

Abstract
Cactophilic Drosophila species provide a valuable model to study gene–environment interactions and ecological adaptation. Drosophila buzzatii and Drosophila mojavensis are two cactophilic species that belong to the repleta group, but have very different geographical distributions and primary host plants. To investigate the genomic basis of ecological adaptation, we sequenced the genome and developmental transcriptome of D. buzzatii and compared its gene content with that of D. mojavensis and two other noncactophilic Drosophila species in the same subgenus. The newly sequenced D. buzzatii genome (161.5 Mb) comprises 826 scaffolds (>3 kb) and contains 13,657 annotated protein-coding genes. Using RNA sequencing data of five life-stages we found expression of 15,026 genes, 80% protein-coding genes, and 20% noncoding RNA genes. In total, we detected 1,294 genes putatively under positive selection. Interestingly, among genes under positive selection in the D. mojavensis lineage, there is an excess of genes involved in metabolism of heterocyclic compounds that are abundant in Stenocereus cacti and toxic to nonresident Drosophila species. We found 117 orphan genes in the shared D. buzzatii–D. mojavensis lineage. In addition, gene duplication analysis identified lineage-specific expanded families with functional annotations associated with proteolysis, zinc ion binding, chitin binding, sensory perception, ethanol tolerance, immunity, physiology, and reproduction. In summary, we identified genetic signatures of adaptation in the shared D. buzzatii–D. mojavensis lineage, and in the two separate D. buzzatii and D. mojavensis lineages. Many of the novel lineage-specific genomic features are promising candidates for explaining the adaptation of these species to their distinct ecological niches.
Keywords: cactophilic Drosophila, genome sequence, ecological adaptation, positive selection, orphan genes, gene duplication
Introduction
Drosophila species are saprophagous insects that feed and breed on a variety of fermenting plant materials, chiefly fruits, flowers, slime fluxes, decaying bark, leaves and stems, cactus necroses, and fungi (Carson 1971). These substrates include bacteria and yeasts that decompose the plant tissues and contribute to the nutrition of larvae and adults (Starmer 1981; Begon 1982). Only two species groups use cacti as their primary breeding site: repleta (Oliveira et al. 2012) and nannoptera (Lang et al. 2014). Both species groups originated at the virilis–repleta radiation, 20–30 Ma (Throckmorton 1975; Morales-Hojas and Vieira 2012; Oliveira et al. 2012) but adapted independently to the cactus niche. The “cactus-yeast-Drosophila system” in arid zones provides a valuable model to investigate gene–environment interactions and ecological adaptation from genetic and evolutionary perspectives (Barker and Starmer 1982; Barker et al. 1990). Rotting cacti provide relatively abundant, predictable, and long-lasting resources that can sustain very large Drosophila populations. For instance, a single saguaro rot may weigh up to several tons, last for many months, and sustain millions of Drosophila larvae and adults (Breitmeyer and Markow 1998). On the other hand, cacti are usually found in arid climates with middle to high temperatures that may impose desiccation and thermal stresses (Loeschcke et al. 1997; Hoffmann et al. 2003; Rajpurohit et al. 2013). Finally, some cacti may contain allelochemicals that can be toxic for Drosophila (see below). Thus, adaptation to use cacti as breeding sites must have entailed a fairly large number of changes in reproductive biology, behavior, physiology, and biochemistry (Markow and O’Grady 2008).
We have sequenced the genome and developmental transcriptome of Drosophila buzzatii to carry out a comparative analysis with those of Drosophila mojavensis, Drosophila virilis, and Drosophila grimshawi (Drosophila 12 Genomes Consortium et al. 2007). Drosophila buzzatii and D. mojavensis are both cactophilic species that belong to the mulleri subgroup of the repleta group (Wasserman 1992; Oliveira et al. 2012), although they have very different geographical distributions and host plants (fig. 1). Drosophila buzzatii is a subcosmopolitan species which is found in four of the six major biogeographic regions (David and Tsacas 1980). This species is originally from Argentina and Bolivia but now has a wide geographical distribution that includes other regions of South America, the Old World, and Australia (Carson and Wasserman 1965; Fontdevila et al. 1981; Hasson et al. 1995; Manfrin and Sene 2006). It chiefly feeds and breeds in rotting tissues of several Opuntia cacti but can also occasionally use columnar cacti (Hasson et al. 1992; Ruiz et al. 2000; Oliveira et al. 2012). The geographical dispersal of Opuntia by humans in historical times is considered the main driver of the world-wide expansion of D. buzzatii (Fontdevila et al. 1981; Hasson et al. 1995).
Fig. 1.—

(a) Phylogenetic relationship of fruit fly species considered in our comparative analysis and their host preference. (b) Geographical distribution of cactophilic species D. buzzatii (red) and D. mojavensis (green) in America.
On the other hand, D. mojavensis is endemic to the deserts of Southwestern United States and Northwestern Mexico. Its primary host plants are Stenocereus gummosus (pitaya agria) in Baja California and Stenocereus thurberi (organ pipe) in Arizona and Sonora, but uses also Ferocactus cylindraceous (California barrel) in Southern California and Opuntia sp. in Santa Catalina Island (Fellows and Heed 1972; Heed and Mangan 1986; Ruiz and Heed 1988; Etges et al. 1999). The ecological conditions of the Sonoran Desert are extreme (dry, arid, and hot), as attested by the fact that only four Drosophila species are endemic (Heed and Mangan 1986). In addition, D. mojavensis chief host plants, pitaya agria and organ pipe, are chemically complex and contain large quantities of triterpene glycosides, unusual medium-chain fatty acids, and sterol diols (Kircher 1982; Fogleman and Danielson 2001). These allelochemicals are toxic to nonresident Drosophila species, decreasing significantly larval performance (Fogleman and Kircher 1986; Ruiz and Heed 1988; Fogleman and Armstrong 1989; Frank and Fogleman 1992). In addition, host plant chemistry and fermentation byproducts affect adult epicuticular hydrocarbons and mating behavior (Havens and Etges 2013) as well as expression of hundreds of genes (Matzkin et al. 2006; Etges et al. 2015; Matzkin 2014).
As a first step to understand the genetic bases of ecological adaptation, here we compare the genomes of the two cactophilic species with those of two noncactophilic species of the Drosophila subgenus: D. virilis that belongs to the virilis species group and D. grimshawi that belongs to the picture wing group of Hawaiian Drosophila (fig. 1). The lineage leading to the common ancestor of D. buzzatii and D. mojavensis after diverging from D. virilis (#3 in fig. 1) represents the lineage that adapted to the cactus niche (likely Opuntia; Oliveira et al. 2012), whereas the lineages leading to D. buzzatii (#1) and D. mojavensis (#2) adapted to the specific niche of each species. We carried out a genome-wide scan for 1) genes under positive selection, 2) lineage-specific genes, and 3) gene-duplications in the three lineages (fig. 1). Based on the results of our comparative analyses, we provide a list of candidate genes that might play a meaningful role in the ecological adaptation of these fruit flies.
Materials and Methods
We sequenced the genome of a highly inbreed D. buzzatii strain, st-1 (Betran et al. 1998). DNA was extracted from male and female adults (Piñol et al. 1988; Milligan 1998). Reads were generated with three different sequencing platforms (supplementary fig. S2 and table S12, Supplementary Material online). The assembly of the genome was performed in three stages (supplementary table S13, Supplementary Material online): Preassembly (Margulies et al. 2005), scaffolding (Boetzer et al. 2011), and gapfilling (Nadalin et al. 2012). In each step, a few chimeric scaffolds were identified and split. The final assembly, named Freeze 1, contains 826 scaffolds greater than 3 kb and N50 and N90 index are 30 and 158, respectively. The distribution of read depth in the preassembly showed a Gaussian distribution with a prominent mode centered at approximately 22× (supplementary fig. S3, Supplementary Material online). CG content is approximately 35% overall, approximately 42% in gene regions (including introns) and reaches approximately 52% in exons (supplementary table S14, Supplementary Material online). Unidentified nucleotides (N’s) represent approximately 9% overall, approximately 4% in gene regions, and 0.004% in exons. Sequence quality was assessed by comparing Freeze 1 with five Sanger sequenced bacterial artificial chromosomes (BACs) (Negre et al. 2005; Prada 2010; Calvete et al. 2012) and with Illumina genomic and RNA sequencing (RNA-Seq) reads (supplementary fig. S4, Supplementary Material online). Quality assessments gave an overall error rate of approximately 0.0005 and a PHRED quality score of approximately Q33 (supplementary tables S15 and S16, Supplementary Material online). An overall proportion of segregating sites of approximately 0.1% was estimated (supplementary table S17, Supplementary Material online).
The genome size of two D. buzzatii strains, st-1 and j-19, was estimated by Feulgen Image Analysis Densitometry. The genome size of D. mojavensis 15081-1352.22 strain (193,826,310 bp) was used as reference (Drosophila 12 Genomes Consortium et al. 2007). Testicles from anesthetized males were dissected in saline solution and fixed in acetic-alcohol 3:1. Double preparations of D. mojavensis and D. buzzatii were made by crushing the fixed testicles in 50% acetic acid. Following Ruiz-Ruano et al. (2011), the samples were stained by Feulgen reaction and images obtained by optical microscopy were analyzed with the pyFIA software (supplementary fig. S5 and table S18, Supplementary Material online).
The 826 scaffolds in Freeze 1 were assigned to chromosomes by aligning their sequences with the D. mojavensis genome using MUMmer (Delcher et al. 2003). In addition, the 158 scaffolds in the N90 index were mapped, ordered, and oriented (supplementary fig. S1, Supplementary Material online) using conserved linkage (Schaeffer et al. 2008), in situ hybridization, and additional information (González et al. 2005; Guillén and Ruiz 2012). To estimate the number of rearrangements between D. buzzatii and D. mojavensis, their chromosomes were compared using GRIMM (Tesler 2002; Delprat A, Guillén Y, Ruiz A, in preparation). Genes in the Hox gene complex (HOM-C) and five other gene complexes were searched in silico in the D. buzzatii genome and manually annotated using available information (Negre et al. 2005), the annotated D. mojavensis and Drosophila melanogaster genomes, and the RNA-seq data generated for D. buzzatii (Negre B, Muyas F, Guillén Y, Ruiz A, in preparation). Transposable elements (TEs) were annotated with RepeatMasker using a comprehensive TE library compiled from FlyBase (St Pierre et al. 2014), Repbase (Jurka et al. 2005), and RepeatModeler. Tandem Repeats Finder version 4.04 (Benson 1999) was used to identify satellite DNAs (satDNAs).
For the RNA-Seq experiments, RNA from frozen samples (embryos, larvae, pupae, adult males, and adult females) was processed using the TruSeq RNA sample preparation kit provided by Illumina. We used a Hi-Seq2000 Illumina Sequencer to generate nonstrand-specific paired-end approximately 100 bp reads from poly(A) + RNA. Between 60 and 89 million reads were generated per sample. A total of approximately 286 million filtered reads were mapped to Freeze 1 with TopHat (Trapnell et al. 2009) representing approximately 180 × coverage of the total genome size (supplementary table S19, Supplementary Material online). Transcripts were assembled with Cufflinks (Trapnell et al. 2010) using Annotation Release 1 as reference (see below).
Protein-coding genes (PCGs) were annotated combining with EVidence Modeler (EVM; Haas et al. 2008) the results of different predictors: Augustus (Stanke and Waack 2003), SNAP (Korf 2004), N-SCAN (Korf et al. 2001), and Exonerate (Slater and Birney 2005). The EVM set contained 12,102 gene models. We noticed that orthologs for a considerable number of D. mojavensis PCGs were absent from this data set. Thus, we used the Exonerate predictions to detect another 1,555 PCGs not reported by EVM (Poptsova and Gogarten 2010). Altogether, we predicted a total of 13,657 PCG models in the D. buzzatii reference genome (Annotation Release 1). Features of these models are given in supplementary table S20, Supplementary Material online. The RSD (reciprocal smallest distance) algorithm (Wall and Deluca 2007) was used to identify 9,114 1:1 orthologs between D. mojavensis and D. buzzatii. Orthology relationships among the four species in the Drosophila subgenus (fig. 1) were inferred from D. buzzatii–D. mojavensis list of orthologs and the OrthoDB catalog (version 6; Kriventseva et al. 2008). To test for positive selection, we compared different codon substitution models using the likelihood ratio test (LRT). We run two pairs if site models (SM) on the orthologs set between D. buzzatii and D. mojavensis: M7 versus M8 and M1a versus M2a (Yang 2007). Then, we used branch-site models (BSM) to test for positive selection in three lineages (fig. 1): D. mojavensis lineage, D. buzzatii lineage, and the lineage that led to the two cactophilic species (D. buzzatii and D. mojavensis). We run Venny software (Oliveros 2007) to create a Venn diagram showing shared selected genes among the different models. We identified genes that are only present in the two cactophilic species, D. mojavensis and D. buzzatii, by blasting the amino acid sequences from the 9,114 1:1 orthologs between D. mojavensis and D. buzzatii (excluding misannotated genes) against all the proteins from the remaining 11 Drosophila species available in FlyBase protein database, excluding D. mojavensis (St Pierre et al. 2014).
