

Abstract
We use full mitochondrial genomes to test the robustness of the phylogeny of the Octocorallia, to determine the evolutionary pathway for the five known mitochondrial gene rearrangements in octocorals, and to test the suitability of using mitochondrial genomes for higher taxonomic-level phylogenetic reconstructions. Our phylogeny supports three major divisions within the Octocorallia and show that Paragorgiidae is paraphyletic, with Sibogagorgia forming a sister branch to the Coralliidae. Furthermore, Sibogagorgia cauliflora has what is presumed to be the ancestral gene order in octocorals, but the presence of a pair of inverted repeat sequences suggest that this gene order was not conserved but rather evolved back to this apparent ancestral state. Based on this we recommend the resurrection of the family Sibogagorgiidae to fix the paraphyly of the Paragorgiidae.
This is the first study to show that in the Octocorallia, mitochondrial gene orders have evolved back to an ancestral state after going through a gene rearrangement, with at least one of the gene orders evolving independently in different lineages. A number of studies have used gene boundaries to determine the type of mitochondrial gene arrangement present. However, our findings suggest that this method known as gene junction screening may miss evolutionary reversals.
Additionally, substitution saturation analysis demonstrates that while whole mitochondrial genomes can be used effectively for phylogenetic analyses within Octocorallia, their utility at higher taxonomic levels within Cnidaria is inadequate. Therefore for phylogenetic reconstruction at taxonomic levels higher than subclass within the Cnidaria, nuclear genes will be required, even when whole mitochondrial genomes are available.
Keywords: Octocorallia, deep-sea corals, soft corals, cnidarian phylogenetics, gene rearrangement, substitution saturation
Introduction
Octocorals, a group of corals commonly known as sea fans, sea whips, sea pens, and soft corals, play a key role in forming structures in a number of habitats including shallow water reefs, deep seamounts, and submarine canyons (Genin et al. 1986; Hecker 1990; Stocks 2004). They act as hosts for a variety of invertebrates and fishes, including some key deep-water fisheries species (Genin et al. 1992; Jones et al. 1994; Rogers 1994; Probert et al. 1997; Stocks 2004; DeVogelaere et al. 2005; Leverette and Metaxas 2005; Mortensen and Buhl-Mortensen 2005; Baco 2007; Buhl-Mortensen et al. 2010; Roberts et al. 2010; Baillon et al. 2012).
Deep-sea corals are slow growing, long lived, and existing evidence suggests that many are recruitment limited (Grigg 1988; Krieger 2001; Roark et al. 2006, 2009; Sun et al. 2010). Thus, they are very vulnerable to anthropogenic impacts and slow to recover from them (Williams et al. 2010). Anthropogenic activities that are known or likely to have large impacts on octocorals include fisheries (Koslow et al. 2001; Clark and Rowden 2009), deep-sea mining for cobalt-rich manganese crusts (Hein 2002; Hein et al. 2009), and climate change and ocean acidification (Guinotte et al. 2006).
Recent reviews of seamount fauna and deep-sea corals have concluded that the global deficiency of scientific expertise in morphological taxonomy is a significant impediment to the understanding of deep-sea coral diversity, coral biogeography, conservation, and seamount ecology (Morgan et al. 2006; Parrish and Baco 2007; Rogers et al. 2007). Likewise, in the past decade, molecular phylogenetic analyses of the anthozoan subclass Octocorallia have shown that the current taxonomic classification of these organisms, based on morphology, needs to be revised (Berntson et al. 2001; Sanchez et al. 2003; McFadden et al. 2006, 2010; Herrera et al. 2010; Brockman and McFadden 2012).
Until recently, the majority of phylogenetic analyses of octocorals have been based on a few mitochondrial genes or nuclear genes or a combination of both (Berntson et al. 2001; Sanchez et al. 2003; McFadden et al. 2006, 2010; Herrera et al. 2010); but recent studies are increasingly using whole mitochondrial genomes, revealing five different gene orders in octocorals (Brugler and France 2008; Uda et al. 2011; Brockman and McFadden 2012; Figueroa and Baco 2014). One of these gene orders is shared by most octocorals, while the other four alternative orders are only found within one of the three major clades of Octocorallia. Therefore, the widespread phylogenetic distribution of this gene order has led to the assumption that it represents the ancestral arrangement in octocorals (Brugler and France 2008; Uda et al. 2011; Brockman and McFadden 2012; Figueroa and Baco 2014). Whole mitochondrial genomes, which in octocorals contain 14 protein-coding genes, provide better resolution of the tree topology in these organisms (Uda et al. 2011). In general, molecular phylogenetic studies agree with the three major clades proposed by McFadden et al. (2006) based on sequences from two mitochondrial genes (nad2 and mutS). One of these major clades is composed of the scleraxonians Coralliidae and Paragorgiidae and the alcyoniina Anthomastus, along with several other genera mostly belonging to the family Alcyoniidae (McFadden et al. 2006; Brockman and McFadden 2012; Figueroa and Baco 2014). These three families are among the most abundant octocoral families in the deep sea (Baco 2007) and thus improving their taxonomy is a high priority.
Thus the goal of our study was to improve our understanding of the relationships within this Anthomastus–Corallium clade, as well as the evolution of the gene orders within this clade. We sequenced the whole mitochondrial genome of two morphospecies of Anthomastus and the paragorgiid Sibogagorgia cauliflora, all three presumably members of McFadden et al.’s (McFadden et al. 2006) Anthomastus–Corallium clade. We also sequenced the whole mitochondrial genome of the primnoid Narella hawaiinensis, a member of McFadden et al. (2006) Calcaxonia–Pennatulacea clade is the sister branch to the Anthomastus–Corallium clade (McFadden et al. 2006; Brockman and McFadden 2012; Figueroa and Baco 2014).
In the process of examining the phylogenetic relationships among these families, we also have the opportunity to gain a better understanding of the utility of whole mitochondrial genomes for unraveling phylogenetics at higher taxonomic levels within the Cnidaria. Recent phylogenetic reconstructions based on whole mitochondrial genomes have suggested that Anthozoa is a paraphyletic group, with Octocorallia branching as a sister clade to the Medusozoa and not the Hexacorallia (Shao et al. 2006; Kayal and Lavrov 2008; Lavrov et al. 2008; Park et al. 2012; Kayal et al. 2013). This observation disagrees with current morphological classification and with phylogenetic reconstructions based on nuclear markers, which strongly support a monophyletic Anthozoa comprised of the Octocorallia and Hexacorallia (France et al. 1996; Odorico and Miller 1997; Berntson et al. 1999; Won et al. 2001; Collins 2002; Daly et al. 2007).
Thus another goal of our analysis is to use the newly sequenced mitochondrial genomes from recently collected specimens of Octocorallia in conjunction with mitochondrial genomes found in GenBank for other Anthozoa, Medusozoa, and Porifera for phylogenetic analyses at three different taxonomic levels: Within subclass Octocorallia, within class Anthozoa, and within the phylum Cnidaria. Thus, phylogenetic analyses were used to achieve three main objectives: 1) To elucidate the internal topology of the Anthomastus–Corallium clade, 2) to test the robustness of the phylogeny of Octocorallia proposed by McFadden et al. (2006), and 3) to test the suitability of mitochondrial genomes to be used in higher order phylogenetic reconstructions within Cnidaria.
Materials and Methods
Collections
For this study, we used four octocoral specimens: Two distinct morphotypes of the genus Anthomastus (one collected from Necker Ridge in the northern Central Pacific and a second morphotype from Derickson Seamount, just south of the Aleutian Islands); a specimen of S. cauliflora (also from Derickson Seamount); and a specimen of N. hawaiinensis (collected from Pioneer Bank in the Northwestern Hawaiian Islands). Samples from Hawaii and Necker were collected using the Pisces IV or V submersible, and from Derickson using the ROV Jason II. Corals were placed in insulated bioboxes for return to the surface and subsamples were frozen at −80 °C. The remainder of each specimen was deposited at the Smithsonian. United States National Museum (USNM)#s for each specimen are listed in table 1.
Table 1.
