

Abstract
The Hedgehog (Hh) signaling pathway is critical for embryonic development. In adult tissues, Hh signaling is relatively quiescent with the exception of roles in tissue maintenance and repair. Aberrant activation of Hh signaling is implicated in multiple aspects of transformation including the maintenance of the cancer stem cell (CSC) phenotype. Pre-clinical studies indicate that CSCs from many tumor types are sensitive to Hh pathway inhibition and that Hh-targeted therapeutics block many aspects of transformation attributed to CSCs including, drug resistance, relapse and metastasis. However, to date, Hh inhibitors, specifically those targeting Smoothened (such as Vismodegib, BMS-833923, Saridegib (IPI-926), Sonidegib/Erismodegib (LDE225), PF-04449913, LY2940680, LEQ 506 and TAK-441) have demonstrated good efficacy as monotherapy in patients with basal cell carcinoma and medulloblastoma, but have shown limited activity in other tumor types. This lack of success is likely due to many factors including a lack of patient stratification in early trials, crosstalk between Hh and other oncogenic signaling pathways that can modulate therapeutic response, and a limited knowledge of Hh pathway activation mechanisms in CSCs from most tumor types. Here we discuss Hh signaling mechanisms in the context of human cancer, particularly in the maintenance of the CSC phenotype, and consider new therapeutic strategies that hold the potential to expand considerably the scope and therapeutic efficacy of Hh-directed anti-cancer therapy.
Background
Hedgehog (Hh) is a highly conserved developmental pathway involved in organogenesis, stem cell maintenance, and tissue repair/regeneration. Aberrant Hh pathway activation controls multiple aspects of tumorigenesis including initiation, progression and relapse, at least in part, by driving a cancer stem cell (CSC) phenotype. Mutational Hh pathway activation drives tumor formation in several tumor types, and many other tumors exhibit epigenetic Hh pathway activation. Small-molecule Hh inhibitors have been used as monotherapy and in combined modalities for cancer treatment. To date, however, Hh inhibitors have enjoyed limited success clinically. Here, we discuss oncogenic Hh signaling mechanisms and highlight new therapeutic strategies that may enhance the clinical efficacy and expand the effective use of Hh inhibitors to new tumor types.
The canonical Hh signaling pathway
Core Hh signaling components include the Hh ligands (sonic Hh (Shh); Indian Hh, (Ihh) and Desert Hh, (Dhh)), the trans-membrane receptor proteins Patched 1 and 2 (PTCH1 and PTCH2), the G-protein-coupled receptor-like protein Smoothened (SMO) and the glioma-associated oncogene transcription factors 1–3 (GLI1, GLI2 and GLI3) (reviewed in (1) (Fig. 1). Primary cilia localize these components to activate or repress signaling (2). Canonical Hh signaling is activated when Hh ligand binds PTCH to relieve PTCH-mediated SMO inhibition at the base of the primary cilium (3). SMO then translocates to the ciliumtip (4), driving a signaling cascade that results in nuclear GLI translocation and activation. GLI activates transcription of context-specific genes regulating self-renewal, cell fate, survival, angiogenesis, epithelial-mesenchymal transition and cell invasion (reviewed in (5)). As Hh transcriptional targets, GLI1 and PTCH establish a feedback loop that regulates Hh signaling (6).
Figure 1.


Schematic of Hedgehog (Hh) signaling in Vertebrates. A) In the absence of Hh ligand, Patched (PTCH) prevents SMO localization to the primary cilium and GLI is suppressed by a protein complex composed of Costal2 (Cos2), Fused (Fu), Suppressor of Fused (SUFU) that promotes Protein Kinase A (PKA), Glycogen Synthase Kinase 3 (GSK3) and Casein Kinase 1 (CK1)- mediated GLI phosphorylation and partial proteosomal processing of GLI into a repressor form that inhibits expression of Hh target genes. B) Hh ligand requires N-terminal palmitoylation mediated by Hh acyltransferase (HHAT) to be activated and released into the extracellular space by Dispatched (Disp). The Hh pathway is activated upon Hh ligand binding to PTCH and the co-receptors Growth Arrest Specific 1 (GAS1), CAM-related/down-regulated by oncogenes (CDO), brother of CDO (BOC) which relieves PTCH-mediated inhibition of SMO. Upon activation, SMO translocations to the primary cilia where it disrupts the repressor protein complex resulting in GLI translocation to the nucleus and activation of GLI-mediated transcription of gene targets that maintain a CSC phenotype. The Hh pathway can be therapeutically targeted by: 1) SMO inhibition (Vismodegib, BMS-833923, IPI-926, LDE225, PF-04449913, LY2940680, LEQ 506, TAK-441 and cyclopamine; 2) receptor-ligand disruption (5E1 anti-Hh ligand antibody and robotnikinin); 3) inhibition of ligand processing (HHAT inhibitor RU-SKI 43); or 4) inhibition of GLI activity (GANT58, GANT61 and HPI 1-4).
Several accessory proteins promote or suppress Hh pathway activity (Fig. 1). Hh ligands are synthesized as precursors that undergo autocatalytic cleavage, addition of a carboxy-terminal cholesterol moiety, and amino-terminal palmitoylation mediated by Skinny Hh/Hh acyltransferase (Ski/Hhat) to produce mature ligand, whose secretion is facilitated by the transmembrane transporter-like protein Dispatched (Disp) (1). Growth Arrest Specific 1 (GAS1), CAM-related/down-regulated by oncogenes (CDO), brother of CDO (BOC) and Glypican-3 (GPC3) are co-receptors that facilitate ligand binding to PTCH (1), whereas Hedgehog Interacting Peptide (HhIP) represses signaling by sequestering Hh ligand (7). Protein kinase A (PKA), glycogen synthase 3β (GSK3β), casein kinase I (CK1), Skip–Cullin–Fbox (SCF) protein, βTransducin repeat Containing Protein (βTrCP), and a suppressor complex comprised of Fused kinase (Fu), Suppressor of Fused (Sufu) and Costal2 (Cos2) regulate GLI expression, stability and localization (reviewed in (1)). Alterations in one or more of these modulatory mechanisms can lead to pathway deregulation and cancer.