For gene duplication analysis (DNA- and RNA-mediated duplications), we used annotated PCGs from the four species of the Drosophila subgenus (see supplementary methods, Supplementary Material online). Briefly, we ran all-against-all BLASTp and selected hits with alignment length extending over at least 50% of both proteins and with amino acid identity of at least 50%. Markov Cluster Algorithm (Enright et al. 2002) was used to cluster retained proteins into gene families. The data set was further modified to include additional family members based on sequence coverage and to exclude family members with internal stop codons and matches to TEs. Gene counts for each family from the four species were analyzed with an updated version of CAFE (CAFE 3.1 provided by the authors; Han et al. 2013) to identify lineage-specific expansions. The sets of CAFE-identified expanded families in the D. buzzatii and D. mojavensis genomes were examined for the presence of lineage-specific duplications. Families that included members with ds <0.4 were examined manually and lineage-specific duplications were inferred when no hits were found in the syntenic region of the genome with a missing copy. Drosophila buzzatii-specific RNA-mediated duplications were identified by examining intron-less and intron-containing gene family members. A duplicate was considered a retrocopy if its sequence spanned all introns of the parental gene. The number of families identified by CAFE as expanded along the internal cactophilic branch was reduced by considering only those families that were also found in expanded category after rerunning the analysis with a less stringent cutoff (35% amino acid identity, 50% coverage). The overlapping set of expanded families was manually examined to verify the absence of D. buzzatii and D. mojavensis new family members in the D. virilis genome. Functional annotation (i.e., Gene Ontology [GO] term) for all expanded families was obtained using the DAVID annotation tool (Huang et al. 2009a, 2009b). For genes without functional annotation in DAVID, annotations of D. melanogaster orthologs were used. An extended version of these methods is given as supplementary methods, Supplementary Material online.
Results
Features of the D. buzzatii Genome
Genome Sequencing and Assembly
We sequenced and de novo assembled the genome of D. buzzatii line st-1 using shotgun and paired-end reads from 454/Roche, mate-pair and paired-end reads from Illumina, and Sanger BAC-end sequences (∼22× total expected coverage; see Materials and Methods for details). We consider the resulting assembly (Freeze 1) as the reference D. buzzatii genome sequence (table 1). This assembly comprises 826 scaffolds greater than 3 kb long with a total size of 161.5 Mb. Scaffold N50 and N90 indexes are 30 and 158, respectively, whereas scaffold N50 and N90 lengths are 1.38 and 0.16 Mb, respectively (table 1). Quality controls (see Materials and Methods) yielded a relatively low error rate of approximately 0.0005 (PHRED quality score Q = 33). For comparison, we also assembled the genome of the same line (st-1) using only four lanes of short (100 bp) Illumina paired-end reads (∼76× expected coverage) and the SOAPdenovo software (Luo et al. 2012). This resulted in 10,949 scaffolds greater than 3 kb long with a total size of 144.2 Mb (table 1). All scaffolds are available for download from the Drosophila buzzatii Genome Project web page (http://dbuz.uab.cat, last accessed January 7, 2015). This site also displays all the information generated in this project (see below).
Table 1.
Summary of Assembly Statistics for the Genome of Drosophila buzzatii
| Assembly | Freeze 1 | SOAPdenovo |
|---|---|---|
| Number of scaffolds (>3 kb) | 826 | 10,949 |
| Coverage | ∼22× | ∼76× |
| Assembly size (bp) | 161,490,851 | 144,184,967 |
| Scaffold N50 index | 30 | 2,035 |
| Scaffold N50 length (bp) | 1,380,942 | 18,900 |
| Scaffold N90 index | 158 | 7,509 |
| Scaffold N90 length (bp) | 161,757 | 5,703 |
| Contig N50 index | 1,895 | 2,820 |
| Contig N50 length (bp) | 17,678 | 3,101 |
Genome Size and Repeat Content
The genome sizes of two D. buzzatii strains, st-1 and j-19, were estimated by Feulgen Image Analysis Densitometry on testis cells (Ruiz-Ruano et al. 2011) using D. mojavensis as reference. Integrative Optical Density values were 21% (st-1) and 25% (j-19) smaller than those for D. mojavensis. Thus, taking 194 Mb (total assembly size) as the genome size of D. mojavensis (Drosophila 12 Genomes Consortium et al. 2007) we estimated the genome sizes for D. buzzatii st-1 and j-19 lines as 153 and 146 Mb, respectively.
To assess the TE content of the D. buzzatii genome, we masked the 826 scaffolds of Freeze 1 assembly using a library of TEs compiled from several sources (see Materials and Methods). We detected a total of 56,901 TE copies covering approximately 8.4% of the genome (table 2). The most abundant TEs seem to be Helitrons, LINEs, long terminal repeat (LTR) retrotransposons, and TIR transposons that cover 3.4%, 1.6%, 1.5%, and 1.2% of the genome, respectively (table 2). In addition, we identified tandemly repeated satDNAs with repeat units longer than 50 bp (Melters et al. 2013) (see Materials and Methods). The two most abundant tandem repeat families are the pBuM189 satellite (Kuhn et al. 2008) and the DbuTR198 satellite, a novel family with repeat units 198 bp long (table 3). The remaining tandem repeats had sequence similarity to integral parts of TEs, such as the internal tandem repeats of the transposon Galileo (de Lima LG, Svartman M, Ruiz A, Kuhn GCS, in preparation).
Table 2.
Transposable Element Content of Drosophila buzzatii Genome
| Class | Order | Annotated Base Pair | Genome Coverage (%) |
|---|---|---|---|
| I (retrotransposons) | LTR | 2,366,439 | 1.47 |
| DIRS | 55 | 0.00 | |
| LINE | 2,541,645 | 1.57 | |
| II (DNA transposons) | TIR | 2,017,167 | 1.25 |
| Helitron | 5,531,009 | 3.42 | |
| Maverick | 189,267 | 0.12 | |
| Unknown | 973,759 | 0.60 | |
| Total | 13,619,341 | 8.43 |
Note.—The classification follows Wicker et al. (2007).
Table 3.
Satellite DNAs Identified in the Drosophila buzzatii Genome
| Tandem repeat Family | Repeat Length | GC Content (%) | Genome Coverage (%)a | Consensus Sequenceb | Distribution |
|---|---|---|---|---|---|
| pBuM189 | 189 | 29 | 0.039 | GCAAAAGACTCCGTCAATTA GAAAACAAAAAATGTTATAGTTTTGAGGATTAACC GGCAAAAACCGTATTATTTGTTATAT GATTTCTGTATGGAATACCGTTTTAGAA GCGTCTTTTATCGTATTACTCAGATATATCT TAAGATTTAGCATAATCTAAGAACTTTT TGAAATATTCACATTTGTCCA | D. buzzatii cluster species D. mojavensis |
| DbuTR198 | 198 | 34 | 0.027 | AAGGTAGAAAGGTAGTTGGTGAGATAAACCAGAAAAA GAGCTAAAAACGGCTAAAAACGGCTAGAAAATAGCCA GAAAGGTAGATTGAACATTAATGGGCAAATGG ATGGATAAATAAGACTGGTCATCATCCAA TGAACAGAATCATGATTAAGAGATAGAAATA TGATTAGAAAGTAGGATAGAAAGGTTAGAAAG | D. buzzatii |
aGenome fraction was calculated assuming a genome size of 163,547,398 bp (version 1 freeze of all contigs).
bConsensus sequence generated after clustering TRF results (see Materials and Methods).
Chromosomal Rearrangements
The basic karyotype of D. buzzatii is similar to that of the Drosophila genus ancestor and consists of six chromosome pairs: Four pairs of equal-length acrocentric autosomes, one pair of “dot” autosomes, a long acrocentric X, and a small acrocentric Y (Ruiz and Wasserman 1993). Because no interchromosomal reorganizations between D. buzzatii and D. mojavensis have previously been found (Ruiz et al. 1990; Ruiz and Wasserman 1993) all 826 scaffolds were assigned to chromosomes by BLASTn against the D. mojavensis genome. In addition, the 158 scaffolds in the N90 index were mapped to chromosomes, ordered, and oriented (supplementary fig. S1, Supplementary Material online; Delprat A, Guillén Y, Ruiz A, in preparation) using conserved linkage (Schaeffer et al. 2008) and additional information (González et al. 2005; Guillén and Ruiz 2012). A bioinformatic comparison of D. buzzatii and D. mojavensis chromosomes confirmed that chromosome 2 differs between these species by ten inversions (2m, 2n, 2z7, 2c, 2f, 2g, 2h, 2q, 2r, and 2s), chromosomes X and 5 differ by one inversion each (Xe and 5g, respectively), and chromosome 4 is homosequential as previously described (Ruiz et al. 1990; Ruiz and Wasserman 1993; Guillén and Ruiz 2012). In contrast, we find that chromosome 3 differs by five inversions instead of the expected two that were previously identified by cytological analyses (Ruiz et al. 1990). These three additional chromosome 3 inversions seem to be specific to the D. mojavensis lineage (Delprat A, Guillén Y, Ruiz A, in preparation). One of these inversions, 3f2, is polymorphic in natural populations of D. mojavensis, but, conflicting with previous reports (Ruiz et al. 1990; Schaeffer et al. 2008), appears to be homozygous in the sequenced strain. This has been corroborated by the cytological reanalysis of its polytene chromosomes (Delprat et al. 2014).
Many developmental genes are arranged in gene complexes each comprising a small number of functionally related genes. We checked the organization of six of these gene complexes in the D. buzzatii genome: HOM-C, Achaete–scute complex, Iroquois complex, NK homeobox gene cluster (NK-C), Enhancer of split complex, and Bearded complex (Brd-C) (Negre B, Muyas F, Guillén Y, Ruiz A, in preparation). Hox genes were arranged in a single complex in the Drosophila genus ancestor (Hughes and Kaufman 2002). However, this HOM-C suffered two splits (caused by chromosomal inversions) in the lineage leading to the repleta species group (Negre et al. 2005). In order to fully characterize HOM-C organization in D. buzzatii, we manually annotated all Hox genes and located them in three scaffolds (2, 5, and 229) of chromosome 2 (Negre B, Muyas F, Guillén Y, Ruiz A, in preparation). The analysis of these scaffolds revealed that only two clusters of Hox genes are present. The distal cluster contains proboscipedia, Deformed, Sex combs reduced, Antennapedia and Ultrabithorax, whereas the proximal cluster contains labial, abdominal A and Abdominal B. This is precisely the same HOM-C organization observed in D. mojavensis (Negre and Ruiz 2007). Therefore, there seem to be no additional rearrangements of the HOM-C in D. buzzatii besides those already described in the genus Drosophila (Negre and Ruiz 2007). The other five developmental gene complexes contain 4, 3, 6, 13, and 6 functionally related genes, respectively (Lai et al. 2000; Garcia-Fernàndez 2005; Irimia et al. 2008; Negre and Simpson 2009). All these complexes seem largely conserved in the D. buzzatii genome with few exceptions (Negre B, Muyas F, Guillén Y, Ruiz A, in preparation). The gene slouch is separated from the rest of the NK-C in D. buzzatii and also in all other Drosophila species outside of the melanogaster species group; in addition, the gene Bearded, a member of the Brd-C, is seemingly absent from the D. buzzatii and D. mojavensis genomes, although it is present in D. virilis and D. grimshawi. On the other hand, genes flanking the complexes are often variable, presumably due to the fixation of chromosomal inversions with breakpoints in the boundaries of the complexes.