Specimens used in this Study
| Subphylum | Subclass | Species | USNM No. | Genbank Acession No. | Sequence From |
|---|---|---|---|---|---|
| Anthozoa | Hexacorallia | Acropora tenuis | NC_003522 | GenBank | |
| Anthozoa | Hexacorallia | Agaricia humilis | NC_008160 | GenBank | |
| Anthozoa | Hexacorallia | Anacropora matthai | NC_006898 | GenBank | |
| Anthozoa | Hexacorallia | Astrangia sp. JVK-2006 | NC_008161 | GenBank | |
| Anthozoa | Hexacorallia | Chrysopathes formosa | NC_008411 | GenBank | |
| Anthozoa | Hexacorallia | Colpophyllia natans | NC_008162 | GenBank | |
| Anthozoa | Hexacorallia | Discosoma sp. CASIZ 168915 | NC_008071 | GenBank | |
| Anthozoa | Hexacorallia | Discosoma sp. CASIZ 168916 | NC_008072 | GenBank | |
| Anthozoa | Hexacorallia | Euphyllia ancora | NC_015641 | GenBank | |
| Anthozoa | Hexacorallia | Fungiacyathus stephanus | NC_015640 | GenBank | |
| Anthozoa | Hexacorallia | Goniopora columna | NC_015643 | GenBank | |
| Anthozoa | Hexacorallia | Lophelia pertusa | NC_015143 | GenBank | |
| Anthozoa | Hexacorallia | Madracis mirabilis | NC_011160 | GenBank | |
| Anthozoa | Hexacorallia | Metridium senile | NC_000933 | GenBank | |
| Anthozoa | Hexacorallia | Metridium senile | NC_000933 | GenBank | |
| Anthozoa | Hexacorallia | Montastraea annularis | NC_007224 | GenBank | |
| Anthozoa | Hexacorallia | Montastraea faveolata | NC_007226 | GenBank | |
| Anthozoa | Hexacorallia | Montastraea franksi | NC_007225 | GenBank | |
| Anthozoa | Hexacorallia | Montipora cactus | NC_006902 | GenBank | |
| Anthozoa | Hexacorallia | Mussa angulosa | NC_008163 | GenBank | |
| Anthozoa | Hexacorallia | Nematostella sp. JVK-2006 | NC_008164 | GenBank | |
| Anthozoa | Hexacorallia | Pavona clavus | NC_008165 | GenBank | |
| Anthozoa | Hexacorallia | Pocillopora damicornis | NC_009797 | GenBank | |
| Anthozoa | Hexacorallia | Pocillopora eydouxi | NC_009798 | GenBank | |
| Anthozoa | Hexacorallia | Polycyathus sp. | NC_015642 | GenBank | |
| Anthozoa | Hexacorallia | Porites okinawensis | NC_015644 | GenBank | |
| Anthozoa | Hexacorallia | Porites porites | NC_008166 | GenBank | |
| Anthozoa | Hexacorallia | Ricordea florida | NC_008159 | GenBank | |
| Anthozoa | Hexacorallia | Savalia savaglia | NC_008827 | GenBank | |
| Anthozoa | Hexacorallia | Savalia savaglia | NC_008827 | GenBank | |
| Anthozoa | Hexacorallia | Seriatopora caliendrum | NC_010245 | GenBank | |
| Anthozoa | Hexacorallia | Seriatopora hystrix | NC_010244 | GenBank | |
| Anthozoa | Hexacorallia | Siderastrea radians | NC_008167 | GenBank | |
| Anthozoa | Hexacorallia | Stylophora pistillata | NC_011162 | GenBank | |
| Anthozoa | Octocorallia | Acanella eburnean | EF672731 | GenBank | |
| Anthozoa | Octocorallia | Anthomastus sp. | 1171062 | KM015352 | This study |
| Anthozoa | Octocorallia | Anthomastus sp. | 1081145 | KM015353 | This study |
| Anthozoa | Octocorallia | Briareum asbestinum | NC_008073 | GenBank | |
| Anthozoa | Octocorallia | Calicogorgia granulosa | GU047880 | GenBank | |
| Anthozoa | Octocorallia | Corallium japonicum | AB595189 | GenBank | |
| Anthozoa | Octocorallia | Dendronephthya castanea | GU047877 | GenBank | |
| Anthozoa | Octocorallia | Dendronephthya gigantea | NC_013573 | GenBank | |
| Anthozoa | Octocorallia | Dendronephthya mollis | HQ694725 | GenBank | |
| Anthozoa | Octocorallia | Dendronephthya putteri | HQ694726 | GenBank | |
| Anthozoa | Octocorallia | Dendronephthya suensoni | GU047878 | GenBank | |
| Anthozoa | Octocorallia | Echinogorgia complexa | HQ694727 | GenBank | |
| Anthozoa | Octocorallia | Euplexaura crassa | HQ694728 | GenBank | |
| Anthozoa | Octocorallia | Hemicorallium imperiale | 1072448 | KC782352 | Figueroa and Baco (2014) |
| Anthozoa | Octocorallia | Hemicorallium imperiale | 1072449 | KC782355 | Figueroa and Baco (2014) |
| Anthozoa | Octocorallia | Hemicorallium laauense | KC782348 | Figueroa and Baco (2014) | |
| Anthozoa | Octocorallia | Keratoisinidae sp. | EF622534 | GenBank | |
| Anthozoa | Octocorallia | Narella hawaiinensis | 1072109 | KM015351 | This study |
| Anthozoa | Octocorallia | Paragorgia sp. | 1075769 | KC782349 | Figueroa and Baco (2014) |
| Anthozoa | Octocorallia | Paragorgia sp. | 1075761 | KC782350 | Figueroa and Baco (2014) |
| Anthozoa | Octocorallia | Paragorgia sp. | 1072362 | KC782351 | Figueroa and Baco (2014) |
| Anthozoa | Octocorallia | Paragorgia sp. | 1072339 | KC782354 | Figueroa and Baco (2014) |
| Anthozoa | Octocorallia | Paragorgia sp. | 1075741 | KC782356 | Figueroa and Baco (2014) |
| Anthozoa | Octocorallia | Paraminabea aldersladei | JX508792 | GenBank | |
| Anthozoa | Octocorallia | Pleurocorallium kishinouyei | 1072441 | KC782353 | Figueroa and Baco (2014) |
| Anthozoa | Octocorallia | Pleurocorallium konojoi | NC015406 | GenBank | |
| Anthozoa | Octocorallia | Pleurocorallium secundum | KC782347 | Figueroa and Baco (2014) | |
| Anthozoa | Octocorallia | Pseudopterogorgia bipinnata | NC_008157 | GenBank | |
| Anthozoa | Octocorallia | Renilla muelleri | JX023273.1 | GenBank | |
| Anthozoa | Octocorallia | Sarcophyton glaucum | AF063191 | GenBank | |
| Anthozoa | Octocorallia | Scleronephthya gracillimum | GU047879 | GenBank | |
| Anthozoa | Octocorallia | Sibogagorgia cauliflora | 1122229 | KM015354 | This study |
| Anthozoa | Octocorallia | Sinularia peculiaris | NC_018379 | GenBank | |
| Anthozoa | Octocorallia | Stylatula elongate | NC_018380 | GenBank | |
| Medusozoa | Hydrozoa | Clava multicornis | NC_016465 | GenBank | |
| Medusozoa | Hydrozoa | Craspedacusta sowerbyi | JN593332 | GenBank | |
| Medusozoa | Hydrozoa | Craspedacusta sowerbyi | NC_018537 | GenBank | |
| Medusozoa | Hydrozoa | Cubaia aphrodite | NC_016467 | GenBank | |
| Medusozoa | Hydrozoa | Hydra magnipapillata | NC_008411 | GenBank | |
| Medusozoa | Hydrozoa | Hydra oligactis | NC_008071 | GenBank | |
| Medusozoa | Hydrozoa | Laomedea flexuosa | NC_016463 | GenBank | |
| Medusozoa | Scyphozoa | Aurelia aurita | HQ694729 | GenBank | |
| Medusozoa | Scyphozoa | Aurelia aurita | NC_008446 | GenBank | |
| Medusozoa | Scyphozoa | Cassiopea frondosa | NC_016466 | GenBank | |
| Medusozoa | Scyphozoa | Chrysaora quinquecirrha | HQ694730 | GenBank | |
| Porifera | Demospongiae | Agelas schmidti | NC_010213 | GenBank | |
| Porifera | Demospongiae | Amphimedon compressa | NC_010201 | GenBank | |
| Porifera | Demospongiae | Aplysina fulva | NC_010203 | GenBank | |
| Porifera | Demospongiae | Igernella notabilis | NC_010216 | GenBank | |
| Porifera | Demospongiae | Oscarella carmela | NC_009090 | GenBank |
DNA Extraction, PCR, Sequencing and Assembly
Total genomic DNA was extracted from each specimen using Qiagen’s DNeasy Blood and Tissue Kit. Complete mitochondrial genomes of each specimen were obtained using a series of overlapping polymerase chain reactions (PCRs) using previously published primers sets (Park et al. 2012; Figueroa and Baco 2014) (table 2). The following thermocycling conditions were used: 96 °C for 2 min, 35 cycles at 94 °C for 1 min, 48 °C for 1 min, 72 °C for 1 min, and a final step at 72 °C for 5 min. The PCR fragments were sent for sequencing at the University of Washington High Throughput Genomics Center for both the forward and reverse strands.
Table 2.