Hh signaling in cancer
Both ligand-dependent and-independent mechanisms result in aberrant Hh pathway activation in cancer. Germline or somatic loss-of-function PTCH or SUFU, and gain-of-function SMO, mutations constitutively activate ligand-independent Hh signaling and drive basal cell carcinoma (BCC), medulloblastoma (MB), rhabdomyosarcoma and meningioma tumor development (8-11). GLI1 amplification occurs in glioblastoma and rhabdomyosarcoma, and activating mutations in GLI1 and GLI3 are evident in pancreatic adenocarcinomas (12-14), although the function of these mutations is not fully explored. Pallister-Hall syndrome, characterized by formation of benign hypothalamic hamartomas, is caused by a frameshift GLI3 mutation that generates a C-terminal truncated protein resembling physiologically-generated GLI3 repressor (15).
Hh signaling can also drive the transformed phenotype through autocrine or paracrine ligand-dependent mechanisms. Autocrine activation ensues when Hh ligand produced by tumor cells activates Hh signaling in the same or neighboring tumor cells to stimulate survival and tumor growth. Autocrine Hh pathway activation occurs in lung, pancreas, stomach, colon, skin, prostate, breast and brain cancers (16-23). In these tumors, SMO inhibitors block tumor cell growth in the absence of stromal cells. Paracrine Hh pathway activation occurs when tumor cells secrete Hh ligands that induce Hh activation in stromal cells, which then promote tumor growth by producing angiogenic factors (i.e. IGF and VEGF) and IL6 and Wnt signaling activation (24-27). Paracrine Hh signaling occurs in pancreatic, lung, esophageal, gastric, colon, lymphomas, multiple myelomas and prostate cancers (25, 27-34). Reverse paracrine Hh signaling has also been described in lymphomas and multiple myelomas, in which Hh ligand produced in bone marrow stroma activates Hh signaling in adjacent tumor cells (35).
Hh pathway activation in cancer stem cells
Lineage tracing studies have demonstrated the existence of a sub-population of tumor cells exhibiting stem-like properties (36-38). These tumor-initiating or cancer stem cells (CSCs) exhibit self-renewal, enhanced tumor initiation, and differentiation into transiently-amplifying cells that populate the bulk tumor. These cells function in tumor maintenance, metastasis, relapse and chemoresistance. Hh signaling drives CSC maintenance in lung, breast, pancreas, colon, glioblastoma, multiple myeloma, and chronic myeloid leukemia (CML) (16, 18, 20, 22, 39-42). Hh signaling is selectively activated in CSCs compared to bulk tumor cells from these tumor types (18, 20, 22, 41, 42), and directly drives the CSC phenotype by regulating expression of CSC markers aldehyde dehydrogenase, BMI1, WNT2 and CD44 (20, 27, 43). Pharmacologic or genetic Hh inhibition in these tumor types decreases self-renewal, tumor growth and metastasis (16, 18, 20, 22, 39-42). Hh signaling also regulates ABCG2 and MDR expression, suggesting a role in the chemo-resistance characteristic of CSCs (44-48).
Clinical-Translational Advances
Hh pathway inhibitors
Four major modes of Hh inhibition have been exploited therapeutically: 1) SMO inhibition; 2) receptor-ligand disruption; 3) inhibition of ligand processing; and 4) GLI inhibition (Fig. 1). Cyclopamine, a naturally-occurring SMO inhibitor, established Hh as a viable therapeutic target (49, 50). Though cyclopamine is not clinically useful due to its low potency and bioavailability, more potent and specific SMO inhibitors Vismodegib, BMS-833923, Saridegib (IPI-926), Sonidegib/Erismodegib (LDE225), PF-04449913, LY2940680, LEQ 506 and TAK-441 (Fig. 1) have been developed and evaluated clinically (Table 1, ClinicalTrials.gov). SMO inhibitors are particularly effective against MBs and BCCs harboring SMO or PTCH mutations, and FDA approval of Vismodegib for advanced BCC solidified Hh as a bona fide therapeutic target. Hh signaling has also been blocked by disrupting Hh ligand-PTCH interactions (Fig. 1). The Hh ligand monoclonal antibody 5E1, and the macrocyclic small molecule Robotnikinin, inhibit Hh:PTCH interactions and exhibit anti-tumor activity (51, 52). Small molecule inhibitors of Ski/HHAT, an enzyme that catalyzes a key step in Hh ligand processing, have recently been developed. HHAT inhibitors block Hh palmitoylation and prevent pathway activation (53). Agents such as GANT58/GANT61 and HPI 1-4 act by blocking GLI processing, activation and/or transcriptional activity (54, 55). These agents may be particularly useful in treating tumors exhibiting ligand-independent Hh pathway activation. Although proof-of-concept has been demonstrated with 5E1, Robotnikinin, HHAT and GLI inhibitors in vitro, further testing is required before these agents can be clinically evaluated.
Table 1.
Preclinical and clinical response to SMO inhibition in various tumor types and their chromosome 3q26 amplification status.