PCG Content
We used a combination of ab initio and similarity-based algorithms in order to reduce the high false-positive rate associated with de novo gene prediction (Wang et al. 2003; Misawa and Kikuno 2010) as well as to avoid the propagation of false-positive predicted gene models when closely related species are used as references (Poptsova and Gogarten 2010). A total of 13,657 PCGs were annotated in the D. buzzatii genome (Annotation Release 1). These PCG models contain a total of 52,250 exons with an average of 3.8 exons per gene. Gene expression analyses provided transcriptional evidence for 88.4% of these gene models (see below). The number of PCGs annotated in D. buzzatii is lower than the number annotated in D. mojavensis (14,595, Release 1.3), but quite close to the number annotated in D. melanogaster (13,955, Release 5.56), one of the best-known eukaryotic genomes (St Pierre et al. 2014). However PCGs in both D. buzzatii and D. mojavensis genomes tend to be smaller and contain fewer exons than those in the D. melanogaster genome (supplementary table S1, Supplementary Material online), which suggests that the annotation in the two cactophilic species might be incomplete. After applying several quality filters, a total of 12,977 high confidence protein-coding sequences (CDS) were selected for further analysis (see Materials and Methods).
Developmental Transcriptome
To characterize the expression profile throughout D. buzzatii development, we performed RNA-Seq experiments using samples from five different stages: Embryos, larvae, pupae, adult females, and adult males. Gene expression levels were calculated based on fragments per kilobase of exon per million fragments mapped (FPKM) values. PCG models that did not show evidence of transcription (FPKM < 1) were classified as nonexpressed PCGs, whereas transcribed regions that did not overlap with any annotated PCG model were tentatively considered noncoding RNA (ncRNA) genes (fig. 2a). We detected expression (FPKM > 1) of 26,455 transcripts and 15,026 genes, 12,066 (80%) are PCGs and 2,960 (20%) are ncRNA genes. The number of expressed genes (PCGs + ncRNA) increases through the life cycle with a maximum of 12,171 in adult males (fig. 2a and supplementary table S2, Supplementary Material online), a pattern similar to that found in D. melanogaster (Graveley et al. 2011). In addition, we observed a clear sex-biased expression in adults: Males express 1,824 more genes than females. Previous studies have attributed this sex-biased gene expression mainly to the germ cells, indicating that the differences between ovary and testis are comparable to those between germ and somatic cells (Parisi et al. 2004; Graveley et al. 2011).
Fig. 2.—
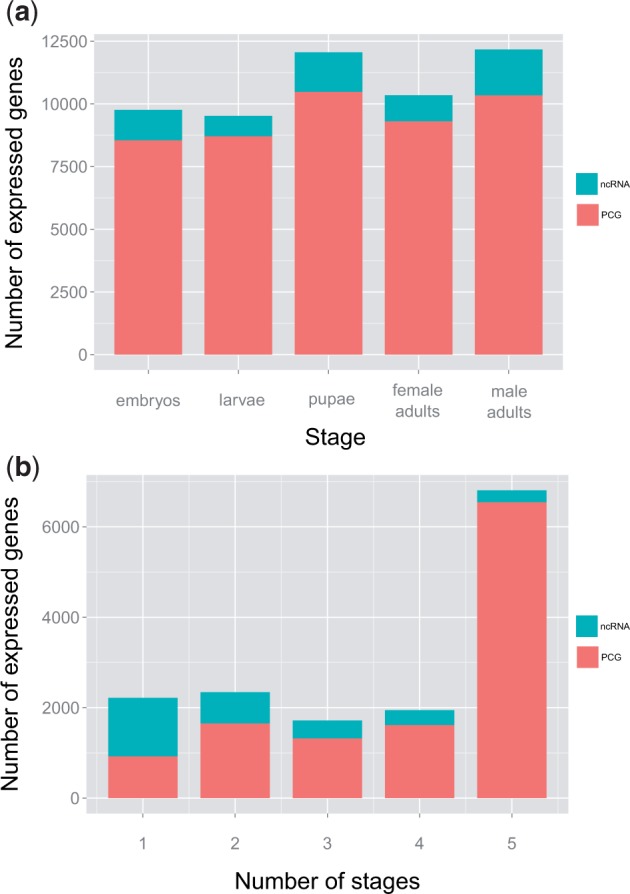
Developmental expression profile of D. buzzatii genes. (a) Number of expressed PCGs (red) and ncRNA genes (blue) along five developmental stages. (b) Classification of PCGs and ncRNA genes according to the number of stages where they are expressed.
We assessed expression breadth for each gene simply as the number of developmental stages with evidence of expression (fig. 2b and supplementary table S2, Supplementary Material online). Expression breadth is significantly different (P < 0.001) for PCGs and ncRNA genes. A total of 6,546 expressed PCGs (54.2%) are constitutively expressed (i.e., we observed expression in the five stages), but only 260 of ncRNA genes (8.8%) are constitutively expressed (supplementary table S2, Supplementary Material online). In contrast, 925 expressed PCGs (7.7%) and 1,292 ncRNA genes (43.6%) are expressed only in one stage. Mean expression breadth was 3.9 for PCGs and 2.2 for ncRNA genes. Adult males show more stage-specific genes (844 genes) compared with adult females (137 genes).
PCGs with no expression in this study (FPKM < 1) might be expressed at a higher level in other tissues or times, or they might be inducible under specific conditions that we did not test (Weake and Workman 2010; Etges et al. 2015; Matzkin 2014). We also must expect that some remaining fraction of gene models will be false positives (Wang et al. 2003). However, because we used a combination of different annotation methods to reduce the proportion of false-positives, we expect this proportion to be very small. On the other hand, transcribed regions that do not overlap with any annotated PCG models are likely ncRNA genes although we cannot discard that some of them might be false negatives, that is, genes that went undetected by our annotation methods perhaps because they contain small open reading frames (Ladoukakis et al. 2011). One observation supporting that most of them are in fact ncRNA genes is that their expression breadth is quite different from that of PCGs and a high fraction of them are stage-specific genes. In most Drosophila species, with limited analyses of the transcriptome (Celniker et al. 2009), few ncRNA genes have been annotated. In contrast, in D. melanogaster with a very well-annotated genome, 2,096 ncRNA genes have been found (Release 5.56, FlyBase). Thus, the number of ncRNA found in D. buzzatii is comparable to that of D. melanogaster.
Website
A website (http://dbuz.uab.cat, last accessed January 7, 2015) has been created to provide free access to all information and resources generated in this work. It includes a customized browser (GBrowse; Stein et al. 2002) for the D. buzzatii genome incorporating multiple tracks for gene annotations with different gene predictors, for expression levels and transcript annotations for each developmental stage, and for repeat annotations. It contains also utilities to download contigs, scaffolds, and data files and to carry out Blast searches against all D. buzzatii contigs and scaffolds.
Lineage-Specific Analyses
We set up to analyze three lineages for several aspects that could reveal genes involved in adaptation to the cactophilic niche. These lineages are denoted as #1, #2, and #3, respectively, in figure 1: D. buzzatii lineage, D. mojavensis lineage, and cactophilic lineage (i.e., lineage shared by D. buzzatii and D. mojavensis). We searched for genes under positive selection, duplicated genes, and orphan genes in those lineages.
Genes under Positive Selection
We first searched for genes evolving under positive selection during the divergence between D. buzzatii and D. mojavensis, using codon substitution models implemented in the PAML 4 package (Yang 2007). Two pairs of different SM were compared by the LRT, M1a versus M2a and M7 versus M8 (see Materials and Methods). In each case, a model that allows for sites with ω > 1 (positive selection) is compared with a null model that considers only sites with ω < 1 (purifying selection) and ω = 1 (neutrality). At P < 0.001, the first comparison (M1a vs. M2a) detected 915 genes whereas the second comparison (M7 vs. M8) detected 802 genes. Comparison of the two gene sets allowed us to detect 772 genes present in both, and this was taken as the final list of genes putatively under positive selection using SM (supplementary table S3, Supplementary Material online).
Next, we used BSM from PAML 4 package (Yang 2007) to search for genes under positive selection in the phylogeny of the four Drosophila subgenus species, D. buzzatii, D. mojavensis, D. virilis, and D. grimshawi (fig. 1). Orthologous relationships among the four species were inferred from D. buzzatii–D. mojavensis list of orthologs and the OrthoDB catalog (see Materials and Methods). A total of 8,328 unequivocal 1:1:1:1 orthologs were included in the comparison of a BSM allowing sites with ω > 1 (positive selection) and a null model that does not. We selected three branches to test for positive selection (the foreground branches): D. buzzatii lineage, D. mojavensis lineage, and cactophilic lineage (denoted as #1, #2, and #3 in fig. 1). The number of genes putatively under positive selection detected at P < 0.001 in the three branches was 350, 172, and 458, respectively (supplementary table S3, Supplementary Material online). These genes only partially overlap those previously detected in the D. buzzatii–D. mojavensis comparison using SM (fig. 3). Although 69.4% and 55.8% of the genes putatively under positive selection in the D. buzzatii and D. mojavensis lineages were also detected in the D. buzzatii–D. mojavensis comparison, only 22.3% of the genes detected in the cactophilic lineage were present in the previous list (fig. 3). Thus, the total number of genes putatively under positive selection is 1,294.
Fig. 3.—
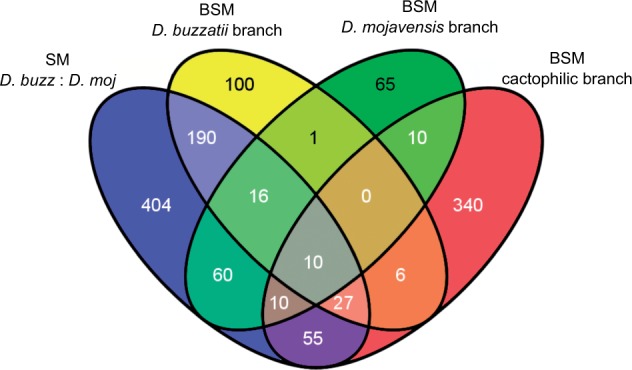
Venn diagram showing the number of genes putatively under positive selection detected by two different methods, SM and BSM using three different lineages as foreground branches.
We looked for functional categories overrepresented among the candidate genes reported by both SM and BSM (table 4). We first performed a GO enrichment analysis with the 772 candidate genes uncovered by SM comparing D. mojavensis and D. buzzatii orthologs using DAVID tools (Huang et al. 2007). Two molecular functions show higher proportion than expected by chance (relative to D. mojavensis genome) within the list of candidate genes: Antiporter activity and transcription factor activity. With respect to the biological process, regulation of transcription is the only overrepresented category. A significant enrichment in Src Homology-3 domain was observed. This domain is commonly found within proteins with enzymatic activity and it is associated with protein binding function.
Table 4.
GO Analysis of Putative Genes under Positive Selection Detected by Both SM and BSM
| Codon substitution Models | Lineage (Branch Number) | Number of Candidates | GO enrichment |
|||||
|---|---|---|---|---|---|---|---|---|
| Molecular Function |
Biological Process |
Interpro Domain |
||||||
| ID | Fold Enrichment | ID | Fold Enrichment | ID | Fold Enrichment | |||
| SM | Drosophila buzzatii versus Drosophila mojavensis | 772 | Antiporter activity | 1.77 | Regulation of transcription | 4.90 | Src homology-3 domain | 1.60 |
| Transcription factor activity | 1.56 | |||||||
| BSM | D. buzzatii #1 | 350 | DNA binding | 1.36 | Regulation of transcription DNA dependent | 1.36 | Immunoglobulin-like | 1.33 |
| Phosphate metabolic process | 0.72 | |||||||
| D. mojavensis #2 | 172 | Dopamine beta-monooxigenase activity | 2.35 | Heterocycle catabolic process | 2.35 | DOMON (DOpamine beta-MOnooxygenase N-terminal domain) | 2.35 | |
| Cation transport | 0.98 | |||||||
| Histidine family amino acid catabolic process | 2.35 | |||||||
| Cactophilic #3 | 458 | Zinc ion binding | 2.01 | Cytoeskeleton organization | 1.67 | Zinc finger, PHD-type | 1.93 | |
| Transition metal ion binding | 2.01 | Regulation of transcription DNA dependent | 1.06 | Proteinase inhibitor I1 kazal | 2.20 | |||
| DNA binding | 1.66 | |||||||
Note.—Only categories showing an enrichment with a P value less than 1.0e-03 are included.