Primers Used for this Study
| Forward | Primer | Reverse | Primer | Start | End | Size (bp) | Overlap |
|---|---|---|---|---|---|---|---|
| 1F | ATGAACAAATATCTTACACG | 1R | ATAARTGCTGRAATAAAAT | 1 | 699 | 698 | 162 |
| 2F | ACAACATTTTTTGATCCT | 2R | GCTAAACCCAAGAAATG | 667 | 1,290 | 623 | 32 |
| 3F | ACAGGTTATAGTTATAATGA | 3R | GTCTGCTGGCACTTAGTTAG | 1,223 | 1,860 | 637 | 67 |
| 4F | CTGGTCGAAGATGCGTAGTA | 4R | TGTGCTAACACTGGGTTAGA | 1,743 | 2,500 | 757 | 117 |
| 5F | TATGCGCTACATTCTCCTAT | 5R | CACACTTCATAGCTAATCAT | 2405 | 3,128 | 723 | 95 |
| ssRNA-F1 | CTGCGTTTAATACGTACTTGGC | 6R | YACTGCATCTAAACCTATCA | 2,680 | 3,591 | 911 | 448 |
| 7F | ATTCTAGGAATGGGCTGC | 7R | GACATTTGTCCCCAAGGTAA | 3,509 | 4,126 | 617 | 82 |
| 8F | ATATTTTAAGAGACGTTAAT | 8R | CTCTACTGGATTAGCCCCTA | 3,964 | 4,726 | 762 | 162 |
| 9Fa | ATCCTTTAGTAACTCCTG | msh2806R | TAACTCAGCTTGAGAGTATGC | 4,501 | 5,088 | 587 | 225 |
| 9Fa | ATCCTTTAGTAACTCCTG | msh3101R | GATATCACATAAGATAATTCCG | 4,527 | 5,354 | 827 | 561 |
| 10F | YTRCTTCAAATGGGGTTTCC | mutS-3458R | TSGAGCAAAAGCCACTCC | 5,268 | 5,731 | 463 | 86 |
| 10F | YTRCTTCAAATGGGGTTTCC | mutS-6088Ra | TGTGATAGGGTTGAGAAG | 5,268 | 5,900 | 632 | 463 |
| 10F | YTRCTTCAAATGGGGTTTCC | 10R | AGAATTGTAACACTCGGG | 5,268 | 5,939 | 671 | 632 |
| mutS-F5 | ATTTAATTAAGAATCTCCAACTTCC | mutS-6979a | TATTAATGGGTGTCGGAG | 5,932 | 6,937 | 1,005 | 7 |
| mutS-6818Fa | CTAAGCTATTTTTWCCCC | mutS-R2 | TCTAAAGACTCATTAAGATAAACCC | 6,918 | 7,875 | 957 | 19 |
| 13R | CTGTTTCCAAGCCTACTT | 13F | CTATTTTAGGYTGGAAGAGA | 7,861 | 8,623 | 762 | 14 |
| 14R | TTTCCTCTTGAGACAGTA | 14F | ACTGGTGTAGTAAGACTA | 8,516 | 9,219 | 703 | 107 |
| octo2-H | CGATAAGAACTCTCCGACAATA | 15F | CAACTGAATGGCCGCGGTAA | 9,134 | 9,601 | 467 | 85 |
| octo1-L | AGACCCTATCGAGCTTTACTGG | nd2-R1 | GTTCAAGCTCTCCTGTGGAGCC | 9,343 | 10,394 | 1051 | 258 |
| nd2-1418R | ACATCGGGAGCCCACATA | 16S-647F | ACACAGCTCGGTTTCTATCTACAA | 9,772 | 10,552 | 780 | 622 |
| 16R | GCACGATAGATAATAGCGCA | 16F | TGGTGACACAGCTCGGTT | 9,791 | 10,590 | 799 | 761 |
| 17R | ATATTTGTTATTACTAAAGG | 17F | ATTRTTATTTAAAGTATCTG | 10,527 | 11,153 | 626 | 63 |
| 18R | TCCCAACCRATAAATARTTG | 18F | GTTTTTAACTAARTGGTATR | 11,043 | 11,709 | 666 | 110 |
| 19R | GCATGAATRATTGAGCCTGC | 19F | ATTCTACAAGTTATATGAGA | 11,605 | 12,323 | 718 | 104 |
| 20R | TATCATTAATGCATAATTAA | 20F | AGTTTATATCAYYTACTAAC | 12,299 | 13,051 | 752 | 24 |
| 21R | AACATTAAACTGAGCCGACT | 21F | TGTCTCTTATCGTACTATAG | 13,005 | 13,653 | 648 | 46 |
| 22R | TTTTATTATTAGTTAACCTTCATC | nad4-F3 | TTTTATTATTAGTTAACCTTCATC | 13,514 | 14,179 | 665 | 139 |
| 22R | GTACTAGTWGAAAAAGCAGC | nd4-13343Fa | AATAGGTTGGTTTGAGGG | 13,514 | 14,300 | 786 | 665 |
| co3bam567F | GCTGCTAGTTGGTATTGGCAT | 23F | ATGGTRTTTACTTTAGCTAA | 14,264 | 14,787 | 523 | 36 |
| 23R | GCTGCTAGTTGGTATTGGCA | 23F | ATGGTRTTTACTTTAGCTAA | 14,274 | 14,835 | 561 | 513 |
| 24R | TATCACCCTTATCATYTAGT | 24F | CTAAGARCCCCACCARTAAA | 14,772 | 15,508 | 736 | 63 |
| 25R | TCWACAGCTAAYAAGGGAAC | 25F | TGAAAATATARTACTGAGCC | 15,468 | 16,063 | 595 | 40 |
| siro-cox2-F1 | AGGCCCACTCTGTATATTTC | atp6-R2 | ATGTAGATTTAGAGTATCATTAATRTA | 15,588 | 16,291 | 703 | 475 |
| 26R | CATTAGSTATTAAAATGGAT | 26F | GTAAATACRTAGGGAAATAG | 15,524 | 16,597 | 1,073 | 767 |
| cox2-16530Fa | CCCCTAAAGATCACCACA | nd42599F | GCCATTATGGTTAACTATTAC | 16,582 | 17,397 | 815 | 15 |
| 27F | GAGTGATTAGCGCCACATAA | 27R | GGAGCCTATATCCTTGRGAT | 16,681 | 17,468 | 787 | 716 |
| REVNRnd6a | ATCGTTAGCGGGACATTATCAATT | coII-8068F | CCATAACAGGACTAGCAGCATC | 17,207 | 17,995 | 788 | 261 |
| nd6-F | TCCTTAGGAATAGTTGGAGCTAG | nd3-2126R | CACATTCATAGACCGACACTT | 17,935 | 18,600 | 665 | 60 |
| siro-nad6-R1 | ATTGCCCCTATGTTAGTTCTAG | 28R | CCAATCATTACTGGCATTAC | 18,304 | 233 | 982 | 296 |
| nd6-F REV | CTAGCTCCAACTATTCCTAAGGA | New NCR2R | ATGATCATCTCCTAACATACTACC | 18,774 | 162 | 585 | 162 |
| 9Fb | ATCCTTTAGTAACTCCTG | COII-8068F | CCATAACAGGACTAGCAGCATC | 4,531 | 5,123 | 593 | — |
| msh2806Rb | TAACTCAGCTTGAGAGTATGC | RevNrND6 | ATCGTTAGCGGGACATTATCAATT | 17,209 | 18,037 | 829 | — |
Note.—Unless otherwise noted, sequence numbers are based on mt genomes with konojoi gene arrangement, starting with cox1.
All primers are from previous research (Brugler and France 2008; Uda et al. 2011; Park et al. 2012, Figueroa and Baco 2014).
aPrimer pairs used for mt genomes with konojoi arrangement only.
bPrimer pairs used for mt genomes with japonicum arrangement only.
The overlapping PCR fragments were assembled using the software CLC Main Workbench 6.7.1 (CLC Bio, Aarhus, Denmark). Sequence quality was assessed by base quality scores and by visually inspecting each chromatogram. Annotation of each mitochondrial genome was done by alignment to all octocoral genomes available in GenBank (table 1) with the aid of the software CLC Main Workbench. The mt genomes were scanned for transfer ribonucleic acids (tRNAs) using the program tRNA scan-SE by Lowe and Eddy (1997).
Substitution Saturation Analysis
A hierarchical substitution saturation analysis was performed at varying taxonomic levels to determine the potential phylogenetic signal contained in the nucleotide sequences of the mitochondrial genomes. There were three steps to this analysis. First, transitions and transversions were plotted against divergence based on general time reversible (GTR) distances (a GTR model was selected as the best fitting evolutionary model by our phylogenetic analysis, see next section). Second, the statistical tests presented by Steel et al. (1993) were used to determine how many sequences in each data set were phylogenetically informative. And third, saturation indices were calculated using the method by Xia et al. (2003) to determine whether the genomes have experienced substitution saturation. All three steps were carried out with the software package DAMBE (Xia and Xie 2001). This analysis was repeated for five groupings of the overall data set: Octocorallia only, Hexacorallia only, Anthozoa (Octocorallia + Hexacorallia), Cnidaria (Anthozoa + Medusozoa), and Cnidaria + Porifera.
Phylogenetic Analysis
In addition to the four specimens used in this study, 82 mitochondrial genomes were obtained from GenBank and included in the phylogenetic analysis: 30 Octocorallia, 33 Hexacorallia, 7 Hydrozoa, 4 Scyphozoa, and 5 Porifera (table 1). The sequences for each gene and ribosomal RNA were aligned with MUSCLE (Edgar 2004) and then sequentially concatenated. The alignment was visually inspected for optimality. All phylogenetic analyses were performed with MEGA v5.05 (Tamura et al. 2011) using maximum-likelihood (ML) methods with bootstrap values from 10,000 replicates. A GTR model with gamma distribution and invariant sites (GTR + G + I) was selected by MEGA v5.05 as the best fitting model of molecular evolution based on the Akaike Information Criterion. Bayesian analyses were performed with MrBayes 3.1 (Ronquist and Huelsenbeck 2003) using a GTR + G + I model of evolution as selected by MrModeltest 2.2 (Nylander 2004). The chains were carried out for 5,000,000 generations, sampling every 500th generation. After inspecting the trace files generated by the Bayesian Markov chain Monte Carlo (MCMC) runs, we determined that the initial 25% (2,500) of sampled generations would be omitted.
For the phylogenetic reconstruction of Octocorallia, all 14 protein-coding genes, including the mutS gene, and 2 RNAs were used. For the phylogenetic reconstructions of both Anthozoa and Cnidaria, only 13 protein-coding genes were used. This is because the mutS gene is only found in octocorals and therefore could not be used in phylogenies above this taxonomic level. The two RNAs were also not included because they varied so much among higher taxa that homologous regions could not be accurately aligned.
Testing Phylogenetic Robustness
Because our inferences on gene order evolution within the Octocorallia rely heavily on their phylogeny, additional analyses were performed on this group to test the robustness of the reconstructed phylogeny. Starting with the alignment, the visual inspection for optimality was compared with alignment optimization using the software GBLOCKS 0.91b (Castresana 2000) using default settings with “Allowed GAP positions” set to “All.” The ML and Bayesian analyses, as described above, were repeated with the alignment selected by GBLOCKS. Because multiple coding genes were used, a partitioned phylogenetic analysis was also performed using PartitionFinder v1.1.1 (Lanfear et al. 2014) and RAxML v8.0.0 (Stamatakis 2014). To find the optimal ML tree with RAxML, 20 independent searches were performed with 1,000 bootstrap replicates. Data blocks were defined by each gene and codon position for the 14 protein-coding genes. Codon positions were not used for the two RNAs. Finally, four additional, independent Bayesian analyses were run using MrBayes 3.1 (Ronquist and Huelsenbeck 2003) with a GTR + G + I model of evolution as selected by MrModeltest 2.2 (Nylander 2004). The chains were carried out for 1,000,000 generations, sampling every 100th generation. The software AWTY (Wilgenbusch et al. 2004) was then used to test for convergence of the MCMC runs.
Results
Mitochondrial Genomes
Four new octocoral mitochondrial genomes were obtained. All four have similar lengths, from shortest to longest: 18,716 bp (Anthomastus sp. USNM# 1171062), 18,838 bp (N. hawaiinensis USNM# 1072109), 18,913 bp (Anthomastus sp. USNM# 1081145), and 19,044 bp (S. cauliflora USNM# 1122229). All 4 mt genomes contain 14 protein-coding genes (atp6, atp8, cox 1–3, cob, nad 1–6, nad4L, and mutS), 2 ribosomal RNAs (12s and 16s), and 1 transfer RNA. The A + T content in all four mt genomes is similar, ranging from 62.2% to 63.3%. The nucleotide lengths of all genes are similar for all four species.