| Cancer type | Response pre-clinically | Response Clinically Clinicaltrials.gov NCT # | 3q26 status % with copy # gains | Reference |
|---|---|---|---|---|
| Basal cell carcinoma | Suppressed proliferation, induced apoptosis and regression of lesions. | Antitumor activity in metastatic and locally advanced BCC; Vismodegib FDA approved for advanced BCC | (74, 75) | |
| Breast | Decreased proliferation, tumor growth and metastasis. | NCT01576666; recruiting patients | 25% (TCGA) | (31, 76) |
| Cervical | Decreased cell proliferation and survival. | 77% (TCGA) | (77) | |
| Chronic myelogenous leukemia | Sensitized cells to chemotherapy. Prolonged survival in leukemia mouse model. Decreased tumorigenic potential of leukemic stem cell population. | NCT01456676; recruiting NCT01218477; completed, no results reported NCT01357655; ongoing, not recruiting | (40, 78, 79) | |
| Colorectal | Blocked cell growth in vitro and growth of xenograft tumors in vivo. Decreased recurrence and metastases. | NTC00636610; Vismodegib does not add to the efficacy of FOLFOX, FOLFIRI, or bevacizumab NCT01576666; recruiting patients | 11% (TCGA) | (22, 27, 80, 81) |
| Esophageal | Decreased cell growth and induced apoptosis. | NCT00909402; completed, no results reported | 53% | (82, 83) |
| Gastric | Decreased cell growth and induced apoptosis. | NCT00982592; Addition of Vismodegib to FOLFOX did not improve PFS in an unselected population NCT00909402; completed, no results reported NCT01576666; recruiting patients | 35% (TCGA) | (84, 85) |
| Gliomas | Decreased self-renewal of CSCs and potentiated the anti-proliferative effect of conventional chemotherapy. | NCT01576666; recruiting patients | 14% (TCGA) | (39) |
| Head and Neck | Decreased colony formation in primary tumor cells ex vivo. | 74% (TCGA) | (86) | |
| Hepatocellular | Blocked proliferation and invasion in vitro and xenograft tumors in vivo. | 17% (TCGA) | (87) | |
| Kidney | Decreased cell growth and caused tumor regression in vivo. | 15% (TCGA) | (88) | |
| Lung | Suppressed growth of small-cell lung cancer cells in vitro and in vivo. Prevented small-cell lung cancer tumor recurrence after chemotherapy treatment. Inhibited growth of lung squamous cell carcinoma CSCs. | NCT01579929; recruiting NCT01722292; recruiting | 84% LSCC (TCGA) 32% LAC (TCGA) 27% SCLC | (18, 23, 89) |
| Lymphoma | Induced apoptosis and inhibits growth of cancer cells in mice. | 29% (TCGA) | (29) | |
| Medulloblastoma | Antitumor activity in mouse models. | NCT00939484, sustained response in 15% of patients | (74, 90-92) | |
| Melanoma | Reduced proliferation, prevented recurrence and lung metastasis. | 20% | (21) | |
| Neuroblastoma | Reduced proliferation, induced apoptosis and blocked tumorigenicity. | (93, 94) | ||
| Neuro-endocrine | Blocked cell growth in vitro. | (95) | ||
| Ovarian | Decreased proliferation, mobility and invasiveness. Induced cancer cell dedifferentiation and apoptosis in vitro and decreased tumor growth in vivo. | NTC00739661; No clinically meaningful improvement in progression-free survival for vismodegib versus placebo | 83%, serous (TCGA) | (96-99) |
| Pancreatic | Blocked growth, migration, invasion, colony formation, the cancer stem cell population, tumor growth and metastasis. | NCT01096732; terminated NCT00878163; Active, not recruiting NCT01576666; recruiting NCT01537107; recruiting | 20% (TCGA) | (100, 101) |
| Prostate | Prevented tumour growth in xenograft model. | NCT01163084; ongoing, not recruiting | 15% (TCGA) | (54) |
| Sarcoma | Decreased rhabdosarcoma cell proliferation, induced apoptosis and blocked tumor growth in vivo. Decreased osteocarcinoma cell growth in vitro and tumor growth in vivo. | NCT01154452; recruiting | 13% (TCGA) | (102, 103) |
| Uterine | Decreased cell growth in vitro. | 64% (TCGA) | (104) |
Targeting Hh in CSCs
CSCs are emerging therapeutic targets whose efficient elimination may offer longer lasting, potentially curative outcomes in cancer patients. Hh pathway inhibition is a promising approach to therapeutically-target CSCs. Cyclopamine preferentially inhibits pancreatic CSCs but not bulk tumor cells (41), and GLI or SMO gene-silencing, or cyclopamine, decreases glioblastoma CSC proliferation, survival and self-renewal (16, 39). shRNA-mediated knockdown of HHAT or GLI1, or treatment with SMO inhibitor LDE225, blocks growth of lung squamous cell carcinoma (LSCC) CSCs in vitro and tumor formation in vivo (18). Cyclopamine or 5E1 antibody reduces multiple myeloma CSC self-renewal and induces terminal differentiation (56). Likewise, Smo inhibition reduces expansion of Bcr-Abl-positive leukemic stem cells in vivo and delays relapse in a mouse CML model (40). Interestingly, inhibition is independent of Bcr-Abl mutation status, indicating that imatinib-resistant leukemic stem cells may retain responsiveness to Hh inhibition.
Drug combinations therapeutically target Hh in CSCs
SMO inhibitors are extremely effective against BCC and MB tumors that harbor driver Hh pathway mutations (Table 1). However, despite promising preclinical results, SMO inhibitors have yielded little or no clinical benefit in tumors not harboring pathway mutations (Table 1). The poor clinical performance of SMO inhibitors beyond BCC and MB may be due, at least in part, to crosstalk between Hh and EGFR, RAS/MEK/ERK, PI3K/AKT/mTOR, NOTCH and/or WNT oncogenic signaling pathways (reviewed in (57)). Hh and EGFR pathways can activate each other, and cooperate to induce GLI1 transcriptional targets and promote tumor growth (58). Oncogenic KRAS/MEK/ERK signaling promotes tumorigenesis through paracrine, SMO-independent regulation of GLI1 expression, phosphorylation, degradation, nuclear localization and activation (59). The PI3K/AKT/mTOR pathway also regulates Hh signaling through GLI phosphorylation, nuclear localization and activation (60). In addition, Shh mediates epithelial-to-mesenchymal transition and metastasis through PI3K/AKT/mTOR activation (61), whereas GLI1 appears to suppress the WNT pathway in colon cancer cells (62). Thus, crosstalk between Hh and other oncogenic signaling pathways may significantly alter clinical response to Hh pathway inhibition and limit efficacy.
Clinical efficacy of Hh inhibitors may be significantly enhanced through rational patient stratification based on advanced knowledge of Hh signaling mechanisms in specific subsets of tumors. For instance, tumors in which Hh signaling is active in CSCs, but not bulk tumor cells, are unlikely to respond effectively to Hh inhibitors as monotherapy. Furthermore, the emergence of drug-resistant SMO mutations is a key factor limiting the efficacy of SMO inhibitors as monotherapy (63-65). Given these limitations, current strategies for therapeutically-targeting the Hh pathway are being reevaluated to include strategic combinations of SMO inhibitors with other therapeutic modalities.
Several preclinical studies report success combining SMO inhibitors and conventional cytotoxic anti-tumor agents. For instance, combined SMO inhibitor IPI-926 and gemcitabine blocks tumor growth in a mouse pancreatic cancer model through gemcitabine-mediated cytotoxicity and IPI-926-mediated CSC inhibition (45). In glioblastoma CSC xenografts, SMO inhibitors enhance the effects of temozolomide (66), and in a mouse CML model cyclopamine enhances the effects of Bcr-Abl inhibitor nilotinib and increases survival by targeting leukemic stem cells (40). Combined docetaxel, NOTCH inhibitor, and cyclopamine inhibits growth of docetaxel-resistant prostate CSCs (67), and combined EGFR and SMO inhibition has proven effective in preclinical prostate, BCC and glioblastoma models (57). Several clinical trials of SMO inhibition combined with other therapeutics are currently underway (Table 1). Results from these trials will provide a key indication of whether use of SMO inhibitors can be effectively extended beyond BCC and MB through use of strategic drug combinations.
