

Abstract
Mammalian and avian genomes are characterized by a substantial spatial heterogeneity of GC-content, which is often interpreted as reflecting the effect of local GC-biased gene conversion (gBGC), a meiotic repair bias that favors G and C over A and T alleles in high-recombining genomic regions. Surprisingly, the first fully sequenced nonavian sauropsid (i.e., reptile), the green anole Anolis carolinensis, revealed a highly homogeneous genomic GC-content landscape, suggesting the possibility that gBGC might not be at work in this lineage. Here, we analyze GC-content evolution at third-codon positions (GC3) in 44 vertebrates species, including eight newly sequenced transcriptomes, with a specific focus on nonavian sauropsids. We report that reptiles, including the green anole, have a genome-wide distribution of GC3 similar to that of mammals and birds, and we infer a strong GC3-heterogeneity to be already present in the tetrapod ancestor. We further show that the dynamic of coding sequence GC-content is largely governed by karyotypic features in vertebrates, notably in the green anole, in agreement with the gBGC hypothesis. The discrepancy between third-codon positions and noncoding DNA regarding GC-content dynamics in the green anole could not be explained by the activity of transposable elements or selection on codon usage. This analysis highlights the unique value of third-codon positions as an insertion/deletion-free marker of nucleotide substitution biases that ultimately affect the evolution of proteins.
Keywords: third-codon positions, phylogeny, karyotype
Introduction
It is a well-known fact that the base composition of DNA sequences varies greatly between and within genomes but the reasons and evolutionary significance of these variations are still in large part mysterious. In mammalian and avian genomes, a significant heterogeneity of local GC-content has been reported at the approximately 100-kb scale (Bernardi 2000; Lander et al. 2001). After decades of controversy (Bernardi 1993; Fryxell and Zuckerkandl 2000; Eyre-Walker and Hurst 2001; Belle et al. 2002; Chojnowski et al. 2007), it is now widely accepted that this intragenomic variation is caused in the first place by GC-biased gene conversion (gBGC), a recombination-associated segregation distortion that favors GC over AT alleles and results in an increased GC-content in high-recombining regions (Eyre-Walker 1993; Galtier et al. 2001; Marais 2003; Montoya-Burgos et al. 2003; Meunier and Duret 2004; Webster et al. 2005; Dreszer et al. 2007; Duret and Arndt 2008; Duret and Galtier 2009; Munch et al. 2014).
The discovery of gBGC had important consequences regarding population and functional genomics, especially in mammals, in which gBGC has been shown to 1) mimic the effects of adaptive evolution, thus confounding positive selection event inferences (Webster and Smith 2004; Galtier and Duret 2007; Berglund et al. 2009; Ratnakumar et al. 2010); and 2) generate a substantial load of deleterious mutations, thus affecting the fitness of individuals and populations (Galtier et al. 2009; Glémin 2010; Necşulea et al. 2011; Capra et al. 2013). The gBGC hypothesis has also the merit of reconciling compositional and karyotypic evolutionary patterns. GC-content is negatively correlated with chromosome length in several vertebrate species (Lander et al. 2001; Kuraku et al. 2006; Goodstadt et al. 2007; Matsubara et al. 2012), which is expected under a gBGC scenario due to the higher per megabase recombination rate in small compared with long chromosomes resulting from the occurrence of at least one (and rarely more) event of cross-over per chromosome arm per meiosis (Lawrie et al. 1995; Li and Freudenberg 2009). In many respects, the focus in studies of GC-content variation in mammals and birds has therefore shifted to questions related to the origin, evolution, mechanism, and genomic impact of gBGC (e.g., Romiguier et al. 2010; Nabholz et al. 2011; Axelsson et al. 2012; Lartillot 2013a, 2013b; Mugal et al. 2013).
Interestingly, the genomes of teleost fishes and amphibians are much less heterogeneous than those of mammals and birds regarding base composition (Bernardi Gia. and Bernardi Gio. 1990; Costantini et al. 2009), despite the evidence for substantial within-genome variation in recombination rate in these groups (e.g., Kai et al. 2011; Lien et al. 2011; Ninwichian et al. 2012). This conspicuous difference in GC-content landscape between taxa casts some doubts about the generality of the gBGC mechanism across vertebrates. Rather, this observation suggests that gBGC appeared in an ancestral amniote and was inherited by both mammals and birds (Duret and Galtier 2009), or has evolved independently in each of the two groups of warm-blooded vertebrates. However, such a (relatively) recent origin of gBGC within vertebrates appears uneasy to reconcile with the numerous pieces of evidence supporting a wide prevalence of gBGC across many eukaryotic taxa (Beye et al. 2006; Glémin et al. 2006; Mancera et al. 2008; Escobar et al. 2011; Katzman et al. 2011; Kent et al. 2012; Pessia et al. 2012), which speaks in favor of an ancient origin of this molecular mechanism.
Reptiles stand as the key group to progress with the issue of gBGC and GC-content evolution in vertebrates. The paraphyletic reptiles form with birds the clade of Sauropsida, which is the sister group of mammals. Nonavian sauropsids include more than 7,500 extant species, including approximately 7,200 squamates (lizards and snakes), approximately 300 turtles, approximately 30 crocodilians, and the 2 tuataras, with a wide variety of morphological, ecological, and genomic traits (Shine 2005; Organ et al. 2008; Janes et al. 2010). Given their phylogenetic position, reptiles are crucial for our understanding of the evolution of genomic landscapes and the origin of gBGC in vertebrates.
Reptiles have long been neglected in genomic studies, so that the study of base composition in this group has first relied on just a limited number of protein-coding sequences (CDS). These early analyses suggested that the distribution of GC-content at third-codon positions of CDS (GC3) in crocodilians and turtles was quite similar to that of mammals and birds (Hughes et al. 1999; Chojnowski et al. 2007; Chojnowski and Braun 2008). Consistent with these reports, the genome of the western-painted turtle Chrysemys picta showed a substantial level of GC-content heterogeneity—although not as strong as in human or chicken (Shaffer et al. 2013). These results are consistent with the hypothesis of a unique origin of gBGC and GC-content heterogeneity in an ancestral amniote. In squamates, the evidence from coding sequence GC3 was scarcer (Fortes et al. 2007). Very surprisingly, the complete genome sequence of the green anole lizard Anolis carolinensis revealed a highly homogeneous genomic GC-content (Alföldi et al. 2011). GC-content heterogeneity in A. carolinensis was even weaker than in the amphibian Xenopus tropicalis, as demonstrated by the detailed study of Fujita et al. (2011). The lack of GC-rich regions in, especially, the microchromosomes of A. carolinensis led to the suggestion that gBGC was no longer at work in this lineage (Fujita et al. 2011). Consistently, the recently published python snake (Python molurus bivittatus) genome was also compositionally quite homogeneous, albeit more heterogeneous than the green anole (Castoe et al. 2013).
