

Abstract
Acute die-offs of amphibian populations worldwide have been linked to the emergence of viral and fungal diseases. Inter and intraspecific immunogenetic differences may influence the outcome of infection. Toll-like receptors (TLRs) are an essential component of innate immunity and also prime acquired defenses. We report the first comprehensive assessment of TLR gene variation for urodele amphibians. The Lissotriton newt TLR repertoire includes representatives of 13 families and is compositionally most similar to that of the anuran Xenopus. Both ancient and recent gene duplications have occurred in urodeles, bringing the total number of TLR genes to at least 21. Purifying selection has predominated the evolution of newt TLRs in both long (∼70 Ma) and medium (∼18 Ma) timescales. However, we find evidence for both purifying and positive selection acting on TLRs in two recently diverged (2–5 Ma) allopatric evolutionary lineages (Lissotriton montandoni and L. vulgaris graecus). Overall, both forms of selection have been stronger in L. v. graecus, while constraint on most TLR genes in L. montandoni appears relaxed. The differences in selection regimes are unlikely to be biased by demographic effects because these were controlled by means of a historical demographic model derived from an independent data set of 62 loci. We infer that TLR genes undergo distinct trajectories of adaptive evolution in closely related amphibian lineages, highlight the potential of TLRs to capture the signatures of different assemblages of pathogenic microorganisms, and suggest differences between lineages in the relative roles of innate and acquired immunity.
Keywords: amphibia, Approximate Bayesian Computation, Lissotriton, positive selection, Toll-like receptors
Introduction
Plant and invertebrate genes involved in detection of pathogens tend to evolve rapidly. They are characterized by pervasive positive selection in Drosophila (Sackton et al. 2007) and by high gene turnover in multilocus families coupled with transient balancing selection in plants (Tiffin and Moeller 2006; Boller and He 2009). The need for high genetic diversity of innate sensors may have been reduced in vertebrates by transferring the role of the pathogen-specific response to the molecules of the adaptive immune system (Wlasiuk et al. 2009; Wlasiuk and Nachman 2010). However, as evidenced by the diversity of the vertebrate innate immune system (Akira et al. 2006; Hansen et al. 2011; Kawai and Akira 2011), rapid recognition of conserved molecular patterns associated with pathogens is still essential.
Toll-like receptors (TLRs) are pattern recognition receptors that sense conserved pathogen associated molecular patterns (PAMPs; Akira et al. 2006; Kawai and Akira 2010). TLRs are of paramount importance for the innate immune response and also provide a crucial link between innate and adaptive immune systems (Iwasaki and Medzhitov 2010; Kawai and Akira 2010). Vertebrates usually have a limited number of TLRs specialized in recognition of various PAMPs (Palti 2011; Keestra et al. 2013). These TLR genes form several families (Roach et al. 2005; Temperley et al. 2008) and representatives of most families appear indispensable for an effective immune response (Medzhitov 2001; Takeda et al. 2003).
Evolutionary rates in various TLR families have been reported as similar and slow (Roach et al. 2005; Temperley et al. 2008). Within the molecules, the extracellular part, consisting of multiple leucine-rich repeats (LRR), is involved in pathogen recognition and evolves faster than the intracellular Toll/interleukin-1 receptor (TIR) domain responsible for intracellular signaling (Mikami et al. 2012). However, the image of a static, functionally conserved and slowly evolving TLR may be oversimplified as suggested by interspecific differences in ligand recognition (Werling et al. 2009; Palti 2011) and by major differences in TLR repertoires among vertebrate groups (Roach et al. 2005; Temperley et al. 2008). Moreover, observations of multiple instances of recurrent and episodic positive selection occurring in various taxonomic groups on relatively short timescales (Wlasiuk et al. 2009; Wlasiuk and Nachman 2010; Alcaide and Edwards 2011; Tschirren et al. 2011) are inconsistent with slowly evolving and strongly conserved TLR genes. Associations between TLR polymorphism and susceptibility to infection have been demonstrated in rodent (Tschirren et al. 2013) and human (Netea et al. 2012) populations, again suggesting that episodes of positive selection in TLR genes may be prompted by changes in PAMP or in the composition of pathogen communities. Thus, TLR genes are substantially conserved, but episodic positive as well as transient balancing selection are not uncommon and variation segregating in populations may be of adaptive significance (Tschirren et al. 2012, 2013).
A detailed study of TLR polymorphism in human populations revealed variable levels of constraint, abundant polymorphism and recent positive selection at some loci (Ferrer-Admetlla et al. 2008; Barreiro et al. 2009; Mukherjee et al. 2009, 2014; Wlasiuk and Nachman 2010). The most constrained human TLRs are 03, 07, 08, and 09 which act as sensors of pathogen nucleic acids (NAs). Barreiro et al. (2009) suggested that less constrained TLRs, in which missense or even nonsense mutations are tolerated, may be promising targets of transient positive/balancing selection because polymorphisms segregating in populations for longer periods of time should be available for selection if environmental conditions or pathogen pressure change. They also hypothesized that changes in pathogen pressure may render some innate immune receptor genes temporarily redundant. Such fluctuations in selection regimes could potentially explain the loss of certain TLR genes observed during vertebrate evolution and may also facilitate the evolution of altered ligand specificity.
Empirical evaluations of TLR polymorphism within and divergence between closely related species have been performed only in a handful of mammals and birds (Palermo et al. 2009; Downing et al. 2010; Grueber et al. 2012; Tschirren et al. 2012). The most comprehensive analyses include studies of humans (Barreiro et al. 2009) and apes (Wlasiuk and Nachman 2010; Quach et al. 2013), which demonstrated substantial interspecific differences in the strength of purifying selection acting on TLRs. Similar information is currently lacking for other vertebrate groups and for TLR genes absent in amniotes.
The only amphibians for which TLRs have been characterized so far are clawed frogs of the genus Xenopus. These anurans exhibit a TLR repertoire somewhat intermediate to that of fish and terrestrial vertebrates (Ishii et al. 2007). No data on the TLRs of urodele amphibians, a group which diverged from anurans about 300 Ma (Roelants et al. 2007), have been reported so far. This is unfortunate as amphibians are in a catastrophic decline partly attributed to emerging infectious diseases (Cheng et al. 2011; Fisher et al. 2012). Resistance to the most important of these diseases, chytridiomycosis, is contingent both on genetic differences among species (Woodhams et al. 2007) and variation within species (Savage and Zamudio 2011). TLRs, by providing a link between the innate and acquired branches of the immune system, may be particularly relevant to understanding the factors that affect susceptibility to chytridiomycosis (Richmond et al. 2009). Moreover, TLRs have been shown to mediate antifungal immunity in other vertebrates (Roeder et al. 2004; Luther and Ebel 2006). Given the acute need for comprehensive information on amphibian immunogenetics, we characterize the TLR repertoire and variation in urodeles with a main focus on newts of the genus Lissotriton. We use transcriptome sequences combined with high-throughput amplicon sequencing to identify, characterize, and assess polymorphism in newt TLR genes. We then identify selection pressures acting on the urodele TLR repertoire at long-term, medium-term, and recent evolutionary time scales. Specifically, we address the following questions: 1) what is the repertoire of TLR genes in urodeles and how does it compare to that of other vertebrate groups? 2) what is the extent of selective constraint of individual TLR genes in urodeles? Are there differences in constraint between amphibians and mammals, especially in NA-sensing and nonNA-sensing genes? 3) is there evidence of recent positive selection or polymorphisms of adaptive significance segregating in TLR genes in distinct evolutionary lineages of Lissotriton newts? 4) do overall patterns of selection on TLRs differ between lineages when controlling for historical demography?
Materials and Methods
Identification of the TLR Repertoire in the newt Transcriptome
Transcripts of TLR genes were identified in transcriptome assembled from liver and spleen mRNA of 14 newts. Liver transcriptomes of six Lissotriton montandoni and six L. vulgaris and spleen transcriptomes of two L. vulgaris were sequenced. Sequencing libraries were prepared using the TruSeq RNA kit starting from total RNA and sequenced with HiSeq2000 (Illumina). In total 471 million pairs (on average 33.6 ± [SD] 9.6 million per sample) of 100 bp reads were used for assembly with Trinity (Grabherr et al. 2011). A database of TLR protein sequences compiled from a diverse set of vertebrate species was used to identify TLR transcripts (using BLASTx algorithm) in the assembled transcriptome. The two Lissotriton species are closely related; due to ongoing hybridization and incomplete lineage sorting, they share a large number of polymorphisms, and in most gene trees the species are not reciprocally monophyletic (Zieliński et al. 2013; Pabijan M, Zieliński P, Babik W, unpublished data). Therefore we did not attempt to reconstruct TLR transcripts for each species; instead we used the assembled contig as a representative L. montandoni/vulgaris TLR-coding sequence for comparisons with other vertebrates. We did not analyze noncoding parts of the transcripts but took advantage of this information to design primers for an assessment of TLR polymorphism.