A similar GO enrichment analysis was carried out with candidate genes found using BSM in each of the three targeted branches. The 350 candidate genes in D. buzzatii lineage show a significant enrichment in DNA-binding function. DNA-dependent regulation of transcription and phosphate metabolic processes were also overrepresented. We also found a significant enrichment in the Ig-like domain, involved in functions related to cell–cell recognition and immune system. The 172 candidate genes in D. mojavensis lineage show a significant excess of genes related to the heterocycle catabolic process (P = 5.9e-04). Interestingly, the main hosts of D. mojavensis (columnar cacti) contain large quantities of triterpene glycosides, which are heterocyclic compounds. Among the candidate genes in the branch leading to the two cactophilic species, there are three overrepresented molecular functions related to both metal and DNA binding. The GO terms with the highest significance in the biological process category are cytoskeleton organization and, once again, regulation of transcription.
Using the RNA-Seq data we determined the expression profiles of all 1,294 genes putatively under positive selection. A total of 1,213 (93.7%) of these genes are expressed in at least one developmental stage (supplementary table S2, Supplementary Material online). A comparison of expression level and breadth between candidate and noncandidate genes revealed that genes putatively under positive selection are expressed at a lower level (Χ2 = 84.96, P < 2e-16) and in fewer developmental stages (Χ2 = 26.99, P < 2e-6) than the rest.
Orphan Genes in the Cactophilic Lineage
To detect orphan genes in the cactophilic lineage, we blasted the amino acid sequences encoded by 9,114 D. buzzatii genes with D. mojavensis 1:1 orthologs against all proteins from the 12 Drosophila genomes except D. mojavensis available in FlyBase (St Pierre et al. 2014). We found 117 proteins with no similarity to any predicted Drosophila protein (cutoff value of 1e-05) and were considered to be encoded by putative orphan genes. We focused on the evolutionary dynamics of these orphan genes by studying their properties in comparison to the remaining 8,997 1:1 orthologs (fig. 4). We observed that median dn of orphan genes was significantly higher than that of nonorphan genes (dnorphan = 0.1291; dnnonorphan = 0.0341; W = 846,254, P < 2.2e-16) and the same pattern was observed for ω (ωorphan = 0.4253, ωnonorphan = 0.0887, W = 951,117, P < 2.2e-16). However, median ds of orphan genes is somewhat lower than that for the rest of genes (dsorphan = 0.3000, dsnonorphan = 0.4056, W = 406,799, P = 2.4e-05).
Fig. 4.—
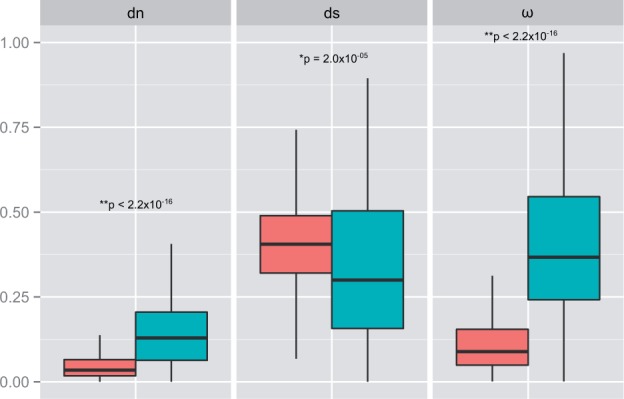
Patterns of divergence in orphan and nonorphan genes. Orphan genes (blue) have significantly higher dn and ω values compared with that of nonorphan genes (red). Nonorphan genes show significantly higher ds.
We found 19 of the 117 orphan genes in the list of candidate genes detected in the D. buzzatii–D. mojavensis comparison (see above). This proportion (16.3%) was significantly higher than that found in nonorphan 1:1 orthologs (753/8,997 = 8.4%), which indicates an association between gene lineage-specificity and positive selection (Fisher exact test, two-tailed, P < 0.0001). The 19 orphan genes included in the candidate gene group are not associated with any GO category. As a matter of fact, information about protein domains was found for only two of these genes (GYR and YLP motifs in both cases: GI20994 and GI20995). These results should be viewed cautiously as newer genes are functionally undercharacterized and GO databases are biased against them (Zhang et al. 2012). We also compared the protein length between orphan and nonorphan gene products. Our results showed that orphan genes are shorter (W = 68,825.5, P < 2.2e-16) and have fewer exons than nonlineage-specific genes (W = 201,068, P < 2.2e-16).
RNA-Seq data allowed us to test for expression of orphan genes. From the 117 gene candidates, 82 (70%) are expressed at least in one of the five analyzed developmental stages. A comparison of the expression profiles between orphan and the rest of 1:1 orthologous genes showed that the expression breadth of orphans is different from that of nonorphans (Χ2 = 101.4, P < 0.001): Most orphan genes are expressed exclusively in one developmental stage with mean expression breadth of 2.56 (vs. 3.94 for nonorphans).
Gene Duplications
The annotated PCGs from four species of the Drosophila subgenus were used to study gene family expansions in the D. buzzatii, D. mojavensis, and cactophilic lineages (fig. 1). Proteins that share 50% identity over 50% of their length were clustered into gene families using Markov Cluster Algorithm. After additional quality filters (see Materials and Methods), the final data set consisted of a total of 56,587 proteins from four species clustered into 19,567 families, including single-gene families (supplementary tables S4–S7, Supplementary Material online).
Considering the D. buzzatii genome alone (supplementary table S4, Supplementary Material online), we find 11,251 single-copy genes and 1,851 duplicate genes (14%) clustered in 691 gene families. Among D. buzzatii gene families, about 70% of families have two members and the largest family includes 16 members (supplementary table S4, Supplementary Material online). Among single-copy genes, 1,786 genes are only present in the D. buzzatii lineage. This number decreases only to 1,624 when proteins are clustered into families with a less stringent cutoff of 35% identity and 50% coverage. Such lineage-specific single-copy genes have been found in all the 12 Drosophila genomes that have been analyzed, including D. mojavensis (Hahn et al. 2007), and although traditionally they have been viewed as annotation artifacts, many of these genes may be either de novo or fast-evolving genes (Reinhardt et al. 2013; Palmieri et al. 2014).
Lineage-specific expansions were identified by analyzing the gene count for each family from the four species using CAFE3.1 (see Materials and Methods). This analysis detected expansions of 86 families along the D. buzzatii lineage. However, 15 families increased in size as the result of extra copies added to the data set after taking into account high sequence coverage. The expansions of these families cannot be confirmed with the current genome assembly. The remaining families were analyzed further in order to confirm D. buzzatii-specific duplications. To do that, we first selected gene families with members that have ds < 0.4 (median ds for D. mojavensis–D. buzzatii orthologs) and then manually examined syntenic regions in D. mojavensis genome. Although this approach might miss some true lineage-specific expansions, it reduces the possibility of including old families into the expansion category that might have been misclassified as a result of incomplete gene annotation in the genomes under study or independent loss of family members in different lineages. Of the 30 gene families whose members had ds < 0.4, we confirmed the expansion of 20 families (supplementary table S8, Supplementary Material online). In 12 of the 20 families, new family members are found on the same scaffold in close proximity suggesting unequal crossing over or proximate segmental duplication as the mechanisms for duplicate formation. The remaining eight families contain dispersed duplicates found in different scaffolds. Six of these families expanded through retroposition, the RNA-mediated duplication mechanism that allows insertion of reverse-transcribed mRNA nearly anywhere in the genome. In most cases, family expansions are due to addition of a new single copy in the D. buzzatii lineage (in 25 of total 35 families). Two families that expanded the most, with up to 5 (Family 95) and 9 (Family 126) new members, encode various peptidases involved in protein degradation. Other expanded families are associated with a broad range of functions, including structural proteins of insect cuticle and chorion, enzymes involved in carbohydrate and lipid metabolism, proteins that function in immune response, and olfactory receptors. In addition, Family 128 encodes female reproductive peptidases (Kelleher and Markow 2009) and it appears that new family members have been acquired independently in D. buzzatii and D. mojavensis lineages (supplementary table S11, Supplementary Material online).
We find six families in D. buzzatii that expanded through retroposition in the 11 Myr since the split between D. buzzatii and D. mojavensis (supplementary table S9, Supplementary Material online). This gives a rate of 0.55 retrogenes/Myr, which is consistent with previous estimates of functional retrogene formation in Drosophila of 0.5 retrogenes/Myr (Bai et al. 2007). The expression of all but one retrogene is supported by RNA-Seq data, with no strong biases in expression between the sexes. Four retrogenes are duplicates of ribosomal proteins, and the parental genes from two of these families (RpL37a and RpL30) have been previously shown to generate retrogenes in other Drosophila lineages (Bai et al. 2007; Han and Hahn 2012). Frequent retroposition of ribosomal proteins could be explained by the high levels of transcription of ribosomal genes although other Drosophila lineages do not show a bias in favor of retroduplication of ribosomal proteins (Bai et al. 2007; Han and Hahn 2012). The remaining two retrogenes include the duplicate of Caf1, protein that is involved in histone modification, and the duplicate of VhaM9.7-b, a subunit of ATPase complex.
CAFE analysis identified 127 families that expanded along the D. mojavensis lineage. Of these families, 86 contain members with ds < 0.4. Further examination of syntenic regions confirmed expansion of only 17 families (supplementary table S8, Supplementary Material online). New members in two families (Families 1121 and 1330) are found in different scaffolds and originated through RNA-mediated duplications. These instances have been previously identified as D. mojavensis-specific retropositions (Han and Hahn 2012). Members of expanded families encode proteins that function in proteolysis, peptide and ion transport, aldehyde and carbohydrate metabolism, as well as sensory perception (supplementary table S11, Supplementary Material online). At least 4 of the 17 expanded families play a role in reproductive biology: Proteases of Family 128 with three new members have been shown to encode female reproductive peptidases (Kelleher and Markow 2009), and members of three additional families (Families 187, 277, and 1234) encode proteins that are found in D. mojavensis accessory gland proteome (Kelleher et al. 2009).
There are 20 gene families that expanded along the cactophilic branch, that is, before the split between D. buzzatii and D. mojavensis (see Materials and Methods; supplementary table S10, Supplementary Material online). Most families (16 of 20) have expanded through tandem or nearby segmental duplication and are still found within the same scaffold. The remaining families with dispersed duplicates included one retrogene, the duplicate of T-cp1, identified previously in D. mojavensis lineage (Han and Hahn 2012). The extent of per-family expansions in the cactophilic lineage is modest, with two new additional members found in four families and a single new copy in the remaining families. Members of the most expanded families encode guanylate cyclases that are involved in intracellular signal transduction, peptidases, and carbon–nitrogen hydrolases. Members of other families include various proteins with metal-binding properties as well as proteins with a role in vesicle and transmembrane transport (supplementary table S11, Supplementary Material online). We also see expansion of three families (Family 775, Family 776, and Family 800) with functions related to regulation of juvenile hormone (JH) levels (see Discussion).
Discussion
The D. buzzatii Genome
Drosophila is a leading model for comparative genomics, with 24 genomes of different species already sequenced (Adams et al. 2000; Drosophila 12 Genomes Consortium et al. 2007; Zhou et al. 2012; Zhou and Bachtrog 2012; Fonseca et al. 2013; Ometto et al. 2013; Chen et al. 2014). However, only five of these species belong to the species-rich Drosophila subgenus, and only one of these species, D. mojavensis, is a cactophilic species from the large repleta species group. Here we sequenced the genome and transcriptome of D. buzzatii, another cactophilic member of the repleta group, to investigate the genomic basis of adaptation to this distinct ecological niche. Using different sequencing platforms and a three-stage de novo assembly strategy, we generated a high quality genome sequence that consists of 826 scaffolds greater than 3 kb (Freeze 1). A large portion (>90%) of the genome is represented by 158 scaffolds with a minimum size of 160 kb that have been assigned, ordered, and oriented in the six chromosomes of the D. buzzatii karyotype. As expected, the assembly is best for chromosome 2 (because of the use of Sanger generated BAC-end sequences) and worst for chromosome X (because of the three-fourth representation of this chromosome in adults of both sexes). The quality of our Freeze 1 assembly compares favorably with the assembly generated using only Illumina reads and the SOAPdenovo assembler, and with those of other Drosophila genomes generated using second-generation sequencing platforms (Zhou et al. 2012; Zhou and Bachtrog 2012; Fonseca et al. 2013; Ometto et al. 2013; Chen et al. 2014), although our Freeze 1 does not attain the quality of the 12 Drosophila genomes generated using Sanger only (Drosophila 12 Genomes Consortium et al. 2007).