The above-reviewed literature on reptile GC-content variation is based on various kinds of data and methods, separately applied to distinct species. The important group of squamates, furthermore, has been insufficiently sampled so far. This calls for a synthetic analysis of the evolution of base composition in reptiles and vertebrates in a phylogenetic context. This is the very goal of this study, in which we focus on GC3 as a marker of the evolutionary dynamic of base composition. The third-codon positions of CDS have several merits: 1) Coding sequence data are available in a large panel of reptilian species, whereas complete reptilian genomes are still scarce; 2) third-codon positions are only moderately affected by natural selection; 3) they are essentially immune from insertions and deletions, which are strongly counterselected in CDS; 4) consequently, they can be aligned across even distantly related species. Third-codon positions thus offer a unique opportunity to analyze long-term patterns of nucleotide substitution at nearly neutral markers.
We built a data set of CDS data from 44 representative species of vertebrates including the newly sequenced transcriptomes of seven sauropsids (five reptiles and two birds) and one amphibian. We report that reptiles, and particularly the green anole, exhibit a level of GC3-heterogeneity comparable to that of mammals and birds, revealing a conflicting signal between third-codon positions and noncoding DNA. We also report a significant impact of chromosome and genome size on GC3 dynamics in vertebrates, including the green anole. Our results suggest that gBGC is of ancient origin in vertebrates and has impacted the neutral nucleotide substitution process in all vertebrate lineages, even though this is not always reflected by a strong GC-heterogeneity in the genomic landscapes of extant species.
Materials and Methods
Sample Collection, RNA Extraction, and Sequencing
Five reptilian, two avian, and one amphibian species were sampled for this study: The golden tegu Tupinambis teguixin, the green iguana Iguana iguana, the Boa constrictor, the Jesus Christ lizard Basiliscus plumifrons, the Nile crocodile Crocodylus niloticus, the great tit Parus major, the emperor penguin Aptenodytes forsteri, and the fire salamander Salamandra salamandra (supplementary table S1, Supplementary Material online). One individual from each species was collected in France from the Pierrelatte zoo (Nile crocodile), the Montpellier zoo (other reptiles), or in nature (great tit: Montpellier; emperor penguin: Terre Adélie, Antarctic; fire salamander: Banyuls). RNA was extracted from tail (fire salamander), pectoral muscle (emperor penguin), or blood (other species) samples using standard and modified protocols as described in Chiari and Galtier (2011) and Gayral et al. (2011). Nonnormalized cDNA libraries were prepared and sequenced on a Hiseq 2000 (Illumina, Inc.) to produce 100-bp reads. Reads were trimmed for low quality (phred quality score < 30) and minimum size (60 bp). All reads are available on the ncbi website (http://www.ncbi.nlm.nih.gov/, last accessed January 13, 2015) through the BioProject PRJNA268920.
Transcriptome Assembly and Coding Sequence Prediction
In 15 additional reptile species, RNA sequencing (RNA-seq) reads or contigs were obtained from the literature: Podarcis sp., Phrynops hilarii, Caretta caretta, Emys orbicularis, Chelonoidis nigra, Caiman crocodilus, Alligator mississipiensis (assembled from National Center for Biotechnology Information [NCBI] SRA SRX012365) from Chiari et al. 2012, Pelodiscus sinensis (NCBI SRA DRX001551), Sphenodon punctatus (Miller et al. 2012), Chamaeleo chamaeleon (Bar-Yaacov et al. 2013), Pogona vitticeps (Tzika et al. 2011), Elaphe guttata (Tzika et al. 2011), Thamnophis elegans (Schwartz et al. 2010), Ophiophagus hannah (NCBI SRA SRX365144), and A. carolinensis (NCBI SRA SRR391650). Four representatives mammals (Homo sapiens, Mus musculus, Monodelphis domestica: Perry et al. 2012, and Bos taurus: NCBI SRA SRX477519) as well as two birds (Gallus gallus: NCBI SRA SRX191158 and Anas platyrhynchos: NCBI SRA SRX255765). and one basal sarcopterygian (Protopterus annectens: Chiari et al. 2012) species were also retrieved from public databases.
De novo transcriptome assembly was performed with a combination of Abyss and Cap3 programs following the strategy B of Cahais et al. (2012). Open reading frames (ORFs) were predicted using the program getORF included in the EMBOSS package. ORFs shorter than 200 bp were discarded. This transcriptome-based data set includes 30 species of which 20 reptiles (supplementary table S2, Supplementary Material online).
Annotated CDS from Complete Genomes
In addition, we also built a data set of CDS annotated from complete genomes—the genome-based data set (supplementary table S3, Supplementary Material online). Complete sets of CDS were retrieved from Ensembl using the biomart tool for 21 vertebrate species of which seven sauropsids (two reptiles and five birds), six mammals, one amphibian and seven bony fish, eight of these species being also present in the transcriptomic data set (A. carolinensis, Pe. sinensis, G. gallus, Ana. platyrhynchos, H. sapiens, M. musculus, Bos taurus, and Mo. domestica). When several transcripts were obtained for a single gene, we retained the longest one. All identified mRNA from the complete genome of the western painted turtle Chr. picta (Shaffer et al. 2013) were also added to this data set.
Species name abbreviations are used in figures 1 and 3 (ALL, Alligator mississippiensis; ANA, Ana. platyrhynchos; ANO, A. carolinensis; APT, Aptenodytes patagonicus; BAS, B. plumifrons; BOA, Boa constrictor; BOS, Bos taurus; CAI, Cai. crocodilus; CAR, Car. caretta; CHA, Cha. chamaeleon; CHE, Che. nigra; CHR, Chr. picta; CRO, Cr. niloticus; DAN, Danio rerio; ELA, El. guttata; EMY, Em. orbicularis; FIC, Ficedula albicollis; GAD, Gadus morhua; GAL, G. gallus; GAS, Gasterosteus aculateus; HOM, H. sapiens; IGU, I. iguana; LAT, Latimeria chalumnae; MEL, Meleagris gallopavo; MON, Mo. domestica; MUS, M. musculus; MYO, Myotis lucifugus; OPH, O. hannah; ORN, Ornithorhynchus anatinus; ORY, Oryzias latipes; PAR, Parus caeruleus; PEL, Pe. sinensis; PHR, P. hilarii; POD, Podarcis sp; POG, Po. vitticeps; PRO, Pr. annectens; SAL, S. salamandra; SPH, Sp. punctatus; TAE, Taeniopygia guttata; TAK, Takifugu rubripes; TET, Tetraodon nigroviridis; THA, Th. elegans; TUP, T. teguixin; XEN, X. tropicalis).
Fig. 1.—

Distribution of GC3 across genes and species in vertebrates. (A) Relationship between across-genes GC3 average and standard deviation: Genome-based data set; each dot is for a species; blue, bony fish; purple, amphibians; red, mammals; light green, birds; dark green, nonavian reptiles; for species name abbreviations, see Materials and Methods. (B) Relationship between across-genes GC3 average and standard deviation: Transcriptome-based data set; same legend as above. (C) Relationship between GC3 average of the 10% poorest (bottom-most regression) and 10% richest (uppermost regression) genes and GC3 average. (D) Distribution of gene GC3 in green anole (dark green), chicken (light green), human (red), and clawed frog (purple): Genome-based data set.