Transcripts of a number of TLR genes were identified in the newt transcriptome. Some of them were highly divergent from TLRs of other vertebrates. A phylogeny of vertebrate TLR proteins including those inferred from newt transcripts was used to assign newt genes to TLR families. This phylogeny included all major vertebrate TLR families, with several representatives from each to facilitate alignment. We searched the transcriptome derived from multiple tissues of the red-spotted newt (Notophthalmus viridescens) (Abdullayev et al. 2013) for orthologs of Lissotriton TLR genes and included them in the phylogenetic analysis if complete open reading frames (ORFs) could be identified. Alignment was done with MAFFT (Katoh et al. 2002) using the L-INS-i strategy and the BLOSUM45 substitution matrix. The best fitting model of amino acid substitution, the WGA model including rate variation among sites and a nonzero proportion of invariable sites (WGA + G + I + F), was selected in ProtTest 3.3 (Darriba et al. 2011). Phylogenetic trees were constructed with the neighbor-joining method with 1,000 bootstrap replicates from a matrix of Jones–Taylor–Thornton distances in MEGA5 (Tamura et al. 2011) and using model-based Bayesian inference in MrBayes 3.2.2 (Ronquist and Huelsenbeck 2003). The Bayesian analysis was run for 106 steps with trees sampled every 200 steps and the first 25% of sampled trees discarded as burn-in; two analyses run in parallel, each consisting of one cold and three heated chains, converged on the same posterior distribution as evidenced by the standard deviation (SD) of split frequencies less than 0.01. An additional phylogenetic analysis was performed for the TLR05 family, which includes members lacking TIR and transmembrane domains; these genes in fish encode a soluble TLR05S form (Tsujita et al. 2004; Palti 2011). Alignment, model selection (also resulting in the WGA + G + I + F model) and phylogenetic analyses were performed as described above. Protein domains were identified using SMART (Letunic et al. 2012).
The selection landscape has been reported to differ between “cell surface” versus “endosomal” (Barreiro et al. 2009) or “viral” versus “nonviral” (Wlasiuk and Nachman 2010) TLRs. To test this hypothesis in newts, we had to modify the criteria of classification because not all newt TLRs could be unambiguously assigned to defined classes: protein (profilin) sensing TLR12 localizes into the endosomal compartment (Yarovinsky 2014), endosomal TLR13 senses bacterial rRNA (Oldenburg et al. 2012) while dsRNA-sensing TLR22 is present on the cell surface (Matsuo et al. 2008). As the core of the hypothesis is the difference in patterns of selection between NA sensing and other TLRs, we classify newt TLRs into two groups according to their ligands: NA-sensing (NA: TLR03, 07, 08, 09, 13, 21, 22) or nonNA-sensing TLRs (nonNA: TLR01, 02, 05, 05 L, 12, 14) (Palti 2011; Oldenburg et al. 2012; Keestra et al. 2013; Koblansky et al. 2013; Pietretti and Wiegertjes 2014; Rauta et al. 2014). The ligand for TLR14 remains unknown, but this gene falls into the TLR01/02 family and therefore was tentatively classified as nonNA. The TLR19 ligand is also unknown, but this gene is so divergent from other TLR families that we did not assign NA/nonNA status.
TLR Expression in Liver and Spleen
TLRs were identified from deep-sequenced transcriptomes of two organs: liver and spleen. Although only two spleen transcriptomes were available, it was nevertheless possible to meaningfully compare the level of expression of various TLRs between organs. Analysis was performed using a pipeline distributed within the Trinity package. First, it uses RSEM (Li and Dewey 2011) to calculate transcript abundance based on reads mapped to TLRs with Bowtie (Langmead et al. 2009). Next, differentially expressed genes were identified with edgeR (Robinson et al. 2010), a Bioconductor package (Gentleman et al. 2004), with trimmed mean of M-values normalization, which accounts for differences between samples in the number of reads. We used a 10−3 false discovery rate and 4-fold change threshold to identify tissue-biased genes.
Polymorphism and Divergence of newt TLR Genes
Examination of TLR polymorphism and divergence within and between closely related species was facilitated by the genomic structure of the vertebrate TLR genes: in most genes the entire or an overwhelming majority of the coding region is composed of a single exon. We could therefore design primers for amplification of TLR genes from genomic DNA in overlapping 800–1,000 bp fragments (supplementary table S1 and fig. S1, Supplementary Material online). Each polymerase chain reaction (PCR) was performed in 15 µl and contained 7.5 µl of HotStarTaq PCR Master Mix (Qiagen), 1.0 µM of each primer and 50–100 ng of genomic DNA. The following PCR protocol was used: 95°C/15 min, 35 × (94°C/30 s, 55°C/30 s, 72°C/60 s), 72°C/10 min. Amplified fragments were pooled in an equimolar fashion. For each newt, a fragment library was prepared using the Nextera XT kit (Illumina) according to the manufacturer’s protocol. Indexed libraries were sequenced on a MiSeq (Illumina), producing 2 × 150 bp reads. Mapping of reads to reference, single nucleotide polymorphism-calling and physical reconstruction of haplotypes were done as described in Zieliński et al. (2014), resulting in phase-resolved haplotypes (alleles) for most heterozygous individuals. In some cases certain positions could not be physically phased; these were phased computationally using PHASE (Stephens et al. 2001; Stephens and Donnelly 2003); physically phased alleles and homozygous individuals were provided as known haplotypes.
To test the mode of natural selection operating on newt TLR genes, we performed analyses of polymorphism and divergence using two complementary sampling schemes (supplementary fig. S2, Supplementary Material online). A single L. helveticus individual was used as an outgroup. In the first scheme 16 L. montandoni and L. vulgaris individuals were selected, representative of genetic diversity of both species (Babik et al. 2005; Nadachowska and Babik 2009; Nadachowska-Brzyska et al. 2012). This species-wide data set was useful for some selection tests such as the McDonald–Kreitman (MK) test, because high overall variation increases power. Due to strong genetic structure in Lissotriton, this sampling strategy was not well suited for neutrality tests based on site-frequency spectra (Städler et al. 2009). Therefore we also used a population sampling strategy, involving two genetically distinct groups which we consider as regional populations: 1) L. vulgaris graecus from Albania, Greece and Macedonia and 2) L. montandoni from the southern part of its range. Genetic variation within these regional populations is substantial but differentiation between demes is limited (Pabijan et al. forthcoming; Zieliński et al. 2013). Newts form discrete demes corresponding to breeding ponds, such demes undergo extinction and recolonization, and thus the regional population can be considered a metapopulation (Marsh and Trenham 2001; Smith and Green 2005). It has been shown (Wakeley 1999, 2004; Wakeley and Aliacar 2001) that if one gene copy per locus is sampled per deme, the ancestral process producing such sample is identical to the unstructured coalescent process, if time is rescaled appropriately. We approximated this optimal strategy by sampling multiple localities, with one individual sampled per locality. For the species-wide data set we sequenced almost entire coding sequences of TLR genes (on average 2.4 kb per gene); for the population data set we focused on the more variable external domain (∼1.5 kb).
Basic measures of polymorphism and divergence including the number of segregating sites (S), number of alleles, overall (π), synonymous (πS) and nonsynonymous (πN) nucleotide diversities, Tajima’s D and Fay and Wu’s H were calculated in DnaSP 5.1 (Librado and Rozas 2009) and mstatspop (http://bioinformatics.cragenomica.es/numgenomics/people/sebas/software/software.html, last accessed December 20, 2014). For completeness, we report values of D and H for synonymous, nonsynonymous and all sites combined, but test significance only for synonymous and nonsynonymous sites. Statistical significance of D and H was tested by comparing the observed values to the null distributions obtained through 100,000 coalescent simulations in ms (Hudson 2002) under an appropriate demographic model (supplementary tables S2 and S3, Supplementary Material online). The demographic model was selected and its parameters estimated using sequences of 62 markers located in 3′-untranslated regions (UTR) of transcripts (Zieliński et al. 2014) within the Approximate Bayesian Computation (ABC) framework; details are given in supplementary material, Supplementary Material online. The null distributions of D and H for each TLR were obtained using synonymous and nonsynonymous mutation rates estimated by comparison with L. helveticus which diverged approximately 18.4 Ma (Pabijan et al. forthcoming) and a single recombination rate estimated in the ABC analysis (supplementary table S3, Supplementary Material online).
Modes and Strength of Natural Selection Acting on newt TLR Genes
The maximum-likelihood estimate of the ratio of nonsynonymous to synonymous divergence between TLR sequences of L. vulgaris/L. montandoni and Notophthalmus was calculated in PAML 4.7 (Yang 2007). The effects of nonsynonymous substitutions were predicted using PROVEAN (Choi et al. 2012). SNiPRE (Eilertson et al. 2012), an MK-like method utilizing information from multiple genes to detect selection was used to estimate the measure of constraint f (the proportion of nonlethal nonsynonymous mutations). This analysis was done using species-wide data set. The standard MK test was calculated using DnaSP on both species-wide and population data sets.
The Hudson–Kreitman Aguadé (HKA) test assesses heterogeneity of the ratio of intraspecific polymorphism to interspecific divergence; significant heterogeneity indicates that some of the investigated genes have been affected by differential selection. Because HKA is known to be sensitive to population structure (Ingvarsson 2004), we applied this test only to the population data set. The multilocus HKA test was calculated in the program HKA (https://bio.cst.temple.edu/∼hey/software/software.htm); significance was assessed with 10,000 coalescent simulations. Because the HKA test was significant in both lineages, we used the maximum-likelihood version of the test (Wright and Charlesworth 2004) to determine which genes are under selection. Likelihood-ratio tests were used to compare the null neutral model to models in which a particular gene is affected by selection. Tests were done in the program MLHKA (http://wright.eeb.utoronto.ca/programs/) with chain length of 200,000 steps.