Two gene arrangements were observed (fig. 1), both species of Anthomastus have the same arrangement as that discovered by Uda et al. (2011) in Corallium japonicum, further referred to as the “japonicum” arrangement; while N. hawaiinensis and S. cauliflora both have what is assumed to be the ancestral arrangement in octocorals (McFadden et al. 2006; Uda et al. 2011; Brockman and McFadden 2012) (fig. 1). In all 4 mitochondrial genomes, 7 of the genes either overlap or do not have a spacer between them, with the rest separated by a total of 12 intergenic spacers, ranging in size from 14 to 396 bp. Within the spacers, the two Anthomastus mt genomes and the Sibogagorgia mt genome have one pair of an inverted repeat sequence (fig. 2), identified previously in the mitochondrial genomes of C. japonicum and Pleurocorallium konojoi (Uda et al. 2011). In Anthomastus, these inverted repeat sequences are found in the intergenic regions between cob and cox2 genes and mutS and nad4L genes; while in Sibogagorgia, they are found in the intergenic regions between cob and nad6 genes, nad4L and mutS genes, and cox1 and cox2 genes (fig. 2).
Fig. 1.—

Mitochondrial gene arrangement based on Medina et al. (2006), Brugler and France (2008), Park et al. (2012), Uda et al. (2011), Figueroa and Baco (2014), and this study. Arrows show direction of replication. Thicker line shows heavy strand, thinner line shows light strand. (A) Presumed octocoral ancestral mt gene arrangement; (B) japonicum mt gene arrangement; and (C) konojoi mt gene arrangement. Taxa that have been shown to have these arrangements are listed within each arrangement. *Although Sibogagorgia cauliflora has the presumed ancestral gene order, it is not because it was conserved in this lineage but rather it reversed back from a different arrangement to this ancestral state, as explained in the text.
Fig. 2.—

Alignment of inverted repeat sequences present in all the Corallium, Paragorgia, Anthomastus, and Sibogagorgia mitochondrial genomes. These were first identified by Uda et al. (2011) and they occur in the intergenic spacers where gene inversions took place leading to the japonicum and konojoi mt gene arrangement. Panel A corresponds to spacer a and Panel B to spacer b shown in figures 5 and 7.
Substitution Saturation Analysis
Plots of transitions and transversions versus divergence based on GTR distances (fig. 3) show a linear relationship for the Octocorallia, with transitions always greater than transversions. For the Hexacorallia, the relationship between transversions and divergence is linear, while the relationship between transitions and divergence starts out linear and then levels off at higher divergences. Also, at these higher divergences transversions begin to surpass transitions. For the Anthozoa (Hexacorallia + Octocorallia) and the Cnidaria (Hexacorallia +Octocorallia + Medusozoa), the relationship between transitions and transversions versus divergence is comparable with that described above for the Hexacorallia. One exception is that in the Cnidaria transversions start to level off at higher divergences and transitions begin to lose their linear relationship and are surpassed by transversions at a lower divergence. When the Porifera are added to the Cnidaria data set (not shown in figure), the relationships are similar to that of the Cnidaria; however, the linearity of the relationship for both transitions and transversions is lost at even lower divergence levels.
Fig. 3.—

Transitions (s) and transversions (v) compared with GTR distance for four data sets: within the subclass Octocorallia, within the subclass Hexacorallia, within the class Anthozoa, and within the phylum Cnidaria (Anthozoa + Medusozoa).
The results for the substitution saturation index defined by Xia et al. (2003) are shown in figure 4. The test, as implemented by DAMBE, calculates a critical index for a symmetrical and an asymmetrical tree and compares it with the observed index (Iss). If the Iss observed value is higher than the Iss critical values, then the sequences will fail to recover the true phylogenetic relationships. The index shows that for the Octocorallia the observed Iss is lower than either of the critical values. For all the remaining data sets Hexacorallia, Anthozoa, Cnidaria, and Cnidaria + Porifera, the Iss observed is higher than either of the critical values. The statistical test by Steel et al. (1993) as implemented in DAMBE gives each sequence a φ score from 0 to 1 based on how phylogenetically informative that sequence is relative to what can be expected by chance. A score below 0.04 is considered as lacking phylogenetic information (Xia and Lemey 2009). These test results are summarized in figure 4 and show that for Octocorallia and Hexacorallia all the sequences are phylogenetically informative. For the Anthozoa only 21% of the sequences are phylogenetically informative, for the Cnidaria only 10%, and for the Cnidaria + Porifera only 13%.
Fig. 4.—

Substitution saturation tests for six data sets as implemented by DAMBE, based on Xia et al. (2003) and Steel et al. (1993). Graph shows Iss observed and Iss critical for both symmetrical and asymmetrical tree. If Iss observed is higher than Iss critical, then it means that the sequences have high substitution saturation and will fail to recover the phylogenetic signal. All differences are significant. The P value for all comparisons is 0.0000 except for the Iss observed versus Iss asymmetrical in Octocorallia, were the P value is 0.029. Below the graph is the average φ value from Steel et al.’s test for each data set. A value of less than 0.04 is considered to lack a phylogenetic signal (Xia and Lemey 2009). Sequences above this threshold are considered phylogenetically informative and are shown as a percentage.
One way to deal with possible substitution saturation is to translate nuclear sequences to amino acid sequences, and then reverse translate them back to nucleotide sequences using a universal code. This effectively gets rid of synonymous substitutions and it is a method used by Park et al (2012) for their study on cnidarian divergence times using whole mitochondrial genomes. Another option is to reconstruct phylogenies using the amino acid sequences themselves after translating nuclear sequences. This method was utilized by Kayal et al. (2012) for reconstructing the phylogeny of the Cnidaria. As part of our analysis, we used the amino acid alignment from Kayal et al. (2012) and reverse translated the alignment following the same procedures as Park et al. (2012). We performed both saturation tests on this data set. There was a marked improvement with respect to Iss scores for the Xia et al. (2003) test compared with our Cnidaria and Cnidaria + Porifera data set (fig. 4). But, it only passes the test if the tree is symmetrical while still failing the test if the resulting tree is asymmetrical. Although the Xia et al. (2003) substitution saturation test did show some improvement, the test by Steel et al (1993) showed that all the sequences in this new data set were lacking phylogenetic information and therefore any tree recovered could statistically be due to chance.
Octocorallia Phylogenetic Analysis
A total of 34 octocoral mitochondrial genomes were used in the octocoral phylogenetic analysis, using all 14 protein-coding sequences and the 2 ribosomal RNAs. Our original alignment was very similar to the alignment selected by GBLOCKS 0.91b where 98% of the original 18,398 bp were retained. Phylogenetic analyses were performed on both alignments and they yielded identical results. The same tree topology was obtained with both ML and Bayesian methods (five independent Bayesian analyses) and both methods resulted in well-supported branches (fig. 5). Analyses using the software AWTY showed convergence of all MCMC runs. All runs yielded identical topology and branch support. Both analyses were performed unrooted; once the tree was obtained, it was then redrawn with Briareum asbestinium as the root because this species is considered to be basal in the Octocorallia (McFadden et al. 2006; Brockman and McFadden 2012; Park et al. 2012). PartitionFinder v1.1.1 (Lanfear et al. 2014) divided the data into six partitions. The partitioned phylogenetic analysis performed with RAxML included 20 independent searches for the optimum ML tree with 1,000 bootstrap replicates. This also yielded the same phylogenetic tree with similar support for all branches with only one exception. Our original tree shows that the species Euplaxaura crassa and Pseudopterogogia bipinnata are sister taxa, while the partitioned analysis collapses this clade.
Fig. 5.—

Octocoral phylogenetic tree inferred by ML, based on all mitochondrial protein-coding genes and RNAs. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. Tree topology inferred by Bayesian methods is identical except for Hemicorallium imperiale USNM# 1072449 branches with Hemicorallium imperiale USNM# 1072448 in the Bayesian topology and with Hemicorallium laauense in the ML topology. Branch values correspond to bootstrap support for ML (first) and Bayesian posterior probabilities (second) for the nonpartitioned data. The third branch value corresponds to bootstrap support for ML as determined by RAxML with the partitioned data. *Support values less than 0.70. Clade numbers are labeled to correspond to the clade designations in McFadden et al. (2006). Coloring corresponds to the different mitochondrial gene orders as shown to the right of the phylogeny. The corresponding genes in each numbered box are given in the bottom panel of the diagram.
The tree shows two main clades, which we will refer to as Clade A and Clade B. Clade A contains members of the suborders Alcyoniina and Holaxonia and includes the same groups of taxa which fall into Clade 1 of McFadden et al. (2006). All members of the Clade A have the presumed ancestral octocoral mitochondrial gene arrangement. Clade B contains the other members of the order Pennatulacea and of the suborders Alcyoniina, Calcaxonia, and Scleraxonia. All four alternate gene arrangements are found in the members of Clade B. This clade splits into two clear subclades, Clade B(2), containing the Pennatulacea and Calcaxonia, and corresponding to Clade 2 of McFadden et al. (2006), and the second, Clade B(3) containing the Scleraxonia and the two Alcyoniina Paraminabea and Anthomoastus, corresponding to Clade 3 of McFadden et al. (2006). In Clade B(3), Paraminabea branches out first, then Anthomastus forms a sister branch with Paragorgiidae and Coralliidae. The Paragorgiidae is a paraphyletic taxon, because Sibogagorgia does not group with the Paragorgia, but rather forms a sister branch to the Coralliidae. The Coralliidae have two main branches, one leading to Corallium and Hemicorallium, all with the japonicum mitochondrial gene arrangement, while the other leading to Pleurocorallium, which have the konojoi mitochondrial gene arrangement.
Anthozoa and Cnidaria Phylogenetic Analysis
A total of 78 mitochondrial genomes were used for the phylogenetic reconstruction of the Anthozoa, 67 Anthozoa (34 Octocorallia and 33 Hexacorallia), and 11 Medusozoa (7 Hydrozoa and 4 Schyphozoa). Unlike the Octocorallia phylogenetic analysis, only 13 protein-coding genes were concatenated and aligned. The Octocorallian mutS gene was excluded as it is not present in any other taxa and the two RNAs were also excluded due to high levels of variation in large gaps in alignments above the subclass level, making it difficult for homologous regions to be aligned. Both ML and Bayesian methods resulted in similar tree topology with well-supported branches (fig. 6A). The medusozoans were included in this analysis as an outgroup for the Anthozoa and they form a distinct clade that divides into two branches, one containing the Hydrozoa and the other the Schyphozoa. There was no support for an Anthozoan clade. Instead, the Octocorallia and the Hexacorallia branched independently. The internal branching of the Octocorallia is similar to that of the previous analysis but some resolution has been lost including the collapse of some branches (tree not shown).