Fig. 3.—

Influence of karyotype on GC3 in vertebrates. (A) Relationship between log(C-value) and mean GC3. (B) Relationship between log(chromosome size standard deviation) and GC3 standard deviation. Species from the genome-based data set are color-filled. Species from the transcriptome-based data set are represented in a colored ring and were not used in linear regression analysis. Color code: Same as figure 1.
Orthologous Genes and Alignments
A set of orthologous sequences was built with the OrthoMCL software (Li et al. 2003) on amino acid-translated sequences with default parameters. We run the program only using the 21 reptiles of this study to maximize the number of shared clusters for these species. We selected among all returned orthologous clusters the ones including at least 18 of the 21 reptilian species and no more than four ORFs per cluster for a particular species. When several ORFs were returned in a cluster for a single species, we retained the longest one. This procedure resulted in a total of 1,025 genes.
Orthologous genes from all other vertebrates taken from Ensembl (19 species) were added to the above defined clusters thanks to the Ensembl predictions of orthology with A. carolinensis and Pe. sinensis. Each cluster was then aligned with the MACSE program (Ranwez et al. 2011), which aligns based on amino acids but allows for frameshifts at the nucleotide level when this results in a significant alignment improvement. Finally, alignments were restricted to third-codon positions having less than 40% missing data, leading to a concatenated alignment of 500 kb for 40 species.
Phylogeny and Divergence Dates
The topology of the phylogenetic tree for our set of vertebrate species was adapted from reference phylogenies: Near et al. (2012) for actinopterygii (ray-finned fishes), Meredith et al. (2011) for mammals, McCormack et al. (2013) for birds, Man et al. (2011) for crocodilians, Guillon et al. (2012) for testudines, and Pyron et al. (2013) for squamata.
Divergence dates were retrieved from the TimeTree of Life database (http://www.timetree.org, last accessed December 18, 2014 ) that combines both paleontological and molecular dating estimates. When divergence dates were inconsistent with the topology (older nodes being assigned an earlier divergence), all the concerned nodes were placed at the most ancient date and when no date was available (Emys–Chrysemys and Anolis–Iguana ancestral nodes), we used the mean between the two neighboring nodes. The effect of phylogenetic dependence on correlations was tested through the method of phylogenetically independent contrasts (Felsenstein 1985) with the “ape” R package, using divergence dates as branch lengths.
Ancestral GC3 Estimation
Ancestral GC3 was estimated for all nodes of the tree separately for each of the 1,025 genes using the NHML program (Galtier and Gouy 1998), implemented in the bpp_ML programs (Dutheil and Boussau 2008). This method uses a nonhomogeneous and nonstationary Markov model of nucleotide evolution to estimate branch-specific GC-content in a maximum-likelihood framework. This program has been used and tested in a large variety of studies (Romiguier et al. 2010, 2013; Fujita et al. 2011).
C-Value and Karyotypes
We retrieved all available C-values from the Animal Genome Size database (Gregory et al. 2007) as a proxy for genome size. When several measures were available for a given species, we took their mean, and the mean of the genera if the species was not available. C-values were obtained for 20 of the 22 species of the genome-based data set.
Karyotypic information provided by Ensembl was used for the 11 species for which it was available and the standard deviation in chromosome length was calculated (excluding the mitochondrial genome). To improve the sampling, karyotypic heterogeneity was also obtained in five additional species by measuring chromosome size from karyotype pictures (A. mississipiensis: Valleley et al. 1994; Pe. sinensis: Sato and Ota 2001; El. guttata: Baker et al. 1971; X. tropicalis: Uno et al. 2013, and Pr. annectens: Omer and Abukashawa 2012). For comparative purpose, in each species the size of each chromosome was divided by the size of the longest one.
For the green anole and the chicken, which both exhibit a clear distinction in size between micro- and macrochromosomes, we also calculated the mean GC3 value of genes associated with each type of chromosomes using the chromosomal assignation of genes provided by Ensembl. Microchromosomes to which less than ten genes were assigned were not considered.
Results
Diversity of GC3 Patterns in Vertebrates
To evaluate in what extent nonavian sauropsids exhibit GC-heterogeneity in their CDS, we calculated the mean and standard deviation of GC3 across genes in 21 reptilian species and compared them with 23 other vertebrate species. For comparability purposes, two distinct data sets were built. The first one, thereafter referred as the genome-based data set, included species for which the (almost) entire set of CDS was available thanks to annotations from fully sequenced genomes; the second one, called the transcriptome-based data set, included species whose CDS were assembled from RNA sequencing and might contain only a fraction of the total gene set and/or partial CDS. The median of analyzed genes among species was 15,289 in the genome-based data set and 9,707 in the transcriptome-based data set. Eight species for which both full genomes and RNA-seq data are available belong to the two data sets.
A similar picture was observed for the two data sets (fig. 1A and B). Consistent with the literature, birds and mammals were found to be more GC3-heteregeneous than both amphibian and bony fish. Among mammals, the mouse and the opossum were the most GC3-homogeneous, consistent with previous reports. The three reptile species of the genome-based data set (green anole, painted turtle, and Chinese softshell turtle) harbored a level of GC3-heterogeneity similar to that of birds and mammals, and clearly above the heterogeneity of nonamniote vertebrates (fig. 1A). In the reptile-rich transcriptomic-gene data set, reptile species again occupied the same range of GC3 and GC3-heterogeneity as birds and mammals (fig. 1B) with no strong effect of taxonomy—species from major clades (squamata, crocodilia, or testudines) did not gather into clusters. Finally, reptiles exhibited a strong correlation between mean and standard deviation of GC3 (r2 = 0.64, P < 1 × 10−4; fig. 1B), suggesting that the forces acting on GC richness simultaneously affect the GC-heterogeneity in this clade—a pattern that had already been observed within mammals (Romiguier et al. 2010).
Under the gBGC hypothesis, a relation between mean GC3 and GC3 standard deviation is expected if genes are differentially affected by gBGC depending on their local recombination rate. Using the transcriptome-based data set, we indeed observed that the GC-poor and GC-rich fractions of genes, here represented by the 10% GC3-poorest and richest genes, behaved very differently. The former varied only slightly among species, whereas the latter was highly variable and strongly correlated to the global mean GC3 (r2 = 0.80, P < 1 × 10−12; fig. 1C). This demonstrates that the process of GC3-increase (or decrease) in amniotes does not apply uniformly across the genome but rather concerns in the first place a subset of the genes or genomic regions. No significant relation between mean GC3 and GC3 standard deviation was detected in nonamniote vertebrates, with for example actinopterygians covering a wide range of mean GC3 but exhibiting a very narrow range of GC3-heterogeneity (fig. 1A).
Within reptiles, the green anole A. carolinensis revealed a striking homogeneity in its GC-content at the genomic scale (Fujita et al. 2011). Surprisingly, no such homogeneity was observed here at the coding sequence level: The green anole even appears to be among the most GC3-heterogeneous reptiles in the transcriptome-based data set (fig. 1B). This GC3-heterogeneity in the green anole is confirmed by a plot of its GC3-content gene distribution which much more resembles the one of the chicken than the one of the clawed frog X. tropicalis (fig. 1D; supplementary fig. S3, Supplementary Material online, for individual species GC3 distribution). Patterns of GC3 variation in reptiles therefore do not differ in any obvious way from those of birds and mammals, in which gBGC is documented.