Drosophila buzzatii is a subcosmopolitan species that has been able to colonize four of the six major biogeographical regions (David and Tsacas 1980). Only two other repleta group species (Drosophila repleta and Drosophila hydei) have reached such widespread distribution. Invasive species are likely to share special genetic traits that enhance their colonizing ability (Parsons 1983; Lee 2002). From an ecological point of view we would expect colonizing species to be r-strategists with a short developmental time (Lewontin 1965). Because there is a correlation between developmental time and genome size (Gregory and Johnston 2008), colonizing species are also expected to have a small genome size (Lavergne et al. 2010). The genome size of D. buzzatii was estimated in our assembly as 161 Mb and by cytological techniques as 153 Mb, approximately 20% smaller than the D. mojavensis genome. The genome size of a second D. buzzatii strain, estimated by cytological techniques, is even smaller, 146 Mb. However, the relationship between genome size and colonizing ability does not hold in the Drosophila genus at large. Although colonizing species such as D. melanogaster and Drosophila simulans have relatively small genomes, specialist species with a narrow distribution such as Drosophila sechelia and Drosophila erecta also have small genomes. On the other hand, Drosophila ananassae, Drosophila malerkotliana, Drosophila suzuki, D. virilis, and Zaprionus indianus are also colonizing Drosophila species but have relatively large genomes (Nardon et al. 2005; Bosco et al. 2007; Drosophila 12 Genomes Consortium et al. 2007; Gregory and Johnston 2008). Further, there seems to be little difference in genome size between original and colonized populations within species (Nardon et al. 2005). Seemingly, other factors such as historical or chance events, niche dispersion, genetic variability, or behavioral shifts are more significant than genome size in determining the current distribution of colonizing species (Markow and O’Grady 2008).
TE content in the D. buzzatii genome was estimated as 8.4% (table 2), a relatively low value compared with that of D. mojavensis, 10–14% (Ometto et al. 2013; Rius et al., in preparation). These data agree well with the smaller genome size of D. buzzatii because genome size is positively correlated with the contribution of TEs (Kidwell 2002; Feschotte and Pritham 2007). However, TE copy number and coverage estimated in D. buzzatii (table 2) must be taken cautiously. Coverage is surely underestimated due to the difficulties in assembling repeats, in particular with short sequence reads, whereas the number of copies may be overestimated due to copy fragmentation (Rius N, Guillén Y, Kapusta A, Feschotte C, Ruiz A, in preparation). The contribution of satDNAs (table 3) is also an underestimate and further experiments are required for a correct assessment of this component (de Lima LG, Svartman M, Ruiz A, Kuhn GCS, in preparation). However, we identified the pBuM189 satDNA as the most abundant tandem repeat of D. buzzatii. Previous in situ hybridization experiments revealed that pBuM189 copies are located in the centromeric region of all chromosomes, except chromosome X (Kuhn et al. 2008). Thus, pBuM189 satellite is likely the main component of the D. buzzatii centromere. Interestingly, a pBuM189 homologous sequence has recently been identified as the most abundant tandem repeat of D. mojavensis (Melters et al. 2013). Although the chromosome location in D. mojavensis has not been determined, the persistence of pBuM189 as the major satDNA in D. buzzatii and D. mojavensis may reflect a possible role for these sequences in centromere function (Ugarković 2009).
Chromosome Evolution
The chromosomal evolution of D. buzzatii and D. mojavensis has been previously studied by comparing the banding pattern of the salivary gland chromosomes (Ruiz et al. 1990; Ruiz and Wasserman 1993). Drosophila buzzatii has few fixed inversions (2m, 2n, 2z7, and 5g) when compared with the ancestor of the repleta group. In contrast, D. mojavensis showed ten fixed inversions (Xe, 2c, 2f, 2g, 2h, 2q, 2r, 2s, 3a, and 3d), five of them (Xe, 2q, 2r, 2s, and 3d) exclusive to D. mojavensis and the rest shared with other cactophilic Drosophila (Guillén and Ruiz 2012). Thus, the D. mojavensis lineage appears to be a derived lineage with a relatively high rate of rearrangement fixation. Here, we compared the organization of both genomes corroborating all known inversions in chromosomes X, 2, 4, and 5. In D. mojavensis chromosome 3, however, we found five inversions instead of the two expected (Delprat A, Guillén Y, Ruiz A, in preparation). One of the three additional inversions is the polymorphic inversion 3f2 (Ruiz et al. 1990). This inversion has previously been found segregating in Baja California and Sonora (Mexico) and is homozygous in the strain of Santa Catalina Island (California) that was used to generate the D. mojavensis genome sequence (Drosophila 12 Genomes Consortium et al. 2007). Previously, the Santa Catalina Island population was thought to have the standard (ancestral) arrangements in all chromosomes, like the populations in Southern California and Arizona (Ruiz et al. 1990; Etges et al. 1999). The presence of inversion 3f2 in Santa Catalina Island is remarkable because it indicates that the flies that colonized this island came from Baja California and are derived instead of ancestral with regard to the rest of D. mojavensis populations (Delprat et al. 2014). The other two additional chromosome 3 inversions are fixed in the D. mojavensis lineage and emphasize its rapid chromosomal evolution. Guillén and Ruiz (2012) analyzed the breakpoint of all chromosome 2 inversions fixed in D. mojavensis and concluded that the numerous gene alterations at the breakpoints with putative adaptive consequences point directly to natural selection as the cause of D. mojavensis rapid chromosomal evolution. The four fixed chromosome 3 inversions provide an opportunity for further testing this hypothesis (Delprat A, Guillén Y, Ruiz A, in preparation).
Candidate Genes under Positive Selection and Orphan Genes
Several methods have been developed to carry out genome-wide scans for genes evolving under positive selection (Nielsen 2005; Anisimova and Liberles 2007; Vitti et al. 2013). We used here a rather simple approach based on the comparison of the nonsynonymous substitution rate (dn) with the synonymous substitution rate (ds) at the codon level (Yang et al. 2000; Wong et al. 2004; Zhang et al. 2005; Yang 2007). Genes putatively under positive selection were detected on the basis of statistical evidence for a subset of codons where replacement mutations were fixed faster than mutation at silent sites. Four species of the Drosophila subgenus (fig. 1) were employed to search for genes under positive selection using SM and BSM. We restricted the analysis to this subset of the Drosophila phylogeny to avoid the saturation of synonymous substitutions expected with phylogenetically very distant species (Bergman et al. 2002; Larracuente et al. 2008), and also because these are the genomes with the highest quality available (Schneider et al. 2009). A total of 1,294 candidate genes were detected with both SM and BSM, which represents approximately 14% of the total set of 1:1 orthologs between D. mojavensis and D. buzzatii. Positive selection seems pervasive in Drosophila (Sawyer et al. 2007; Singh et al. 2009; Sella et al. 2009; Mackay et al. 2012) and, using methods similar to ours, it has been estimated that 33% of single-copy orthologs in the melanogaster group have experienced positive selection (Drosophila 12 Genomes Consortium et al. 2007). The smaller fraction of genes putatively under positive selection in our analyses may be due to the fewer lineages considered in our study. In addition, both studies may be underestimating the true proportion of positively selected genes because only 1:1 orthologs were included in the analyses and genes that evolve too fast may be missed by the methods used to establish orthology relationships (Bierne and Eyre-Walker 2004). At any rate, the 1,294 candidate genes found here should be evaluated using other genomic methods for detecting positive selection, for example, those comparing levels of divergence and polymorphism (Vitti et al. 2013). Furthermore, functional follow-up tests will be necessary for a full validation of their adaptive significance (Lang et al. 2012).
BSM allowed us to search for positively selected genes in the three-targeted lineages (D. buzzatii, D. mojavensis, and cactophilic branch). We then performed GO enrichment analyses in order to identify potential candidates for environmental adaptation given the ecological properties of both cactophilic species (table 4). The most interesting result of this analysis is that genes putatively under positive selection in D. mojavensis branch are enriched in genes involved in heterocyclic catabolic processes. Four candidate D. mojavensis genes, GI19101, GI20678, GI21543 and GI22389, that are orthologous to D. melanogaster genes nahoda, CG5235, slgA and knk, respectively, participate in these processes and might be involved in adaptation of D. mojavensis to the Stenocereus cacti, plants with particularly large quantities of heterocyclic compounds (see Introduction). A difficulty with this interpretation is the fact that the D. mojavensis genome sequence was generated using a strain from Santa Catalina Island where D. mojavensis inhabits Opuntia cactus (Drosophila 12 Genomes Consortium et al. 2007). However, the evidence indicates that the ancestral D. mojavensis population is the agria-inhabiting Baja California population and that the Mainland Sonora population split from Baja California approximately 0.25 Ma whereas the Mojave Desert and Mainland Sonora populations diverged more recently, approximately 0.125 Ma (Smith et al. 2012). Moreover, the presence of inversion 3f2 in the Santa Catalina Island population suggests that the flies that colonized this island came from Baja California populations, where this inversion is currently segregating, and not from the Mojave Desert, where this inversion is not present (Delprat et al. 2014). This is compatible with mitochondrial DNA sequence data (Reed et al. 2007) although in contrast to other data (Machado et al. 2007). Finally, the transcriptional profiles of the four D. mojavensis subpopulations reveal only minor gene expression differences between individuals from Santa Catalina Island and Baja California (Matzkin and Markow 2013).
Orphan genes are genes with restricted taxonomic distribution. Such genes have been suggested to play an important role in phenotypic and adaptive evolution in multiple species (Domazet-Lošo and Tautz 2003; Khalturin et al. 2009; Chen et al. 2013). The detection of orphan genes is highly dependent on the availability of sequenced and well-annotated genomes of closely related species, and the total number of lineage-specific genes tend to be overestimated (Khalturin et al. 2009). We were as conservative as possible by considering only high-confidence 1:1 orthologs in two species, D. buzzatii and D. mojavensis. The result is a set of 117 orphans in the cactophilic lineage.
We observe that orphan genes clearly show a different pattern of molecular evolution compared with that of older conserved genes. Orphans exhibit a higher dn that can be attributed to more beneficial mutations fixed by positive selection or to lower constraint, or both (Cai and Petrov 2010; Chen et al. 2010). However, as the number of genes putatively under positive selection within the set of orphan genes is higher than expected by chance, we suggest that the elevated dn likely reflects adaptive evolution.
Orphans also have fewer exons and encode shorter proteins than nonorphans. This observation has been reported in multiple eukaryotic organisms such as yeasts (Carvunis et al. 2012), fruitflies (Domazet-Lošo and Tautz 2003) and primates (Cai and Petrov 2010), and it is further supported by a positive correlation between protein length and sequence conservation (Lipman et al. 2002) (see above). We did not find expression support for all the orphan genes detected. This suggests to us that either orphans are more tissue- or stage-specific than nonorphans (Zhang et al. 2012) or we are actually detecting artifactual CDS that are not expressed. However, given the patterns of sequence evolution of orphan genes, we favor the first explanation for the majority of them. Collectively, all these results support the conclusion that orphan genes evolve faster than older genes, and that they experience lower levels of purifying selection and higher rates of adaptive evolution (Chen et al. 2010).
It has been widely reported that younger genes have lower expression levels than older genes on average (Cai and Petrov 2010; Tautz and Domazet-Lošo 2011; Zhang et al. 2012). Here, we observe that orphan genes that are being transcribed are less expressed than nonorphans (Kruskal test, Χ2 = 9.37, P = 0.002). One of the proposed hypotheses to explain these observations is that genes that are more conserved are indeed involved in more functions (Pál et al. 2006; Tautz and Domazet-Lošo 2011).