Fig. 6.—

Phylogenetic trees for the Anthozoa (A, unrooted) and Cnidaria + Porifera (B, unrooted and C, rooted by the Porifera) inferred by ML, based on all mitochondrial protein-coding genes, excluding RNAs and mutS. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. Tree topology inferred by Bayesian methods is identical. Branch values correspond to bootstrap support for ML (first) and Bayesian posterior probabilities (second). The number of taxa in each branch is shown in parenthesis.
The same mt genomes used in the Anthozoa analysis were used for the Cnidarian analysis with the addition of five Porifera as the outgroup. The mutS gene and the two RNAs were also omitted in this analysis. The phylogenetic reconstruction shows four distinct and well-supported clades: 1) The Porifera, 2) the Medusozoans, 3) the Hexacorallia, and 4) the Octocorallia (fig. 6B). When the tree is redrawn, using the Porifera as the root for the Cnidaria, the Hexacorallia form the first derived branch for this group, while the Medusozoans and Octocorallia form a second branch (fig. 6C).
Discussion
Phylogeny of the Octocorallia
Our phylogenetic reconstruction supports two major clades within the Octocorallia (fig. 5). One includes the Alcyoniina and Holaxonia, while the other divides into two branches, one composed of Pennatulacea and Calcaxonia, and the second with Anthomastus, Paragorgiidae, and Coralliidae. This largely agrees with the phylogeny proposed by McFadden et al. (2006) except that in their study the basal relationships between these three clades remain inconclusive, while here they are more supported. Their phylogenetic analysis is based on two mitochondrial genes, nad2 and mutS. When using maximum parsimony and Bayesian methods, their phylogeny show Clades A(1) Alcyoniina and Holaxonia and B(3) Anthomastus–Paragorgiidae–Coralliidae as sister clades, while their reconstruction using ML shows Clades B(2) Pennatulacea and Calcaxonia and B(3) Anthomastus–Paragorgiidae–Coralliidae as sister branches. Our phylogenetic reconstruction supports the latter. Using the entire mitochondrial genome provides robust support for an independent Clade A(1) Alcyoniina–Holaxonia. Clades B(2) Pennatulacea–Calcaxonia and B(3) Anthomastus–Paragorgiidae–Coralliidae have strong support as sister clades in the Bayesian analysis. Our ML analysis recovers the same relationships, but in this case the support for a sister relationship between Clades B(2) and B(3) is weaker.
The phylogenetic relationships within Clades A(1) Alcyoniina–Holaxonia and B(2) Pennatulacea–Calcaxonia are discussed at length by McFadden et al. (2006). The number of full mitochondrial genomes available for members of these two clades is limited, 12 for Clade A(1) and 5 for Clade B(2), when compared with the number of taxa used in McFadden et al. (2006) where there are 73 for the former and 24 for the latter. Therefore it will suffice to say that our limited data set for these two clades is congruent with that of McFadden et al. (2006) and we will defer further discussion to their study and the sequencing of further mt genomes. In the case of Clade B(3) Anthomastus–Paragorgiidae–Coralliidae, our study includes 16 members, while McFadden et al. (2006) only has 3. Our study shows that full mitochondrial genomes work well in resolving the phylogeny within this clade. Paraminabea is the basal member of this clade, followed by Anthomastus. In a recent taxonomic revision of Anthomastus based on morphology, it was suggested that this genus should be divided into at least three genera, Anthomastus, Heteropolypus, and Pseudoanthomastus (Molodtsova 2013). We support this taxonomic revision because Anthomastus ritteri, which has been revised by Molodstova (2013) as Heteropolypus ritteri, has the presumed octocoral ancestral gene order (Brockman and McFadden 2012), while our two morphospecies of what are presumably Anthomastus have a japonicum gene order. This genetic information supports at least two distinct lineages. Genetic support for the third lineage will have to wait until the full mitochondrial genomes of members of all three revised genera are sequenced.
After Anthomastus, the next branch in Clade B(3) is composed of Paragorgia. Paragorgia was erroneously thought to be a sister branch to the Coralliidae (Brockman and McFadden 2012; Uda et al. 2013; Figueroa and Baco 2014), but our results clearly show that the sister branch to the Coralliidae is Sibogagorgia. Both Paragorgia and Sibogagorgia currently belong to the family Paragorgiidae. Our phylogenetic analyses show that Paragorgia and Sibogagorgia are two independent lineages, making the Paragorgiidae a paraphyletic group. We propose that to fix this taxonomic inadequacy, the family Sibogagorgiidae, as suggested by Verseveldt (1942), should be resurrected for Sibogagorgia. Sibogagorgia was also found to be highly divergent in the analyses by Herrera et al (2010) based on mitochondrial genes. A less favorable alternative to make Paragorgiidae monophyletic would be to subsume the Coralliidae into the Paragorgiidae. The last branch in Clade B(3) has the members of the Coralliidae. The Coralliidae are clearly composed of three lineages, which support the recent split of Corallium into three genera, Corallium, Hemicorallium, and Pleurocorallium (Ardila et al. 2012; Figueroa and Baco 2014).
Mitochondrial Gene Order: Evidence of Reversal to an Ancestral State
The four mitochondrial genomes of N. hawaiinensis, S. cauliflora, and the two morphospecies of Anthomastus have the same compositional elements as the mitochondrial genomes of all 29 species of octocorals that have been published to date (fig. 1). There are five different gene arrangements that have been identified in the Octocorallia (Beaton et al. 1998; Brugler and France 2008; Uda et al. 2011; Brockman and McFadden 2012; Park et al. 2012). Our study shows that Anthomastus has the same mitochondrial gene arrangement as the one discovered in Paracorallium japonicum by Uda et al. (2011) and also shared by at least three species of Corallium (Figueroa and Baco 2014). Both N. hawaiinensis and S. cauliflora have the presumed ancestral mitochondrial gene order. However, despite having the presumed ancestral gene order, the presence of a pair of inverted repeat sequences in the spacer regions of S. cauliflora suggest that this apparent ancestral mitochondrial gene arrangement was not conserved in this species but rather evolved back to its ancestral state after going through a rearrangement (fig. 7).
Fig. 7.—

Theoretical origin of inverted repeat sequences a and b in Sibogagorgia. There are two possible scenarios. In Scenario A, the konojoi arrangement arises first, creating the two inverted repeat sequences; these are conserved in the subsequent evolution of the japonicum arrangement and in the return to an ancestral state in Sibogagorgia. In Scenario B, the japonicum arrangement arises first creating the inverted repeats; these are conserved in the subsequent evolution of the japonicum arrangement and in the return to an ancestral state in Sibogagorgia.
The inverted repeat sequences were first identified by Uda et al. (2011) in the mitochondrial genomes of both P. japonicum and Corallium konojoi and have since been identified in several other species of Corallium and Paragorgia (Figueroa and Baco 2014). The origin of these inverted repeat sequences are discussed in detail in Uda et al. (2011). The authors suggest two possible pathways for the origin of these inverted repeat sequences, one is going from a presumed ancestral mitochondrial gene arrangement to a japonicum arrangement, and the other is going from the presumed ancestral arrangement to a konojoi arrangement. Either pathways result in inversions leading to the inverted repeat sequences in the intergenic spacer regions that carry part of the gene to which they were previously adjacent. Uda et al. (2011) clearly show that the only way these inverted repeat spacer sequences can form is to go through either the konojoi or japonicum rearrangements. Therefore because these inverted repeat sequences are present in the mitochondrial genome of S. cauliflora, which has the presumed ancestral gene order, it suggests that the gene arrangement in this taxon is not an indication of a conserved ancestral state but rather that the gene order evolved back to the ancestral state from either a konojoi or a japonicum arrangement.
This is the first observation that shows that in the Octocorallia, mitochondrial gene arrangement is not only diverse but it can evolve back to an ancestral state. This has important implications for genetic studies that use gene boundaries to determine the type of mitochondrial gene arrangement present and then use that information for classification or phylogenetic purposes. This practice of testing gene boundaries has been referred to as “gene junction screening” (Brockman and McFadden 2012). If this were done with S. cauliflora, it would show that it has the ancestral gene arrangement and lead to the erroneous conclusion that Sibogagorgia is basal to Paragorgia and Coralliidae because those taxa have derived mitochondrial gene arrangements. But this is not the case, by analyzing the complete mitochondrial genome, including intergenic spacers, it is clear that the gene arrangement in S. cauliflora is also derived and has evolved back to an ancestral gene order. Therefore we recommend for future studies of gene rearrangements not to rely exclusively on gene junction screening as it will miss reversals to ancestral states.
Evolution of Mitochondrial Gene Arrangements
Our phylogenetic analysis shows that within the Clade B of the Octocorallia, mitochondrial gene order has changed at least six times. The first change occurs in the basal branch of this clade from the presumed ancestral gene order to the unique order shared by Keratoisidinae sp. and Acanella eburnea (fig. 5). The second change comes in the basal branch for Clade B(3) (fig. 5), going from the presumed ancestral gene order to the unique arrangement found in Paraminabea aldersladei. Paraminabea aldersladei is the sister branch to the rest of the members of Clade B(3) where presumably the ancestral gene order was maintained. From this point, there are three equally plausible scenarios for the evolution of the japonicum and konojoi gene order and the return to an ancestral state in Sibogagorgia (fig. 8).