Estimation of Ancestral GC3-Content
The observation that nonavian sauropsids do not express a distinctive behavior in terms of GC3 patterns suggests that GC3-heterogeneity could be an ancestral feature of amniotes. To clarify the origin and the dynamic of GC3-heterogeneity in amniotes, we extracted a set of 1,025 orthologous genes shared by our 21 reptilian species and an additional 19 vertebrate species and reconstructed the evolutionary dynamic of GC3 through a phylogenetic approach using a nonhomogeneous model of sequence evolution (fig. 2; supplementary figs. S1 and S2, Supplementary Material online, for mean and equilibrium GC3-content reconstruction).
Fig. 2.—

Reconstruction of GC3-heterogeneity in vertebrates. An alignment of 1,025 orthologous genes was used. GC3-heterogeneity is shown along branches with a color code from green (low) to yellow and red (high). The green anole (Anolis carolinensis) position is indicated by an asterisk. The tree is a chronogram with branch lengths in million years.
Consistent with the above-stated hypothesis, the ancestral amniote was predicted to have been strongly GC3-heterogeneous: The estimated standard deviation of GC3 in this ancestor is similar to that of human. According to our reconstruction, this ancestral heterogeneity has been preserved, or even reinforced, in some lineages (e.g., passerine birds, sphenodon, platypus, nonrodent placental mammals), but was eroded to various extents in other groups, and particularly in squamates, whose ancestor is predicted to be GC3-homogeneous. The green anole A. carolinensis was not more GC3-eroded than its squamate relatives, and even showed a slight trend toward increased GC3-heterogeneity in its terminal branch. Interestingly, the ancestral tetrapode was also predicted by our analysis to have been highly GC3-heterogeneous, which would imply an even earlier emergence of this feature than previously thought, and a subsequent erosion in amphibians.
Influence of the Karyotype on Vertebrate GC3 Patterns
In search for a mechanism responsible for the evolution of GC3-heterogeneity in amniotes, we investigated a potential effect of karyotype, based on the prediction that shorter chromosomes should exhibit a higher GC3-content under the gBGC hypothesis (Goodstadt et al. 2007; Matsubara et al. 2012). We used the C-value of a genome as a proxy for its mean chromosome size, thus assuming that large genomes tend to contain large chromosomes and should therefore display a lower average GC3. Despite this simplification, we observed a strong and significantly positive correlation between C-value and mean GC3 across vertebrates (r2 = 0.50, P < 1 × 10−3) (fig. 3A). This relationship was robust to the control for phylogenetic dependence by the method of independent contrasts (Felsenstein 1985) (r2 = 0.41, P = 0.003). The C-value/GC3 relationship was particularly strong when the analysis was restricted to bony fish (Pr. annectens included, r2 = 0.80, P = 0.002). This probably results from the fact that the fish species we analyzed have a similar number (around n = 22) of chromosomes that are very homogeneous in size within-species, making the C-value a very good proxy of average chromosome length. This result suggests that gBGC has been at work in all major vertebrate clades including nontetrapods, for which the wide range of mean GC3 could be explained by the large among-species differences in genome size.
Using only species for which a full karyotype was available, we investigated the impact of variance in chromosome length on GC3-heterogeneity. We observed that the more heterogeneous karyotypes exhibit a higher heterogeneity in GC3-content (r2 = 0.58, P = 0.002; fig. 3B), a relationship that was robust to phylogenetic control (r2 = 0.36, P = 0.03). Again, this observation reinforces the hypothesis of gBGC at work in all vertebrate lineages and provides a plausible explanation to the absence of GC3-heterogeneity in fish, which seems to be a consequence of their peculiar karyotypic structure.
Impact of Chromosome Size on GC-Content in the Green Anole
Finally, we investigated in more details the particular case of the green anole for which coding and genomic regions have returned a conflicting signal regarding GC-content. We observed that in the green anole GC3 decreases with chromosome length in a way essentially similar to the chicken pattern (fig. 4), and that microchromosomes (size < 10 Mb, mean GC3 = 59.9%) are significantly higher in GC3 than macrochromosomes (size > 80 Mb, mean GC3 = 47.2%, Student test: P < 2.2e-16). This contrasts with the pattern observed for genomic GC-content, which shows no significant difference between chromosomes in A. carolinensis, but a substantial one in chicken (Fujita et al. 2011; fig. 4).
Fig. 4.—
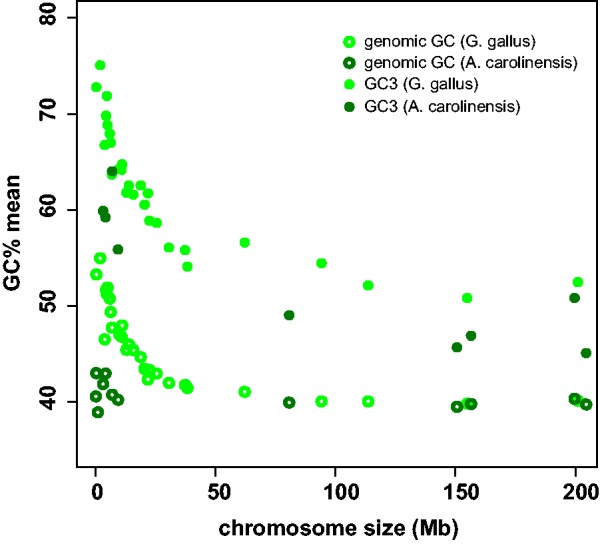
Relation between chromosome size, genomic GC-content, and GC3 in the green anole and the chicken.
In order to investigate the recent dynamic of GC3 evolution in A. carolinensis we considered the GC3* statistic, which corresponds to the equilibrium GC3 value toward which the species has been evolving. GC3* was estimated through the previously described phylogenetic analysis of 1,025 orthologous CDS using a nonhomogeneous model of sequence evolution (fig. 2). Two categories of genes were made depending on their location on either micro- or macrochromosomes. The average GC3 in microchromosomes was predicted to be increasing (mean GC3 for the 1,025 genes: 49.9%, GC3*: 58.4%), whereas macrochromosomes appeared to be at equilibrium (GC3: 44.6%, GC3*: 45.0%), again consistent with the hypothesis of active gBGC in the green anole.