The distribution of selection coefficients within the TLR genes was investigated using a population genetics-phylogenetics approach implemented in the program gammamap (Wilson et al. 2011). This method is embedded in a Bayesian framework and models evolution of codons to explain both observed intraspecific polymorphism and interspecific divergence. A sliding window approach is used to model variation in selection pressures along genes and infer the posterior probability of positive selection for each codon. The analysis was performed on sequence data from L. montandoni and L. v. graecus, and a single outgroup (L. helveticus) sequence. Prior distributions on all model parameters were chosen as in Wilson et al. (2011). We assumed lineage-specific priors for the distribution of fitness effects (DFE), sliding window smoothing parameter (p), branch length (T), and transition:transversion ratio (κ). An uninformative, symmetric Dirichelet prior was applied on DFE, where 12 fitness classes ranging from effectively inviable to beneficial (γ = −500, −100, −50, −10, −5, −1, 0, 1, 5, 10, 50, 100) are equally likely. We employed a uniform prior on p within interval (0, 1), and improper log-uniform priors on κ and T. The population-scaled mutation parameter θ was assigned a separate log-normal prior distribution for each lineage and each gene with hyperparameters mean μ and SD σ. The mean μ was given an improper uniform prior and SD σ was given a log-normal prior with mean 0 and SD 2. The posterior distributions of selection coefficients for each codon (γ) together with all other parameters were estimated jointly in six separate MCMC runs, each 3,000,000 steps long, with values recorded every 50 steps. Mixing conditions for each parameter were adjusted beforehand. We removed the first 300,000 steps as burn-in and compared chains for convergence. The final estimates were obtained by merging values from all chains together.
Results
TLR Repertoire and Expression in newts
Sixteen TLR genes were identified in the Lissotriton transcriptome (fig. 1). In most cases assignment to TLR families was straightforward. However, the classification of two genes (TLR12 and TLR19) should be regarded as tentative because of high sequence divergence from other vertebrate genes (fig. 1). Representatives of all major vertebrate TLR families except TLR04 were found in the Lissotriton transcriptome. The TLR repertoire is similar to that of Xenopus. Notable differences include the lack of divergent TLR14 paralogs, and the presence of TLR19 which has so far been detected only in fish. Xenopus has only four genes in the TLR11 (sensu Roach et al. 2005) family, whereas Lissotriton has six (fig. 1). TLR09 and TLR22 underwent duplication in urodeles before the divergence of Lissotriton and Notophthalmus approximately 70 Ma. Full-length ORFs of 11 and partial ORFs of four apparent orthologs were identified in the Notophthalmus transcriptome, making TLR12 the only TLR gene not detected in the available Notophthalmus assembly (table 1). A short gene from the TLR05 family (TLR05L), lacking the transmembrane and TIR domains, is present in the newt transcriptome. Phylogenetic analysis demonstrated that it is not orthologous to the teleost soluble TLR05S (supplementary fig. S3, Supplementary Material online).
Fig. 1.—

A Bayesian tree showing the relationships among vertebrate TLRs. Amphibian TLRs are color coded by species.
Table 1.
Synonymous and Nonsynonymous Divergence between Notophthalmus and Lissotriton TLR Genes
| Gene | ncodons | cds | dN | dS | dN/dS |
|---|---|---|---|---|---|
| TLR01 | 786 | 1.00 | 0.056 | 0.142 | 0.39 |
| TLR02 | 799 | 1.00 | 0.069 | 0.184 | 0.37 |
| TLR03 | 901 | 1.00 | 0.057 | 0.186 | 0.31 |
| TLR05 | 888 | 1.00 | 0.089 | 0.170 | 0.52 |
| TLR05L | 664 | 1.00 | 0.064 | 0.162 | 0.40 |
| TLR07 | 1,041 | 0.99 | 0.048 | 0.211 | 0.22 |
| TLR08 | 534 | 0.50 | 0.076 | 0.169 | 0.45 |
| TLR09A | 1,038 | 1.00 | 0.056 | 0.154 | 0.37 |
| TLR09B | 1,034 | 1.00 | 0.079 | 0.147 | 0.54 |
| TLR12 | 0 | 0.00 | — | — | — |
| TLR13 | 566 | 0.59 | 0.114 | 0.160 | 0.71 |
| TLR14 | 819 | 1.00 | 0.050 | 0.167 | 0.30 |
| TLR19 | 955 | 1.00 | 0.084 | 0.204 | 0.41 |
| TLR21 | 707 | 0.73 | 0.083 | 0.316 | 0.26 |
| TLR22A | 871 | 0.91 | 0.067 | 0.146 | 0.46 |
| TLR22B | 956 | 1.00 | 0.059 | 0.151 | 0.39 |
Note.—ncodons, the length of the Lissotriton-Nothophthalmus alignment in codons; cds, proportion of the coding sequence covered by alignment; dN, maximum likelihood (ML) estimate of nonsynonymous divergence; dS, ML estimate of synonymous divergence.
Domain structure of newt TLR proteins is generally similar to that of Xenopus (fig. 2). Remarkably, neither newt nor frog has a SMART-identifiable transmembrane domain in TLR07. Newts do not have a transmembrane domain in TLR22, a feature shared with fugu TLR22 and 23 proteins but not with Xenopus TLR22. The presence of three transmembrane domains (fig. 2) is a unique feature of newt TLR12.
Fig. 2.—

Comparison of the domain structure between newt and frog TLRs. TLR19 is absent in Xenopus and zebrafish structure is provided instead; abbreviations of protein domains: LRR NT, Leucine-rich repeat, N-terminal; LRR, Leucine-rich repeat; LRR CT, LRR C-terminal.
All TLR genes were expressed in both liver and spleen although expression level differed widely across genes and organs; the differences spanned almost three orders of magnitude (supplementary fig. S4, Supplementary Material online). Significant differences between organs were detected for five TLR genes: 05, 05L, and 22A had higher expression in liver, whereas TLR12 and 19 had higher expression in spleen (supplementary fig. S4, Supplementary Material online).
Variation and the Nature of Selection Acting on TLR in Lissotriton newts
The presence of more than two expressed variants per individual and high within-individual variation indicate that five genes (TLR05, 08 12, 14, and 19) have recently been duplicated in the Lissotriton lineage (data not shown). The duplicated copies did not assemble into separate contigs, presumably because of high sequence similarity. We were thus unable to sort out the paralogs and we do not present an analysis of polymorphism for these genes, but use the assembled transcript sequences to assess long-term evolutionary constraint.
The extent of constraint was estimated at three evolutionary scales: 1) long-term, by calculating Lissotriton–Notophthalmus dN/dS, 2) medium-term, by using L. montandoni/vulgaris polymorphism data in relation to divergence from L. helveticus, and 3) recent, by employing population genetic and phylogenetic methods in two regional newt populations and controlling for the effects of historical demography. The dN/dS ranged from 0.22 to 0.52, with TLR13 (for which <60% coding sequence was available for dN/dS calculation) a clear outlier at 0.71 (table 1). No difference in dN/dS was detected between the NA and nonNA TLRs (Welch t-test, t10.5 = 0.4022, P = 0.70, TLR13 excluded). At a medium-term evolutionary scale the range of constraint (f, proportion of nonlethal nonsynonymous mutations as calculated by SNiPRE) was relatively narrow (table 2) and did not differ between the NA and nonNA TLRs (t4.9 = 1.2246, P = 0.28). A positive correlation between dN/dS and f indicates that the overall constraint has been similar across the timescales (r = 0.765, t8 = 3.3605, P = 0.0099).
Table 2.