Emerging insights into Hh pathway regulation in CSCs may lead to even more effective combination strategies for targeting Hh signaling in these cells. In this regard, the atypical Protein Kinase C iota (PKCι) is an oncogene (68, 69) (reviewed in (70) (71) that has emerged as a major regulator of Hh pathway activity in BCC and LSCC (18). In BCC, PKCι regulates GLI1 in a SMO-independent fashion to promote BCC tumor growth (71), suggesting that PKCι inhibition may be an alternative approach to treating BCC tumors with acquired resistance to SMO inhibitors. In LSCC, PKCι and SOX2, both of which are lineage-specific lung oncogenes, cooperate to drive a CSC phenotype through Hh pathway activation (18). Amplification of chromosome 3q26, which occurs in ∼70% of LSCCs, results in the co-amplification and co-overexpression of PKCι and SOX2 which cooperate to drive cell-autonomous Hh signaling in LSCC CSCs (18) (Fig. 2A). Mechanistically, PKCι phosphorylates SOX2, a transcription factor that functions in stem cell maintenance, and controls SOX2-mediated transcriptional activation of HHAT, resulting in increased levels of mature, palmitoylated Hh ligand and Hh pathway activation that drives the LSCC CSC phenotype (18). PKCι also activates Rac1/MEK/ERK signaling in LSCC cells to transcriptionally-regulate Matrix Metalloproteinase 10 (MMP10). This PKCι/Rac1/MEK/ERK/MMP10 signaling axis is required for both CSC maintenance and transformed growth of bulk LSCC cells (Fig. 2A) (reviewed in (70). Thus, PKCι drives both CSC and bulk tumor cell growth suggesting that combined PKCι and Hh inhibition may be a particularly effective therapeutic intervention strategy in LSCC. Indeed, combined treatment with the selective PKCι inhibitor auranofin (ANF) (18) and the SMO inhibitor LDE225 causes synergistic inhibition of LSCC CSC expansion and viability (Fig. 2B). ANF inhibits expression of PKCι-dependent transcriptional targets HHAT, GLI1 and MMP10 (18, 72), whereas LDE225 causes decreased GLI1, consistent with on-target effects of these agents. Combined ANF and LDE225 caused a more pronounced inhibition of downstream effectors when compared to either agent alone (Fig. 2C), consistent with the observed synergistic growth inhibition. Since the PKCι-SOX2-Hh signaling axis is driven by chromosome 3q26 amplification, combined PKCι and SMO inhibitor may represent a particularly effective treatment strategy for LSCCs harboring chromosome 3q26 amplification. These results have implications well beyond LSCC because many other tumor types harbor this genetic alteration. Indeed, chromosome 3q26 amplification is the most prevalent genetic copy number gain alteration in human cancers, occurring in approximately 15% of human tumors (73). As a result, PKCι and SOX2 co-amplification and co-overexpression is observed in significant percentages of bladder, breast, cervical, esophageal, head and neck, kidney, lung adenocarcinoma, lung squamous cell carcinoma, serous ovarian, stomach and uterine cancers (Table 1, (reviewed in (70)). Thus, a large patient population, identifiable by tumor-specific 3q26 amplification, is likely to exhibit active PKCι/SOX2/Hh signaling, a stem-like phenotype driven by Hh pathway activation, and responsiveness to combined ANF/SMO inhibitor treatment. Early phase clinical trials are being actively pursued to evaluate this novel therapeutic strategy.
Figure 2.

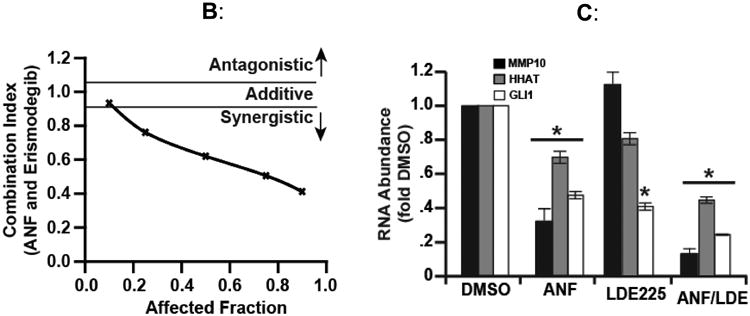
Combined inhibition of PKCι and Hh signaling inhibits the CSC phenotype. A) PKCι regulates multiple signaling pathways that maintain both CSC and bulk tumor cells and drives tumor initiation, growth, survival, invasion, metastasis and chemoresistance. In lung squamous cell carcinoma (LSCC) cells harboring 3q26 amplification, PKCι phosphorylates SOX2 and regulates SOX2-mediated transcriptional activation of HHAT leading to autocrine Hh pathway activation. This PKCι/SOX2/Hh signaling axis is required to maintain a CSC phenotype. PKCι also forms an oncogenic complex with the polarity protein PAR6 and the guanine nucleotide exchange factor Epithelial Cell Transforming Sequence 2 (ECT2) that functions to drive a RAC1/PAK/MEK/ERK signaling cascade that transcriptional upregulates Matrix Metalloproteinase 10 (MMP10) to promote tumorigenicity of both bulk tumor and cancer stem cells. B) The PKCι inhibitor, Auranofin and the SMO inhibitor, LDE225 synergistically block the growth of LSCC CSCs. C) PKCι and Hh inhibition transcriptionally downregulate their respective transcriptional targets HHAT, GLI1 and MMP10 and combined inhibition of PKCι and Hh further downregulates expression of these gene targets. DMSO, dimethyl sulfoxide.