Discussion
Evolution of GC3 in Amniotes and Vertebrates
Analyzing thousands of CDS from 44 species of vertebrates, we showed that the across-genes mean and variance of GC3 in nonavian sauropsids, although they vary between species, are essentially similar to those observed in mammals and birds. The GC3 variance in particular is generally higher in reptiles than in the fish and amphibian species we analyzed. This is true of squamates, turtles, crocodilians, and tuataras—the four main lineages of reptiles. Interestingly, the green anole is no exception: The distribution of GC3 in A. carolinensis is similar to the one observed in the chicken G. gallus. This is a surprising result given the very distinctive genomic patterns that were reported in these two species as far as genomic sequences are concerned: Chicken is a typical GC-heterogeneous species (Hillier et al. 2004), whereas the green anole is a highly GC-homogeneous one (Alföldi et al. 2011; Fujita et al. 2011). Figure 1 also demonstrates a positive relationship between the mean and variance of GC3 and a much stronger contribution of high-GC3 than low-GC3 genes to the between-species variation. This indicates that GC3-enrichment in GC3-rich genomes does not occur uniformly, but only affects a fraction of the genes. This is consistent with the idea of a spatially heterogeneous GC-increasing process—such as gBGC. A similar relationship has been previously reported in seed plants (Serres-Giardi et al. 2012).
When we considered chromosomal locations, we found that in the green anole, just like in chicken, the average GC3 and GC3* of genes located on microchromosomes are significantly higher than that of genes located on macrochromosomes (fig. 4), as expected under the gBGC hypothesis. Genome size and chromosome size generally affect the dynamics of gene GC3 in vertebrates (fig. 3), and this is true of A. carolinensis too. Again, this result is in apparent conflict with the report by Fujita et al. (2011) of a highly similar average genomic GC-content in micro- versus macrochromosomes. We are aware of five recent articles that compared GC-content between micro- and macrochromosomes in reptiles. Two of them used genomic, mostly noncoding data and detected no significant differences in A. carolinensis (Alföldi et al. 2011) and the central bearded dragon Po. vitticeps (Young et al. 2013). The other three studies used third-codon positions and detected a significant excess of GC-content in microchromosomes in A. carolinensis (this study), the four-striped rat snake Elaphe quadrivirgata (Matsubara et al. 2012), and the Chinese softshell turtle Pe. sinensis (Kuraku et al. 2006). Clearly, the two kinds of data yield contradicting pictures.
Our phylogenetic reconstruction of former coding sequence base compositions suggests that both the amniote ancestor and the tetrapod ancestor harbored a substantial amount of GC3-heterogeneity across genes (fig. 2). This is consistent with the hypothesis that the karyotype of ancestral amniotes and tetrapods did include microchromosomes, as suggested by ancestral chromosome reconstructions (Uno et al. 2012). We suggest that GC3 might serve as a useful marker of ancestral karyotypic structure in vertebrates, and possibly in other groups, given its tight relationship with chromosome size and our capacity to reliably trace its evolution through phylogenetic methods.
According to our phylogenetic analysis, the ancestral genome-wide heterogeneity in GC3 would have independently eroded in several lineages of tetrapods, such as marsupials and muridae, as previously documented (Romiguier et al. 2010). A similar erosion process is here predicted to have occurred in early amphibians and in early squamates, thus impacting the level of between-genes GC3-heterogeneity in these groups (fig. 2). However, it is noteworthy that the current average and variance of GC3 in A. carolinensis are higher than that of its predicted recent ancestors, which is again at odds with the hypothesis of an interruption of GC-increasing molecular processes in this lineage (Fujita et al. 2011).
gBGC, Transposable Elements, and the GC3 versus Genomic GC Discrepancy
Various explanations have been proposed to explain the highly reduced genomic GC-content heterogeneity in A. carolinensis—weakened conversion bias, increased genetic drift, homogeneous recombination map—all of them invoking an arrest of the effective gBGC in this genome (Alföldi et al. 2011; Fujita et al. 2011). However, if this hypothesis was true, we would expect third-codon positions to be similarly homogenized—which is not the case. The existence of many GC3-rich genes in the green anole genome, and especially in the presumably high-recombining microchromosomes, does not seem easy to reconcile with the hypothesis of an arrest of gBGC in this lineage. In contrast, our results suggest that gBGC is probably still active in A. carolinensis, despite the homogeneity of genomic GC-content in this species.
Several hypothesis might be considered to account for the discrepancy between GC3 and genomic GC-content in A. carolinensis. First, it might be that third-codon positions are affected by specific evolutionary processes, such as selection on synonymous codon usage, or particularly strong gBGC. Second, it might be that the genomic distribution of recombination hot spots in A. carolinensis is different from that of other amniotes and concentrated in genic or exonic regions, resulting in a coding sequence-specific GC-bias. Finally, the difference between GC3 and noncoding GC-content might be the consequence of insertions and deletions (indels), which affect noncoding DNA but are strongly counterselected in CDS. It should be noted that these hypotheses are not mutually exclusive.
To further explore these hypotheses, we correlated the GC-content of genes to coding sequence GC3 in the chicken and the green anole using Ensembl data. We detected a significant correlation between GC3 and genic GC-content in both species, albeit weaker in the green anole (chicken: r2 = 0.75, n = 7,350 genes; green anole: r2 = 0.35, n = 6,388 genes). We also correlated GC3 to GC-content at first (GC1) and second (GC2) positions of CDS, and again obtained highly significant correlation coefficients (GC3–GC1: r2 = 0.50, GC3–GC2: r2 = 0.26 for the green anole; GC3–GC1: r2 = 0.54, GC3–GC2: r2 = 0.33 for the chicken). These results demonstrate that the GC-bias we report in A. carolinensis is not restricted to third-codon positions but affects surrounding sites as well, rejecting the hypothesis that selection on codon usage is the main driver of GC3 in A. carolinensis.
Unlike CDS, the noncoding DNA of vertebrates undergoes frequent indels, and particularly frequent insertion of transposable elements (TE). The base composition of non-CDS is therefore affected not only by the nucleotide substitution process but also by the influx of elements whose GC-content typically differs from the substitutional equilibrium. Compared with birds and mammals, the green anole genome is characterized by an intense TE activity, as demonstrated by the large number of distinct families of relatively young repeated elements reported in this species (Alföldi et al. 2011). We therefore hypothesized that TE insertion could be a major driver of noncoding base composition in the green anole, acting to homogenize the GC-content landscape across chromosomes. To test this hypothesis, we retrieved the complete chromosomal sequences of the green anole from Ensembl, masking or not masking the repeated elements. Analyzing nonoverlapping windows of 3 kb with less than 20% missing data, we did not detect any difference in genomic GC-content heterogeneity between the masked and unmasked data set. In particular, microchromosomes and macrochromosomes were still indistinguishable in terms of GC-content after masking repeated elements. Therefore, if the discrepancy between GC3 and noncoding GC-content had to be explained by the process of insertions and deletions, this is apparently through nontransposable-element indels. Further data sets and analyses—for example, polymorphism data—will be necessary to investigate deeper the mystery of the GC3/noncoding GC-content discrepancy in this species.
Our analysis of existing and newly generated CDS suggests that gBGC is the main driver of GC3 evolution in all amniote and vertebrate lineages, not only mammals and birds. Interestingly, the effect of gBGC does not seem to impact the noncoding fraction of the genome to the same extent in all taxa: The correlation between GC3 and noncoding GC-content is high in some species (e.g., human and chicken), but low in others (e.g., green anole; Fujita et al. 2011). Following Elhaik et al. (2009), we therefore conclude that GC3 is not always a good proxy for genomic GC-content—and clearly one should not rely on GC3 to characterize the dynamics of noncoding base composition in A. carolinensis. On the other hand, we suggest that one should not rely on genome-wide patterns of GC-content to draw conclusions on the process of nucleotide substitution. The A. carolinensis example points to indel-free third-codon positions is a unique source of information regarding compositional biases of the nucleotide substitution process ultimately affecting the evolution of proteins.
