracellular matrix-derived chemoattractant in chronic neutrophilic inflammation. J Immunol. 2008;180:5662–5669. doi: 10.4049/jimmunol.180.8.5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Weathington NM, et al. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nature Med. 2006;12:317–323. doi: 10.1038/nm1361. [DOI] [PubMed] [Google Scholar]
- 121.Muto J, et al. Hyaluronan digestion controls DC migration from the skin. J Clin Invest. 2014;124:1309–1319. doi: 10.1172/JCI67947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Salmon H, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. 2012;122:899–910. doi: 10.1172/JCI45817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kim S, et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102–106. doi: 10.1038/nature07623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wolf K, Friedl P. Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends Cell Biol. 2011;21:736–744. doi: 10.1016/j.tcb.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 125.Tozluoglu M, et al. Matrix geometry determines optimal cancer cell migration strategy and modulates response to interventions. Nature Cell Biol. 2013;15:751–762. doi: 10.1038/ncb2775. [DOI] [PubMed] [Google Scholar]
- 126.Bergers G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nature Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lee S, Jilani SM, Nikolova GV, Carpizo D, Iruela-Arispe ML. Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. J Cell Biol. 2005;169:681–691. doi: 10.1083/jcb.200409115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ferrara N. Binding to the extracellular matrix and proteolytic processing: two key mechanisms regulating vascular endothelial growth factor action. Mol Biol Cell. 2010;21:687–690. doi: 10.1091/mbc.E09-07-0590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chen TT, et al. Anchorage of VEGF to the extracellular matrix conveys differential signaling responses to endothelial cells. J Cell Biol. 2010;188:595–609. doi: 10.1083/jcb.200906044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Phillips JJ, et al. Heparan sulfate sulfatase SULF2 regulates PDGFRα signaling and growth in human and mouse malignant glioma. J Clin Invest. 2012;122:911–922. doi: 10.1172/JCI58215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Grafe I, et al. Excessive transforming growth factor-β signaling is a common mechanism in osteogenesis imperfecta. Nature Med. 2014;20:670–675. doi: 10.1038/nm.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Alcaraz LB, et al. Tenascin-X promotes epithelial-to-mesenchymal transition by activating latent TGF-β. J Cell Biol. 2014;205:409–428. doi: 10.1083/jcb.201308031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Watt FM, Huck WT. Role of the extracellular matrix in regulating stem cell fate. Nature Rev Mol Cell Biol. 2013;14:467–473. doi: 10.1038/nrm3620. [DOI] [PubMed] [Google Scholar]
- 134.Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nature Rev Cancer. 2005;5:675–688. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- 135.Ridky TW, Chow JM, Wong DJ, Khavari PA. Invasive three-dimensional organotypic neoplasia from multiple normal human epithelia. Nature Med. 2010;16:1450–1455. doi: 10.1038/nm.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yurchenco PD. Basement membranes: cell scaffoldings and signaling platforms. Cold Spring Harb Perspect Biol. 2011;3:a004911. doi: 10.1101/cshperspect.a004911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Seif-Naraghi SB, et al. Safety and efficacy of an injectable extracellular matrix hydrogel for treating myocardial infarction. Sci Transl Med. 2013;5:173ra25. doi: 10.1126/scitranslmed.3005503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lee B, et al. Perlecan domain V is neuroprotective and proangiogenic following ischemic stroke in rodents. J Clin Invest. 2011;121:3005–3023. doi: 10.1172/JCI46358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Martino MM, et al. Growth factors engineered for super-affinity to the extracellular matrix enhance tissue healing. Science. 2014;343:885–888. doi: 10.1126/science.1247663. [DOI] [PubMed] [Google Scholar]
- 140.Giger RJ, Hollis ER, 2nd, Tuszynski MH. Guidance molecules in axon regeneration. Cold Spring Harb Perspect Biol. 2010;2:a001867. doi: 10.1101/cshperspect.a001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Busch SA, Silver J. The role of extracellular matrix in CNS regeneration. Curr Opin Neurobiol. 2007;17:120–127. doi: 10.1016/j.conb.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 142.Alilain WJ, Horn KP, Hu H, Dick TE, Silver J. Functional regeneration of respiratory pathways after spinal cord injury. Nature. 2011;475:196–200. doi: 10.1038/nature10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Back SA, et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nature Med. 2005;11:966–972. doi: 10.1038/nm1279. [DOI] [PubMed] [Google Scholar]
- 144.Barry-Hamilton V, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nature Med. 2010;16:1009–1017. doi: 10.1038/nm.2208. This study demonstrates that inhibiting LOXL2 using a monoclonal antibody decreases fibrosis in models of lung and liver fibrosis and reduces metastasis in xenografted tumours. [DOI] [PubMed] [Google Scholar]
- 145.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 146.Devy L, Dransfield DT. New strategies for the next generation of matrix-metalloproteinase inhibitors: selectively targeting membrane-anchored MMPs with therapeutic antibodies. Biochem Res Int. 2011;2011:191670. doi: 10.1155/2011/191670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zeisberg M, et al. Stage-specific action of matrix metalloproteinases influences progressive hereditary kidney disease. PLoS Med. 2006;3:e100. doi: 10.1371/journal.pmed.0030100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346–354. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 149.Miyamoto H, et al. Tumor-stroma interaction of human pancreatic cancer: acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas. 2004;28:38–44. doi: 10.1097/00006676-200401000-00006. [DOI] [PubMed] [Google Scholar]
- 150.Sethi T, et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nature Med. 1999;5:662–668. doi: 10.1038/9511. [DOI] [PubMed] [Google Scholar]
- 151.Eke I, Storch K, Krause M, Cordes N. Cetuximab attenuates its cytotoxic and radiosensitizing potential by inducing fibronectin biosynthesis. Cancer Res. 2013;73:5869–5879. doi: 10.1158/0008-5472.CAN-13-0344. [DOI] [PubMed] [Google Scholar]
- 152.Henderson NC, et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nature Med. 2013;19:1617–1624. doi: 10.1038/nm.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nature Rev Cancer. 2009;9:285–293. doi: 10.1038/nrc2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kaplan RN, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Oskarsson T, et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nature Med. 2011;17:867–874. doi: 10.1038/nm.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Malanchi I, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2011;481:85–89. doi: 10.1038/nature10694. References 156 and 157 show that ECM components are crucial for providing proliferative, survival and stemness signals during tumour metastasis. [DOI] [PubMed] [Google Scholar]
- 157.Ghajar CM, et al. The perivascular niche regulates breast tumour dormancy. Nature Cell Biol. 2013;15:807–817. doi: 10.1038/ncb2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pietras A, et al. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell. 2014;14:357–369. doi: 10.1016/j.stem.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Reticker-Flynn NE, et al. A combinatorial extracellular matrix platform identifies cell-extracellular matrix interactions that correlate with metastasis. Nature Commun. 2012;3:1122. doi: 10.1038/ncomms2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ishizuya-Oka A, Hasebe T. Establishment of intestinal stem cell niche during amphibian metamorphosis. Curr Top Dev Biol. 2013;103:305–327. doi: 10.1016/B978-0-12-385979-2.00011-3. [DOI] [PubMed] [Google Scholar]