Perspective
Hh inhibitors have been successfully employed as monotherapy for BCC and MB tumors harboring Hh pathway mutations. However, therapeutic response in these tumors may be limited by the challenge of acquired SMO resistance. SMO inhibitors have been less successful in other tumor types, probably due to many complicating factors including a lack of patient stratification in early phase trials, crosstalk between Hh and other signaling pathways, the complexity of Hh signaling in CSCs, bulk tumor cells and tumor microenvironment, and a lack of in-depth knowledge of Hh pathway activation mechanisms in specific tumor subtypes. Tumors not harboring Hh pathway mutations are unlikely to respond to Hh inhibitors alone. However, combining Hh inhibitors with chemotherapeutics or other targeted agents coupled with appropriate patient stratification paradigms provide new opportunities for more effective Hh-based therapy. One promising strategy involves combined Hh and PKCι inhibitor therapy. PKCι activates a novel PKCι-SOX2-Hh signaling axis in CSCs from LSCC tumors harboring chromosome 3q26 amplification, and these cells exhibit synergistic response to combined SMO/PKCι inhibition. The high prevalence of chromosome 3q26 copy number gains, and the resulting co-amplification of PKCι and SOX2, in many tumor types (∼15% of human tumors) raises the exciting possibility that combined Hh and PKCι inhibitor therapy will prove effective in the large target patient population whose tumors harbor chromosome 3q26 copy number gains and a CSC phenotype driven by PKCXι-SOX2-Hh pathway activation.
Acknowledgments
The authors thank Katharyn Brennan and colleagues in the Fields laboratory for helpful suggestions and critical review of the manuscript.
Grant Support: Work in the Fields laboratory was supported by grants from the NIH/NCI (R01CA081436 and R21CA151250), the V Foundation for Cancer Research, the James and Esther King Biomedical Research Program (1KG-05-33971), and the Mayo Clinic Center for Individualized Medicine (to A.P. Fields) and an NIH Research Supplement to Promote Diversity in Health-Related Research Award from the NCI (to V. Justilien). A.P. Fields is the Monica Flynn Jacoby Professor of Cancer Research, an endowment fund that provides partial support for the investigator's research program.
Footnotes
Disclosure of Potential Conflicts of Interest: A.P. Fields reports receiving a commercial research grant from Teva Pharmaceuticals. No potential conflicts of interest were disclosed by the other author.
Disclaimer: The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
References
- 1.Briscoe J, Therond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013;14:416–29. doi: 10.1038/nrm3598. [DOI] [PubMed] [Google Scholar]
- 2.Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11:331–44. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–6. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 4.Rohatgi R, Milenkovic L, Corcoran RB, Scott MP. Hedgehog signal transduction by Smoothened: pharmacologic evidence for a 2-step activation process. Proc Natl Acad Sci U S A. 2009;106:3196–201. doi: 10.1073/pnas.0813373106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hui CC, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol. 2011;27:513–37. doi: 10.1146/annurev-cellbio-092910-154048. [DOI] [PubMed] [Google Scholar]
- 6.Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–87. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- 7.Chuang PT, McMahon AP. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature. 1999;397:617–21. doi: 10.1038/17611. [DOI] [PubMed] [Google Scholar]
- 8.Belyea B, Kephart JG, Blum J, Kirsch DG, Linardic CM. Embryonic signaling pathways and rhabdomyosarcoma: contributions to cancer development and opportunities for therapeutic targeting. Sarcoma. 2012;2012:406239. doi: 10.1155/2012/406239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caro I, Low JA. The role of the hedgehog signaling pathway in the development of basal cell carcinoma and opportunities for treatment. Clin Cancer Res. 2010;16:3335–9. doi: 10.1158/1078-0432.CCR-09-2570. [DOI] [PubMed] [Google Scholar]
- 10.Clark VE, Erson-Omay EZ, Serin A, Yin J, Cotney J, Ozduman K, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. 2013;339:1077–80. doi: 10.1126/science.1233009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Romer J, Curran T. Targeting medulloblastoma: small-molecule inhibitors of the Sonic Hedgehog pathway as potential cancer therapeutics. Cancer Res. 2005;65:4975–8. doi: 10.1158/0008-5472.CAN-05-0481. [DOI] [PubMed] [Google Scholar]
- 12.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kinzler KW, Bigner SH, Bigner DD, Trent JM, Law ML, O'Brien SJ, et al. Identification of an amplified, highly expressed gene in a human glioma. Science. 1987;236:70–3. doi: 10.1126/science.3563490. [DOI] [PubMed] [Google Scholar]
- 14.Roberts WM, Douglass EC, Peiper SC, Houghton PJ, Look AT. Amplification of the gli gene in childhood sarcomas. Cancer Res. 1989;49:5407–13. [PubMed] [Google Scholar]
- 15.Kang S, Graham JM, Jr, Olney AH, Biesecker LG. GLI3 frameshift mutations cause autosomal dominant Pallister-Hall syndrome. Nat Genet. 1997;15:266–8. doi: 10.1038/ng0397-266. [DOI] [PubMed] [Google Scholar]
- 16.Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W, et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25:2524–33. doi: 10.1634/stemcells.2007-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425:846–51. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 18.Justilien V, Walsh MP, Ali SA, Thompson EA, Murray NR, Fields AP. The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer Cell. 2014;25:139–51. doi: 10.1016/j.ccr.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431:707–12. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 20.Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–71. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, et al. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc Natl Acad Sci U S A. 2007;104:5895–900. doi: 10.1073/pnas.0700776104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P, et al. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol Med. 2009;1:338–51. doi: 10.1002/emmm.200900039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422:313–7. doi: 10.1038/nature01493. [DOI] [PubMed] [Google Scholar]