Supplementary Material
Supplementary tables S1–S3 and figures S1–S3 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank the GBE Editorial Board for accepting to consider this submission accompanied with reviews. They thank four anonymous reviewers, who had done the job for another journal. They do not thank the Editorial Board of this other journal for rejecting our submission based on the review of another manuscript. They are grateful to Pierrelatte zoo “La ferme aux crocodiles,” Montpellier zoo, Cédric Libert, Benjamin Rey, Ylenia Chiari, Claire Doutrelan, and Philippe Peret for their help with sampling. They also thank Laurent Duret for useful advice and the Montpellier Bioinformatics & Biodiversity platform for support regarding computational aspects. This work was supported by European Research Council advanced grant 232971 (PopPhyl) and Agence Nationale de la Recherche grant ANR-10-BINF-01-01 (Ancestrome).
Literature Cited
- Alföldi J, et al. The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature. 2011;477:587–591. doi: 10.1038/nature10390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson E, Webster MT, Ratnakumar A, Ponting CP, Lindblad-Toh K. Death of PRDM9 coincides with stabilization of the recombination landscape in the dog genome. Genome Res. 2012;22:51–63. doi: 10.1101/gr.124123.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RJ, Bull JJ, Mengden GA. Chromosomes of Elaphe subocularis (Reptilia: Serpentes), with the description of an in vivo technique for preparation of snake chromosomes. Experientia. 1971;27:1228–1229. [Google Scholar]
- Bar-Yaacov D, Bouskila A, Mishmar D. The first Chameleon transcriptome: comparative genomic analysis of the OXPHOS system reveals loss of COX8 in Iguanian lizards. Genome Biol Evol. 2013;5:1792–1799. doi: 10.1093/gbe/evt131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belle EMS, Smith N, Eyre-Walker A. Analysis of the phylogenetic distribution of isochores in vertebrates and a test of the thermal stability hypothesis. J Mol Evol. 2002;55:356–363. doi: 10.1007/s00239-002-2333-1. [DOI] [PubMed] [Google Scholar]
- Berglund J, Pollard KS, Webster MT. Hotspots of biased nucleotide substitutions in human genes. PLoS Biol. 2009;7:e26. doi: 10.1371/journal.pbio.1000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi G. The vertebrate genome: isochores and evolution. Mol Biol Evol. 1993;10:186–204. doi: 10.1093/oxfordjournals.molbev.a039994. [DOI] [PubMed] [Google Scholar]
- Bernardi G. Isochores and the evolutionary genomics of vertebrates. Gene. 2000;241:3–17. doi: 10.1016/s0378-1119(99)00485-0. [DOI] [PubMed] [Google Scholar]
- Bernardi Gia, Bernardi Gio Compositional transitions in the nuclear genomes of cold-blooded vertebrates. J Mol Evol. 1990;31:282–293. doi: 10.1007/BF02101123. [DOI] [PubMed] [Google Scholar]
- Beye M, et al. Exceptionally high levels of recombination across the honey bee genome. Genome Res. 2006;16:1339–1344. doi: 10.1101/gr.5680406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahais V, et al. Reference-free transcriptome assembly in non-model animals from next-generation sequencing data. Mol Ecol Resour. 2012;12:834–845. doi: 10.1111/j.1755-0998.2012.03148.x. [DOI] [PubMed] [Google Scholar]
- Capra JA, Hubisz MJ, Kostka D, Pollard KS, Siepel A. A model-based analysis of GC-biased gene conversion in the human and chimpanzee genomes. PLoS Genet. 2013;9:e1003684. doi: 10.1371/journal.pgen.1003684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoe TA, et al. The Burmese python genome reveals the molecular basis for extreme adaptation in snakes. Proc Natl Acad Sci U S A. 2013;110:20645–20650. doi: 10.1073/pnas.1314475110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiari Y, Cahais V, Galtier N, Delsuc F. Phylogenomic analyses support the position of turtles as the sister group of birds and crocodiles (Archosauria) BMC Biol. 2012;10:1–14. doi: 10.1186/1741-7007-10-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiari Y, Galtier N. RNA extraction from sauropsids blood: evaluation and improvement of methods. Amphib-Reptil. 2011;32:136–139. [Google Scholar]
- Chojnowski JL, Braun EL. Turtle isochore structure is intermediate between amphibians and other amniotes. Integr Comp Biol. 2008;48:454–462. doi: 10.1093/icb/icn062. [DOI] [PubMed] [Google Scholar]
- Chojnowski JL, et al. Patterns of vertebrate isochore evolution revealed by comparison of expressed mammalian, avian, and crocodilian genes. J Mol Evol. 2007;65:259–266. doi: 10.1007/s00239-007-9003-2. [DOI] [PubMed] [Google Scholar]
- Costantini M, Cammarano R, Bernardi G. The evolution of isochore patterns in vertebrate genomes. BMC Genomics. 2009;10:146. doi: 10.1186/1471-2164-10-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreszer TR, Wall GD, Haussler D, Pollard KS. Biased clustered substitutions in the human genome: the footprints of male-driven biased gene conversion. Genome Res. 2007;17:1420–1430. doi: 10.1101/gr.6395807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Arndt PF. The impact of recombination on nucleotide substitutions in the human genome. PLoS Genet. 2008;4:e1000071. doi: 10.1371/journal.pgen.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Galtier N. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 2009;10:285–311. doi: 10.1146/annurev-genom-082908-150001. [DOI] [PubMed] [Google Scholar]
- Dutheil J, Boussau B. Non-homogeneous models of sequence evolution in the Bio++ suite of libraries and programs. BMC Evol Biol. 2008;8:255. doi: 10.1186/1471-2148-8-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhaik E, Landan G, Graur D. Can GC content at third-codon positions be used as a proxy for isochore composition? Mol Biol Evol. 2009;26:1829–1833. doi: 10.1093/molbev/msp100. [DOI] [PubMed] [Google Scholar]
- Escobar JS, Glémin S, Galtier N. GC-biased gene conversion impacts ribosomal DNA evolution in vertebrates, angiosperms, and other eukaryotes. Mol Biol Evol. 2011;28:2561–2575. doi: 10.1093/molbev/msr079. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A. Recombination and mammalian genome evolution. Proc R Soc Lond B Biol Sci. 1993;252:237–243. doi: 10.1098/rspb.1993.0071. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A, Hurst LD. The evolution of isochores. Nat Rev Genet. 2001;2:549–555. doi: 10.1038/35080577. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Phylogenies and the comparative method. Am Nat. 1985;125:1–15. [Google Scholar]
- Fortes GG, Bouza C, Martínez P, Sánchez L. Diversity in isochore structure among cold-blooded vertebrates based on GC content of coding and non-coding sequences. Genetica. 2007;129:281–289. doi: 10.1007/s10709-006-0009-2. [DOI] [PubMed] [Google Scholar]