Polymorphism in 11 TLR Genes in a Species-Wide Sample of Lissotriton montandoni/vulgaris, Data for 62 UTR Are Also Shown
| Gene | 2N | nsites | cds | n alleles | S | π | πN | πS | NI | MK P-val | f Lo95 | f | f Hi95 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TLR01 | 32 | 2,358 | 1.00 | 25 | 160 | 0.0146 | 0.0105 | 0.0293 | 0.93 | 1.000 | 0.23 | 0.28 | 0.35 |
| TLR02 | 32 | 2,313 | 0.96 | 21 | 87 | 0.0090 | 0.0066 | 0.0174 | 0.74 | 0.451 | 0.23 | 0.30 | 0.41 |
| TLR03 | 32 | 1,776 | 0.66 | 26 | 132 | 0.0136 | 0.0074 | 0.0351 | 0.46 | 0.086 | 0.17 | 0.22 | 0.27 |
| TLR05L | 32 | 1,782 | 0.89 | 24 | 146 | 0.0117 | 0.0068 | 0.0271 | 0.28 | 0.063 | 0.19 | 0.25 | 0.30 |
| TLR07 | 32 | 3,045 | 0.97 | 27 | 171 | 0.0109 | 0.0044 | 0.0331 | 0.69 | 0.348 | 0.16 | 0.21 | 0.25 |
| TLR09A | 32 | 3,090 | 0.99 | 23 | 118 | 0.0080 | 0.0047 | 0.0194 | 0.96 | 1.000 | 0.20 | 0.26 | 0.34 |
| TLR09B | 30 | 2,083 | 0.67 | 19 | 119 | 0.0121 | 0.0089 | 0.023 | 0.40 | 0.075 | 0.25 | 0.31 | 0.40 |
| TLR13 | 32 | 2,502 | 0.88 | 26 | 181 | 0.0100 | 0.0067 | 0.0209 | 1.68 | 0.303 | 0.22 | 0.27 | 0.33 |
| TLR21 | 32 | 2,841 | 0.98 | 26 | 215 | 0.0141 | 0.0077 | 0.0347 | 0.99 | 1.000 | 0.18 | 0.23 | 0.28 |
| TLR22A | 32 | 2,787 | 0.98 | 23 | 149 | 0.0097 | 0.0054 | 0.0242 | 0.95 | 1.000 | 0.20 | 0.25 | 0.30 |
| TLR22B | 32 | 2,026 | 0.71 | 26 | 145 | 0.0141 | 0.0084 | 0.0334 | 1.12 | 1.000 | 0.22 | 0.28 | 0.36 |
| TLR average | 32 | 2,418 | 0.88 | 24.2 | 147.5 | 0.0116 | 0.0070 | 0.0271 | 0.83 | 0.26 | |||
| 62 UTR average | 32 | 499 | 17.7 | 37.8 | 0.0146 |
Note.—2N, number of gene copies sampled; nsites, the length of alignment; cds, proportion of the coding sequence covered by alignment; S, number of segregating sites; π, overall nucleotide diversity; πN, nucleotide diversity at nonsynonymous sites; πS nucleotide diversity at synonymous sites; NI, neutrality index; MK P-val, McDonald and Kreitman Fisher’s exact test P-value (Lissotriton helveticus was used as an outgroup); f Lo95, lower credible limit for constraint calculated in SNiPRE; f, estimate of constraint; f Hi 95%, upper credible limit for constraint.
Below, we present analyses of polymorphism and divergence based both on species-wide and population data sets (for L. montandoni and L. v. graecus). TLR genes exhibited comparable variation in the species-wide data set and their average π was about 0.8 of that for 62 3′-UTRs (table 2 and supplementary fig. S5, Supplementary Material online). No differences in nucleotide diversity between the NA and nonNA TLRs were detected (t3.063 = 0.113, P = 0.917).
The MK test did not detect adaptive evolution for any TLR in the species-wide data set (table 2), but in population data sets significant results were obtained for TLR05L in L. montandoni (P = 0.035) and TLR09B in L. v. graecus (P = 0.006), indicating that either power was increased in relation to the species-wide data set or that selection pressures have varied in the history of the two evolutionary lineages. The multilocus HKA test was significant in both lineages (L. montandoni P < 0.0001, L. v. graecus P = 0.007). In individual tests two genes exhibited an excess of divergence: TLR05L in L. montandoni (P = 0.006) and TLR21 in L. v. graecus (P = 0.001) and one gene, TLR07, showed a marginally significant excess of polymorphism (P = 0.04) in L. v. graecus (table 3).
Table 3.
Polymorphism in 11 TLR Genes in Population-Level Samples of Two Evolutionary Lineages: Lissotriton montandoni (Lm, 38 gene copies sampled) and L. vulgaris graecus (Lvg, 34 gene copies sampled), Data for 62 UTR Are Also Shown
| Gene | nsites | cds | n alleles |
S |
π |
πS |
πN |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lm | Lvg | Lm | Lvg | Lm | Lvg | Lm | Lvg | Lm | Lvg | |||
| TLR01 | 1,626 | 0.69 | 13 | 18 | 65 | 39 | 0.0112 | 0.0047 | 0.0220 | 0.0113 | 0.0081 | 0.0028 |
| TLR02 | 1,656 | 0.69 | 10 | 12 | 23 | 35 | 0.0024 | 0.0031 | 0.0074 | 0.0047 | 0.0009 | 0.0027 |
| TLR03 | 1,620 | 0.60 | 17 | 19 | 65 | 47 | 0.0097 | 0.0037 | 0.0289 | 0.0079 | 0.0041 | 0.0025 |
| TLR05L | 1,463 | 0.73 | 5 | 18 | 5 | 41 | 0.0008 | 0.0051 | 0.0019 | 0.0139 | 0.0004 | 0.0024 |
| TLR07 | 1,527 | 0.49 | 22 | 17 | 44 | 58 | 0.0067 | 0.0065 | 0.0143 | 0.0195 | 0.0045 | 0.0026 |
| TLR09A | 1,683 | 0.54 | 7 | 14 | 27 | 39 | 0.0056 | 0.0048 | 0.0097 | 0.0112 | 0.0034 | 0.0029 |
| TLR09B | 1,452 | 0.47 | 14 | 16 | 15 | 26 | 0.0017 | 0.0046 | 0.0029 | 0.0105 | 0.0013 | 0.0028 |
| TLR13 | 1,479 | 0.52 | 16 | 14 | 46 | 48 | 0.0092 | 0.0047 | 0.0168 | 0.0067 | 0.0068 | 0.0040 |
| TLR21 | 1,611 | 0.55 | 25 | 11 | 82 | 15 | 0.0124 | 0.0017 | 0.0283 | 0.0033 | 0.0074 | 0.0015 |
| TLR22A | 1,723 | 0.60 | 9 | 16 | 47 | 47 | 0.0095 | 0.0041 | 0.0221 | 0.0130 | 0.0058 | 0.0015 |
| TLR22B | 1,387 | 0.48 | 18 | 18 | 53 | 43 | 0.0095 | 0.0070 | 0.0219 | 0.0151 | 0.0058 | 0.0047 |
| TLR average | 1,566 | 0.58 | 14.2 | 15.7 | 42.9 | 39.8 | 0.0071 | 0.0045 | 0.0160 | 0.0106 | 0.0044 | 0.0028 |
| 62 UTR average | 499 | 7 | 9.5 | 0.0044 | 0.0052 | |||||||
Note.—nsites, the length of alignment; cds, proportion of the coding sequence covered by alignment; S, number of segregating sites; π, overall nucleotide diversity; πN, nucleotide diversity at nonsynonymous sites; πS nucleotide diversity at synonymous sites.
The distribution of selection coefficients for 11 TLR genes as estimated by the population genetics-phylogenetics gammamap method differed substantially between L. montandoni and L. v. graecus, although the 95% credible intervals estimated for each fitness class were wide in both species (fig. 3). The proportion of strongly constrained codons (−500 ≤ γ ≤ −5) was higher in L. v. graecus than in L. montandoni (86% vs. 68%). The frequencies of neutral and beneficial mutations in L. montandoni exceeded those in L. v. graecus, with the starkest contrast among neutral and nearly neutral mutations (−1 ≤ γ ≤ 1, 24% vs. 9%). Despite overall constraint, adaptive evolution may have occurred at individual codons. Following Quach et al. (2013), we considered codons as evolving under positive selection if the proportion of simulated positive gamma values (γ > 0) exceeded 0.75. In total 33 codons under positive selection were inferred in L. v. graecus, compared with only 15 in L. montandoni (supplementary table S4, Supplementary Material online). Overall, the number of positively selected codons per gene was low, ranging from zero in TLR01 in both lineages to 8 in TLR09B in L. v. graecus and only four were predicted to have a radical effect on protein function (supplementary table S4, Supplementary Material online). In five genes (TLR02, 09A, 09B, 21, 22B) positively selected codons were detected in both lineages. In addition, positive selection was inferred in four genes (TLR03, 07, 13, 22A) only in L. v. graecus, and in one gene (TLR05L) in L. montandoni. Remarkably, only a single codon was identified as positively selected in both lineages (codon 294 of TLR21, in which a nonsynonymous polymorphism shared with the outgroup species segregates in both lineages). Nonsense mutations segregated at appreciable frequencies in TLR09B in L. montandoni, the more common of these mutations produced a stop codon in position 48, indicating that the respective allele does not encode a functional protein.
Fig. 3.—

The distribution of fitness effects of new mutations in 11 Lissotriton TLR genes.
In the population data sets TLR nucleotide diversity tended to be higher in L. montandoni (πmon= 0.0072, πgre = 0.0045, t12.61 = 2.027, P = 0.064), whereas for 62 3′-UTRs it was similar in both lineages (πmon = 0.0044, πgre = 0.0052, t121.9 = 1.079, P = 0.283) (table 3 and fig. 4). Tajima’s D in TLR genes was significantly higher in L. montandoni (Dmon = 0.20, Dgre = −0.96, t18.28 = −3.827, P = 0.0012), whereas no differences were detected in UTRs (Dmon = −0.95, Dgre = −0.92, t120.9 = 0.175, P = 0.086) (table 4 and fig. 4). In each lineage Tajima’s D values at synonymous and nonsynonymous sites of most genes were remarkably similar and in most cases did not depart from expectations under the inferred demographic model (supplementary tables S2 and S3, Supplementary Material online; table 4 and fig. 4). The few significant D values indicated an excess of high frequency variants (table 4). Significantly negative Fay and Wu’s H, signaling an excess of high-frequency derived alleles consistent with an incomplete selective sweep was detected in one L. montandoni gene (TLR22B, only synonymous sites), whereas three (TLR03, 07, 13) L. v. graecus genes exhibited this pattern (table 4). The same three genes were identified by the gammamap method as targets of positive selection in L. v. graecus.