- 24.Ingram WJ, Wicking CA, Grimmond SM, Forrest AR, Wainwright BJ. Novel genes regulated by Sonic Hedgehog in pluripotent mesenchymal cells. Oncogene. 2002;21:8196–205. doi: 10.1038/sj.onc.1205975. [DOI] [PubMed] [Google Scholar]
- 25.Mills LD, Zhang Y, Marler RJ, Herreros-Villanueva M, Zhang L, Almada LL, et al. Loss of the transcription factor GLI1 identifies a signaling network in the tumor microenvironment mediating KRAS oncogene-induced transformation. J Biol Chem. 2013;288:11786–94. doi: 10.1074/jbc.M112.438846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pola R, Ling LE, Silver M, Corbley MJ, Kearney M, Blake Pepinsky R, et al. The morphogen Sonic hedgehog is an indirect angiogenic agent upregulating two families of angiogenic growth factors. Nat Med. 2001;7:706–11. doi: 10.1038/89083. [DOI] [PubMed] [Google Scholar]
- 27.Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455:406–10. doi: 10.1038/nature07275. [DOI] [PubMed] [Google Scholar]
- 28.Becher OJ, Hambardzumyan D, Fomchenko EI, Momota H, Mainwaring L, Bleau AM, et al. Gli activity correlates with tumor grade in platelet-derived growth factor-induced gliomas. Cancer Res. 2008;68:2241–9. doi: 10.1158/0008-5472.CAN-07-6350. [DOI] [PubMed] [Google Scholar]
- 29.Dierks C, Grbic J, Zirlik K, Beigi R, Englund NP, Guo GR, et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat Med. 2007;13:944–51. doi: 10.1038/nm1614. [DOI] [PubMed] [Google Scholar]
- 30.Hegde GV, Peterson KJ, Emanuel K, Mittal AK, Joshi AD, Dickinson JD, et al. Hedgehog-induced survival of B-cell chronic lymphocytic leukemia cells in a stromal cell microenvironment: a potential new therapeutic target. Mol Cancer Res. 2008;6:1928–36. doi: 10.1158/1541-7786.MCR-08-0142. [DOI] [PubMed] [Google Scholar]
- 31.O'Toole SA, Machalek DA, Shearer RF, Millar EK, Nair R, Schofield P, et al. Hedgehog overexpression is associated with stromal interactions and predicts for poor outcome in breast cancer. Cancer Res. 2011;71:4002–14. doi: 10.1158/0008-5472.CAN-10-3738. [DOI] [PubMed] [Google Scholar]
- 32.Theunissen JW, de Sauvage FJ. Paracrine Hedgehog signaling in cancer. Cancer Res. 2009;69:6007–10. doi: 10.1158/0008-5472.CAN-09-0756. [DOI] [PubMed] [Google Scholar]
- 33.Tian H, Callahan CA, DuPree KJ, Darbonne WC, Ahn CP, Scales SJ, et al. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc Natl Acad Sci U S A. 2009;106:4254–9. doi: 10.1073/pnas.0813203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilkinson SE, Furic L, Buchanan G, Larsson O, Pedersen J, Frydenberg M, et al. Hedgehog signaling is active in human prostate cancer stroma and regulates proliferation and differentiation of adjacent epithelium. Prostate. 2013;73:1810–23. doi: 10.1002/pros.22720. [DOI] [PubMed] [Google Scholar]
- 35.Scales SJ, de Sauvage FJ. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci. 2009;30:303–12. doi: 10.1016/j.tips.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 36.Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–6. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Driessens G, Beck B, Caauwe A, Simons BD, Blanpain C. Defining the mode of tumour growth by clonal analysis. Nature. 2012;488:527–30. doi: 10.1038/nature11344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, et al. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science. 2012;337:730–5. doi: 10.1126/science.1224676. [DOI] [PubMed] [Google Scholar]
- 39.Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17:165–72. doi: 10.1016/j.cub.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell. 2008;14:238–49. doi: 10.1016/j.ccr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 41.Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M, et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 2007;67:2187–96. doi: 10.1158/0008-5472.CAN-06-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci U S A. 2007;104:4048–53. doi: 10.1073/pnas.0611682104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoon C, Park do J, Schmidt B, Thomas NJ, Lee HJ, Kim TS, et al. CD44 Expression Denotes a Subpopulation of Gastric Cancer Cells in Which Hedgehog Signaling Promotes Chemotherapy Resistance. Clin Cancer Res. 2014;20:3974–88. doi: 10.1158/1078-0432.CCR-14-0011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Huang FT, Zhuan-Sun YX, Zhuang YY, Wei SL, Tang J, Chen WB, et al. Inhibition of hedgehog signaling depresses self-renewal of pancreatic cancer stem cells and reverses chemoresistance. Int J Oncol. 2012;41:1707–14. doi: 10.3892/ijo.2012.1597. [DOI] [PubMed] [Google Scholar]
- 45.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sims-Mourtada J, Izzo JG, Ajani J, Chao KS. Sonic Hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene. 2007;26:5674–9. doi: 10.1038/sj.onc.1210356. [DOI] [PubMed] [Google Scholar]
- 47.Singh S, Chitkara D, Mehrazin R, Behrman SW, Wake RW, Mahato RI. Chemoresistance in prostate cancer cells is regulated by miRNAs and Hedgehog pathway. PLoS ONE. 2012;7:e40021. doi: 10.1371/journal.pone.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steg AD, Katre AA, Bevis KS, Ziebarth A, Dobbin ZC, Shah MM, et al. Smoothened antagonists reverse taxane resistance in ovarian cancer. Mol Cancer Ther. 2012;11:1587–97. doi: 10.1158/1535-7163.MCT-11-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Binns W, Keeler RF, Balls LD. Congenital deformities in lambs, calves, and goats resulting from maternal ingestion of Veratrum californicum: hare lip, cleft palate, ataxia, and hypoplasia of metacarpal and metatarsal bones. Clin Toxicol. 1972;5:245–61. doi: 10.3109/15563657208991003. [DOI] [PubMed] [Google Scholar]