- Fryxell KJ, Zuckerkandl E. Cytosine deamination plays a primary role in the evolution of mammalian isochores. Mol Biol Evol. 2000;17:1371–1383. doi: 10.1093/oxfordjournals.molbev.a026420. [DOI] [PubMed] [Google Scholar]
- Fujita MK, Edwards SV, Ponting CP. The Anolis lizard genome: an amniote genome without isochores. Genome Biol Evol. 2011;3:974–984. doi: 10.1093/gbe/evr072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N, Duret L. Adaptation or biased gene conversion? Extending the null hypothesis of molecular evolution. Trends Genet. 2007;23:273–277. doi: 10.1016/j.tig.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Galtier N, Duret L, Glémin S, Ranwez V. GC-biased gene conversion promotes the fixation of deleterious amino acid changes in primates. Trends Genet. 2009;25:1–5. doi: 10.1016/j.tig.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Galtier N, Gouy M. Inferring pattern and process: maximum-likelihood implementation of a nonhomogeneous model of DNA sequence evolution for phylogenetic analysis. Mol Biol Evol. 1998;15:871–879. doi: 10.1093/oxfordjournals.molbev.a025991. [DOI] [PubMed] [Google Scholar]
- Galtier N, Piganeau G, Mouchiroud D, Duret L. GC-content evolution in mammalian genomes: the biased gene conversion hypothesis. Genetics. 2001;159:907–911. doi: 10.1093/genetics/159.2.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayral P, et al. Next-generation sequencing of transcriptomes: a guide to RNA isolation in nonmodel animals. Mol Ecol Resour. 2011;11:650–661. doi: 10.1111/j.1755-0998.2011.03010.x. [DOI] [PubMed] [Google Scholar]
- Glémin S. Surprising fitness consequences of GC-biased gene conversion: I. Mutation load and inbreeding depression. Genetics. 2010;185:939–959. doi: 10.1534/genetics.110.116368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glémin S, Bazin E, Charlesworth D. Impact of mating systems on patterns of sequence polymorphism in flowering plants. Proc Biol Sci. 2006;273:3011–3019. doi: 10.1098/rspb.2006.3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodstadt L, Heger A, Webber C, Ponting CP. An analysis of the gene complement of a marsupial, Monodelphis domestica: evolution of lineage-specific genes and giant chromosomes. Genome Res. 2007;17:969–981. doi: 10.1101/gr.6093907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory TR, et al. Eukaryotic genome size databases. Nucleic Acids Res. 2007;35:D332–D338. doi: 10.1093/nar/gkl828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillon J, Hulin V, Girondot M. A large phylogeny of turtles (Testudines) using molecular data. Contrib Zool. 2012;81:147–158. [Google Scholar]
- Hillier LW, et al. International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- Hughes S, Zelus D, Mouchiroud D. Warm-blooded isochore structure in Nile crocodile and turtle. Mol Biol Evol. 1999;16:1521–1527. doi: 10.1093/oxfordjournals.molbev.a026064. [DOI] [PubMed] [Google Scholar]
- Janes DE, Organ CL, Fujita MK, Shedlock AM, Edwards SV. Genome evolution in Reptilia, the sister group of mammals. Annu Rev Genomics Hum Genet. 2010;11:239–264. doi: 10.1146/annurev-genom-082509-141646. [DOI] [PubMed] [Google Scholar]
- Kai W, et al. Integration of the genetic map and genome assembly of fugu facilitates insights into distinct features of genome evolution in teleosts and mammals. Genome Biol Evol. 2011;3:424–442. doi: 10.1093/gbe/evr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzman S, Capra JA, Haussler D, Pollard KS. Ongoing GC-biased evolution is widespread in the human genome and enriched near recombination hot spots. Genome Biol Evol. 2011;3:614–626. doi: 10.1093/gbe/evr058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent CF, Minaei S, Harpur BA, Zayed A. Recombination is associated with the evolution of genome structure and worker behavior in honey bees. Proc Natl Acad Sci U S A. 2012;109:18012–18017. doi: 10.1073/pnas.1208094109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraku S, et al. cDNA-based gene mapping and GC3 profiling in the soft-shelled turtle suggest a chromosomal size-dependent GC bias shared by sauropsids. Chromosome Res. 2006;14:187–202. doi: 10.1007/s10577-006-1035-8. [DOI] [PubMed] [Google Scholar]
- Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lartillot N. Interaction between selection and biased gene conversion in mammalian protein-coding sequence evolution revealed by a phylogenetic covariance analysis. Mol Biol Evol. 2013a;30:356–368. doi: 10.1093/molbev/mss231. [DOI] [PubMed] [Google Scholar]
- Lartillot N. Phylogenetic patterns of GC-biased gene conversion in placental mammals and the evolutionary dynamics of recombination landscapes. Mol Biol Evol. 2013b;30:489–502. doi: 10.1093/molbev/mss239. [DOI] [PubMed] [Google Scholar]
- Lawrie NM, Tease C, Hultén MA. Chiasma frequency, distribution and interference maps of mouse autosomes. Chromosoma. 1995;104:308–314. doi: 10.1007/BF00352262. [DOI] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Freudenberg J. Two-parameter characterization of chromosome-scale recombination rate. Genome Res. 2009;19:2300–2307. doi: 10.1101/gr.092676.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien S, et al. A dense SNP-based linkage map for Atlantic salmon (Salmo salar) reveals extended chromosome homeologies and striking differences in sex-specific recombination patterns. BMC Genomics. 2011;12:615. doi: 10.1186/1471-2164-12-615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man Z, Yishu W, Peng Y, Xiaobing W. Crocodilian phylogeny inferred from twelve mitochondrial protein-coding genes, with new complete mitochondrial genomic sequences for Crocodylus acutus and Crocodylus novaeguineae. Mol Phylogenet Evol. 2011;60:62–67. doi: 10.1016/j.ympev.2011.03.029. [DOI] [PubMed] [Google Scholar]
- Mancera E, Bourgon R, Brozzi A, Huber W, Steinmetz LM. High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature. 2008;454:479–485. doi: 10.1038/nature07135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais G. Biased gene conversion: implications for genome and sex evolution. Trends Genet. 2003;19:330–338. doi: 10.1016/S0168-9525(03)00116-1. [DOI] [PubMed] [Google Scholar]
- Matsubara K, et al. Intra-genomic GC heterogeneity in sauropsids: evolutionary insights from cDNA mapping and GC(3) profiling in snake. BMC Genomics. 2012;13:604. doi: 10.1186/1471-2164-13-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JE, et al. A phylogeny of birds based on over 1,500 loci collected by target enrichment and high-throughput sequencing. PLoS One. 2013;8:e54848. doi: 10.1371/journal.pone.0054848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith RW, et al. Impacts of the Cretaceous Terrestrial Revolution and KPg extinction on mammal diversification. Science. 2011;334:521–524. doi: 10.1126/science.1211028. [DOI] [PubMed] [Google Scholar]