Fig. 4.—
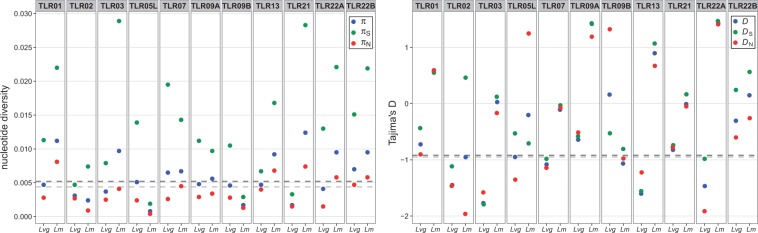
Nucleotide diversity and Tajima’s D in population data sets; Lm, Lissotriton montandoni, Lvg, L. vulgaris graecus; π, overall, πS, synonymous and πN, nonsynonymous nucleotide diversity; D, overall, DS, synonymous and DN, nonsynonymous Tajima’s D.
Table 4.
Results of Neutrality Tests and Values of Tajima’s D and Fay and Wu’s H Statistics in Lissotriton montandoni (Lm, 38 gene copies sampled) and L.vulgaris graecus (Lvg, 34 gene copies sampled)
| MK P-val |
HKA P-val |
D |
DS |
DN |
H |
HS |
HN |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lm | Lvg | Lm | Lvg | Lm | Lvg | Lm | Lvg | Lm | Lvg | Lm | Lvg | Lm | Lvg | Lm | Lvg | |
| TLR01 | 0.806 | 0.644 | 0.273 | 0.968 | 0.57 | −0.74 | 0.52 | −0.45 | 0.57 | −0.94 | 0.75 | −0.98 | −0.11 | −2.50 | 1.05 | 1.63 |
| TLR02 | 1.000 | 0.621 | 0.108 | 0.695 | −0.93 | −1.41 | 0.50 | −1.08 | −1.93 | −1.45 | −6.15 | −4.03 | −3.25 | −1.46 | −2.95 | −3.72 |
| TLR03 | 0.264 | 0.824 | 0.471 | 0.752 | 0.00 | −1.79 | 0.10 | −1.77 | −0.19 | −1.63 | 4.14 | −16.66 | 3.49 | −11.46* | 0.55 | −5.14 |
| TLR05L | 0.035 | 0.170 | 0.006 | 0.359 | −0.16 | −0.94 | −0.72 | −0.52 | 1.25 | −1.34 | 0.66 | 0.19 | 0.32 | −0.55 | 0.24 | 0.74 |
| TLR07 | 0.139 | 0.741 | 0.152 | 0.041 | −0.06 | −1.07 | 0.00 | −0.99 | −0.10 | −1.10 | 2.13 | −6.45 | 0.11 | −3.08 | 1.97 | −3.26* |
| TLR09A | 0.435 | 1.000 | 0.165 | 0.735 | 1.38 | −0.61 | 1.39* | −0.60 | 1.18 | −0.56 | −4.38 | 2.44 | −1.46 | 0.02 | −1.88 | 1.98 |
| TLR09B | 0.112 | 0.006 | 0.115 | 0.305 | −1.06 | 0.15 | −0.84 | −0.53 | −1.01 | 1.29* | 0.84 | −1.81 | −0.20 | −1.54 | 0.76 | −0.33 |
| TLR13 | 0.361 | 0.576 | 0.206 | 0.234 | 0.85 | −1.63 | 1.04 | −1.58 | 0.63 | −1.20 | 3.20 | −11.04 | 1.01 | −5.43* | 2.19 | −5.28* |
| TLR21 | 0.843 | 0.452 | 0.594 | 0.001 | 0.01 | −0.83 | 0.12 | −0.76 | −0.07 | −0.74 | −5.17 | −1.78 | −1.21 | −0.93 | −2.20 | 1.13 |
| TLR22A | 0.818 | 0.198 | 0.685 | 0.743 | 1.43 | −1.45 | 1.43* | −1.01 | 1.37* | −1.92 | 1.91 | −7.97 | 0.94 | −3.30 | 0.99 | −4.04 |
| TLR22B | 0.723 | 1.000 | 0.140 | 0.123 | 0.15 | −0.26 | 0.58 | 0.25 | −0.25 | −0.62 | −7.10 | −0.65 | −4.23* | 2.95 | −0.89 | 0.23 |
| TLR average | 0.20 | −0.96 | 0.37 | −0.82 | 0.13 | −0.93 | −0.83 | −4.43 | −0.42 | −2.48 | −0.01 | −1.46 | ||||
Note.—MK P-val, McDonald and Kreitman Fisher’s exact test P-value; HKA P-val, maximum-likelihood HKA test P-value; D, Tajima’s D value for all sites; DS, Tajima’s D value for synonymous sites; DN Tajima’s D value for nonsynonymous sites; H, Fay and Wu’s H value for all sites; HS, Fay and Wu’s H value for synonymous sites; HN, Fay and Wu’s H value for nonsynonymous sites; *, P < 0.01; P-values are provided only for DS, DN, HS, and HN, for D two-sided, for H one-sided.
Discussion
This work is the first analysis of the repertoire and evolutionary dynamics of TLR genes in urodele amphibians and one of the few studies employing population genetic methods to understand processes shaping variation in these genes, while controlling for nonequilibrium demography. Overall, we found that amid substantial evolutionary conservation episodes of lineage-specific adaptation have involved individual codons of some TLR genes, potentially providing a more efficient response to pathogen assault that may vary in space and time. Available data from several systems indicate that lineage-specific bouts of adaptive evolution may dominate the dynamics of vertebrate TLRs (Tschirren et al. 2011, 2012; Quach et al. 2013; Grueber et al. 2014). Thus, although adaptive changes in TLR genes may be subtle in comparison to other components of the immune system (Piertney and Oliver 2006; Downing et al. 2010; Quintana-Murci and Clark 2013), these genes should be incorporated into research aiming to understand associations between genetic variation and susceptibility to pathogens in natural populations (Tschirren et al. 2013).
The Evolution of the Urodele TLR Repertoire
Substantial differences in the composition of the TLR family occur between fish and amniotes (Roach et al. 2005; Temperley et al. 2008; Palti 2011). Amphibians have been suggested to possess an intermediate TLR repertoire which may reflect their association with both aquatic and terrestrial environments (Ishii et al. 2007; Oshiumi et al. 2008). The data so far have been restricted to Xenopus (Ishii et al. 2007). Our study shows that TLR composition in urodeles and anurans is broadly similar. The only major family present in newts but absent in Xenopus is TLR19, otherwise found only in some fish (Palti 2011), and newts express representatives of all families present in Xenopus. No TLR04 orthologs have been found in the newt transcriptome, leaving TLR04 the only major vertebrate TLR family not detected in urodeles. TLR04 plays a crucial role in the mammalian response against diverse microbes, especially gram-negative bacteria (Akira et al. 2006). In fish TLR04 is functionally different from the mammalian counterpart and present only in some species (Palti 2011; Kanwal et al. 2014). The presence of TLR04 in Xenopus has been controversial (Roach et al. 2005; Ishii et al. 2007), but TLR04 genes derived from automated prediction are present in the most recent versions of the Xenopus laevis and Xenopus tropicalis genome assemblies (http://www.xenbase.org/). TLR04 has also been reported in the transcriptome of another anuran, Bombina maxima (Zhao et al. 2014), and, based on indirect data, also in frogs (Nikolaeva et al. 2012). Thus the evidence regarding the presence and nature of amphibian TLR04 is somewhat equivocal. A short amphibian gene from the TLR05 family lacking the TIR domain (TLR05L) is not orthologous to the teleost soluble TLR05S (Muñoz et al. 2013), but is unique to some tetrapods, being found in turtles (supplementary fig. S3, Supplementary Material online) and the anole lizard (Muñoz et al. 2013). It is currently unknown whether TLR05L binds flagellin and modulates a flagellin-mediated immune response, as has been demonstrated for teleost TLR05S (Muñoz et al. 2013). The expression of newt TLR05L was much higher in liver than in spleen, similar to that of fish TLR05S.