- 50.Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–9. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 51.Maun HR, Wen X, Lingel A, de Sauvage FJ, Lazarus RA, Scales SJ, et al. Hedgehog pathway antagonist 5E1 binds hedgehog at the pseudo-active site. J Biol Chem. 2010;285:26570–80. doi: 10.1074/jbc.M110.112284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stanton BZ, Peng LF, Maloof N, Nakai K, Wang X, Duffner JL, et al. A small molecule that binds Hedgehog and blocks its signaling in human cells. Nat Chem Biol. 2009;5:154–6. doi: 10.1038/nchembio.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petrova E, Rios-Esteves J, Ouerfelli O, Glickman JF, Resh MD. Inhibitors of Hedgehog acyltransferase block Sonic Hedgehog signaling. Nat Chem Biol. 2013;9:247–9. doi: 10.1038/nchembio.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lauth M, Bergstrom A, Shimokawa T, Toftgard R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci U S A. 2007;104:8455–60. doi: 10.1073/pnas.0609699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hyman JM, Firestone AJ, Heine VM, Zhao Y, Ocasio CA, Han K, et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc Natl Acad Sci U S A. 2009;106:14132–7. doi: 10.1073/pnas.0907134106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Merchant AA, Matsui W. Targeting Hedgehog--a cancer stem cell pathway. Clin Cancer Res. 2010;16:3130–40. doi: 10.1158/1078-0432.CCR-09-2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brechbiel J, Miller-Moslin K, Adjei AA. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat Rev. 2014;40:750–9. doi: 10.1016/j.ctrv.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 58.Eberl M, Klingler S, Mangelberger D, Loipetzberger A, Damhofer H, Zoidl K, et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol Med. 2012;4:218–33. doi: 10.1002/emmm.201100201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lauth M, Toftgard R. Hedgehog signaling and pancreatic tumor development. Adv Cancer Res. 2011;110:1–17. doi: 10.1016/B978-0-12-386469-7.00001-3. [DOI] [PubMed] [Google Scholar]
- 60.Riobo NA, Lu K, Ai X, Haines GM, Emerson CP., Jr Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc Natl Acad Sci U S A. 2006;103:4505–10. doi: 10.1073/pnas.0504337103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoo YA, Kang MH, Lee HJ, Kim BH, Park JK, Kim HK, et al. Sonic hedgehog pathway promotes metastasis and lymphangiogenesis via activation of Akt, EMT, and MMP-9 pathway in gastric cancer. Cancer Res. 2011;71:7061–70. doi: 10.1158/0008-5472.CAN-11-1338. [DOI] [PubMed] [Google Scholar]
- 62.Akiyoshi T, Nakamura M, Koga K, Nakashima H, Yao T, Tsuneyoshi M, et al. Gli1, downregulated in colorectal cancers, inhibits proliferation of colon cancer cells involving Wnt signalling activation. Gut. 2006;55:991–9. doi: 10.1136/gut.2005.080333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Buonamici S, Williams J, Morrissey M, Wang A, Guo R, Vattay A, et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med. 2010;2:51ra70. doi: 10.1126/scitranslmed.3001599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dijkgraaf GJ, Alicke B, Weinmann L, Januario T, West K, Modrusan Z, et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011;71:435–44. doi: 10.1158/0008-5472.CAN-10-2876. [DOI] [PubMed] [Google Scholar]
- 65.Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326:572–4. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ferruzzi P, Mennillo F, De Rosa A, Giordano C, Rossi M, Benedetti G, et al. In vitro and in vivo characterization of a novel Hedgehog signaling antagonist in human glioblastoma cell lines. Int J Cancer. 2012;131:E33–44. doi: 10.1002/ijc.27349. [DOI] [PubMed] [Google Scholar]
- 67.Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco R, et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch-and hedgehog-dependent tumor-initiating cells. Cancer Cell. 2012;22:373–88. doi: 10.1016/j.ccr.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Regala RP, Weems C, Jamieson L, Khoor A, Edell ES, Lohse CM, et al. Atypical protein kinase C iota is an oncogene in human non-small cell lung cancer. Cancer Res. 2005;65:8905–11. doi: 10.1158/0008-5472.CAN-05-2372. [DOI] [PubMed] [Google Scholar]
- 69.Zhang L, Huang J, Yang N, Liang S, Barchetti A, Giannakakis A, et al. Integrative genomic analysis of protein kinase C (PKC) family identifies PKCiota as a biomarker and potential oncogene in ovarian carcinoma. Cancer Res. 2006;66:4627–35. doi: 10.1158/0008-5472.CAN-05-4527. [DOI] [PubMed] [Google Scholar]
- 70.Parker PJ, Justilien V, Riou P, Linch M, Fields AP. Atypical protein kinase Ciota as a human oncogene and therapeutic target. Biochem Pharmacol. 2014;88:1–11. doi: 10.1016/j.bcp.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Atwood SX, Li M, Lee A, Tang JY, Oro AE. GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature. 2013;494:484–8. doi: 10.1038/nature11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Frederick LA, Matthews JA, Jamieson L, Justilien V, Thompson EA, Radisky DC, et al. Matrix metalloproteinase-10 is a critical effector of protein kinase Ciota-Par6alpha-mediated lung cancer. Oncogene. 2008;27:4841–53. doi: 10.1038/onc.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–33. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164–72. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- 75.Williams JA, Guicherit OM, Zaharian BI, Xu Y, Chai L, Wichterle H, et al. Identification of a small molecule inhibitor of the hedgehog signaling pathway: effects on basal cell carcinoma-like lesions. Proc Natl Acad Sci U S A. 2003;100:4616–21. doi: 10.1073/pnas.0732813100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kubo M, Nakamura M, Tasaki A, Yamanaka N, Nakashima H, Nomura M, et al. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004;64:6071–4. doi: 10.1158/0008-5472.CAN-04-0416. [DOI] [PubMed] [Google Scholar]
- 77.Samarzija I, Beard P. Hedgehog pathway regulators influence cervical cancer cell proliferation, survival and migration. Biochem Biophys Res Commun. 2012;425:64–9. doi: 10.1016/j.bbrc.2012.07.051. [DOI] [PubMed] [Google Scholar]
- 78.Queiroz KC, Ruela-de-Sousa RR, Fuhler GM, Aberson HL, Ferreira CV, Peppelenbosch MP, et al. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene. 2010;29:6314–22. doi: 10.1038/onc.2010.375. [DOI] [PubMed] [Google Scholar]
- 79.Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458:776–9. doi: 10.1038/nature07737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Berlin J, Bendell JC, Hart LL, Firdaus I, Gore I, Hermann RC, et al. A randomized phase II trial of vismodegib versus placebo with FOLFOX or FOLFIRI and bevacizumab in patients with previously untreated metastatic colorectal cancer. Clin Cancer Res. 2013;19:258–67. doi: 10.1158/1078-0432.CCR-12-1800. [DOI] [PubMed] [Google Scholar]