Fig. 8.—
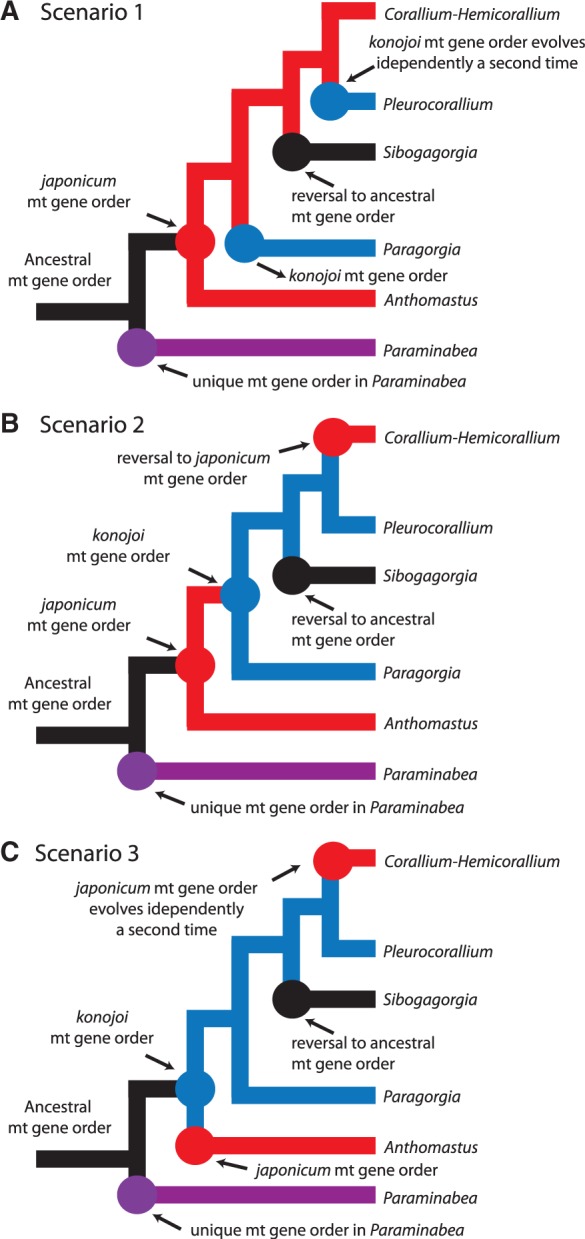
Three possible evolutionary pathways for the different mitochondrial gene orders found in Clade B(3) of the Octocorallia. All three scenarios are equally plausible in terms of the number of evolutionary steps needed. The tree is not drawn to scale. Arrows point to nodes where a particular gene order evolved. Branches are color coded for the gene orders, black for ancestral, purple for Paraminabea, red for japonicum, and blue for konojoi. Panel A shows Scenario 1 where the japonicum arrangement evolves first and it is conserved throughout going back to an ancestral state in Sibogagorgia and with the konojoi order evolving twice independently. Panel B shows Scenario 2 where the japonicum order also evolves first, but it is not conserved. Instead, the konojoi order evolves right and it is conserved afterwards, going back to an ancestral state in Sibogagorgia and with the japonicum order evolving independently a second time in the Corallium–Hemicorallium clade. Panel C shows Scenario 3 where the konojoi order evolves first and it is conserved throughout, going back to an ancestral order in Sibogagorgia and with the japonicum order evolving twice independently.
In the first scenario, the third change occurs from the presumed ancestral gene order to the japonicum gene order (fig.8A and B) found in the two morphospecies of Anthomastus. The japonicum gene order is maintained and conserved through to the Corallium and Hemicorallium clade, while the konojoi gene order arises independently twice, once in Paragorgia and a second time in the Pleurocorallium (fig. 8A). In Sibogagorgia, it returns to an ancestral order from a japonicum arrangement (fig. 8A). In the second scenario, the japonicum gene order also evolves first from the presumed ancestral gene order, but then is only conserved in the Anthomastus clade, while the konojoi emerges as ancestral to the remaining branches (fig. 8B) and is therefore conserved in Paragorgia and Pleurocorallium. In this scenario in the Corallium–Hemicorallium clade, the gene order reverses to the japonicum arrangement and Sibogagorgia returns to the ancestral gene order from a konojoi arrangement (fig. 8B).
In the third scenario, the konojoi gene order evolves first (fig. 8C). The konojoi order is maintained throughout the main branch and conserved through to Pleurocorallium. In this case, the japonicum arrangement evolves independently, once in the Anthomastus and a second time in the Corallium–Hemicorallium clade and Sibogagorgia goes back to an ancestral state from a konojoi arrangement.
All three of these possible scenarios have the same number of evolutionary steps and in all three, one of the gene orders, japonicum or konojoi, had to evolve twice. Previous studies have also tried to determine the sequence of evolutionary events leading to these gene arrangements in Clade B (Uda et al. 2011; Brockman and McFadden 2012). Our present study agrees with some of their conclusions but there are several key differences. Uda et al. (2011) suggest two possible mechanisms by which the japonicum and konojoi mt gene orders arose. Their favored mechanism involves tandem duplication by slipped-strand mispairing followed by a random loss of genes and inversion by intramitochondrial recombination. This mechanism leads to the japonicum gene arrangement first and the konojoi arrangement second.
Brockman and McFadden (2012) also lend support to a japonicum mt gene arrangement evolving first, but their proposed mechanism of inversions leading to the japonicum arrangement cannot explain the creation of the inverted repeat sequences observed in all these taxa. They sequenced the full mitochondrial genome of Pa. aldersladei (family Alcyoniidae), discovering the fifth novel gene arrangement in octocorals. Then they proceed to map the five different arrangements onto a phylogeny of the Octocorallia based on two mitochondrial genes (mutS and cox1) and a nuclear gene (28S). Their phylogeny shows that the japonicum gene arrangement evolved first, before the konojoi arrangement, in the branch leading to the Coralliidae and Paragorgiidae. They present Paracorallium (now subsumed into Corallium; Ardila et al. 2012), which has the japonicum gene arrangement, as the sister branch to Paragorgia and C. konojoi and Corallium kishinouyei (the genus Pleurocorallium has been resurrected for these species; Figueroa and Baco 2014), which have the konojoi gene arrangement. Furthermore, they show that Anthomastus is the sister branch to the Coralliidae and Paragorgiidae clade. And by using gene junction screening, they determine that A. ritteri has the presumed ancestral octocoral mitochondrial gene order.
Our analysis agrees with that of Brockman and McFadden (2012) in placing Anthomastus as the sister branch to the Paragorgiidae and Coralliidae, but it differs in that the two morphospecies of Anthomastus used in our study have the japonicum gene arrangement, while the species of Anthomastus used by Brockman and McFadden (2012) has the presumed ancestral gene arrangement. Because Brockman and McFadden (2012) only used gene junction screening to determine the mitochondrial gene arrangement of A. ritteri, the possibility remains that instead of being an example of conserved mitochondrial gene order, this particular species of Anthomastus could have reverted back to the ancestral state as it happened with S. cauliflora. So far, every species of octocoral belonging to McFadden et al.’s (McFadden et al. 2006) Anthomastus–Corallium clade, which also include the Paragorgiidae (Figueroa and Baco 2014), has a derived mitochondrial gene arrangement, except for A. ritteri (Brockman and McFadden 2012). Therefore it would be interesting to sequence the full mitochondrial genome of A. ritteri, because if it truly has a conserved ancestral mitochondrial gene order then it is likely a basal member of this major octocoral clade.
Further research is needed to determine the evolutionary order of the mitochondrial gene arrangement in this Anthomastus–Corallidae–Paragorgiidae clade. Although Uda et al. (2011) and Brockman and McFadden (2012) support a japonicum gene arrangement evolving before the konojoi arrangement, our present research shows that this is not necessarily the case because each major branch in this clade has its own unique arrangement with possible reversals to ancestral states and with at least one of these arrangements evolving in two independent events. Therefore it is very likely that when the full mitochondrial genomes are sequenced from more members of this clade, more unique gene orders will be found and possibly more reversals to ancestral states will also be identified.
Mitochondrial Genomes and Higher Level Phylogenies within Cnidaria
The class Anthozoa consists of two subclasses, the Hexacorallia and the Octocorallia (Daly et al. 2007). The monophyly of Anthozoa is well supported by both morphological and molecular phylogenetic reconstructions based on nuclear genes (France et al. 1996; Odorico and Miller 1997; Berntson et al. 1999; Won et al. 2001; Collins 2002; Daly et al. 2007). However, recent studies based on whole mitochondrial genomes disagree with this observation and suggest that Anthozoa is paraphyletic because in their phylogenetic reconstructions, the Octocorallia is more closely related to the Medusozoa than to the Hexacorallia (Shao et al. 2006; Kayal and Lavrov 2008; Lavrov et al. 2008; Park et al. 2012; Kayal et al. 2013). In our phylogenetic reconstruction, where members of the Porifera were included with the Cnidaria, the resulting unrooted phylogeny shows that the Porifera are a sister branch to the Hexacorallia. If this tree is redrawn and rooted by the Porifera, then the resulting phylogeny appears as if the Hexacorallia are the basal branch to the Cnidaria with the Octocorallia branching later, as a sister clade to the Medusozoa. This is the same pattern observed by those studies that suggest that the Anthozoa is paraphyletic (Shao et al. 2006; Kayal and Lavrov 2008; Lavrov et al. 2008; Park et al. 2012; Kayal et al. 2013). This suggests that the close association between the Octocorallia and the Medusozoa is likely an artifact due to the use of Porifera as a root for the Cnidaria. This observation is further supported by our phylogenetic analysis that only included the Octocorallia, Hexacorallia, and Medusozoa. This phylogeny clearly shows that based on whole mitochondrial genomes, no assertion can be made whether the Octocorallia belong to the Hexacorallia or the Medusozoa. Each of these taxa form an independent well-supported branch.
Because of the mismatch in previous studies between phylogenies based on whole mitochondrial genomes compared with nuclear and morphological data (France et al. 1996; Odorico and Miller 1997; Berntson et al. 1999; Won et al. 2001; Collins 2002; Shao et al. 2006; Daly et al. 2007; Kayal and Lavrov 2008; Lavrov et al. 2008; Park et al. 2012; Kayal et al. 2013), we explored the possibility of saturation in the mitochondrial sequences that have been used for Cnidaria. Our phylogenetic reconstruction and substitution saturation analysis show that whole mitochondrial genomes can be used effectively for phylogenetic analyses of the Octocorallia. However, it appears that the utility of mt genomes at higher taxonomic levels is limited (figs. 3 and 4).