Lineage-specific duplications have occurred in anurans and urodeles, leading to differences in the composition of several TLR families between the amphibian orders. At least 16 TLR genes are expressed in newts, four of them result from duplications (TLR09A-09B and TLR22A-22B) predating the divergence of Lissotriton and Notophthalmus at approximately 70 Ma (Roelants et al. 2007). Duplications have also been common in the more recent history of the Lissotriton TLR genes, as evidenced by closely related paralogs in five of them, which brings the total number of TLR genes to at least 21. Due to high sequence similarity, examination of variation in these TLR genes would require a separate study employing dedicated methods. Such studies are desirable as they may shed light on the fates of TLR paralogs immediately following duplication: whether both copies are commonly maintained by selection or if one typically undergoes pseudogenization. Gene duplication and to a lesser extent, gene conversion among paralogs within families, feature prominently in the evolution of vertebrate TLR genes. Mammals and birds differ in this respect from fish. Although in the former duplication and gene conversion are restricted mainly to the TLR01 family (Huang et al. 2011), in the latter duplications are common, particularly in fish-specific TLRs, resulting in multigene families sometimes composed of many similar paralogs (Star et al. 2011; Sundaram et al. 2012; Quiniou et al. 2013; Pietretti et al. 2014). Duplications in fish seem lineage-specific and short lived, as their patchy phylogenetic distribution suggests. The data from newts reveal a similar pattern with recent duplications in four of five major TLR families. The propensity for retention of duplicated TLR copies may be related to the efficiency of other components of the immune system as has been suggested in the case of extreme duplications of TLR in the Atlantic cod, a fish species which lacks a functional major histocompatibility complex (MHC) class II pathway (Star et al. 2011; Sundaram et al. 2012).
Constraint and Lineage-Specific Adaptation in newt TLR Genes
Urodele TLR genes are substantially constrained at the long-term evolutionary scale, and the extent of constraint in individual genes is similar to that reported for birds (Alcaide and Edwards 2011; Grueber et al. 2014). Thus purifying selection has predominated the evolution of urodele TLR genes, similarly as in other vertebrates (Roach et al. 2005; Mikami et al. 2012). However, several studies found signatures of recurrent or episodic positive selection affecting a relatively minor (<1–5%) fraction of codons in birds and mammals (Wlasiuk et al. 2009; Wlasiuk and Nachman 2010; Alcaide and Edwards 2011; Areal et al. 2011; Grueber et al. 2014). With data from only two species, we did not have enough power to test for individual codons under positive selection, but flag this as a priority for future studies with more data.
We used the extensive data on polymorphism and divergence of TLR genes to test for selection in the more recent evolutionary history of Lissotriton newts, that is, since the origin of the L. vulgaris lineage at approximately 18 Ma (Pabijan et al. forthcoming). At this scale constraint remains strong; no differences were detected between NA-sensing and nonNA-sensing TLR genes. Stronger purifying selection in the former versus more positive selection and less constraint in the latter were reported in primates and birds (Barreiro et al. 2009; Wlasiuk and Nachman 2010; Huang et al. 2011). Conservation of NA-sensing TLRs has been ascribed to functional constraint, that is, proteins encoded by these genes must recognize motives and types of foreign NA without triggering autoimmune reactions. This may well be true for specific clades, but a broader analysis of mammal TLR did not confirm differences in the rate of adaptive evolution between viral and nonviral TLR genes (Areal et al. 2011). Likewise, we did not find this pattern in newts.
Our results suggest substantial differences in selection regimes between the two closely related lineages L. montandoni and L. v. graecus; their divergence has been estimated at approximately 7 Ma (supplementary table S3, Supplementary Material online). Natural selection operating on TLR genes has been stronger in L. v. graecus and, interestingly, both purifying and positive selection seem more prominent in this lineage compared with L. montandoni in which selection appears somewhat relaxed. Despite the overwhelming signal of purifying selection, positive selection was detected by at least one method in 9 of 11 genes in L. v. graecus, but only in 6 L. montandoni genes. Also over twice as many codons evolved adaptively in the former lineage compared with the latter. Recent positive selection in L. v. graecus is emphasized by concomitant signals of incomplete sweeps in the Fay and Wu test and positive selection inferred by the gammamap method in three genes. Qualitatively similar differences in overall TLR selection landscapes have been reported between humans and other great apes and attributed to differences in demographic history (Quach et al. 2013). It is unlikely that our inference of selection has been severely distorted by demographic factors in newts because effective population sizes and demographic histories of both lineages were similar as estimated from an independent data set using ABC modeling. Therefore the strength of selection may signify differences between lineages in the overall importance of TLRs for an effective immune response. Because MHC class II variation is lower in L. v. graecus compared with L. montandoni (Nadachowska-Brzyska et al. 2012), it is tempting to speculate that the innate components of the L. v. graecus immune system may be relatively more important in fighting pathogen assault. Interestingly, a recent study reported drastic differences between species of the genus Lissotriton in susceptibility to an emerging urodele fungal pathogen Batrachochytrium salamandrivorans (Martel et al. 2014) which may suggest substantial interspecific immunogenetic differences.
The strongest and most consistent signatures of positive selection were observed in TLR05L of L. montandoni. This gene exhibits an excess of fixed replacement substitutions, an excess of divergence, and two codons with fixed substitutions identified by gammamap as targets of positive selection. Although direct evidence is lacking, it is likely that TLR05L modulates, together with TLR05, a flagellin-mediated immune response (see above). TLR05 is a common target of positive selection (Wlasiuk et al. 2009) that may be driven by coevolution with bacteria which, in some bacterial species, leads to variation in flagellin sequences resulting in the evasion of recognition by hosts (Andersen-Nissen et al. 2005). Thus positive selection in L. montandoni TLR05L may reflect adaptation to changing pathogen pressure. In this context it would be interesting to investigate TLR05 variation and test for recent selection in this gene which we were unable to do because of the presence of closely related paralogs.
The MK test and gammamap detected signatures of positive selection acting on TLR09B in L. v. graecus. TLR09 senses mainly unmethylated CpG-rich bacterial DNA and DNA:RNA hybrids (Bauer et al. 2001; Yeh et al. 2013; Rigby et al. 2014). Although TLR09 is generally strongly constrained in mammals, a few codons under positive selection have been reported (Wlasiuk and Nachman 2010; Areal et al. 2011) and adaptive evolution of this gene may be more frequent in fish (Chen et al. 2008; Zhu et al. 2013). The importance of TLR09 in fish antibacterial immunity is suggested by two observations. First, in zebrafish two distantly related proteins, TLR09 and TLR21, mediate CpG oligonucleotide activity, but have different recognition profiles (Yeh et al. 2013). Second, the only species in which TLR09 duplication has been reported so far is cod, which lacks MHC class II signaling but has five TLR09 paralogs; expansion of the TLR09 family may reflect its importance for antibacterial immunity in this species (Star et al. 2011). In contrast to birds and mammals with either TLR09 or TLR21, amphibians have both receptors, thus the role of these receptors in antibacterial response may be similar to that of fish. Because CpG DNA from various bacteria differ in their potential to activate TLR09 (Bauer et al. 2001), coevolution between host TLR and bacterial pathogens appears likely. The two urodele TLR09 paralogs have been maintained by selection for at least tens of millions of years, so possibly these genes specialized in recognition of various bacterial DNA PAMPs. Contrary to the situation observed in L. v. graecus, relaxation of selection and some functional redundancy of L. montandoni TLR09B is suggested by the relatively high frequency (∼15%) of a nonsense (premature stop codon) mutation. Patterns consistent with recent positive selection were also detected in TLR21 of L. v. graecus: an excess of divergence, low variation, and six replacement substitutions fixed by selection as inferred by gammamap. Again, relaxation of selection in L. montandoni is more likely: TLR21 is the most variable of all TLR genes and nonsynonymous polymorphisms are still segregating in all codons identified as positively selected by gammamap. Hence, L. v. graecus and L. montandoni likely differ in the response to bacterial DNA mediated by TLR09 and TLR21 and such differences may reflect variable selection pressure exerted by pathogens on TLR genes in closely related evolutionary lineages.
Conclusions
In this study, we characterized urodele TLR genes which encode a crucial component of the innate immune system and provide a link to adaptive immunity. The TLR repertoires of two major amphibian groups, urodeles and anurans, are broadly similar and intermediate between those of fish and amniotes. Gene duplications feature prominently in the evolution of urodele TLR. Both old duplications, apparently maintained by long-term selection and very recent duplications are present. TLR genes examined for polymorphism and divergence in Lissotriton newts exhibit a comparable degree of conservation and evolutionary constraint. However, the rate and targets of adaptive evolution differ substantially between two recently diverged Lissotriton lineages. These differences were not an artifact of nonequilibrium demography because demographic effects were accounted for through ABC modeling using an independent data set of 62 loci. Short-term selective pressures thus vary both between closely related lineages and among TLR genes, and may indicate differences in the relative roles of innate and acquired immunity. TLR variation may play an important role in the response to changing pathogen pressure, and these genes are a promising target for studies aiming to link genetic variation to pathogen susceptibility in amphibians, a vertebrate group severely threatened by emerging infectious diseases.
Supplementary Material
Supplementary material, tables S1–S4, and figures S1–S5 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank D. Cogalniceanu, S-D. Covaciu-Marcov, and M. Liana for help in sampling and K. Nadachowska-Brzyska for help in sampling and with ABC modeling. This work was supported by the Polish National Science Center [UMO-2012/04/A/NZ8/00662 to W.B.] and by Jagiellonian University [DS/WBiNoZ/INoS/762/13]. P.Z. was supported by EU resources under the European Social Fund: Creating English interdisciplinary graduate program in the area of ecology and enhancing the teaching potential of the academic staff at the Institute of Environmental Sciences of the Jagiellonian University. The study was performed with the financial support of the Foundation for Polish Science.