- 81.Mazumdar T, DeVecchio J, Shi T, Jones J, Agyeman A, Houghton JA. Hedgehog signaling drives cellular survival in human colon carcinoma cells. Cancer Res. 2011;71:1092–102. doi: 10.1158/0008-5472.CAN-10-2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ma X, Sheng T, Zhang Y, Zhang X, He J, Huang S, et al. Hedgehog signaling is activated in subsets of esophageal cancers. Int J Cancer. 2006;118:139–48. doi: 10.1002/ijc.21295. [DOI] [PubMed] [Google Scholar]
- 83.Zaidi AH, Komatsu Y, Kelly LA, Malhotra U, Rotoloni C, Kosovec JE, et al. Smoothened inhibition leads to decreased proliferation and induces apoptosis in esophageal adenocarcinoma cells. Cancer Invest. 2013;31:480–9. doi: 10.3109/07357907.2013.820317. [DOI] [PubMed] [Google Scholar]
- 84.Cohen DJ, Christos PJ, Kindler HL, Catenacci DVT, Bekaii-Saab TB, Tahiri S, et al. Vismodegib (V), a hedgehog (HH) pathway inhibitor, combined with FOLFOX for first-line therapy of patients (pts) with advanced gastric and gastroesophageal junction (GEJ) carcinoma: A New York Cancer Consortium led phase II randomized study. J Clin Oncol. 2013;31 suppl; abstr 4011. [Google Scholar]
- 85.Ma X, Chen K, Huang S, Zhang X, Adegboyega PA, Evers BM, et al. Frequent activation of the hedgehog pathway in advanced gastric adenocarcinomas. Carcinogenesis. 2005;26:1698–705. doi: 10.1093/carcin/bgi130. [DOI] [PubMed] [Google Scholar]
- 86.Mozet C, Stoehr M, Dimitrova K, Dietz A, Wichmann G. Hedgehog targeting by cyclopamine suppresses head and neck squamous cell carcinoma and enhances chemotherapeutic effects. Anticancer Res. 2013;33:2415–24. [PubMed] [Google Scholar]
- 87.Xu Y, Chenna V, Hu C, Sun HX, Khan M, Bai H, et al. Polymeric nanoparticle-encapsulated hedgehog pathway inhibitor HPI-1 (NanoHHI) inhibits systemic metastases in an orthotopic model of human hepatocellular carcinoma. Clin Cancer Res. 2012;18:1291–302. doi: 10.1158/1078-0432.CCR-11-0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dormoy V, Danilin S, Lindner V, Thomas L, Rothhut S, Coquard C, et al. The sonic hedgehog signaling pathway is reactivated in human renal cell carcinoma and plays orchestral role in tumor growth. Mol Cancer. 2009;8:123. doi: 10.1186/1476-4598-8-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Park KS, Martelotto LG, Peifer M, Sos ML, Karnezis AN, Mahjoub MR, et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat Med. 2011;17:1504–8. doi: 10.1038/nm.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee MJ, Hatton BA, Villavicencio EH, Khanna PC, Friedman SD, Ditzler S, et al. Hedgehog pathway inhibitor saridegib (IPI-926) increases lifespan in a mouse medulloblastoma model. Proc Natl Acad Sci U S A. 2012;109:7859–64. doi: 10.1073/pnas.1114718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rohner A, Spilker ME, Lam JL, Pascual B, Bartkowski D, Li QJ, et al. Effective targeting of Hedgehog signaling in a medulloblastoma model with PF-5274857, a potent and selective Smoothened antagonist that penetrates the blood-brain barrier. Mol Cancer Ther. 2012;11:57–65. doi: 10.1158/1535-7163.MCT-11-0691. [DOI] [PubMed] [Google Scholar]
- 92.Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361:1173–8. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mao L, Xia YP, Zhou YN, Dai RL, Yang X, Duan SJ, et al. A critical role of Sonic Hedgehog signaling in maintaining the tumorigenicity of neuroblastoma cells. Cancer Sci. 2009;100:1848–55. doi: 10.1111/j.1349-7006.2009.01262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xu L, Wang X, Wan J, Li T, Gong X, Zhang K, et al. Sonic Hedgehog pathway is essential for neuroblastoma cell proliferation and tumor growth. Mol Cell Biochem. 2012;364:235–41. doi: 10.1007/s11010-011-1222-6. [DOI] [PubMed] [Google Scholar]
- 95.Fendrich V, Waldmann J, Esni F, Ramaswamy A, Mullendore M, Buchholz M, et al. Snail and Sonic Hedgehog activation in neuroendocrine tumors of the ileum. Endocr Relat Cancer. 2007;14:865–74. doi: 10.1677/ERC-07-0108. [DOI] [PubMed] [Google Scholar]
- 96.Bhattacharya R, Kwon J, Ali B, Wang E, Patra S, Shridhar V, et al. Role of hedgehog signaling in ovarian cancer. Clin Cancer Res. 2008;14:7659–66. doi: 10.1158/1078-0432.CCR-08-1414. [DOI] [PubMed] [Google Scholar]
- 97.Chen X, Horiuchi A, Kikuchi N, Osada R, Yoshida J, Shiozawa T, et al. Hedgehog signal pathway is activated in ovarian carcinomas, correlating with cell proliferation: it's inhibition leads to growth suppression and apoptosis. Cancer Sci. 2007;98:68–76. doi: 10.1111/j.1349-7006.2006.00353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kaye SB, Fehrenbacher L, Holloway R, Amit A, Karlan B, Slomovitz B, et al. A phase II, randomized, placebo-controlled study of vismodegib as maintenance therapy in patients with ovarian cancer in second or third complete remission. Clin Cancer Res. 2012;18:6509–18. doi: 10.1158/1078-0432.CCR-12-1796. [DOI] [PubMed] [Google Scholar]
- 99.Liao X, Siu MK, Au CW, Wong ES, Chan HY, Ip PP, et al. Aberrant activation of hedgehog signaling pathway in ovarian cancers: effect on prognosis, cell invasion and differentiation. Carcinogenesis. 2009;30:131–40. doi: 10.1093/carcin/bgn230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Feldmann G, Fendrich V, McGovern K, Bedja D, Bisht S, Alvarez H, et al. An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol Cancer Ther. 2008;7:2725–35. doi: 10.1158/1535-7163.MCT-08-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xu FG, Ma QY, Wang Z. Blockade of hedgehog signaling pathway as a therapeutic strategy for pancreatic cancer. Cancer Lett. 2009;283:119–24. doi: 10.1016/j.canlet.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 102.Hirotsu M, Setoguchi T, Sasaki H, Matsunoshita Y, Gao H, Nagao H, et al. Smoothened as a new therapeutic target for human osteosarcoma. Mol Cancer. 2010;9:5. doi: 10.1186/1476-4598-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tostar U, Toftgard R, Zaphiropoulos PG, Shimokawa T. Reduction of human embryonal rhabdomyosarcoma tumor growth by inhibition of the hedgehog signaling pathway. Genes Cancer. 2010;1:941–51. doi: 10.1177/1947601910385449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Feng YZ, Shiozawa T, Miyamoto T, Kashima H, Kurai M, Suzuki A, et al. Overexpression of hedgehog signaling molecules and its involvement in the proliferation of endometrial carcinoma cells. Clin Cancer Res. 2007;13:1389–98. doi: 10.1158/1078-0432.CCR-06-1407. [DOI] [PubMed] [Google Scholar]