- Meunier J, Duret L. Recombination drives the evolution of GC-content in the human genome. Mol Biol Evol. 2004;21:984–990. doi: 10.1093/molbev/msh070. [DOI] [PubMed] [Google Scholar]
- Miller HC, Biggs PJ, Voelckel C, Nelson NJ. De novo sequence assembly and characterisation of a partial transcriptome for an evolutionarily distinct reptile, the tuatara (Sphenodon punctatus) BMC Genomics. 2012;13:1–12. doi: 10.1186/1471-2164-13-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya-Burgos JI, Boursot P, Galtier N. Recombination explains isochores in mammalian genomes. Trends Genet. 2003;19:128–130. doi: 10.1016/S0168-9525(03)00021-0. [DOI] [PubMed] [Google Scholar]
- Mugal CF, Arndt PF, Ellegren H. Twisted signatures of GC-biased gene conversion embedded in an evolutionary stable karyotype. Mol Biol Evol. 2013;30:1700–1712. doi: 10.1093/molbev/mst067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munch K, Mailund T, Dutheil JY, Schierup MH. A fine-scale recombination map of the human-chimpanzee ancestor reveals faster change in humans than in chimpanzees and a strong impact of GC-biased gene conversion. Genome Res. 2014;24:467–474. doi: 10.1101/gr.158469.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabholz B, Künstner A, Wang R, Jarvis ED, Ellegren H. Dynamic evolution of base composition: causes and consequences in avian phylogenomics. Mol Biol Evol. 2011;28:2197–2210. doi: 10.1093/molbev/msr047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Near TJ, et al. Resolution of ray-finned fish phylogeny and timing of diversification. Proc Natl Acad Sci U S A. 2012;109:13698–13703. doi: 10.1073/pnas.1206625109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Necşulea A, et al. Meiotic recombination favors the spreading of deleterious mutations in human populations. Hum Mutat. 2011;32:198–206. doi: 10.1002/humu.21407. [DOI] [PubMed] [Google Scholar]
- Ninwichian P, et al. Second-generation genetic linkage map of catfish and its integration with the BAC-based physical map. G3. 2012;2:1233–1241. doi: 10.1534/g3.112.003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omer A, Abukashawa S. 2012 Morphometric traits and karyotypic features of the African lungfish (Um Koro) Protopterus annectens annectens (Owen, 1839) and Protopterus aethiopicus aethiopicus aethiopicus (Heckel, 1851) in Sudan. J Fish Poultry Wildl Sci. 1. [Google Scholar]
- Organ CL, Moreno RG, Edwards SV. Three tiers of genome evolution in reptiles. Integr Comp Biol. 2008;48:494–504. doi: 10.1093/icb/icn046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry GH, et al. Comparative RNA sequencing reveals substantial genetic variation in endangered primates. Genome Res. 2012;22:602–610. doi: 10.1101/gr.130468.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessia E, et al. Evidence for widespread GC-biased gene conversion in eukaryotes. Genome Biol Evol. 2012;4:675–682. doi: 10.1093/gbe/evs052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyron RA, Burbrink FT, Wiens JJ. A phylogeny and revised classification of Squamata, including 4161 species of lizards and snakes. BMC Evol Biol. 2013;13:1–53. doi: 10.1186/1471-2148-13-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranwez V, Harispe S, Delsuc F, Douzery EJP. MACSE: Multiple Alignment of Coding SEquences accounting for frameshifts and stop codons. PLoS One. 2011;6:e22594. doi: 10.1371/journal.pone.0022594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnakumar A, et al. Detecting positive selection within genomes: the problem of biased gene conversion. Philos Trans R Soc Lond B Biol Sci. 2010;365:2571–2580. doi: 10.1098/rstb.2010.0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romiguier J, Ranwez V, Douzery EJP, Galtier N. Contrasting GC-content dynamics across 33 mammalian genomes: relationship with life-history traits and chromosome sizes. Genome Res. 2010;20:1001–1009. doi: 10.1101/gr.104372.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romiguier J, Ranwez V, Douzery EJP, Galtier N. Genomic evidence for large, long-lived ancestors to placental mammals. Mol Biol Evol. 2013;30:5–13. doi: 10.1093/molbev/mss211. [DOI] [PubMed] [Google Scholar]
- Sato H, Ota H. Karyotype of the Chinese soft-shelled turtle, Pelodiscus sinensis, from Japan and Taiwan, with chromosomal data for Dogania subplana. Curr Herpetol. 2001;20:19–25. [Google Scholar]
- Schwartz TS, et al. A garter snake transcriptome: pyrosequencing, de novo assembly, and sex-specific differences. BMC Genomics. 2010;11:1–17. doi: 10.1186/1471-2164-11-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serres-Giardi L, Belkhir K, David J, Glémin S. Patterns and evolution of nucleotide landscapes in seed plants. Plant Cell. 2012;24:1379–1397. doi: 10.1105/tpc.111.093674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer HB, et al. The western painted turtle genome, a model for the evolution of extreme physiological adaptations in a slowly evolving lineage. Genome Biol. 2013;14:R28. doi: 10.1186/gb-2013-14-3-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shine R. Life-history evolution in reptiles. Annu Rev Ecol Evol Syst. 2005;36:23–46. [Google Scholar]
- Tzika AC, Helaers R, Schramm G, Milinkovitch MC. Reptilian-transcriptome v1.0, a glimpse in the brain transcriptome of five divergent Sauropsida lineages and the phylogenetic position of turtles. Evodevo. 2011;2:1–17. doi: 10.1186/2041-9139-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno Y, et al. Inference of the protokaryotypes of amniotes and tetrapods and the evolutionary processes of microchromosomes from comparative gene mapping. PLoS One. 2012;7:e53027. doi: 10.1371/journal.pone.0053027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno Y, Nishida C, Takagi C, Ueno N, Matsuda Y. Homoeologous chromosomes of Xenopus laevis are highly conserved after whole-genome duplication. Heredity. 2013;111:430–436. doi: 10.1038/hdy.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valleley EMA, Harrison CJ, Cook Y, Ferguson MWJ, Sharpe PT. The karyotype of Alligator mississippiensis, and chromosomal mapping of the ZFY/X homologue, Zfc. Chromosoma. 1994;103:502–507. doi: 10.1007/BF00337388. [DOI] [PubMed] [Google Scholar]
- Webster MT, Smith NGC. Fixation biases affecting human SNPs. Trends Genet. 2004;20:116–122. doi: 10.1016/j.tig.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Webster MT, Smith NGC, Hultin-Rosenberg L, Arndt PF, Ellegren H. Male-driven biased gene conversion governs the evolution of base composition in human alu repeats. Mol Biol Evol. 2005;22:1468–1474. doi: 10.1093/molbev/msi136. [DOI] [PubMed] [Google Scholar]
- Young MJ, O’Meally D, Sarre SD, Georges A, Ezaz T. Molecular cytogenetic map of the central bearded dragon, Pogona vitticeps (Squamata: Agamidae) Chromosome Res. 2013;21:361–374. doi: 10.1007/s10577-013-9362-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.