Literature Cited
- Abdullayev I, Kirkham M, Björklund ÅK, Simon A, Sandberg R. A reference transcriptome and inferred proteome for the salamander Notophthalmus viridescens. Exp Cell Res. 2013;319:1187–1197. doi: 10.1016/j.yexcr.2013.02.013. [DOI] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Alcaide M, Edwards SV. Molecular evolution of the toll-like receptor multigene family in birds. Mol Biol Evol. 2011;28:1703–1715. doi: 10.1093/molbev/msq351. [DOI] [PubMed] [Google Scholar]
- Andersen-Nissen E, et al. Evasion of Toll-like receptor 5 by flagellated bacteria. Proc Natl Acad Sci U S A. 2005;102:9247–9252. doi: 10.1073/pnas.0502040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Areal H, Abrantes J, Esteves PJ. Signatures of positive selection in Toll-like receptor (TLR) genes in mammals. BMC Evol Biol. 2011;11:368. doi: 10.1186/1471-2148-11-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babik W, et al. Phylogeography of two European newt species—discordance between mtDNA and morphology. Mol Ecol. 2005;14:2475–2491. doi: 10.1111/j.1365-294X.2005.02605.x. [DOI] [PubMed] [Google Scholar]
- Barreiro LB, et al. Evolutionary dynamics of human toll-like receptors and their different contributions to host defense. PLoS Genet. 2009;5:e1000562. doi: 10.1371/journal.pgen.1000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer S, et al. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci U S A. 2001;98:9237–9242. doi: 10.1073/pnas.161293498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boller T, He SY. Innate immunity in plants: an arms race between pattern recognition receptors in plants and effectors in microbial pathogens. Science. 2009;324:742–743. doi: 10.1126/science.1171647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JSC, Wang TY, Tzeng TD, Wang CY, Wang D. Evidence for positive selection in the TLR9 gene of teleosts. Fish Shellfish Immunol. 2008;24:234–242. doi: 10.1016/j.fsi.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Cheng TL, Rovito SM, Wake DB, Vredenburg VT. Coincident mass extirpation of neotropical amphibians with the emergence of the infectious fungal pathogen Batrachochytrium dendrobatidis. Proc Natl Acad Sci U S A. 2011;108:9502–9507. doi: 10.1073/pnas.1105538108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27:1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing T, Lloyd AT, O'Farrelly C, Bradley DG. The differential evolutionary dynamics of avian cytokine and TLR gene classes. J Immunol. 2010;184:6993–7000. doi: 10.4049/jimmunol.0903092. [DOI] [PubMed] [Google Scholar]
- Eilertson KE, Booth JG, Bustamante CD. SnIPRE: selection inference using a poisson random effects model. PLoS Comput Biol. 2012;8:e1002806. doi: 10.1371/journal.pcbi.1002806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Admetlla A, et al. Balancing selection is the main force shaping the evolution of innate immunity genes. J Immunol. 2008;181:1315–1322. doi: 10.4049/jimmunol.181.2.1315. [DOI] [PubMed] [Google Scholar]
- Fisher MC, et al. Emerging fungal threats to animal, plant and ecosystem health. Nature. 2012;484:186–194. doi: 10.1038/nature10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueber CE, Wallis GP, Jamieson IG. Episodic positive selection in the evolution of avian toll-like receptor innate immunity genes. PLoS One. 2014;9:e89632. doi: 10.1371/journal.pone.0089632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueber CE, Wallis GP, King TM, Jamieson IG. Variation at innate immunity Toll-like receptor genes in a bottlenecked population of a New Zealand robin. PLoS One. 2012;7:e45011. doi: 10.1371/journal.pone.0045011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JD, Vojtech LN, Laing KJ. Sensing disease and danger: a survey of vertebrate PRRs and their origins. Dev Comp Immunol. 2011;35:886–897. doi: 10.1016/j.dci.2011.01.008. [DOI] [PubMed] [Google Scholar]
- Huang Y, et al. Molecular evolution of the vertebrate TLR1 gene family—a complex history of gene duplication, gene conversion, positive selection and co-evolution. BMC Evol Biol. 2011;11:149. doi: 10.1186/1471-2148-11-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Ingvarsson PK. Population subdivision and the Hudson-Kreitman-Aguade test: testing for deviations from the neutral model in organelle genomes. Genet Res. 2004;83:31–39. doi: 10.1017/s0016672303006529. [DOI] [PubMed] [Google Scholar]
- Ishii A, Kawasaki M, Matsumoto M, Tochinai S, Seya T. Phylogenetic and expression analysis of amphibian Xenopus Toll-like receptors. Immunogenetics. 2007;59:281–293. doi: 10.1007/s00251-007-0193-y. [DOI] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanwal Z, Wiegertjes GF, Veneman WJ, Meijer AH, Spaink HP. Comparative studies of Toll-like receptor signalling using zebrafish. Dev Comp Immunol. 2014;46:35–52. doi: 10.1016/j.dci.2014.02.003. [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma KI, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Keestra AM, de Zoete MR, Bouwman LI, Vaezirad MM, Van Putten JPM. Unique features of chicken Toll-like receptors. Dev Comp Immunol. 2013;41:316–323. doi: 10.1016/j.dci.2013.04.009. [DOI] [PubMed] [Google Scholar]
- Koblansky AA, et al. Recognition of profilin by Toll-like receptor 12 is critical for host resistance to Toxoplasma gondii. Immunity. 2013;38:119–130. doi: 10.1016/j.immuni.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Doerks T, Bork P. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 2012;40:D302–D305. doi: 10.1093/nar/gkr931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Luther K, Ebel F. Toll-like receptors: recent advances, open questions and implications for aspergillosis control. Med Mycol. 2006;44:219–227. doi: 10.1080/13693780600892972. [DOI] [PubMed] [Google Scholar]
- Marsh DM, Trenham PC. Metapopulation dynamics and amphibian conservation. Cons Biol. 2001;15:40–49. [Google Scholar]
- Martel A, et al. Recent introduction of a chytrid fungus endangers Western Palearctic salamanders. Science. 2014;346:630–631. doi: 10.1126/science.1258268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo A, et al. Teleost TLR22 recognizes RNA duplex to induce IFN and protect cells from birnaviruses. J Immunol. 2008;181:3474–3485. doi: 10.4049/jimmunol.181.5.3474. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- Mikami T, Miyashita H, Takatsuka S, Kuroki Y, Matsushima N. Molecular evolution of vertebrate Toll-like receptors: evolutionary rate difference between their leucine-rich repeats and their TIR domains. Gene. 2012;503:235–243. doi: 10.1016/j.gene.2012.04.007. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Ganguli D, Majumder PP. Global footprints of purifying selection on toll-like receptor genes primarily associated with response to bacterial infections in humans. Genome Biol Evol. 2014;6:551–558. doi: 10.1093/gbe/evu032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Sarkar-Roy N, Wagener DK, Majumder PP. Signatures of natural selection are not uniform across genes of innate immune system, but purifying selection is the dominant signature. Proc Natl Acad Sci U S A. 2009;106:7073–7078. doi: 10.1073/pnas.0811357106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz I, Sepulcre MP, Meseguer J, Mulero V. Molecular cloning, phylogenetic analysis and functional characterization of soluble toll-like receptor 5 in gilthead seabream, Sparus aurata. Fish Shellfish Immunol. 2013;35:36–45. doi: 10.1016/j.fsi.2013.03.374. [DOI] [PubMed] [Google Scholar]
- Nadachowska K, Babik W. Divergence in the face of gene flow: the case of two newts (Amphibia: Salamandridae) Mol Biol Evol. 2009;26:829–841. doi: 10.1093/molbev/msp004. [DOI] [PubMed] [Google Scholar]
- Nadachowska-Brzyska K, Zielinski P, Radwan J, Babik W. Interspecific hybridization increases MHC class II diversity in two sister species of newts. Mol Ecol. 2012;21:887–906. doi: 10.1111/j.1365-294X.2011.05347.x. [DOI] [PubMed] [Google Scholar]
- Netea MG, Wijmenga C, O'Neill LAJ. Genetic variation in Toll-like receptors and disease susceptibility. Nat Immunol. 2012;13:535–542. doi: 10.1038/ni.2284. [DOI] [PubMed] [Google Scholar]
- Nikolaeva S, et al. Frog urinary bladder epithelial cells express TLR4 and respond to bacterial LPS by increase of iNOS expression and L-arginine uptake. Am J Physiol Regul Integr Comp Physiol. 2012;303:R1042–R1052. doi: 10.1152/ajpregu.00045.2012. [DOI] [PubMed] [Google Scholar]
- Oldenburg M, et al. TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance—forming modification. Science. 2012;337:1111–1115. doi: 10.1126/science.1220363. [DOI] [PubMed] [Google Scholar]
- Oshiumi H, Matsuo A, Matsumoto M, Seya T. Pan-vertebrate toll-like receptors during evolution. Curr Genomics. 2008;9:488–493. doi: 10.2174/138920208786241234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabijan M, et al. Forthcoming. The dissection of a Pleistocene refugium: phylogeography of the smooth newt, Lissotriton vulgaris, in the Balkans. J Biogeogr. Advance Access published November 27, 2014, doi: 10.1111/jbi.12449. [Google Scholar]