Different studies have demonstrated that newer genes are more likely to have stage-specific expression than older genes (Zhang et al. 2012). Here, we show that the number of stage-specific expressed orphans is significantly higher than that of older genes. It has been proposed that newer genes tend to be more developmentally regulated than older genes (Tautz and Domazet-Lošo 2011). This means that they contribute most to the ontogenic differentiation between taxa (Chen et al. 2010). In D. buzzatii the vast majority of stage-specific orphan genes are expressed in larvae (15/29), indicating that expression of younger genes is mostly related to stages in which D. buzzatii and D. mojavensis lineages most diverge from each other.
Gene Duplication
The study of gene duplications in the D. buzzatii and D. mojavensis lineages aims at understanding the genetic bases of the ecological specialization associated with colonization of novel cactus habitats. Although we only considered expanded families, it is known that specialization sometimes involves gene losses. For example, D. sechellia and D. erecta, which are specialized to grow on particular substrates, have lost gustatory receptors and detoxification genes (Drosophila 12 Genomes Consortium et al. 2007; Dworkin and Jones 2009). Sometimes the losses are driven by positive selection, as has been suggested in the case of the neverland gene in Drosophila pachea (Lang et al. 2012) where positive selection appears to have favored a novel neverland allele that has lost the ability to metabolize cholesterol. In our study of gene families, the incompleteness of the annotation of D. buzzatii PCGs precludes us from being able to reliably identify gene families that lost family members.
To minimize the possibility of missing gene copies that were potentially collapsed into single genes during D. buzzatii genome assembly, we used sequence coverage to adjust the size of gene families. Two of the families that expanded as a result of this correction encoded chorion genes. However, chorion genes are known to undergo somatic amplifications in ovarian follicle cells (Claycomb and Orr-Weaver 2005), and the use of sequence coverage to correct for “missing” copies can be misleading in these cases. As there is no easy way to verify families that were placed into the expanded category due to high sequence coverage alone, our discussion below is limited to gene duplicates that were annotated in the D. buzzatii genome.
A recent survey of the functional roles of new genes across various taxa offers evidence for the rapid recruitment of new genes into gene networks underlying a wide range of phenotypes including reproduction, behavior, and development (Chen et al. 2013). A number of lineage-specific duplicates identified in our study fit this description, but further experimental confirmation of their functions through loss-of-function studies and characterization of molecular interactions are necessary. Among families that expanded in the D. buzzatii, the D. mojavensis, and the cactophilic lineages, 35% have functional annotations that are similar to those of rapidly evolving families identified in the analysis of the 12 Drosophila genomes (Hahn et al. 2007). These families include genes that are involved in proteolysis, zinc ion binding, chitin binding, sensory perception, immunity, and reproduction. A fraction of these expanded families may reflect physiological adaptations to a novel habitat. For example, given the importance of olfactory perception in recognition of the host cactus plants (Date et al. 2013), the duplication of an olfactory receptor in D. buzzatii may represent an adaptation to cactophilic substrates. Another D. buzzatii family includes ninjurin, a gene involved in tissue regeneration that is one of the components of the innate immune response (Boutros et al. 2002). In D. mojavensis, we also observe the duplication of an odorant receptor and, coinciding with a previous report (Croset et al. 2010), of an ionotropic glutamate receptor that belongs to a novel family of diversified chemosensory receptors (Benton et al. 2009; Croset et al. 2010). An aldehyde dehydrogenase is also duplicated in the D. mojavensis lineage and might reveal a role in detoxification of particular aldehydes and ethanol (Fry and Saweikis 2006). In the D. buzzatii–D. mojavensis lineage, one family contains proteins with the MD-2-related lipid recognition domain involved in pathogen recognition and in D. mojavensis we find a duplicate of a phagosome-associated peptide transporter that is involved in bacterial response in D. melanogaster (Charrière et al. 2010).
Several of the D. mojavensis-specific gene duplicates have been described as male and female reproductive proteins. Unlike the accessory gland proteins of D. melanogaster, the proteome of D. mojavensis accessory glands is rich in metabolic enzymes and nutrient transport proteins (Kelleher et al. 2009). Three of the expanded families include metabolic proteins previously identified as candidate seminal fluid proteins specific to D. mojavensis lineage (Kelleher et al. 2009). We also detect an increase of female reproductive tract proteases as a possible counter adaptation to fast-evolving male ejaculate (Kelleher and Markow 2009).
Three gene families are of particular interest among those that were expanded in the lineages leading to D. buzzatii and D. mojavensis, as they contain duplicates of genes with functions related to the regulation of JH levels. One family includes a new duplicate of JH esterase duplication gene (Jhedup in D. melanogaster). JH esterases are involved in JH degradation (Bloch et al. 2013), although Jhedup has much lower level of JH esterase activity than Jhe (Crone et al. 2007). Another family includes new duplicate that encodes protein with sequence similarity to hemolymph JH-binding protein (CG5945 in D. melanogaster). JH-binding proteins belong to a large gene family regulated by circadian genes and affect circadian behavior, courtship behavior, metabolism, and aging (Vanaphan et al. 2012). This family includes JH-binding proteins that function as carriers of JH through the hemolymph to its target tissues (Bloch et al. 2013). The third family includes a new duplicate of a dopamine synthase gene (ebony in D. melanogaster). ebony is involved in the synthesis of dopamine, and it is known that dopamine levels affect behavior and circadian rhythms through regulation of hormone levels including JH (Rauschenbach et al. 2012). All three duplicates are expressed in D. buzzatii adults. At insect adult stage, JHs play a role in physiology and behavior, and their levels oscillate daily (Bloch et al. 2013). Gene duplications of JH-binding proteins (JHBP), JH esterases (JHE), and ebony may change the timing and levels of active JHs which, in turn, alter the behavior and physiology regulated by JHs. One interesting effect of mutations in circadian rhythm genes, or of direct perturbations of the circadian rhythm, is a reduced ethanol tolerance in D. melanogaster (Pohl et al. 2013). Intriguingly, Jhedup and another gene duplicated in the cactophilic lineage, Sirt2 (a protein deacetylase), have been also shown to affect ethanol tolerance and sensitivity when mutated (Kong et al. 2010). Given that both D. mojavensis and D. buzzatii breed and feed on rotting fruit, a shift in tolerance to ethanol and other cactus-specific compounds is one of the expected adaptations associated with a switch to a cactus host. Future functional studies of these new duplicates are required to understand their role in physiological and behavioral changes associated with a change to a new habitat.
Supplementary Material
Supplementary methods, figures S1–S6, and tables S1–S20 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported by grants BFU2008-04988 and BFU2011-30476 from Ministerio de Ciencia e Innovación (Spain) to A.R., by an FPI fellowship to Y.G. and a PIF-UAB fellowship to N.R, and by the National Institute of General Medical Sciences of the National Institute of Health under award number R01GM071813 to E.B. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Literature Cited
- Adams MD, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Anisimova M, Liberles DA. The quest for natural selection in the age of comparative genomics. Heredity. 2007;99:567–579. doi: 10.1038/sj.hdy.6801052. [DOI] [PubMed] [Google Scholar]
- Bai Y, Casola C, Feschotte C, Betrán E. Comparative genomics reveals a constant rate of origination and convergent acquisition of functional retrogenes in Drosophila. Genome Biol. 2007;8:R11. doi: 10.1186/gb-2007-8-1-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker JSF, Starmer WT. Ecological genetics and evolution: the cactus-yeast-Drosophila model system. Sydney (NSW): Academic Press; 1982. [Google Scholar]
- Barker JSF, Starmer WT, MacIntyre RJ. Ecological and evolutionary genetics of Drosophila. New York: Plenum Press; 1990. [Google Scholar]
- Begon M. Yeasts and Drosophila. In: Ashburner M, Carson HL, Thompson JN Jr, editors. The genetics and biology of Drosophila. Vol. 3b. London: Academic Press; 1982. pp. 3345–3384. [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton R, Vannice KS, Gomez-Diaz C, Vosshall LB. Variant ionotropic glutamate receptors as chemosensory receptors in Drosophila. Cell. 2009;136:149–162. doi: 10.1016/j.cell.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman CM, et al. Assessing the impact of comparative genomic sequence data on the functional annotation of the Drosophila genome. Genome Biol. 2002;3:research0086. doi: 10.1186/gb-2002-3-12-research0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betran E, Santos M, Ruiz A. Antagonistic pleiotropic effect of second-chromosome inversions on body size and early life-history traits in Drosophila buzzatii. Evolution. 1998;52:144–154. doi: 10.1111/j.1558-5646.1998.tb05147.x. [DOI] [PubMed] [Google Scholar]
- Bierne N, Eyre-Walker A. The genomic rate of adaptive amino acid substitution in Drosophila. Mol Biol Evol. 2004;21:1350–1360. doi: 10.1093/molbev/msh134. [DOI] [PubMed] [Google Scholar]
- Bloch G, Hazan E, Rafaeli A. Circadian rhythms and endocrine functions in adult insects. J Insect Physiol. 2013;59:56–69. doi: 10.1016/j.jinsphys.2012.10.012. [DOI] [PubMed] [Google Scholar]
- Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27:578–579. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- Bosco G, Campbell P, Leiva-Neto JT, Markow TA. Analysis of Drosophila species genome size and satellite DNA content reveals significant differences among strains as well as between species. Genetics. 2007;177:1277–1290. doi: 10.1534/genetics.107.075069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros M, Agaisse H, Perrimon N. Sequential activation of signaling pathways during innate immune responses in Drosophila. Dev Cell. 2002;3:711–722. doi: 10.1016/s1534-5807(02)00325-8. [DOI] [PubMed] [Google Scholar]
- Breitmeyer CM, Markow TA. Resource availability and population size in cactophilic Drosophila. Funct Ecol. 1998;12:14–21. [Google Scholar]
- Cai JJ, Petrov DA. Relaxed purifying selection and possibly high rate of adaptation in primate lineage-specific genes. Genome Biol Evol. 2010;2:393–409. doi: 10.1093/gbe/evq019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvete O, González J, Betrán E, Ruiz A. Segmental duplication, microinversion, and gene loss associated with a complex inversion breakpoint region in Drosophila. Mol Biol Evol. 2012;29:1875–1889. doi: 10.1093/molbev/mss067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson HL. 1971 The ecology of Drosophila breeding sites. No. 2. Honolulu (Hawaii): University of Hawaii Foundation Lyon Arboretum Fund. [Google Scholar]
- Carson HL, Wasserman M. A widespread chromosomal polymorphism in a widespread species, Drosophila buzzatii. Am Nat. 1965;99:111–115. [Google Scholar]