It has been shown that when substitution saturation is high, similarity between sequences does not accurately reflect phylogenetic relationships (Steel et al. 1993; Xia et al. 2003; Xia and Lemey 2009). Sequences that have not experienced substantial substitution saturation will show a linear relationship for both transitions and transversions versus sequence divergence; also, transitions will occur more often than transversions (Xia and Xie 2001). This relationship is found in the Octocorallia, but it starts to break down in the Hexacorallia and it deviates even further at higher taxonomic levels with the Anthozoa and Cnidaria. This suggests that at higher taxonomic levels the phylogenetic signal in mitochondrial genomes may be lost due to substitution saturation. The statistical tests proposed by Steel et al. (1993) support this observation. These tests showed that when only the Octocorallia or the Hexacorallia are considered, all the sequences are phylogenetically informative. But, when higher taxonomic levels are considered, such as Anthozoa and Cnidaria, more than 80% of the sequences are no longer phylogenetically informative. This clearly shows that the nucleotide sequences of mitochondrial genomes at the Anthozoan and Cnidarian taxonomic level have experienced full substitution saturation and therefore are no longer phylogenetically informative.
To minimize the problem generated by substitution saturation, nucleotide sequences can be translated into amino acid sequences; then they can be translated back into a nucleotide sequence using a standard genetic code, essentially getting rid of any synonymous substitutions. This was done by Park et al. (2012) when using full mitochondrial genomes to look at Cnidarian evolution using the Porifera as a root. Because the alignment by Park et al. (2012) is not available on an online repository, we used the amino acid alignment from Kayal et al. (2013) and followed the methods of Park et al. (2012) to reverse translate this alignment to a nucleotide alignment. The alignment by Kayal et al. (2013) includes all the sequences used by Park et al. (2012) plus many more obtained in that study. We analyzed this new data set for substitution saturation using the tests developed by Xia et al. (2003) and Steel et al. (1993). Xia’s test showed that the observed saturation index is lower than the critical saturation index if the resulting tree is symmetrical, but it is still higher if the tree is asymmetrical (fig. 4). The phylogeny presented by both Park et al. (2012) and Kayal et al. (2013) is highly asymmetrical, which suggests that despite eliminating synonymous substitutions from the analysis, substitution saturation was still a problem for analyzing the Cnidaria using reverse-translated nucleotide sequences. The inadequacy of these reverse-translated nucleotide sequences for reconstructing the phylogeny of the Cnidaria is further supported by Steel’s test which shows that none of the sequences are phylogenetically informative. Therefore, the nucleotide sequences of mitochondrial genomes should not be used to determine phylogenetic relationships for the Anthozoa or the Cnidaria. Kayal et al. (2013) address the issue of nucleotide saturation by removing the third codon position as well as all codons for arginine, leucine, and serine. Additionally, they use amino acid sequences to reconstruct their phylogeny of Cnidaria using the best evolutionary models available to reduce the effects of saturation. Unfortunately, saturation tests for their nucleotide alignments are not presented and no such tests exist for amino acid alignments. Therefore, although they go through great lengths to compensate for saturation, whether their methods were enough will likely go unanswered until other molecular markers are used to reconstruct the phylogeny of Cnidarians. The dubious association of the Porifera as a sister branch of the Hexacorallia and the resulting appearance of the Octocorallia forming a clade with the Medusozoa could just be an artifact of substitution saturation in the mitochondrial genomes of these taxa. Therefore we recommend that for phylogenetic reconstruction at taxonomic levels higher than subclass within the Cnidaria, nuclear genes will be required, even when whole mitochondrial genomes are available.
Conclusions
Our phylogenetic reconstruction supports two major clades within the Octocorallia. One includes the Alcyoniina and Holaxonia, while the other divides into two branches, one composed of Pennatulacea and Calcaxonia, and the second with Anthomastus, Paragorgiidae, and Coralliidae. Our phylogeny also shows that Paragorgia and Sibogagorgia are two independent lineages, making the Paragorgiidae a paraphyletic group. We propose that to fix this taxonomic inadequacy, the family Sibogagorgiidae should be resurrected.
Our study is the first to show that in the Octocorallia, mitochondrial gene arrangement is not only diverse but it can evolve back to an ancestral state. This has important implications for genetic studies that use gene boundaries to determine the type of mitochondrial gene arrangement present and then use that information for classification or phylogenetic purposes. Therefore we recommend for future studies of gene rearrangements not to rely exclusively on gene junction screening as it will miss reversals to ancestral states.
Further research is needed to determine the evolutionary order of the mitochondrial gene arrangement in the Anthomastus–Corallidae–Paragorgiidae clade. Our study shows that each major branch in this clade has its own unique arrangement with possible reversals to ancestral states and with at least one of these arrangements evolving in two independent events.
Our phylogenetic reconstruction and substitution saturation analysis demonstrates that whole mitochondrial genomes can be used effectively for phylogenetic analyses of the Octocorallia. However, the utility of mt genomes at higher taxonomic levels is limited. Therefore we recommend that for phylogenetic reconstruction at taxonomic levels higher than subclass within the Cnidaria, nuclear genes will be required, even when whole mitochondrial genomes are available.
Acknowledgments
The authors would like to thank the crew of the RV Ka’Imikai-O-Kanaloa and the RV Revelle, as well as the pilots of the Pisces V submersible and the Jason II for their support of collections at sea. Collections were funded by the following grants to A.R.B: NOAA NURC West Coast and Polar Regions grant UAF 040118, NOAA Office of Ocean Exploration grant NA03OAR4600110, and a grant from the Hawaii Undersea Research Laboratory for exploration of Necker in 2011. Laboratory analyses and D.F.F. were supported by startup funds from Florida State University to A.R.B.
Literature Cited
- Ardila N, Giribet G, Sanchez J. A time-calibrated molecular phylogeny of the precious corals: reconciling discrepancies in the taxonomic classification and insights into their evolutionary history. BMC Evol Biol. 2012;12:246. doi: 10.1186/1471-2148-12-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baco AR. Exploration for deep-sea corals on North Pacific seamounts and islands. Oceanography. 2007;20:108–117. [Google Scholar]
- Baillon S, Hamel J, Wareham VE, Mercier A. Deep cold-water corals as nurseries for fish larvae. Front Ecol Environ. 2012;10:351–356. [Google Scholar]
- Beaton MJ, Roger AJ, Cavalier-Smith T. Sequence analysis of the mitochondrial genome of Sarcophyton glaucum: conserved gene order among octocorals. J Mol Evol. 1998;47:697–708. doi: 10.1007/pl00006429. [DOI] [PubMed] [Google Scholar]
- Berntson EA, Bayer FM, McArthur AG, France SC. Phylogenetic relationships within the Octocorallia (Cnidaria: Anthozoa) based on nuclear 18S rRNA sequences. Mar Biol. 2001;138:235–246. [Google Scholar]
- Berntson EA, France SC, Mullineaux LS. Phylogenetic relationships within the class Anthozoa (phylum Cnidaria) based on nuclear 18S rDNA sequences. Mol Phylogenet Evol. 1999;13:417–433. doi: 10.1006/mpev.1999.0649. [DOI] [PubMed] [Google Scholar]
- Brockman SA, McFadden CS. The mitochondrial genome of Paraminabea aldersladei (Cnidaria: Anthozoa: Octocorallia) supports intramolecular recombination as the primary mechanism of gene rearrangement in octocoral mitochondrial genomes. Genome Biol Evol. 2012;4:994–1006. doi: 10.1093/gbe/evs074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugler MR, France SC. The mitochondrial genome of a deep-sea bamboo coral (Cnidaria, Anthozoa, Octocorallia, Isididae): genome structure and putative origins of replication are not conserved among octocorals. J Mol Evol. 2008;67:125–136. doi: 10.1007/s00239-008-9116-2. [DOI] [PubMed] [Google Scholar]
- Buhl-Mortensen L, et al. Biological structures as a source of habitat heterogeneity and biodiversity on the deep ocean margins. Mar Ecol. 2010;31:21–50. [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Clark MR, Rowden AA. Effect of deepwater trawling on the macro-invertebrate assemblages of seamounts on the Chatham Rise, New Zealand. Deep Sea Res Part I. 2009;56:1540–1554. [Google Scholar]
- Collins AG. Phylogeny of Medusozoa and the evolution of cnidarian life cycles. J Evol Biol. 2002;15:418–432. [Google Scholar]
- Daly M, et al. The phylum Cnidaria: a review of phylogenetic patterns and diversity 300 years after Linnaeus. Zootaxa. 2007;182:127–128. [Google Scholar]
- DeVogelaere A. Deep-sea corals and resource protection at the Davidson Seamount, California, U.S.A. In: Freiwald A, Roberts JM, editors. Cold-water corals and ecosystems. Berlin Heidelberg (Germany): Springer; 2005. pp. 1189–1198. [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa DF, Baco AR. Complete mitochondrial genomes elucidate phylogenetic relationships of the deep-sea octocoral families Coralliidae and Paragorgiidae. Deep Sea Res Part II. 2014;99:83–91. [Google Scholar]
- France SC, Rosel PE, Agenbroad JE, Mullineaux LS, Kocher T. DNA sequence variation of mitochondrial large-subunit rRNA provides support for a two-subclass organization of the Anthozoa (Cnidaria) Mol Mar Biol Biotechnol. 1996;5:15–28. [PubMed] [Google Scholar]