- Palermo S, et al. Toll-like receptor 4 genetic diversity among pig populations. Anim Genet. 2009;40:289–299. doi: 10.1111/j.1365-2052.2008.01833.x. [DOI] [PubMed] [Google Scholar]
- Palti Y. Toll-like receptors in bony fish: from genomics to function. Dev Comp Immunol. 2011;35:1263–1272. doi: 10.1016/j.dci.2011.03.006. [DOI] [PubMed] [Google Scholar]
- Piertney SB, Oliver MK. The evolutionary ecology of the major histocompatibility complex. Heredity. 2006;96:7–21. doi: 10.1038/sj.hdy.6800724. [DOI] [PubMed] [Google Scholar]
- Pietretti D, et al. Identification and functional characterization of nonmammalian Toll-like receptor 20. Immunogenetics. 2014;66:123–141. doi: 10.1007/s00251-013-0751-4. [DOI] [PubMed] [Google Scholar]
- Pietretti D, Wiegertjes GF. Ligand specificities of Toll-like receptors in fish: indications from infection studies. Dev Comp Immunol. 2014;43:205–222. doi: 10.1016/j.dci.2013.08.010. [DOI] [PubMed] [Google Scholar]
- Quach H, et al. Different selective pressures shape the evolution of toll-like receptors in human and African great ape populations. Hum Mol Genet. 2013;22:4829–4840. doi: 10.1093/hmg/ddt335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiniou SMA, Boudinot P, Bengtén E. Comprehensive survey and genomic characterization of Toll-like receptors (TLRs) in channel catfish, Ictalurus punctatus: Identification of novel fish TLRs. Immunogenetics. 2013;65:511–530. doi: 10.1007/s00251-013-0694-9. [DOI] [PubMed] [Google Scholar]
- Quintana-Murci L, Clark AG. Population genetic tools for dissecting innate immunity in humans. Nat Rev Immunol. 2013;13:280–293. doi: 10.1038/nri3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauta PR, Samanta M, Dash HR, Nayak B, Das S. Toll-like receptors (TLRs) in aquatic animals: signaling pathways, expressions and immune responses. Immunol Lett. 2014;158:14–24. doi: 10.1016/j.imlet.2013.11.013. [DOI] [PubMed] [Google Scholar]
- Richmond JQ, Savage AE, Zamudio KR, Rosenblum EB. Toward immunogenetic studies of amphibian chytridiomycosis: linking innate and acquired immunity. Bioscience. 2009;59:311–320. [Google Scholar]
- Rigby RE, et al. RNA:DNA hybrids are a novel molecular pattern sensed by TLR9. EMBO J. 2014;33:542–558. doi: 10.1002/embj.201386117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach JC, et al. The evolution of vertebrate Toll-like receptors. Proc Natl Acad Sci U S A. 2005;102:9577–9582. doi: 10.1073/pnas.0502272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder A, et al. Toll-like receptors as key mediators in innate antifungal immunity. Med Mycol. 2004;42:485–498. doi: 10.1080/13693780400011112. [DOI] [PubMed] [Google Scholar]
- Roelants K, et al. Global patterns of diversification in the history of modern amphibians. Proc Natl Acad Sci U S A. 2007;104:887–892. doi: 10.1073/pnas.0608378104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Sackton TB, et al. Dynamic evolution of the innate immune system in Drosophila. Nat Genet. 2007;39:1461–1468. doi: 10.1038/ng.2007.60. [DOI] [PubMed] [Google Scholar]
- Savage AE, Zamudio KR. MHC genotypes associate with resistance to a frog-killing fungus. Proc Natl Acad Sci U S A. 2011;108:16705–16710. doi: 10.1073/pnas.1106893108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Green DM. Dispersal and the metapopulation paradigm in amphibian ecology and conservation: are all amphibian populations metapopulations? Ecography. 2005;28:110–128. [Google Scholar]
- Städler T, Haubold B, Merino C, Stephan W, Pfaffelhuber P. The impact of sampling schemes on the site frequency spectrum in nonequilibrium subdivided populations. Genetics. 2009;182:205–216. doi: 10.1534/genetics.108.094904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Star B, et al. The genome sequence of Atlantic cod reveals a unique immune system. Nature. 2011;477:207–210. doi: 10.1038/nature10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Donnelly P. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73:1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram AY, Kiron V, Dopazo J, Fernandes JM. Diversification of the expanded teleost-specific toll-like receptor family in Atlantic cod, Gadus morhua. BMC Evol Biol. 2012;12:256. doi: 10.1186/1471-2148-12-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- Tamura K, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temperley ND, Berlin S, Paton IR, Griffin DK, Burt DW. Evolution of the chicken Toll-like receptor gene family: a story of gene gain and gene loss. BMC Genomics. 2008;9:62. doi: 10.1186/1471-2164-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffin P, Moeller DA. Molecular evolution of plant immune system genes. Trends Genet. 2006;22:662–670. doi: 10.1016/j.tig.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Tschirren B, Andersson M, Scherman K, Westerdahl H, Råberg L. Contrasting patterns of diversity and population differentiation at the innate immunity gene toll-like receptor 2 (TLR2) in two sympatric rodent species. Evolution. 2012;66:720–731. doi: 10.1111/j.1558-5646.2011.01473.x. [DOI] [PubMed] [Google Scholar]
- Tschirren B, et al. Polymorphisms at the innate immune receptor TLR2 are associated with Borrelia infection in a wild rodent population. Proc R Soc B Biol Sci. 2013;280:20130364. doi: 10.1098/rspb.2013.0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschirren B, Råberg L, Westerdahl H. Signatures of selection acting on the innate immunity gene Toll-like receptor 2 (TLR2) during the evolutionary history of rodents. J Evol Biol. 2011;24:1232–1240. doi: 10.1111/j.1420-9101.2011.02254.x. [DOI] [PubMed] [Google Scholar]
- Tsujita T, et al. Sensing bacterial flagellin by membrane and soluble orthologs of toll-like receptor 5 in rainbow trout (Onchorhynchus mikiss) J Biol Chem. 2004;279:48588–48597. doi: 10.1074/jbc.M407634200. [DOI] [PubMed] [Google Scholar]
- Wakeley J. Nonequilibrium migration in human history. Genetics. 1999;153:1863–1871. doi: 10.1093/genetics/153.4.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley J. Metapopulation models for historical inference. Mol Ecol. 2004;13:865–875. doi: 10.1111/j.1365-294x.2004.02086.x. [DOI] [PubMed] [Google Scholar]
- Wakeley J, Aliacar N. Gene genealogies in a metapopulation. Genetics. 2001;159:893–905. doi: 10.1093/genetics/159.2.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werling D, Jann OC, Offord V, Glass EJ, Coffey TJ. Variation matters: TLR structure and species-specific pathogen recognition. Trends Immunol. 2009;30:124–130. doi: 10.1016/j.it.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Wilson DJ, Hernandez RD, Andolfatto P, Przeworski M. A population genetics-phylogenetics approach to inferring natural selection in coding sequences. PLoS Genet. 2011;7:e1002395. doi: 10.1371/journal.pgen.1002395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlasiuk G, Khan S, Switzer WM, Nachman MW. A history of recurrent positive selection at the toll-like receptor 5 in primates. Mol Biol Evol. 2009;26:937–949. doi: 10.1093/molbev/msp018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlasiuk G, Nachman MW. Adaptation and constraint at toll-like receptors in primates. Mol Biol Evol. 2010;27:2172–2186. doi: 10.1093/molbev/msq104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhams DC, et al. Resistance to chytridiomycosis varies among amphibian species and is correlated with skin peptide defenses. Anim Cons. 2007;10:409–417. [Google Scholar]
- Wright SI, Charlesworth B. The HKA test revisited: a maximum-likelihood-ratio test of the standard neutral model. Genetics. 2004;168:1071–1076. doi: 10.1534/genetics.104.026500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZH. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yarovinsky F. Innate immunity to Toxoplasma gondii infection. Nat Rev Immunol. 2014;14:109–121. doi: 10.1038/nri3598. [DOI] [PubMed] [Google Scholar]
- Yeh DW, et al. Toll-like receptor 9 and 21 have different ligand recognition profiles and cooperatively mediate activity of CpG-oligodeoxynucleotides in zebrafish. Proc Natl Acad Sci U S A. 2013;110:20711–20716. doi: 10.1073/pnas.1305273110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F, et al. Comprehensive transcriptome profiling and functional analysis of the frog (Bombina maxima) immune system. DNA Res. 2014;21:1–13. doi: 10.1093/dnares/dst035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Sun Y, Wang R, Xu T. Evolutionary analysis of TLR9 genes reveals the positive selection of extant teleosts in Perciformes. Fish Shellfish Immunol. 2013;35:448–457. doi: 10.1016/j.fsi.2013.04.043. [DOI] [PubMed] [Google Scholar]
- Zieliński P, et al. No evidence for nuclear introgression despite complete mtDNA replacement in the Carpathian newt (Lissotriton montandoni) Mol Ecol. 2013;22:1884–1903. doi: 10.1111/mec.12225. [DOI] [PubMed] [Google Scholar]
- Zieliński P, Stuglik MT, Dudek K, Konczal M, Babik W. Development, validation and high-throughput analysis of sequence markers in nonmodel species. Mol Ecol Res. 2014;14:352–360. doi: 10.1111/1755-0998.12171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.