- Carvunis A-R, et al. Proto-genes and de novo gene birth. Nature. 2012;487:370–374. doi: 10.1038/nature11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celniker SE, et al. Unlocking the secrets of the genome. Nature. 2009;459:927–930. doi: 10.1038/459927a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrière GM, et al. Identification of Drosophila Yin and PEPT2 as evolutionarily conserved phagosome-associated muramyl dipeptide transporters. J Biol Chem. 2010;285:20147–20154. doi: 10.1074/jbc.M110.115584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Krinsky BH, Long M. New genes as drivers of phenotypic evolution. Nat Rev Genet. 2013;14:645–660. doi: 10.1038/nrg3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhang YE, Long M. New genes in Drosophila quickly become essential. Science. 2010;330:1682–1685. doi: 10.1126/science.1196380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZX, et al. Comparative validation of the D. melanogaster modENCODE transcriptome annotation. Genome Res. 2014;24:1209–1223. doi: 10.1101/gr.159384.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claycomb JM, Orr-Weaver TL. Developmental gene amplification: insights into DNA replication and gene expression. Trends Genet. 2005;21:149–162. doi: 10.1016/j.tig.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Crone EJ, et al. Only one esterase of Drosophila melanogaster is likely to degrade juvenile hormone in vivo. Insect Biochem Mol Biol. 2007;37:540–549. doi: 10.1016/j.ibmb.2007.02.010. [DOI] [PubMed] [Google Scholar]
- Croset V, et al. Ancient protostome origin of chemosensory ionotropic glutamate receptors and the evolution of insect taste and olfaction. PLoS Genet. 2010;6:e1001064. doi: 10.1371/journal.pgen.1001064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Date P, et al. Divergence in olfactory host plant preference in D. mojavensis in response to cactus host use. PLoS One. 2013;8:e70027. doi: 10.1371/journal.pone.0070027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David J, Tsacas L. Cosmopolitan, subcosmopolitan and widespread species: different strategies within the Drosophilid family (Diptera) C R Soc Biogéogr. 1980;57:11–26. [Google Scholar]
- Delcher AL, Salzberg SL, Phillippy AM. Using MUMmer to identify similar regions in large sequence sets. 2003 doi: 10.1002/0471250953.bi1003s00. Curr Protoc Bioinformatics. Chapter 10:Unit 10.3. [DOI] [PubMed] [Google Scholar]
- Delprat A, Etges WJ, Ruiz A. 2014 Reanalysis of polytene chromosomes in Drosophila mojavensis populations from Santa Catalina Island, California, USA. Drosoph Inf Serv. 97:53–57. [Google Scholar]
- Domazet-Lošo T, Tautz D. An evolutionary analysis of orphan genes in Drosophila. Genome Res. 2003;13:2213–2219. doi: 10.1101/gr.1311003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium, et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Dworkin I, Jones CD. Genetic changes accompanying the evolution of host specialization in Drosophila sechellia. Genetics. 2009;181:721–736. doi: 10.1534/genetics.108.093419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etges WJ, et al. Deciphering life history transcriptomes in different environments. Mol Ecol. 2015;24:151–179. doi: 10.1111/mec.13017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etges WJ, Johnson WR, Duncan GA, Huckins G, Heed WB. Ecological genetics of cactophilic Drosophila. In: Robichaux R, editor. Ecology of Sonoran Desert plants and plant communities. Tucson (AZ): University of Arizona Press; 1999. pp. 164–214. [Google Scholar]
- Fellows DP, Heed WB. Factors affecting host plant selection in desert-adapted cactiphilic Drosophila. Ecology. 1972;53:850–858. [Google Scholar]
- Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–368. doi: 10.1146/annurev.genet.40.110405.090448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogleman JC, Armstrong L. Ecological aspects of cactus triterpene glycosides I. Their effect on fitness components of Drosophila mojavensis. J Chem Ecol. 1989;15:663–676. doi: 10.1007/BF01014709. [DOI] [PubMed] [Google Scholar]
- Fogleman JC, Danielson PB. Chemical interactions in the cactus-microorganism-Drosophila model system of the Sonoran Desert. Am Zool. 2001;41:877–889. [Google Scholar]
- Fogleman JC, Kircher HW. Differential effects of fatty acid chain length on the viability of two species of cactophilic Drosophila. Comp Biochem Physiol A Physiol. 1986;83:761–764. [Google Scholar]
- Fonseca NA, et al. Drosophila americana as a model species for comparative studies on the molecular basis of phenotypic variation. Genome Biol Evol. 2013;5:661–679. doi: 10.1093/gbe/evt037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontdevila A, Ruiz A, Alonso G, Ocaña J. Evolutionary history of Drosophila buzzatii. I. Natural chromosomal polymorphism in colonized populations of the Old World. Evolution. 1981;35:148–157. doi: 10.1111/j.1558-5646.1981.tb04867.x. [DOI] [PubMed] [Google Scholar]
- Frank MR, Fogleman JC. Involvement of cytochrome P450 in host-plant utilization by Sonoran Desert Drosophila. Proc Natl Acad Sci U S A. 1992;89:11998–12002. doi: 10.1073/pnas.89.24.11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry JD, Saweikis M. Aldehyde dehydrogenase is essential for both adult and larval ethanol resistance in Drosophila melanogaster. Genet Res. 2006;87:87–92. doi: 10.1017/S0016672306008032. [DOI] [PubMed] [Google Scholar]
- Garcia-Fernàndez J. The genesis and evolution of homeobox gene clusters. Nat Rev Genet. 2005;6:881–892. doi: 10.1038/nrg1723. [DOI] [PubMed] [Google Scholar]
- González J, et al. A BAC-based physical map of the Drosophila buzzatii genome. Genome Res. 2005;15:885–889. doi: 10.1101/gr.3263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, et al. The developmental transcriptome of Drosophila melanogaster. Nature. 2011;471:473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory TR, Johnston JS. Genome size diversity in the family Drosophilidae. Heredity. 2008;101:228–238. doi: 10.1038/hdy.2008.49. [DOI] [PubMed] [Google Scholar]
- Guillén Y, Ruiz A. Gene alterations at Drosophila inversion breakpoints provide prima facie evidence for natural selection as an explanation for rapid chromosomal evolution. BMC Genomics. 2012;13:53. doi: 10.1186/1471-2164-13-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9:R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MW, Han MV, Han S-G. Gene family evolution across 12 Drosophila genomes. PLoS Genet. 2007;3:e197. doi: 10.1371/journal.pgen.0030197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MV, Hahn MW. Inferring the history of interchromosomal gene transposition in Drosophila using n-dimensional parsimony. Genetics. 2012;190:813–825. doi: 10.1534/genetics.111.135947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MV, Thomas GWC, Lugo-Martinez J, Hahn MW. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol Biol Evol. 2013;30:1987–1997. doi: 10.1093/molbev/mst100. [DOI] [PubMed] [Google Scholar]
- Hasson E, et al. The evolutionary history of Drosophila buzzatii. XXVI. Macrogeographic patterns of inversion polymorphism in New World populations. J Evol Biol. 1995;8:369–384. [Google Scholar]
- Hasson E, Naveira H, Fontdevila A. The breeding sites of Argentinian cactophilic species of the Drosophila mulleri complex (subgenus Drosophila-repleta group) Rev Chil Hist Nat. 1992;65:319–326. [Google Scholar]
- Havens JA, Etges WJ. Premating isolation is determined by larval rearing substrates in cactophilic Drosophila mojavensis. IX. Host plant and population specific epicuticular hydrocarbon expression influences mate choice and sexual selection. J Evol Biol. 2013;26:562–576. doi: 10.1111/jeb.12073. [DOI] [PubMed] [Google Scholar]
- Heed WB, Mangan RL. Community ecology of the Sonoran Desert Drosophila. In: Ashburner M, Carson HL, Thompson JN, editors. The genetics and biology of Drosophila. Vol. 3e. London: Academic Press; 1986. pp. 311–345. [Google Scholar]
- Hoffmann AA, Sørensen JG, Loeschcke V. Adaptation of Drosophila to temperature extremes: bringing together quantitative and molecular approaches. J Therm Biol. 2003;28:175–216. [Google Scholar]
- Huang DW, et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:W169–W175. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009a;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009b;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Hughes CL, Kaufman TC. Hox genes and the evolution of the arthropod body plan. Evol Dev. 2002;4:459–499. doi: 10.1046/j.1525-142x.2002.02034.x. [DOI] [PubMed] [Google Scholar]
- Irimia M, Maeso I, Garcia-Fernàndez J. Convergent evolution of clustering of Iroquois homeobox genes across metazoans. Mol Biol Evol. 2008;25:1521–1525. doi: 10.1093/molbev/msn109. [DOI] [PubMed] [Google Scholar]
- Jurka J, et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Kelleher ES, Markow TA. Duplication, selection and gene conversion in a Drosophila mojavensis female reproductive protein family. Genetics. 2009;181:1451–1465. doi: 10.1534/genetics.108.099044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher ES, Watts TD, LaFlamme BA, Haynes PA, Markow TA. Proteomic analysis of Drosophila mojavensis male accessory glands suggests novel classes of seminal fluid proteins. Insect Biochem Mol Biol. 2009;39:366–371. doi: 10.1016/j.ibmb.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Khalturin K, Hemmrich G, Fraune S, Augustin R, Bosch TCG. More than just orphans: are taxonomically-restricted genes important in evolution? Trends Genet. 2009;25:404–413. doi: 10.1016/j.tig.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Kidwell MG. Transposable elements and the evolution of genome size in eukaryotes. Genetica. 2002;115:49–63. doi: 10.1023/a:1016072014259. [DOI] [PubMed] [Google Scholar]
- Kircher HW. Chemical composition of cacti and its relationship to Sonoran Desert Drosophila. In: Barker JSF, Starmer WT, editors. Ecological genetics and evolution: the cactus-yeast-Drosophila model system. Sydney (NSW): Academic Press; 1982. pp. 143–158. [Google Scholar]
- Kong EC, et al. Ethanol-regulated genes that contribute to ethanol sensitivity and rapid tolerance in Drosophila. Alcohol Clin Exp Res. 2010;34:302–316. doi: 10.1111/j.1530-0277.2009.01093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5:59. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I, Flicek P, Duan D, Brent MR. Integrating genomic homology into gene structure prediction. Bioinformatics. 2001;17(Suppl 1):S140–S148. doi: 10.1093/bioinformatics/17.suppl_1.s140. [DOI] [PubMed] [Google Scholar]
- Kriventseva EV, Rahman N, Espinosa O, Zdobnov EM. OrthoDB: the hierarchical catalog of eukaryotic orthologs. Nucleic Acids Res. 2008;36:D271–D275. doi: 10.1093/nar/gkm845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn GCS, Sene FM, Moreira-Filho O, Schwarzacher T, Heslop-Harrison JS. Sequence analysis, chromosomal distribution and long-range organization show that rapid turnover of new and old pBuM satellite DNA repeats leads to different patterns of variation in seven species of the Drosophila buzzatii cluster. Chromosome Res. 2008;16:307–324. doi: 10.1007/s10577-007-1195-1. [DOI] [PubMed] [Google Scholar]
- Ladoukakis E, Pereira V, Magny EG, Eyre-Walker A, Couso JP. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biol. 2011;12:R118. doi: 10.1186/gb-2011-12-11-r118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai EC, Bodner R, Posakony JW. The enhancer of split complex of Drosophila includes four Notch-regulated members of the bearded gene family. Development. 2000;127:3441–3455. doi: 10.1242/dev.127.16.3441. [DOI] [PubMed] [Google Scholar]
- Lang M, et al. Mutations in the neverland gene turned Drosophila pachea into an obligate specialist species. Science. 2012;337:1658–1661. doi: 10.1126/science.1224829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang M, et al. Radiation of the Drosophila nannoptera species group in Mexico. J Evol Biol. 2014;27:575–584. doi: 10.1111/jeb.12325. [DOI] [PubMed] [Google Scholar]
- Larracuente AM, et al. Evolution of protein-coding genes in Drosophila. Trends Genet. 2008;24:114–123. doi: 10.1016/j.tig.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Lavergne S, Muenke NJ, Molofsky J. Genome size reduction can trigger rapid phenotypic evolution in invasive plants. Ann Bot. 2010;105:109–116. doi: 10.1093/aob/mcp271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CE. Evolutionary genetics of invasive species. Trends Ecol Evol. 2002;17:386–391. [Google Scholar]
- Lewontin RC. Selection for colonizing ability. In: Baker HG, Stebbins GL, editors. The genetics of colonizing species. New York: Academic Press; 1965. pp. 77–94. [Google Scholar]
- Lipman DJ, Souvorov A, Koonin EV, Panchenko AR, Tatusova TA. The relationship of protein conservation and sequence length. BMC Evol Biol. 2002;2:20. doi: 10.1186/1471-2148-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeschcke V, Krebs RA, Dahlgaard J, Michalak P. High-temperature stress and the evolution of thermal resistance in Drosophila. EXS. 1997;83:175–190. doi: 10.1007/978-3-0348-8882-0_10. [DOI] [PubMed] [Google Scholar]