- Genin A, Dayton PK, Lonsdale PF, Spiess FN. Corals on seamount peaks provide evidence of current acceleration over deep-sea topography. Nature. 1986;322:59–61. [Google Scholar]
- Genin A, Paull CK, Dilon WP. Anomalous abundances of deep-sea fauna on a rocky bottom exposed to strong currents. Deep Sea Res. 1992;39:293–302. [Google Scholar]
- Grigg RW. Recruitment limitation of a deep benthic hard-bottom octocoral population in the Hawaiian Islands. Mar Ecol Prog Ser. 1988;45:121–126. [Google Scholar]
- Guinotte JM, et al. Will human-induced changes in seawater chemistry alter the distribution of deep-sea scleractinian corals? Front Ecol Environ. 2006;4:141–146. [Google Scholar]
- Hecker B. Variation in megafaunal assemblages on the continental margin south of New England. Deep Sea Res Part A. 1990;37:37–57. [Google Scholar]
- Hein JR. Cobalt-rich ferromanganese crusts: global distribution, composition, origin and research activities. ISA Technical Study. 2002;2:36–89. [Google Scholar]
- Hein JR, Conrad TA, Dunham RE. Seamount characteristics and mine-site model applied to exploration- and mining-lease-block selection for cobalt-rich ferromanganese crusts. Mar Georesour Geotechnol. 2009;27:160–176. [Google Scholar]
- Herrera S, Baco A, Sánchez JA. Molecular systematics of the bubblegum coral genera (Paragorgiidae, Octocorallia) and description of a new deep-sea species. Mol Phylogenet Evol. 2010;55:123–135. doi: 10.1016/j.ympev.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Jones CG, Lawton JH, Shachak M. Organisms as ecosystem engineers. Oikos. 1994;69:373–386. [Google Scholar]
- Kayal E, Lavrov DV. The mitochondrial genome of Hydra oligactis (Cnidaria, Hydrozoa) sheds new light on animal mtDNA evolution and cnidarian phylogeny. Gene. 2008;410:177–186. doi: 10.1016/j.gene.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Kayal E, Roure B, Philippe H, Collins A, Lavrov D. Cnidarian phylogenetic relationships as revealed by mitogenomics. BMC Evol Biol. 2013;13:5. doi: 10.1186/1471-2148-13-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayal E, et al. Evolution of linear mitochondrial genomes in medusozoan cnidarians. Genome Biol Evol. 2012;4:1–12. doi: 10.1093/gbe/evr123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koslow JA, et al. Seamount benthic macrofauna off southern Tasmania: community structure and impacts of trawling. Mar Ecol Prog Ser. 2001;213:111–125. [Google Scholar]
- Krieger KJ. Coral (Primnoa) impacted by fishing gear in the Gulf of Alaska. In: Wilson JHM, et al., editors. Proceedings of the first international symposium on deep-sea corals. Halifax (NS): Ecology Action Centre; 2001. pp. 106–116. [Google Scholar]
- Lanfear R, Calcott B, Kainer D, Mayer C, Stamatakis A. Selecting optimal partitioning schemes for phylogenomic datasets. BMC Evol Biol. 2014;14:82–96. doi: 10.1186/1471-2148-14-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavrov DV, Wang X, Kelly M. Reconstructing ordinal relationships in the Demospongiae using mitochondrial genomic data. Mol Phylogenet Evol. 2008;49:111–124. doi: 10.1016/j.ympev.2008.05.014. [DOI] [PubMed] [Google Scholar]
- Leverette TL, Metaxas A. Predicting habitat for two species of deep-water coral on the Canadian Atlantic continental shelf and slope. In: Freiwald A, Roberts JM, editors. Cold-water corals and ecosystems. Berlin Heidelberg (Germany): Springer; 2005. pp. 467–479. [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden CS, France SC, Sanchez JA, Alderslade P. A molecular phylogenetic analysis of the Octocorallia (Cnidaria: Anthozoa) based on mitochondrial protein-coding sequences. Mol Phylogenet Evol. 2006;41:513–527. doi: 10.1016/j.ympev.2006.06.010. [DOI] [PubMed] [Google Scholar]
- McFadden CS, Sanchez JA, France SC. Molecular phylogenetic insights into the evolution of Octocorallia: a review. Integr Comp Biol. 2010;50:389–410. doi: 10.1093/icb/icq056. [DOI] [PubMed] [Google Scholar]
- Medina M, Collins AG, Takaoka TL, Kuehl JV, Boore JL. Naked corals: skeleton loss in Scleractinia. Proc Nat Acad Sci U S A. 2006;103:9096–9100. doi: 10.1073/pnas.0602444103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molodtsova TN. Deep-sea mushroom soft corals (Octocorallia: Alcyonacea: Alcyoniidae) of the Northern Mid-Atlantic Ridge. Mar Biol Res. 2013;9:488–515. [Google Scholar]
- Morgan LE, Tsao CF, Guinotte JM. Status of deep sea corals in US waters. Bellevue (WA): Marine Conservation Biology Institute; 2006. [Google Scholar]
- Mortensen PB, Buhl-Mortensen L. Deep-water corals and their habitats in The Gully, a submarine canyon off Atlantic Canada. Cold-Water Corals and Ecosystems Erlangen Earth Conference Series. 2005. pp. 247–277. [Google Scholar]
- Nylander JAA. MrModeltest v2. Program distributed by the author. Uppsala (Sweden): Evolutionary Biology Centre, Uppsala University; 2004. [Google Scholar]
- Odorico DM, Miller DJ. Internal and external relationships of the Cnidaria: implications of primary and predicted secondary structure of the 5′-end of the 23S-like rDNA. Proc Biol Sci. 1997;264:77–82. doi: 10.1098/rspb.1997.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, et al. Estimation of divergence times in cnidarian evolution based on mitochondrial protein-coding genes and the fossil record. Mol Phylogenet Evol. 2012;62:329–345. doi: 10.1016/j.ympev.2011.10.008. [DOI] [PubMed] [Google Scholar]
- Parrish FA, Baco AR. State of deep coral ecosystems in the U. S. Pacific Islands region: Hawaii and the U. S. Pacific Territories. In: Lumsden SE, Horigan TF, Bruckner AW, Dorr G, editors. The state of deep coral ecosystems of the United States: 2007. Maryland, USA: NOAA Tech. Memo. CRCP-3, Silver Spring, Maryland, USA; 2007. pp. 165–194. [Google Scholar]
- Probert PK, McKnight DG, Grove SL. Benthic invertebrate bycatch from a deep-water trawl fishery, Chatham Rise, New Zealand. Aquat Conserv Mar Freshwater Ecosyst. 1997;7:27–40. [Google Scholar]
- Roark EB, GuiIderson TP, Dunbar RB, Fallon SJ, Mucciarone DA. Extreme longevity in proteinaceous deep-sea corals. Proc Natl Acad Sci U S A. 2009;106:5204–5208. doi: 10.1073/pnas.0810875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roark EB, Thomas PG, Robert BD, Ingram BL. Radiocarbon-based ages and growth rates of Hawaiian deep-sea corals. Mar Ecol Prog Ser. 2006;327:1–14. [Google Scholar]
- Roberts JM, Wheeler A, Friewald A, Cairns S. Cold-water corals: the biology and geology of deep-sea coral habitats. Cambrige (United Kingdom): Cambrige University Press; 2010. [Google Scholar]
- Rogers AD. The biology of seamounts. Adv Mar Biol. 1994;30:305–354. doi: 10.1016/bs.amb.2018.06.001. [DOI] [PubMed] [Google Scholar]
- Rogers AD, Baco AR, Griffiths A, Hart H, Hall-Spencer T. Corals on seamounts. In: Pitcher TJ, et al., editors. Seamounts: ecology, conservation and management. Fish and aquatic resources series. Oxford (United Kingdom): Blackwell; 2007. pp. 141–169. [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Sanchez JA, McFadden CS, France SC, Lasker HR. Molecular phylogenetic analyses of shallow-water Caribbean octocorals. Mar Biol. 2003;142:975–987. [Google Scholar]
- Shao Z, Graf S, Chaga OY, Lavrov DV. Mitochondrial genome of the moon jelly Aurelia aurita (Cnidaria, Scyphozoa): a linear DNA molecule encoding a putative DNA-dependent DNA polymerase. Gene. 2006;381:92–101. doi: 10.1016/j.gene.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel MA, Lockhart PJ, Penny D. Confidence in evolutionary trees from biological sequence data. Nature. 1993;364:440–442. doi: 10.1038/364440a0. [DOI] [PubMed] [Google Scholar]
- Stocks K. Seamount invertebrates: composition and vulnerability. In: Morato T, Pauly D, editors. Seamounts: biodiversity and fisheries. Vancouver (BC): University of British Columbia; 2004. pp. 17–24. [Google Scholar]
- Sun Z, Hamel J, Edinger E, Mercier A. Reproductive biology of the deep-sea octocoral Drifa glomerata in the Northwest Atlantic. Mar Biol. 2010;157:863–873. [Google Scholar]
- Tamura K, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uda K, et al. Complete mitochondrial genomes of two Japanese precious corals, P. japonicum and C. konojoi (Cnidaria, Octocorallia, Coralliidae): notable differences in gene arrangement. Gene. 2011;476:27–37. doi: 10.1016/j.gene.2011.01.019. [DOI] [PubMed] [Google Scholar]
- Uda K, et al. Complete mitochondrial genomes of the Japanese pink coral (Corallium elatius) and the Mediterranean red coral (Corallium rubrum): a reevaluation of the phylogeny of the family Coralliidae based on molecular data. Comp Biochem Physiol Part D Genomics Proteomics. 2013;8:209–219. doi: 10.1016/j.cbd.2013.05.003. [DOI] [PubMed] [Google Scholar]
- Verseveldt J. Further studies on Octocorallia. Zoologische Mededelingen. 1942;24:159–186. [Google Scholar]
- Wilgenbusch JC, Warren DL, Swofford DL. AWTY: a system for graphical exploration of MCMC convergence in Bayesian phylogenetic inference. 2004. [cited 2005 August 1]. Available from: http://ceb.csit.fsu.edu/awty. [DOI] [PubMed] [Google Scholar]
- Williams A, et al. Seamount megabenthic assemblages fail to recover from trawling impacts. Mar Ecol. 2010;31:183–199. [Google Scholar]
- Won J, Rho B, Song J. A phylogenetic study of the Anthozoa (phylum Cnidaria) based on morphological and molecular characters. Coral Reefs. 2001;20:39–50. [Google Scholar]
- Xia X, Lemey P. Assessing substitution saturation with DAMBE. In: Lemey P, Salemi M, Vandamme AM, editors. The phylogenetic handbook: a practical approach to DNA and protein phylogeny. 2nd. Cambridge (United Kingdom): Cambridge University Press; 2009. pp. 615–630. [Google Scholar]
- Xia X, Xie Z. DAMBE: software package for data analysis in molecular biology and evolution. J Hered. 2001;92:371–373. doi: 10.1093/jhered/92.4.371. [DOI] [PubMed] [Google Scholar]
- Xia X, Xie Z, Salemi M, Chen L, Wang Y. An index of substitution saturation and its application. Mol Phylogenet Evol. 2003;26:1–7. doi: 10.1016/s1055-7903(02)00326-3. [DOI] [PubMed] [Google Scholar]