- Luo R, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado CA, Matzkin LM, Reed LK, Markow TA. Multilocus nuclear sequences reveal intra- and interspecific relationships among chromosomally polymorphic species of cactophilic Drosophila. Mol Ecol. 2007;16:3009–3024. doi: 10.1111/j.1365-294X.2007.03325.x. [DOI] [PubMed] [Google Scholar]
- Mackay TFC, et al. The Drosophila melanogaster genetic reference panel. Nature. 2012;482:173–178. doi: 10.1038/nature10811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfrin MH, Sene FM. Cactophilic Drosophila in South America: a model for evolutionary studies. Genetica. 2006;126:57–75. doi: 10.1007/s10709-005-1432-5. [DOI] [PubMed] [Google Scholar]
- Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markow TA, O’Grady P. Reproductive ecology of Drosophila. Funct Ecol. 2008;22:747–759. [Google Scholar]
- Matzkin LM. Ecological genomics of host shifts in Drosophila mojavensis. Adv Exp Med Biol. 2014;781:233–247. doi: 10.1007/978-94-007-7347-9_12. [DOI] [PubMed] [Google Scholar]
- Matzkin LM, Markow TA. Transcriptional differentiation across the four subspecies of Drosophila mojavensis. In: Pawel M, editor. Speciation: natural processes, genetics and biodiversity. New York: Nova Scientific Publishers; 2013. [Google Scholar]
- Matzkin LM, Watts TD, Bitler BG, Machado CA, Markow TA. Functional genomics of cactus host shifts in Drosophila mojavensis. Mol Ecol. 2006;15:4635–4643. doi: 10.1111/j.1365-294X.2006.03102.x. [DOI] [PubMed] [Google Scholar]
- Melters DP, et al. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 2013;14:R10. doi: 10.1186/gb-2013-14-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan B. Total DNA isolation. In: Hoelzel AR, editor. Molecular genetic analysis of populations: A practical approach. Oxford (UK): IRL Press; 1998. pp. 29–64. [Google Scholar]
- Misawa K, Kikuno RF. GeneWaltz—a new method for reducing the false positives of gene finding. BioData Min. 2010;3:6. doi: 10.1186/1756-0381-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales-Hojas R, Vieira J. Phylogenetic patterns of geographical and ecological diversification in the subgenus Drosophila. PLoS One. 2012;7:e49552. doi: 10.1371/journal.pone.0049552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadalin F, Vezzi F, Policriti A. GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics. 2012;13(Suppl 14):S8. doi: 10.1186/1471-2105-13-S14-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardon C, et al. Is genome size influenced by colonization of new environments in dipteran species? Mol Ecol. 2005;14:869–878. doi: 10.1111/j.1365-294X.2005.02457.x. [DOI] [PubMed] [Google Scholar]
- Negre B, et al. Conservation of regulatory sequences and gene expression patterns in the disintegrating Drosophila Hox gene complex. Genome Res. 2005;15:692–700. doi: 10.1101/gr.3468605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negre B, Ruiz A. HOM-C evolution in Drosophila: is there a need for Hox gene clustering? Trends Genet. 2007;23:55–59. doi: 10.1016/j.tig.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Negre B, Simpson P. Evolution of the achaete-scute complex in insects: convergent duplication of proneural genes. Trends Genet. 2009;25:147–152. doi: 10.1016/j.tig.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Nielsen R. Molecular signatures of natural selection. Annu Rev Genet. 2005;39:197–218. doi: 10.1146/annurev.genet.39.073003.112420. [DOI] [PubMed] [Google Scholar]
- Oliveira DCSG, et al. Monophyly, divergence times, and evolution of host plant use inferred from a revised phylogeny of the Drosophila repleta species group. Mol Phylogenet Evol. 2012;64:533–544. doi: 10.1016/j.ympev.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Oliveros J. VENNY. An interactive tool for comparing lists with Venn diagrams. Madrid (Spain): BioinfoGP CNB-CSIC; 2007. [Google Scholar]
- Ometto L, et al. Linking genomics and ecology to investigate the complex evolution of an invasive Drosophila pest. Genome Biol Evol. 2013;5:745–757. doi: 10.1093/gbe/evt034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pál C, Papp B, Lercher MJ. An integrated view of protein evolution. Nat Rev Genet. 2006;7:337–348. doi: 10.1038/nrg1838. [DOI] [PubMed] [Google Scholar]
- Palmieri N, Kosiol C, Schlötterer C. The life cycle of Drosophila orphan genes. eLife. 2014;3:e01311. doi: 10.7554/eLife.01311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M, et al. A survey of ovary-, testis-, and soma-biased gene expression in Drosophila melanogaster adults. Genome Biol. 2004;5:R40. doi: 10.1186/gb-2004-5-6-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons P. The evolutionary biology of colonizing species. New York: Cambridge University Press; 1983. [Google Scholar]
- Piñol J, Francino O, Fontdevila A, Cabré O. Rapid isolation of Drosophila high molecular weight DNA to obtain genomic libraries. Nucleic Acids Res. 1988;16:2736. doi: 10.1093/nar/16.6.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl JB, et al. Circadian genes differentially affect tolerance to ethanol in Drosophila. Alcohol Clin Exp Res. 2013;37:1862–1871. doi: 10.1111/acer.12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poptsova MS, Gogarten JP. Using comparative genome analysis to identify problems in annotated microbial genomes. Microbiology. 2010;156:1909–1917. doi: 10.1099/mic.0.033811-0. [DOI] [PubMed] [Google Scholar]
- Prada CF. 2010 Evolución cromosómica del cluster Drosophila martensis: origen de las inversiones y reutilización de los puntos de rotura [PhD thesis]. Barcelona (Spain): Universitat Autònoma de Barcelona. [Google Scholar]
- Rajpurohit S, Oliveira CC, Etges WJ, Gibbs AG. Functional genomic and phenotypic responses to desiccation in natural populations of a desert drosophilid. Mol Ecol. 2013;22:2698–2715. doi: 10.1111/mec.12289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauschenbach IY, Bogomolova EV, Karpova EK, Shumnaya LV, Gruntenko NE. The role of D1 like receptors in the regulation of juvenile hormone synthesis in Drosophila females with increased dopamine level. Dokl Biochem Biophys. 2012;446:231–234. doi: 10.1134/S1607672912050134. [DOI] [PubMed] [Google Scholar]
- Reed LK, Nyboer M, Markow TA. Evolutionary relationships of Drosophila mojavensis geographic host races and their sister species Drosophila arizonae. Mol Ecol. 2007;16:1007–1022. doi: 10.1111/j.1365-294X.2006.02941.x. [DOI] [PubMed] [Google Scholar]
- Reinhardt JA, et al. De novo ORFs in Drosophila are important to organismal fitness and evolved rapidly from previously non-coding sequences. PLoS Genet. 2013;9:e1003860. doi: 10.1371/journal.pgen.1003860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz A, Cansian AM, Kuhn GC, Alves MA, Sene FM. The Drosophila serido speciation puzzle: putting new pieces together. Genetica. 2000;108:217–227. doi: 10.1023/a:1004195007178. [DOI] [PubMed] [Google Scholar]
- Ruiz A, Heed WB. Host-plant specificity in the cactophilic Drosophila mulleri species complex. J Anim Ecol. 1988;57:237–249. [Google Scholar]
- Ruiz A, Heed WB, Wasserman M. Evolution of the mojavensis cluster of cactophilic Drosophila with descriptions of two new species. J Hered. 1990;81:30–42. doi: 10.1093/oxfordjournals.jhered.a110922. [DOI] [PubMed] [Google Scholar]
- Ruiz A, Wasserman M. Evolutionary cytogenetics of the Drosophila buzzatii species complex. Heredity. 1993;70:582–596. doi: 10.1038/hdy.1993.85. [DOI] [PubMed] [Google Scholar]
- Ruiz-Ruano FJ, et al. DNA amount of X and B chromosomes in the grasshoppers Eyprepocnemis plorans and Locusta migratoria. Cytogenet Genome Res. 2011;134:120–126. doi: 10.1159/000324690. [DOI] [PubMed] [Google Scholar]
- Sawyer SA, Parsch J, Zhang Z, Hartl DL. Prevalence of positive selection among nearly neutral amino acid replacements in Drosophila. Proc Natl Acad Sci U S A. 2007;104:6504–6510. doi: 10.1073/pnas.0701572104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer SW, et al. Polytene chromosomal maps of 11 Drosophila species: the order of genomic scaffolds inferred from genetic and physical maps. Genetics. 2008;179:1601–1655. doi: 10.1534/genetics.107.086074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, et al. Estimates of positive Darwinian selection are inflated by errors in sequencing, annotation, and alignment. Genome Biol Evol. 2009;1:114–118. doi: 10.1093/gbe/evp012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sella G, Petrov DA, Przeworski M, Andolfatto P. Pervasive natural selection in the Drosophila genome? PLoS Genet. 2009;5:e1000495. doi: 10.1371/journal.pgen.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh ND, Larracuente AM, Sackton TB, Clark AG. Comparative genomics on the Drosophila phylogenetic tree. Annu Rev Ecol Evol Syst. 2009;40:459–480. [Google Scholar]
- Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G, Lohse K, Etges WJ, Ritchie MG. Model-based comparisons of phylogeographic scenarios resolve the intraspecific divergence of cactophilic Drosophila mojavensis. Mol Ecol. 2012;21:3293–3307. doi: 10.1111/j.1365-294X.2012.05604.x. [DOI] [PubMed] [Google Scholar]
- St Pierre SE, Ponting L, Stefancsik R, McQuilton P, FlyBase Consortium FlyBase 102—advanced approaches to interrogating FlyBase. Nucleic Acids Res. 2014;42(Database issue):D780–D788. doi: 10.1093/nar/gkt1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19(Suppl 2):ii215–ii225. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- Starmer WT. A comparison of Drosophila habitats according to the physiological attributes of the associated yeast communities. Evolution. 1981;35:38–52. doi: 10.1111/j.1558-5646.1981.tb04856.x. [DOI] [PubMed] [Google Scholar]
- Stein LD, et al. The generic genome browser: a building block for a model organism system database. Genome Res. 2002;12:1599–1610. doi: 10.1101/gr.403602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D, Domazet-Lošo T. The evolutionary origin of orphan genes. Nat Rev Genet. 2011;12:692–702. doi: 10.1038/nrg3053. [DOI] [PubMed] [Google Scholar]
- Tesler G. GRIMM: genome rearrangements web server. Bioinformatics. 2002;18:492–493. doi: 10.1093/bioinformatics/18.3.492. [DOI] [PubMed] [Google Scholar]
- Throckmorton L. The phylogeny, ecology and geography of Drosophila. In: King R, editor. Handbook of genetics. Vol. 3. New York: Plenum Press; 1975. pp. 421–469. [Google Scholar]
- Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugarković Đ. Centromere-competent DNA: structure and evolution. In: Ugarković Đ, editor. Centromere. Structure and evolution. Progress in molecular and subcellular biology. 2009. Vol. 48. Berlin (Germany): Springer. p. 53–76. [DOI] [PubMed] [Google Scholar]
- Vanaphan N, Dauwalder B, Zufall RA. Diversification of takeout, a male-biased gene family in Drosophila. Gene. 2012;491:142–148. doi: 10.1016/j.gene.2011.10.003. [DOI] [PubMed] [Google Scholar]
- Vitti JJ, Grossman SR, Sabeti PC. Detecting natural selection in genomic data. Annu Rev Genet. 2013;47:97–120. doi: 10.1146/annurev-genet-111212-133526. [DOI] [PubMed] [Google Scholar]
- Wall DP, Deluca T. Ortholog detection using the reciprocal smallest distance algorithm. Methods Mol Biol. 2007;396:95–110. doi: 10.1007/978-1-59745-515-2_7. [DOI] [PubMed] [Google Scholar]
- Wang J, et al. Vertebrate gene predictions and the problem of large genes. Nat Rev Genet. 2003;4:741–749. doi: 10.1038/nrg1160. [DOI] [PubMed] [Google Scholar]
- Wasserman M. Cytological evolution of the Drosophila repleta species group. In: Krimbas CB, Powell JR, editors. Drosophila inversion polymorphism. Boca Raton (FL): CRC Press; 1992. pp. 455–552. [Google Scholar]
- Weake VM, Workman JL. Inducible gene expression: diverse regulatory mechanisms. Nat Rev Genet. 2010;11:426–437. doi: 10.1038/nrg2781. [DOI] [PubMed] [Google Scholar]
- Wicker T, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- Wong WS, Yang Z, Goldman N, Nielsen R. Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics. 2004;168:1041–1051. doi: 10.1534/genetics.104.031153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R, Goldman N, Pedersen AM. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- Zhang YE, Landback P, Vibranovski M, Long M. New genes expressed in human brains: implications for annotating evolving genomes. Bioessays. 2012;34:982–991. doi: 10.1002/bies.201200008. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Bachtrog D. Sex-specific adaptation drives early sex chromosome evolution in Drosophila. Science. 2012;337:341–345. doi: 10.1126/science.1225385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, et al. Deciphering neo-sex and B chromosome evolution by the draft genome of Drosophila albomicans. BMC Genomics. 2012;13:109. doi: 10.1186/1471-2164-13-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.